

Abstract
Background and Purpose:
Glutamate N-methyl-D-aspartate receptors (NMDARs) play a major role in the initiation of ischemic brain damage. However, NMDAR antagonists have no protective effects in stroke patients, possibly because they impair physiological functions of NMDARs. α2δ−1 (encoded by Cacna2d1) is strongly expressed in many brain regions. We determined the contribution of α2δ−1 to NMDAR hyperactivity and brain injury induced by ischemia and reperfusion.
Methods:
Mice were subjected to 90 min of middle cerebral artery occlusion (MCAO) followed by 24 h of reperfusion. Neurological deficits, brain infarct volumes, and calpain/caspase-3 activity in brain tissues were measured. NMDAR activity of hippocampal CA1 neurons was measured in an in vitro ischemic model.
Results:
MCAO increased α2δ−1 protein glycosylation in the cerebral cortex, hippocampus, and striatum. Coimmunoprecipitation showed that ischemia rapidly enhanced the α2δ−1–NMDAR physical interaction in the mouse brain tissue. Inhibiting α2δ−1 with gabapentin, uncoupling the α2δ−1–NMDAR interaction with an α2δ−1 C-terminus–interfering peptide, or genetically ablating Cacna2d1 had no effect on basal NMDAR currents but strikingly abolished oxygen-glucose deprivation-induced NMDAR hyperactivity in hippocampal CA1 neurons. Systemic treatment with gabapentin or α2δ−1 C-terminus–interfering peptide or Cacna2d1 genetic knockout reduced MCAO-induced infarct volumes, neurological deficit scores, and calpain/caspase-3 activation in brain tissues.
Conclusions:
α2δ−1 is essential for brain ischemia-induced neuronal NMDAR hyperactivity, and α2δ−1–bound NMDARs mediate brain damage caused by cerebral ischemia. Targeting α2δ−1–bound NMDARs, without impairing physiological α2δ−1–free NMDARs, may be a promising strategy for treating ischemic stroke.
Keywords: neuroprotection, gabapentinoids, pregabalin, glycosylation, calpain, NMDA receptor
Introduction
Glutamate is the major excitatory neurotransmitter in the central nervous system and plays an essential role in excitatory synaptic transmission and synaptic plasticity. After brain ischemia, however, glutamate accumulates rapidly at synapses, resulting in extensive stimulation of its N-methyl-D-aspartate receptors (NMDARs) that can eventually be toxic to neurons.1,2 Excessive and prolonged activation of NMDARs induces a large amount of Ca2+ influx through the NMDAR channels. Intracellular Ca2+ overload can subsequently trigger several downstream lethal reactions including proteolysis of key cellular proteins, resulting in irreversible neuronal death in the brain.3–6 Thus, NMDARs have long been considered a therapeutic target for ischemic stroke.
Despite abundant preclinical evidence that excessive NMDAR stimulation is a crucial initiating factor for brain damage during ischemic stroke,2,4 NMDAR antagonists have failed to show neuroprotective efficacy in clinical trials or have produced intolerable adverse effects in stroke patients.7–10 It has been recognized that blocking NMDARs totally may be detrimental because these receptors also have important physiological functions.11 For example, the normal activity of NMDARs plays a pivotal role in the maintenance of cortical GABAergic interneurons.12 However, little is known about how brain ischemia leads to neuronal NMDAR hyperactivity. Identifying the molecular mechanism leading to pathological NMDAR hyperactivity during brain ischemia is essential for developing targeted neuroprotective treatments for stroke patients.
α2δ−1 (encoded by Cacna2d1) has been considered a subunit of voltage-gated Ca2+ channels (VGCCs). α2δ−1 is also a binding site of gabapentinoids, including gabapentin and pregabalin, which are clinically used to treat neuropathic pain. We discovered recently that α2δ−1 is a novel interacting protein of NMDARs in the spinal cord and mediates nerve injury-induced synaptic NMDAR hyperactivity.13 α2δ−1 is strongly expressed in many brain regions, including the cerebral cortex and hippocampus, and is preferentially localized in excitatory neurons.14,15 However, whether α2δ−1–bound NMDARs play a role in brain injury caused by ischemia/reperfusion remains unknown. In this study, we used a mouse model of middle cerebral artery occlusion (MCAO) to determine the contribution of α2δ−1 to ischemia/reperfusion-induced brain injury. We showed that α2δ−1 is essential for ischemia-induced neuronal NMDAR hyperactivity and that uncoupling the α2δ−1–NMDAR interaction protects against ischemia/reperfusion-induced brain injury without impairing physiological NMDAR activity. Thus, α2δ−1–bound NMDARs may be targeted for treating ischemic stroke.
Materials and Methods
This article adheres to the American Heart Association Journals implementation of the Transparency and Openness Promotion Guidelines. Details of Methods are available in the online-only Data Supplement. The data that support the findings of this study are available from the corresponding author on reasonable request.
Animal model.
All procedures and protocols were approved by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center. Adult male wild-type (WT) and conventional Cacna2d1 knockout (KO) mice (25–30 g, 8–10 weeks of age) were used in our study. We used the filament model with 90 min of MCAO in mice, as described previously.16,17 The neurological deficits of mice subjected to MCAO were evaluated using a modified Longa test on a scale of 0–5, as reported previously.17,18 The brain coronal sections were stained using 1% 2,3,5-triphenyltetrazolium chloride (TTC), and the stained sections were captured as digital images and quantified for infarct volume.
Brain slice preparation and recording.
