

Abstract
The wide reactivity of the thiol group enables the formation of a variety of reversible, covalent modifications on cysteine residues. S-nitrosylation, like many other post-translational modifications, is site selective, reversible, and necessary for a wide variety of fundamental cellular processes. The overall abundance of s-nitrosylated proteins and reactivity of the nitrosyl group necessitates an enrichment strategy for accurate detection with adequate depth. Herein, a method is presented for the enrichment and detection of endogenous protein s-nitrosylation from complex mixtures of cell or tissue lysate utilizing organomercury resin. Minimal adaptations to the method also support the detection of either s-glutathionylation or s-acylation using the same enrichment platform. When coupled with high accuracy mass spectrometry, these methods enable a site specific level of analysis, facilitating the curation comparable datasets of three separate cysteine post-translational modifications.
Keywords: nitrosylation, glutathionylation, s-acylation, cysteine, signaling, proteomics
Introduction
In general, the detection of most cysteine modifications requires some form of enrichment prior to analysis. The high reactivity and low abundance of most cysteine modifications generally precludes the detection of these modifications directly. Therefore, typical methodology first utilizes a selective reaction with the modified thiol group of the cysteine, followed by resin assisted enrichment and the detection of the captured peptide or protein using appropriate analytical methodology, typically via mass spectrometry or western blot. For the detection of protein s-nitrosylation, the covalent adduction of a nitrosyl group to the cysteine thiol, the organic mercury resin is designed to react directly with nitrosothiols, thus the selective reaction and enrichment are effectively coupled.
Cysteine is uniquely capable of being modified by a number of different modifications. Due to inherent differences in the reactivity of various enrichments methods, it is advantageous to use the same general method to mitigate analytical bias to achieve datasets that are more comparable between different thiol modifications. Therefore, we have adapted the basic protocol with alterations that enable the use of the organomercury resin for the detection of two additional cysteine modifications, s-glutathionylation and s-acylation as well.
Strategic Planning
It is important to determine several factors before beginning. The first consideration is whether the interest is at the protein level or site level. The protocol enables two analysis parameters, one at the protein level (protein capture), in which intact proteins are eluted from the columns, and the other at the site level (peptide capture), in which proteins are digested on the column and peptides are eluted. Since the site specific analysis utilizes on column digestion, protein identity is generally predicated on a single peptide sequence since many proteins are only modified at a single site. It is advisable to have a secondary method (i.e. a complementary protein capture) to achieve a higher confidence protein identification.
The second consideration is if a protein capture is performed, whether the downstream analytical method will use MS or western blotting. MS enables high resolution protein identification for even low levels of expression, but it is expensive and requires additional sample processing following enrichment. Western blotting provides for an easy, semi-quantitative method, that when run with the fraction of protein that is not bound to the column can provide an estimation of occupancy. However, this requires a known protein target that is sufficiently expressed and/or modified at a level to detect with an antibody as well as the existence of antibodies for the protein(s) of interest.
Overall, it is suggested that the investigators determine the precise experimental question they aim to answer prior to beginning this method or its variants.
Basic Protocol 1
Enrichment of S-nitrosylation
This section describes the sample processing that is involved in immobilization and enrichment of s-nitrosylated proteins from tissue using organic mercury resin. Additional processing for downstream analysis of proteins or peptides is described in Basic Protocol 2.
Materials
Homogenization buffer (See Recipe)
Protease Inhibitors
Organic Mercury Resin (See Support Protocol 1 for synthesis and Support Protocol 2 for preparation)
HEN buffer (See Recipe)
Reducing agent (Tris(2-carboxyethyl)phosphine (TCEP), Dithiothreitol (DTT)) or alternatively Mannitol
Acetone
S-Methyl thiomethanesulfonate, (MMTS)
Sodium dodecyl sulfate (SDS)
Loading Buffer (See Recipe)
Wash Buffers (See Recipe)
TissueMizer (Tekmar, Mason, Ohio)
Teflon pestle and a Jumbo Stirrer (Fisher Scientific, Pittsburgh, PA)
Low retention tubes (Axygen, Union City, CA)
UV light source
β-mercaptoethanol
15 mL and 0.5 mL capacity 10 kDa centrifugal concentration filters (Amicon)
Appropriate agarose gel for running protein
Prepare tissue homogenate
Nitrosothiols exhibit reduced stability to light. Thus, tissue homogenization must be performed in tubes shielded from light. Moreover, for tissue rich in connective tissue such as skeletal muscle, heart and lung tissue, additional steps for efficient homogenization are required (see step 1). Sonication of the tissue should be avoided since it impacts the stability of nitrosothiols.