The tissues containing the hippocampus were sliced sagittally (300 µm thick) using a vibratome. Oxygen-glucose deprivation (OGD) was induced by perfusing brain slices with aCSF saturated with 95% N2/5% CO2 and by replacing the 11 mM glucose in aCSF with 11 mM sucrose.19 NMDAR currents of hippocampal CA1 pyramidal neurons were recorded by puff application of 100 µM NMDA directly to the recorded neuron.
Western immunoblotting and coimmunoprecipitation.
These assays were performed using standard methods described in the Supplemental Information.
Results
α2δ−1 and its interaction with NMDARs are essential for the OGD-induced increase in neuronal NMDAR activity
Although increased NMDAR activity has been implicated in neuronal injury during cerebral ischemia, the underlying mechanisms remain unclear. We thus determined the potential role of α2δ−1 in ischemia-induced NMDAR hyperactivity in the brain. Because MCAO causes severe neuronal damage, it is difficult to record NMDAR activity in and around the ischemic region. We therefore used hippocampal brain slices subjected to oxygen-glucose deprivation (OGD) to simulate cerebral ischemia. OGD increases NMDAR activity in hippocampal CA1 pyramidal neurons, which are highly susceptible to ischemic injury.19,20 OGD induction for 5 min induced a large increase in the amplitude of inward NMDAR currents in WT mice (Figure 1A and 1B). Gabapentin binds primarily to α2δ−1 and is a clinically used α2δ−1 inhibitory ligand.21,22 Treatment with gabapentin (30 µM or 100 µM for 60 min) had no effect on the basal puff NMDAR currents of hippocampal CA1 pyramidal neurons before OGD. Strikingly, the OGD-induced increase in the puff NMDAR currents was abolished in the presence of 30 µM or 100 µM gabapentin (Figure 1A and 1B).
Figure 1. α2δ−1 and its interaction with NMDARs are essential for OGD-induced potentiation of NMDAR activity in hippocampal CA1 neurons.
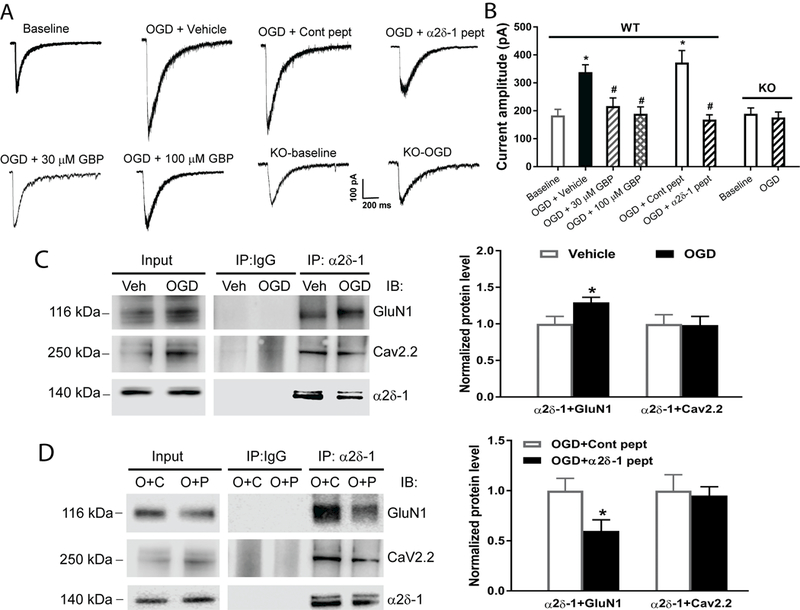
(A and B) Representative recording traces (A) and quantification (B) of puff NMDA-elicited NMDAR currents show the effects of gabapentin (GBP, 30 µM for 60 min, n = 10 neurons; 100 µM for 60 min, n = 12 neurons), vehicle (n = 11 neurons), α2δ−1Tat peptide (1 µM for 60 min, n = 10 neurons), control peptide (cont peptide; 1 µM for 60 min, n = 10 neurons), and Cacna2d1 KO (n = 12 neurons for baseline and OGD groups) on NMDAR activity in hippocampal CA1 neurons subjected to 5 min of OGD. (C) Representative gel images and quantification of co-IP show the effect of OGD on the α2δ−1–GluN1 or α2δ−1–Cav2.2 association in mouse brain tissues (n = 6 mice per group). (D) Original gel images and quantification of co-IP show the effect of α2δ−1Tat peptide (P) and control peptide (C; both 1 µM for 30 min) on the α2δ−1–GluN1 or α2δ−1–Cav2.2 association in mouse brain slices subjected to OGD (O, n = 6 mice per group). Data are shown as means ± SEM. *p < 0.05 compared with the baseline current amplitude before OGD or with the vehicle or control peptide-treated group. #p < 0.05 compared with the OGD+vehicle or OGD+control peptide group.
α2δ−1 interacts with NMDARs predominantly through its C-terminal domain, and a 30–amino-acid peptide (VSGLNPSLWSIFGLQFILLWLVSGSRHYLW) mimicking the C-terminal domain of α2δ−1 fused with Tat protein (YGRKKRRQRRR; α2δ−1Tat peptide) interrupts the α2δ−1–NMDAR interaction in vivo.13 Treatment with α2δ−1Tat peptide (1 µM for 60 min) had no effect on the basal puff NMDAR currents of hippocampal CA1 pyramidal neurons from WT mice. However, OGD failed to increase NMDAR currents in neurons treated with α2δ−1Tat peptide (Figure 1A and 1B). Similar treatment with a control peptide, which was a Tat-fused scrambled peptide (FGLGWQPWSLSFYLVWSGLILSVLHLIRSN), did not alter the OGD-induced increase in the NMDAR currents (Figure 1A and 1B).
We used coimmunoprecipitation (co-IP) to determine whether α2δ−1 physically interacts with GluN1, an obligatory subunit of NMDARs, in mouse brain tissues. Co-IP analysis showed that OGD increased the α2δ−1–GluN1 protein complex level but had no effect on the α2δ−1–Cav2.2 association in membrane extracts from brain tissues (Figure 1C). Treatment with α2δ−1Tat peptide substantially reduced the α2δ−1–GluN1 interaction but had no effect on the α2δ−1–Cav2.2 association in brain tissues subjected to OGD (Figure 1D).
In addition, we used Cacna2d1 KO mice to validate the critical role of α2δ−1 in OGD-induced NMDAR hyperactivity of hippocampal CA1 pyramidal neurons. Cacna2d1 KO mice do not show any evident neurological abnormalities although they exhibit reduced pain and reduced spinal NMDAR activity potentiated by nerve injury.13 The basal NMDAR currents of CA1 neurons were similar in WT and Cacna2d1 KO mice before OGD induction. In contrast to OGD in the WT mice, OGD failed to affect puff NMDAR currents in CA1 pyramidal neurons in Cacna2d1 KO mice (Figure 1A and 1B). Collectively, these data indicate that α2δ−1, by interacting with NMDARs, is essential for ischemia-induced neuronal NMDAR hyperactivity in the brain.
Focal cerebral ischemia/reperfusion increases α2δ−1 protein glycosylation in the brain
To determine whether focal cerebral ischemia/reperfusion affects α2δ−1 protein levels in brain tissues, we induced 90 min of MCAO followed by 24 h of reperfusion in mice to produce stable infarct volume and neurological impairment. Immunoblotting of the forebrain tissues detected two α2δ−1 protein bands at 140 kDa and 110 kDa in sham control mice but a single 140 kDa band in MCAO mice (Figure 2A). The α2δ−1 protein level at 140 kDa in the cerebral cortex, hippocampus, and striatum was significantly higher in the MCAO group than in the sham control group (Figure 2A).
Figure 2. Focal cerebral ischemia increases the protein level and glycosylation of α2δ−1.
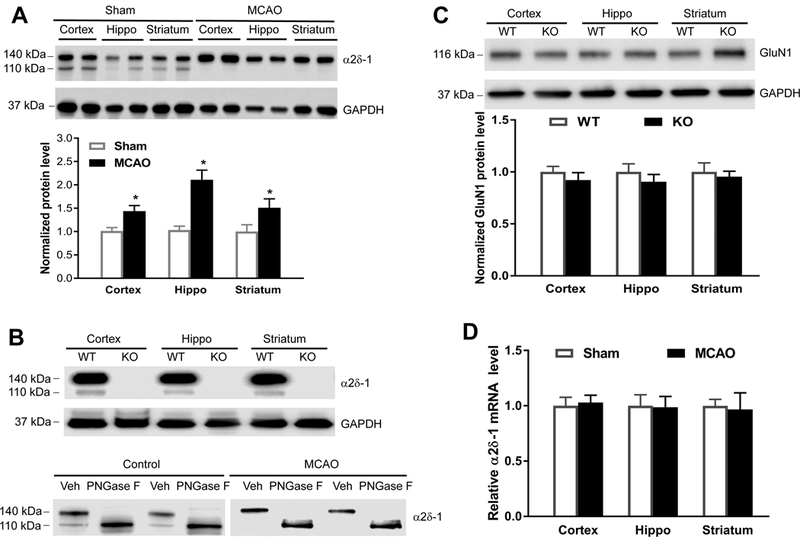
(A) Representative gel images and quantification of α2δ−1 protein levels in the cerebral cortex, hippocampus (Hippo), and striatum from sham control mice and mice subjected to 90 min of MCAO followed by 24 h of reperfusion (n = 8 mice per group). (B) Upper: Representative gel images of α2δ−1 protein bands in different regions of brain tissues from wild-type (WT) and Cacna2d1 knockout (KO) mice; Lower: α2δ−1 protein bands detected in 2 sets of cortical tissue lysates from control and MCAO mice treated with PNGase F or vehicle (Veh). (C) Original gel images and quantification of GluN1 protein levels in brain tissues from WT and Cacna2d1 KO mice (n = 8 mice per group). (D) Quantification of α2δ−1 mRNA levels in the cerebral cortex, hippocampus (Hippo), and striatum in MCAO and sham control mice (n = 6 mice per group). Data are shown as means ± SEM. *p < 0.05 compared with the sham group in the corresponding brain region.
Immunoblotting of brain tissues using Cacna2d1 KO mice confirmed that both 110 kDa and 140 kDa protein bands in control mice are α2δ−1 proteins (Figure 2B). Because α2δ−1 is a highly glycosylated protein,22 we determined whether the MCAO-induced α2δ−1 protein band shift results from increased glycosylation. We treated cortical tissue samples from sham and MCAO mice with 500 units of PNGase F for 60 min.23 In tissues treated with PNGase F, only a single 110 kDa protein brand was detected in both sham and MCAO mice (Figure 2B). The GluN1 protein level in brain tissues was similar in WT and Cacna2d1 KO mice (Figure 2C). The α2δ−1 mRNA level did not differ significantly between sham control and MCAO groups (Figure 2D). These data suggest that cerebral ischemia/reperfusion increases α2δ−1 glycosylation in brain tissues.