1. Tissue is thawed quickly in light protected glass vial containing 2 mL of homogenization buffer with 1× protease inhibitors and chopped into small pieces using a TissueMizer (Tekmar, Mason, Ohio) with power set at 70%. For soft tissue such as the brain, liver and thymus this step is not necessary.
2. Homogenize tissue in 10× (w/v) chilled lysis buffer containing protease inhibitors using a Teflon pestle and a Jumbo Stirrer (Fisher Scientific, Pittsburgh, PA).
3. Cell debris is removed by centrifugation at 13000 × g for 30 minutes at 4°C.
4. Supernatant is transferred carefully into new tubes trying to avoid the lipid layer on the top. Note: Lipids have polar surface which may interfere with agarose beads increasing the nonspecific interactions.
5. Determine protein concentration of the homogenate by Bradford or BCA assay using BSA as standard protein. This will be used to normalize and ensure an adequate protein input in Step 6 below.
Chemical enrichment for S-nitrosylated proteins and peptides
The organic mercury-based chemical enrichment requires two steps; the blocking of reduced thiols and the reaction of nitrosothiols with organic mercury.
Blocking of reduced thiols and generation of negative controls
6. Transfer the protein homogenate to a clean tube. Calculate the volume of homogenate to transfer based on the measured protein concentration from Step 5. Equalize the volume using homogenization buffer.
Note: Good results have been achieved using as little as 3mg protein for whole proteome analysis. For targeted protein analysis, protein input may need to be adjusted based on the expression and/or nitrosylation levels of the protein of interest.
7. Adjust the protein concentration to 0.5 mg/mL with homogenization buffer to facilitate more efficient blocking.
8. Generation of negative controls. It is recommended to couple each experimental sample with a negative control. Divide the sample in half and treat the negative control with a reducing agent such as TCEP or DTT to reduce all thiols. Alternatively, copper catalyzed ascorbate reduction, thiol-bound NO displacement by HgCl2, or photolysis by exposure to UV light for 5 min with mannitol (0.1M) in solution to react with the free NO can be performed to selectively remove nitrosothiols. Any of these methods have been successfully employed to eliminate protein nitrosocysteine in biological samples.
9. Protein homogenate solution is mixed with a 3× volume of chilled acetone and precipitated for 30 minutes at −20 °C and pelleted by centrifugation at 5000 × g for 15 minutes at 4 °C.
10. Pellets are extensively washed with chilled acetone (at least five washes with 5 mL acetone each wash).
12. Pellets are reconstituted in 1-2 mL of 5% SDS-containing HEN buffer.
13. Protein suspensions are transferred to clean tubes and diluted to 2.5% SDS using HEN buffer without SDS at a final protein concentration of 0.5 mg/mL and rocked for 2-3 minutes shielded from light at room temperature.
Note: It is critical that protein pellets are entirely soluble before the addition of MMTS. Heating protein suspensions for few minutes at 50 °C facilitates protein solubility. However, prolonged heating eliminates S-nitrosocysteine. Therefore, if insoluble material remains after 5 minutes at 50°C the samples are centrifuged at 5000 × g for 5 minutes at room temperature and the insoluble pellet is discarded.
14. MMTS at final concentration of 40 mM is added and the samples are vortexed for 30 seconds.
15. Sample are placed for 35 minutes at 50 °C with frequent vortexing (every 5 minutes).
16. Proteins are precipitated with a 3× volume of chilled acetone for 30 minutes at −20 °C.
17. Proteins are pelleted by centrifugation at 3500 × g for 15 minutes at 4 °C.
18. Wash pellets with chilled acetone (See Step 11) and resuspend in the same volume of blocking buffer
19. Transfer protein suspension to clean tubes and repeat the precipitation from steps 16-17 once. During the second precipitation, prepare the organic mercury columns according to Support Protocol 2. With the protein pellet from Step 17, resume with Step 21.
Organic mercury column loading
20. Protein pellets are resuspended into sufficient volume of loading buffer to achieve 1-1.5 mg of protein per mL of organic mercury resin, and then are loaded onto the columns.