α2δ−1 Contributes to MCAO-induced neurological deficits and brain infarct
We next used gabapentin to determine the contribution of α2δ−1 to brain injury and neurological deficits caused by 90 min of MCAO followed by 24 h of reperfusion in WT mice. We administered gabapentin intraperitoneally (i.p.) either before or after initiating MCAO in separate groups of mice (pretreatment and posttreatment groups). For pretreatment, a single dose of gabapentin (100 mg/kg, i.p.) was injected 15 min before MCAO. For posttreatment, two doses of gabapentin (100 mg/kg, i.p.) were injected: one at 30 min after MCAO and another at 6 h after reperfusion. Saline at the same volume as the gabapentin was administered i.p. in a vehicle group that also received MCAO. Both pretreatment and posttreatment with gabapentin significantly improved neurological deficit scores compared with vehicle-treated MCAO mice (Figure 3A). Furthermore, the MCAO-induced brain infarct volume was much smaller in both groups of mice treated with gabapentin than in mice treated with vehicle (Figure 3B). These data suggest that α2δ−1 contributes significantly to ischemia/reperfusion-induced brain damage and neurological deficits.
Figure 3. α2δ−1 critically contributes to brain injury and neurological deficit caused by MCAO.
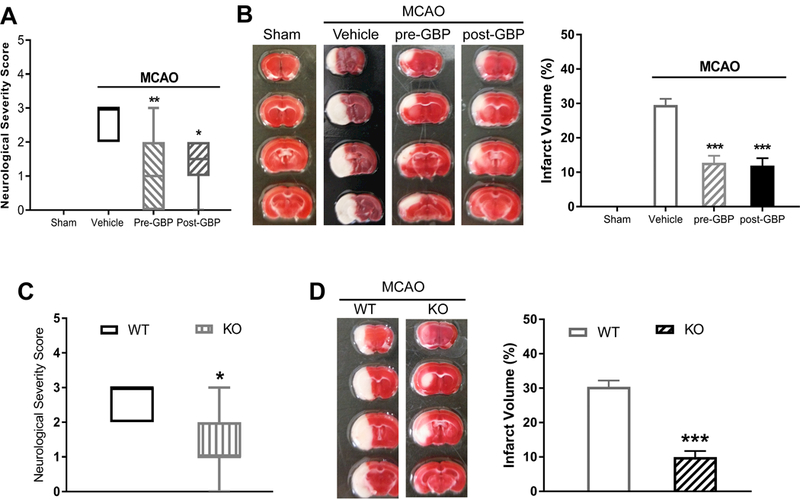
(A) Neurological deficit scores shown in a box-whisker plot in sham control mice (n = 8 mice) and MCAO mice treated with vehicle (n = 9 mice), gabapentin before MCAO (pre-GBP, n = 10 mice), or gabapentin after MCAO (post-GBP, n = 8 mice). (B) Representative TTC staining and quantification of brain infarct volume in sham control mice (n = 8 mice) and MCAO mice treated with vehicle (n = 9 mice), gabapentin before MCAO (n = 10 mice), or gabapentin after MCAO (n = 8 mice). (C) Neurological deficit scores shown in a box-whisker plot in Cacna2d1 knockout (KO) mice and wild-type (WT) mice subjected to MCAO and reperfusion (n = 8 mice per group). (D) Representative TTC staining and quantification of brain infarct volume in Cacna2d1 KO and WT mice (n = 8 mice per group). Data are shown as means ± SEM (neurological deficit score data shown as median ± min-max). *p < 0.05, **p < 0.01, ***p < 0.001 each compared with the vehicle MCAO or WT control group.
α2δ−1 Ablation attenuates MCAO-induced neurological deficits and brain injury
To validate the important role of α2δ−1 in MCAO-induced brain damage, we induced 90 min of MCAO followed by 24 h of reperfusion in Cacna2d1 KO mice. The MCAO-induced neurological deficit score (1.36 ± 0.81 vs. 2.50 ± 0.76, p = 0.0064) and brain infarct volume (11.54 ± 3.07% vs. 30.41 ± 5.08%, p < 0.0001) were much lower in Cacna2d1 KO mice than in WT mice (Figure 3C and 3D). These findings provide unequivocal evidence for the critical role of α2δ−1 in ischemia/reperfusion-induced brain injury and neurological impairment.
α2δ−1–bound NMDARs contribute to MCAO-induced neurological deficits and brain infarct
Using mouse brain tissues, co-IP analysis showed that the α2δ−1 protein was precipitated by an anti-GluN1 antibody, but not by an irrelevant IgG (Figure 4A). To determine whether α2δ−1Tat peptide reduces the α2δ−1–NMDAR interaction in the mouse tissue, we conducted additional co-IP assays using mouse brain slices treated with α2δ−1Tat peptide or a Tat-fused control peptide (1 µM for 60 min). Treatment with α2δ−1Tat peptide caused a large reduction in the α2δ−1–GluN1 protein complex in the brain tissues (Figure 4B). Furthermore, 15 min of MCAO followed by 2 h of reperfusion significantly increased the α2δ−1–GluN1 complex level in mouse brain tissues (Figure 4C).
Figure 4. α2δ−1–bound NMDARs contribute to brain injury and neurological deficit caused by MCAO in the brain.
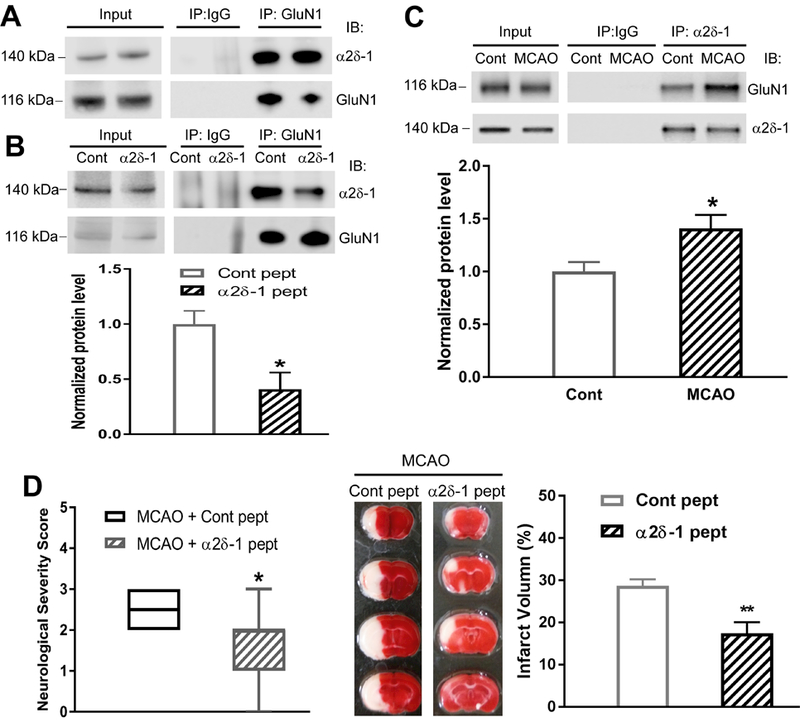
(A) Original co-IP gel images show the interaction between α2δ−1 and GluN1 in two samples from mouse brain tissues. (B) Effect of α2δ−1Tat peptide (1 µM for 60 min) on the α2δ−1–GluN1 protein complex in mouse brain tissues (n = 6 mice per group). (C) Representative gel images and quantification of co-IP analysis show the effect of 15 min of MCAO followed by 2 h of reperfusion on the α2δ−1–GluN1 protein complex level in mouse brain tissues (Cont, sham control; n = 6 samples from 12 mice per group). (D) Neurological severity scores shown in a box-whisker plot (left) and representative TTC staining and quantification (right) of brain infarct volume in MCAO mice treated with α2δ−1Tat peptide or control peptide (both 200 µg/kg, i.p.) at 15 min before MCAO and immediately before reperfusion (n = 8 mice per group). Data are shown as means ± SEM (neurological deficit score data shown as median ± min-max). *p < 0.05, **p < 0.01 compared with the control peptide-treated or sham control group.
We then determined whether the α2δ−1–NMDAR interaction is involved in brain injury and neurological deficits caused by MCAO. In this protocol, we treated MCAO-subjected mice with two injections of α2δ−1Tat peptide or control peptide (both 200 µg/kg, i.p.): one at 15 min before MCAO and another immediately before reperfusion. Treatment with α2δ−1Tat peptide significantly reduced the neurological deficit score increased by MCAO compared with that in control peptide-treated mice (Figure 4D). Furthermore, the brain infarct volume was much less in mice treated with α2δ−1Tat peptide than in mice treated with the control peptide (Figure 4D). These results indicate that α2δ−1–bound NMDARs play a major role in ischemia/reperfusion-induced brain injury and associated neurological deficits.
α2δ−1 and α2δ−1–bound NMDARs mediate MCAO-induced calpain activation in brain tissues
Cerebral ischemia increases calpain activity, and calpain inhibition is neuroprotective against ischemia-induced brain injury.24 Spectrin is highly sensitive to proteolysis by calpain, and the stable αII-spectrin breakdown product (spectrin BD, 150 kDa) is commonly used as a biochemical assay for calpain activation. Immunoblotting analysis showed that compared with the sham control group, 90 min of MCAO followed by 24 h of reperfusion caused a large increase in the spectrin BD level in the cerebral cortex, hippocampus, and striatum in WT mice subjected to MCAO (Figure 5A and 5B). Systemic administration of gabapentin in WT mice subjected to MCAO (posttreatment protocol) and Cacna2d1 KO mice subjected to MCAO significantly reduced the spectrin BD level in all three regions compared with that in vehicle-treated WT mice subjected to MCAO (Figure 5A and 5B).
Figure 5. Gabapentin, α2δ−1Tat peptide, or α2δ−1 genetic deletion reduces MCAO-induced calpain activity in brain tissues.
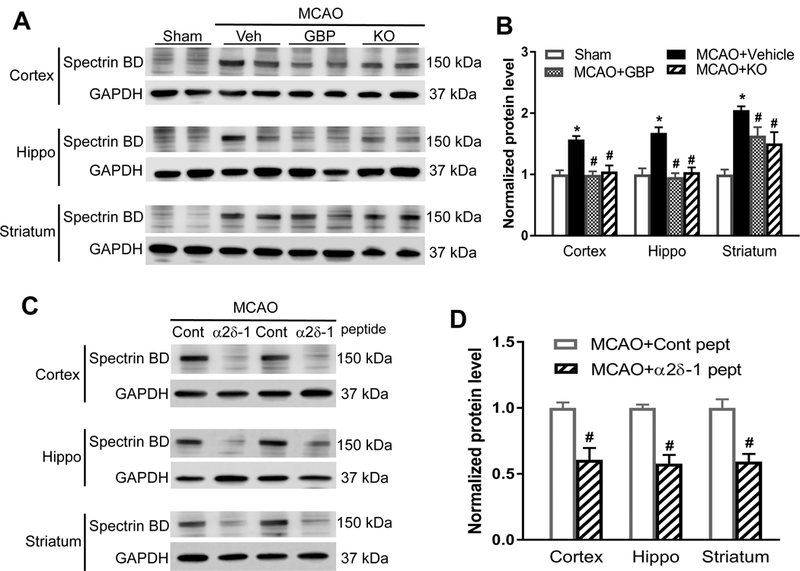
(A and B) Original gel images (A) and quantification (B) of spectrin BD levels in the cerebral cortex, hippocampus (Hippo), and striatum from sham control mice, MCAO mice treated with vehicle (Veh) or gabapentin (GBP), and Cacna2d1 knockout (KO) MCAO mice (n = 8 mice per group). (C and D) Original blotting images (C, two pairs of samples) and quantification (D) of spectrin BD levels in the cerebral cortex, hippocampus, and striatum from MCAO mice treated with α2δ−1Tat peptide or control peptide (n = 8 mice per group). Data are shown as means ± SEM. *p < 0.05 compared with the sham group. #p < 0.05 compared with MCAO+vehicle group or control peptide group in the same brain region.