Note: It is critical that protein pellets are entirely soluble before loading. Heating protein suspensions for few minutes at 50 °C facilitates protein solubility. If insoluble material remains after 5 minutes at 50°C the centrifuge the samples at 5000 × g for 5 minutes at room temperature and discard the insoluble pellet.
21. Protein suspension reacts with organic mercury, shielded from light, for one hour at room temperature (the columns are covered with aluminum foil).
22. Following the incubation, uncap the columns and allow the buffer to flow through.
23. Wash the resin with 50 resin bed volumes of 50 mM Tris-Cl pH 7.5, 0.3M NaCl, 0.5 % SDS
24. Followed by 50 resin bed volumes of 50mM Tris-Cl pH 7.5, 0.3M NaCl, 0.05% SDS.
25. Next, wash the resin with 50 bed volumes of 50mM Tris-Cl pH 7.5, 0.3M NaCl, 1% TritonX-100
26. Followed by 50 bed volumes of 50mM Tris-Cl pH 7.5, 0.3M NaCl, 0.1% TritonX-100, 0.1M Urea
27. Finally, wash the resin with at least 200 bed volumes of H2O.
Note: During the washes with water the resin is mixed gently 4-5 times to facilitate more thorough removal of detergent or denaturing agents.
28. Continue with Basic Protocol 2 for elution of intact proteins for protein identification and for site specific identification via on column digest and peptide elution.
Support Protocol 1
Synthesis of organomercury matrix
This support protocol describes the synthesis of the organic mercury resin from the Affi-gel-10 agarose resin and p-amino-phenylmercuric acetate. Synthesis should be performed ahead of time and the synthesized mercury resin stored in a bottle shielded from light at 4°C until use.
This support protocol describes the preparation of the organic mercury resin columns for enrichment of nitrosylation (or glutathionylation/s-acylation if following one of the alternate protocols). This can be performed at any point prior to loading the samples on the column.
Caution should be taken to select the appropriate columns to support the organic mercury resin. Typically, plastic columns generate molecules that are ionized during electrospray ionization (ESI) and thus may interfere with peptide detection. The use of glass columns (Biorad 7371522) is recommended for experiments that aim to identify site specific S-nitrosylation (peptide capture). Protein capture experiments are typically performed using PD-10 disposable plastic columns).
Materials
Affi-Gel-10 support, 4 × 25 ml (Biorad 153-6046)
Anhydrous Isopropyl alcohol plus for HPLC, 99.9%
Ethanolamine, minimum 98%
N,N-Dimethylformamide (DMF), sequencing grade
p-amino-phenylmercuric-acetate
Vacuum Flask and Vacuum Source
Buchner Funnel
Filter paper
Spatula
Organic mercury resin (See support protocol 1 for synthesis)
Isopropanol
0.1M NaHCO3 pH 8.8
Equilibration buffer
Glass columns (Biorad 7371522) or PD-10 disposable plastic columns (GE Healthcare 17043501)
1. 100 mL of Affi-Gel 10, are thawed for 30 min at room temperature and are transferred to a Buchner funnel.
2. The gel is washed with 300 mL of anhydrous isopropyl alcohol while it is gently mixed. Caution is needed to prevent the gel from drying. A dry gel requires extensive washes with anhydrous isopropyl alcohol until it is fully hydrated. However, if insoluble agarose clumps remain after the washes the synthesis process should stop and a new gel must be used.
3. 210g p-amino-phenyl-mercuric acetate is dissolved in 30 mL of DMF at room temperature. Typically, mercury powder is fully dissolved after 15-20 minutes.
4. The agarose slurry is transferred into a clean dark bottle where the mercury solution is added. The bottle is placed on a stirrer, set at low speed, for at least 4 hours or overnight at room temperature. Note: coupling under anhydrous condition is preferable since there is no hydrolysis of the active esters in the absence of water.
5. Uncoupled active groups are blocked by the addition of 1mL of ethanolamine. The solution is stirred for 1h at room temperature.
6. The gel slurry is then transferred onto a Buchner funnel and washed with 300 mL DMF.
7. The gel is washed with 1000 mL of anhydrous isopropyl alcohol while it is gently mixed.
8. After the final wash, the gel is suspended in 200 mL of anhydrous isopropyl alcohol and the slurry is stored into a dark bottle at 4°C. Resin should be used within 3 months.