Also, compared with the MCAO group treated with control peptide, α2δ−1Tat peptide treatment (two injections of 200 µg/kg, i.p.: one at 15 min before MCAO and another immediately before reperfusion) significantly reduced the spectrin BD level in the cortex, hippocampus, and striatum in WT mice (Figure 5C and 5D). Together, these data suggest that α2δ−1, through its interaction with NMDARs, mediates ischemia/reperfusion-induced calpain activation in brain tissues.
α2δ−1 and α2δ−1–bound NMDARs contribute to MCAO-induced caspase-3 activation in brain tissues
Increased caspase-3 activity usually denotes the cleavage of procaspase-3 to active caspase-3, and inhibition of caspase-3 can protect against ischemic neuronal death.25 Immunoblotting analysis showed that 90 min of MCAO followed by 24 h of reperfusion substantially increased the cleaved caspase-3 level in the cerebral cortex, hippocampus, and striatum in vehicle-treated WT mice compared with WT sham control mice (Figure 6A and 6B). Although systemic treatment with gabapentin in WT mice and the use of Cacna2d1 KO mice significantly attenuated the MCAO-induced increase in the cleaved caspase-3 level in the cerebral cortex and hippocampus, gabapentin and Cacna2d1 KO each only slightly attenuated the MCAO-induced increase in the cleaved caspase-3 level in the striatum (Figure 6A and 6B).
Figure 6. Gabapentin, α2δ−1Tat peptide, or α2δ−1 ablation suppresses MCAO-induced apoptotic signaling in brain tissues.
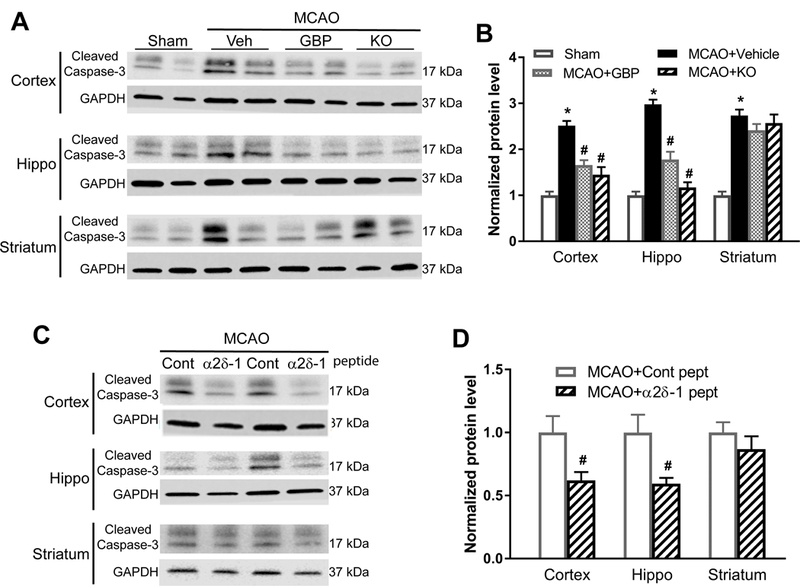
(A and B) Representative blotting images (A) and quantification (B) of cleaved caspase-3 protein levels in the cerebral cortex, hippocampus (Hippo), and striatum from WT sham control mice, WT MCAO mice treated with vehicle (Veh) or gabapentin (GBP), and Cacna2d1 knockout (KO) MCAO mice (n = 8 mice per group). (C and D) Original gel images (C, two pairs of samples) and quantification (D) of cleaved caspase-3 protein levels in the cerebral cortex, hippocampus, and striatum from MCAO mice treated with α2δ−1Tat peptide or control peptide (n = 8 mice per group). Data are shown as means ± SEM. *p < 0.05 compared with the sham group. #p < 0.05 compared with MCAO+vehicle group or control peptide group in the same brain region.
In addition, systemic treatment with α2δ−1Tat peptide in WT MCAO mice significantly reduced the cleaved caspase-3 level in the cerebral cortex and hippocampus, but not in the striatum, compared with control peptide-treated WT MCAO mice (Figure 6C and 6D). These results suggest that α2δ−1–bound NMDARs contribute to ischemia/reperfusion-induced caspase-3 activation in the cortex and hippocampus.
Discussion
A salient finding of our study is that α2δ−1 is essential for ischemia-induced neuronal NMDAR hyperactivity. Although α2δ−1 is commonly considered a VGCC subunit, quantitative proteomic analysis indicates that α2δ−1 has a weak interaction with VGCCs in the brain tissues.26 Also, VGCC currents in brain neurons are similar in WT and Cacna2d1 KO mice.27 In the present study, we found that α2δ−1 physically interacted with NMDARs in the mouse brain and that ischemia increased the glycosylated form of α2δ−1 proteins and the α2δ−1–NMDAR association. Because the hippocampal CA1 area is highly vulnerable to ischemic brain damage via glutamate-mediated excitotoxicity, we used hippocampal slices subjected to OGD to simulate ischemia to identify the molecular mechanism responsible for ischemia-induced NMDAR hyperactivity in CA1 neurons. We showed that inhibiting α2δ−1 with gabapentin, interrupting the α2δ−1–NMDAR interaction with α2δ−1Tat peptide, or ablating α2δ−1 completely blocked OGD-induced NMDAR hyperactivity in hippocampal neurons. In addition, presynaptic α2δ−1-NMDAR complexes may be involved in increased glutamate release during brain ischemia. The OGD-induced NMDAR hyperactivity may involve a rapid activity-dependent α2δ−1–NMDAR interaction and mobilization to the neuronal membrane. Importantly, gabapentin, α2δ−1Tat peptide, or α2δ−1 ablation did not affect basal NMDAR activity. In a heterologous expression system, gabapentin inhibits NMDAR activity only when α2δ−1 is coexpressed.13 Thus, our findings suggest that α2δ−1–bound NMDARs are distinctively involved in brain ischemia-induced abnormal NMDAR hyperactivity.
Another important finding of our study is that α2δ−1–bound NMDARs play a critical role in brain injury caused by ischemia/reperfusion. We found that pretreatment or posttreatment with gabapentin attenuated both the neurological deficits and brain infarct volumes caused by MCAO. Furthermore, these neuroprotective effects were validated in mice devoid of α2δ−1 in our study. It has been reported that gabapentinoids produce neuroprotective effects in rodent models of cerebral ischemia.28,29 The neuroprotective effects of gabapentinoids were previously assumed to be mediated by VGCC inhibition. However, gabapentinoids have little effect on VGCC activity.13,30,31 We showed that α2δ−1Tat peptide, targeting the C-terminal domain of α2δ−1, disrupted the α2δ−1–NMDAR interaction and that α2δ−1Tat peptide treatment effectively decreased the MCAO-induced neurological deficits and brain infarct volumes. In addition to promoting surface trafficking of NMDARs, α2δ−1 can also reduce the Mg2+ block of GluN2A-containing NMDARs.13 Thus, treatment with gabapentinoids or α2δ−1Tat peptide not only attenuates the number of α2δ−1–bound NMDARs on the membrane surface but also restores the Mg2+ block of NMDAR channels to limit Ca2+ influx.
Our study provides new information about the downstream signaling mechanism involved in the neuroprotective effects of inhibiting α2δ−1–bound NMDARs during cerebral ischemia/reperfusion. There is a direct link between the early calcium-mediated calpain activation after cerebral ischemia and the subsequent caspase-3 activation.32 We found that gabapentin, α2δ−1Tat peptide, or α2δ−1 ablation largely normalized the increased calpain and caspase-3 activity in brain tissues induced by ischemia/reperfusion. Interestingly, we found that these interventions had little effect on the increased caspase-3 activity in the ischemic striatum. A limitation of our study is that many of the biochemical measurements were made at 24 h after MCAO when the majority of the neurons are dead and their proteins are undergoing degradation. Because striatal infarction is mostly necrotic and occurs rapidly after MCAO induction,33,34 this may explain why inhibition of α2δ−1–bound NMDARs failed to reverse the increased caspase-3 activity in this location. Our findings suggest that α2δ−1–bound NMDARs play a critical role in the increased calpain/caspase-3 activity in ischemic brain tissues.
Because α2δ−1 is mainly expressed in excitatory neurons,14 gabapentinoids and α2δ−1Tat peptide may selectively inhibit α2δ−1–bound NMDARs expressed in excitatory neurons and spare the NMDARs present in inhibitory neurons. Such a preferential effect on α2δ−1–bound NMDARs by gabapentinoids and α2δ−1Tat peptide may be highly desirable for treating ischemic stroke since this effect does not interfere with basal α2δ−1–free NMDAR function, which may be an endogenous physiological mechanism for neuronal survival in ischemic brain tissues.11,35 Gabapentinoids bind to both α2δ−1 and α2δ−2 in the central nervous system.22 The fact that α2δ−2 is predominantly expressed in inhibitory neurons14 may account for the adverse central nervous system effects of gabapentinoids. Because the amino acid sequence of α2δ−1Tat peptide is unique to α2δ−1, it is conceivable that α2δ−1 C-terminus–interfering peptides or drugs specifically targeting α2δ−1 may have fewer adverse effects than gabapentinoids. Thus, α2δ−1 C-terminus–interfering peptides or inhibitors could circumvent the negative consequences associated with NMDAR antagonists and could be developed as neuroprotective agents for treating ischemic stroke. Preemptive treatment with gabapentinoids or α2δ−1 competing peptides/inhibitors may also offer neuroprotection in patients undergoing major neurological and cardiac surgeries.
In summary, our study provides substantial new evidence that α2δ−1–bound NMDARs play a major role in brain damage caused by ischemia/reperfusion. This new information advances our understanding of the molecular mechanism of neuronal NMDAR hyperactivity during ischemic stroke. α2δ−1–bound NMDARs are a promising target for the development of novel neuroprotective therapies that could have greater therapeutic windows than general NMDAR antagonists.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by the National Institutes of Health (Grants HL131161 and NS101880) and the N.G. and Helen T. Hawkins Endowment (to H.-L.P.).
Footnotes
Disclosures
None.