Preparation and activation of mercury columns
9. Glass or PD-10 disposable columns are initially washed with 10mL of 50% acetonitrile (ACN) followed by extensive washes with water.
10. Equilibrate the resin at room temperature for 20 min.
11. Gel slurry is poured into each column and the storage buffer (isopropyl alcohol) is allowed to flow through the column. The gel bed should be of sufficient volume to achieve a ratio of roughly 1-1.5 mg protein (measured in Step 6 of the Basic Protocol 1) per mL resin.
12. Gel is washed with 10 bed volumes of isopropanol and if necessary is mixed gently to remove air bubbles.
Note: One bed volume is equal to the volume of the resin bed in mL. If the resin bed is 3 mL then 30 mL should be used.
13. The gel is then washed with 20 bed volumes of water. In this step the resin will swell, however, the volume should not be readjusted.
14. Resin is mixed again gently to remove air bubbles.
15. The resin is activated by washing with 20 bed volumes of 0.1M NaHCO3 pH 8.8 followed by 20 bed volumes of equilibration buffer
Basic Protocol 2
Detection of site specific nitrosylation
These steps are used to identify the specific site of nitrosylation for intact proteins and additional steps for protein digestion to isolate peptides. The steps involve the continuation of Basic Protocol 1 and incorporate an overnight on-column trypsin digest. Following wash steps to remove unbound peptides, what remains are peptides that were s-nitrosylated on at least one cysteine within the peptide.
Materials
0.1M NH4HCO3
MS-grade Trypsin
Performic Acid, 1%
Formic Acid
Glass vials with cap
Lyophilizer
Vacuum Concentrator
Reaction with organic mercury for protein Identification
These steps are for the elution and processing of intact protein from the organic mercury columns. This is useful for downstream application using mass spectrometry or western blotting.
1. Following the final wash, position a clean collection tube below the column and add 10 mL of 50mM β-mercaptoethanol in water to elute bound proteins.
2. The eluate can be stored at −80°C or processed immediately.
3. Using 10 kDa cutoff Amicon centrifugal filters with a 15mL capacity, concentrate the eluate according to the manufacturer’s directions until the minimum volume of 500 μL is reached.
4. Transfer the eluate to a smaller 0.5 mL capacity 10 kDa cutoff Amicon centrifugal filter unit and proceed centrifuge according to the manufacturer’s directions until the volume of the eluate is roughly 20 μL.
Note: The concentrated protein eluate can be stored at −80°C until analysis.
5. Combine the protein eluate with sample buffer and run on a standard agarose gel. Based on the experimental needs, the gel can be processed for in-gel trypsin digest and mass spectrometry analysis to identify the s-nitrosylated proteins or if a protein of interest is identified a priori, a standard western blot can be run and probed with the antibody of interest.
On column digestion for detection of site specific nitrosylation in peptides
1. After the final wash with water, the resin is washed with 5 bed volumes of 0.1M NH4HCO3 followed by the addition of one bed volume of 0.1M NH4HCO3 containing 1ug/mL MS-grade Trypsin.
2. Incubate the columns covered for at least 6 hours or overnight at room temperature.
3. After the incubation wash the resin with 40 bed volumes of 1M NH4HCO3 plus 0.3M NaCl.
Note that NH4HCO3 based buffers generate CO2 as the result of their decomposition in H2O. CO2 formation may cause bubbles to form in the resin bed impacting the flow. In this case, the resin is mixed gently and is allowed to settle before being washed with the next buffer.
4. The resin is washed with 40 bed volumes 1M NH4HCO3 followed by 40 bed volumes 0.1M NH4HCO3
5. The resin is then washed with at least 200 bed volumes of water. In this step the resin is mixed gently 3-4 times during the washes.
6. Bound peptides are oxidatively released by incubation with 1% performic acid (equal to bed volume) for 45 minutes at room temperature and collected into glass tubes.
Note: Mild performic acid cleaves the mercury –thiol bond and releases the bound peptides. In addition, it oxidizes cysteine thiols to sulfonic acid thereby creating a unique mass spectroscopic signature that permits site specific identification of the modified cysteine residues
7. Eluates are frozen at −80 °C overnight
8. Lyophilize the peptide eluates. Lyophilized eluates can be stored at −80°C until ready for analysis.
9. To prepare the lyophilized peptides for mass spectrometry analysis, re-suspend in 300 μL 0.1% formic acid and transfer to a clean low retention tube.