References
- 1.Rossi DJ, Oshima T and Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000;403:316–21. [DOI] [PubMed] [Google Scholar]
- 2.Simon RP, Swan JH, Griffiths T and Meldrum BS. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science 1984;226:850–2. [DOI] [PubMed] [Google Scholar]
- 3.Lai TW, Shyu WC and Wang YT. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med 2011;17:266–75. [DOI] [PubMed] [Google Scholar]
- 4.Lipton SA. NMDA receptors, glial cells, and clinical medicine. Neuron 2006;50:9–11. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, et al. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci 1998;18:4914–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neumar RW, Meng FH, Mills AM, Xu YA, Zhang C, Welsh FA, et al. Calpain activity in the rat brain after transient forebrain ischemia. Exp Neurol 2001;170:27–35. [DOI] [PubMed] [Google Scholar]
- 7.Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, et al. Selfotel in acute ischemic stroke: possible neurotoxic effects of an NMDA antagonist. Stroke 2000;31:347–54. [DOI] [PubMed] [Google Scholar]
- 8.Sacco RL, DeRosa JT, Haley EC, Jr., Levin B, Ordronneau P, Phillips SJ, et al. Glycine antagonist in neuroprotection for patients with acute stroke. JAMA 2001;285:1719–28. [DOI] [PubMed] [Google Scholar]
- 9.Albers GW, Atkinson RP, Kelley RE and Rosenbaum DM. Safety, tolerability, and pharmacokinetics of the N-methyl-D-aspartate antagonist dextrorphan in patients with acute stroke. Stroke 1995;26:254–8. [DOI] [PubMed] [Google Scholar]
- 10.Albers GW, Goldstein LB, Hall D and Lesko LM. Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA 2001;286:2673–82. [DOI] [PubMed] [Google Scholar]
- 11.Ikonomidou C and Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 2002;1:383–6. [DOI] [PubMed] [Google Scholar]
- 12.Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T and Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci 2006;26:1604–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RE, et al. The alpha2delta-1-NMDA Receptor Complex Is Critically Involved in Neuropathic Pain Development and Gabapentin Therapeutic Actions. Cell Rep 2018;22:2307–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA, et al. Differential distribution of voltage-gated calcium channel alpha-2 delta subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 2005;491:246–69. [DOI] [PubMed] [Google Scholar]
- 15.Taylor CP and Garrido R. Immunostaining of rat brain, spinal cord, sensory neurons and skeletal muscle for calcium channel alpha2-delta type 1 protein. Neuroscience 2008;155:510–21. [DOI] [PubMed] [Google Scholar]
- 16.Engel O, Kolodziej S, Dirnagl U and Prinz V. Modeling stroke in mice - middle cerebral artery occlusion with the filament model. J Vis Exp 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo Y, Zhou Y, Xiao W, Liang Z, Dai J, Weng X, et al. Interleukin-33 ameliorates ischemic brain injury in experimental stroke through promoting Th2 response and suppressing Th17 response. Brain Res 2015;1597:86–94. [DOI] [PubMed] [Google Scholar]
- 18.Longa EZ, Weinstein PR, Carlson S and Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84–91. [DOI] [PubMed] [Google Scholar]
- 19.Gee CE, Benquet P, Raineteau O, Rietschin L, Kirbach SW, et al. NMDA receptors and the differential ischemic vulnerability of hippocampal neurons. Eur J Neurosci 2006;23:2595–603. [DOI] [PubMed] [Google Scholar]
- 20.Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M and Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 1999;19:Rc39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuller-Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, et al. Targeted disruption of the voltage-dependent calcium channel alpha2/delta-1-subunit. Am J Physiol Heart Circ Physiol 2009;297:H117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marais E, Klugbauer N and Hofmann F. Calcium channel alpha(2)delta subunits-structure and Gabapentin binding. Mol Pharmacol 2001;59:1243–8. [DOI] [PubMed] [Google Scholar]
- 23.Ye ZY, Li DP, Byun HS, Li L and Pan HL. NKCC1 upregulation disrupts chloride homeostasis in the hypothalamus and increases neuronal activity-sympathetic drive in hypertension. J Neurosci 2012;32:8560–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markgraf CG, Velayo NL, Johnson MP, McCarty DR, Medhi S, Koehl JR, Chmielewski PA and Linnik MD. Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke 1998;29:152–8. [DOI] [PubMed] [Google Scholar]
- 25.Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, et al. Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A 2002;99:15188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A 2010;107:14950–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felsted JA, Chien CH, Wang D, Panessiti M, Ameroso D, Greenberg A, et al. Alpha2delta-1 in SF1(+) Neurons of the Ventromedial Hypothalamus Is an Essential Regulator of Glucose and Lipid Homeostasis. Cell Rep 2017;21:2737–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffmann U, Lee JH, Qin T, Eikermann-Haerter K and Ayata C. Gabapentin reduces infarct volume but does not suppress peri-infarct depolarizations. J Cereb Blood Flow Metab 2011;31:1578–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon JS, Lee JH, Son TG, Mughal MR, Greig NH and Mattson MP. Pregabalin suppresses calcium-mediated proteolysis and improves stroke outcome. Neurobiol Dis 2011;41:624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rock DM, Kelly KM and Macdonald RL. Gabapentin actions on ligand- and voltage-gated responses in cultured rodent neurons. Epilepsy Res 1993;16:89–98. [DOI] [PubMed] [Google Scholar]
- 31.Schumacher TB, Beck H, Steinhauser C, Schramm J and Elger CE. Effects of phenytoin, carbamazepine, and gabapentin on calcium channels in hippocampal granule cells from patients with temporal lobe epilepsy. Epilepsia 1998;39:355–63. [DOI] [PubMed] [Google Scholar]
- 32.Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, et al. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia. J Biol Chem 2001;276:10191–8. [DOI] [PubMed] [Google Scholar]
- 33.Garcia JH, Liu KF and Ho KL. Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke 1995;26:636–42; discussion 643. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Chopp M, Jiang N, Zhang ZG and Zaloga C. Induction of DNA fragmentation after 10 to 120 minutes of focal cerebral ischemia in rats. Stroke 1995;26:1252–7. [DOI] [PubMed] [Google Scholar]
- 35.Ikonomidou C, Stefovska V and Turski L. Neuronal death enhanced by N-methyl-D-aspartate antagonists. Proc Natl Acad Sci U S A 2000;97:12885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.