10. Reduce the sample volume to roughly 30 μL by using a vacuum concentrator.
11. Centrifuge the samples at 16000 × g for 20 minutes at 4 °C to remove any precipitate and recover the peptides in the supernatant.
12. Further process the peptide containing supernatant to remove additional contaminants and salts using the preferred method compatible with downstream MS analytical methods.
Note: We have had good results using C18 stage tips (Thermo Electron) following the manufacturer’s recommended procedure.
13. Transfer 20 μL of the peptide suspension to an HPLC vial and analyze via mass spectrometry.
Alternate Protocol 1
Detection of S-glutathionylation
The key feature than enables the use of organomercury resin for the detection of nitrosylation as well as other cysteine modifications is the that it is reactive toward both nitrosothiols and reduced thiols. When detecting nitrosylation, specificity for the modification is derived from the direct reactivity of the organomercury resin with the nitrosothiols in the sample. When detecting other alternative cysteine modifications, specificity is derived through a selective reduction step and subsequent enrichment of the newly reduced thiol. For the detection of glutathionylation; commercially available recombinant glutaredoxin 1 is utilized to perform the selective reduction (Hamnell-Pamment, Lind, Palmberg, Bergman, & Cotgreave, 2005).
Additional materials
N-ethylmaleimide (NEM)
250 μg Recombinant Glutaredoxin 1 (Cayman Chemicals prod no. 11533) resuspended in 250 μL ddH2O for an enzyme concentration of 1 μg/μL.
L-Glutathione, reduced
β-Nicotinamide adenine phosphate dinucleotide, reduced disodium salt hydrate
PD-10 columns prepacked with Sephadex G-25 (GE Healthcare 17-0851-01)
Prepare tissue homogenate
1. Homogenize tissue using the same homogenization buffer from Basic Protocol 1.
2. Centrifuge homogenate at 1500×g for 10 minutes at 4°C, discard pellet.
3. Measure protein concentration of the homogenate using a standard Bradford or BCA protein assay.
Good coverage has been obtained using 6 mg of soluble protein as the input material for whole proteome analysis using mass spectrometry. If the analysis is a targeted protein analysis, the input may be adjusted to fit the abundance of the protein and its modification state.
Unused soluble protein can be stored at −80°C for use in other applications. It is best to only analyze freshly prepared homogenate for cysteine modifications, free thaw cycles can impart artificial oxidation states on cysteine residues.
S-nitrosothiol removal
4. Adjust the soluble protein concentration to 0.5 mg/mL by adding an appropriate volume of HEN buffer with 5% (w/v) SDS, 20 mM NEM, and 0.1M Mannitol added.
If using 6 mg of protein as the input, the final volume will be in 12 mL.
5. Carefully invert to mix so as to avoid excessive bubble formation and transfer to glass petri dish. With the petri dish on a bed of ice, place a UV light over the sample and let stand for 5 min. It is important to get the dish as close to the UV source as possible, as the energy of the light dissipates with distance and nitrosothiol photolysis will be impaired.
6. Following photolysis, pipette protein solution from petri dish into a clean 50 mL conical tube containing 3× volume of chilled acetone. Incubate for 30 min at −20°C.
For example, if the volume of the protein solution is 12 mL, add 36 mL of acetone in the 50 mL conical tube and pipette the 12 mL of protein solution into the acetone.
It is also helpful to vigorously shake or vortex the tubes to aid in precipitation.
7. Centrifuge at 3000 ×g for 10 min at 4°C and discard the supernatant.
8. Wash the pellet 3× by carefully pipetting acetone along the side of the tube and gently swirling. Following the washes, invert the tubes on bench paper to allow excess acetone to dry.
Blocking reduced cysteine residues
9. Resuspend the pellet by adding 1 mL of HEN buffer with 5% SDS (w/v) to the pellet. Break up the pellet with the end of the P1000 filtered pipet tip and repeatedly pipette until the protein is fully in suspension. The suspension should be a clarified solution with a clear to yellow hue, if it is cloudy then continue to pipette with the P1000 or switch to a P200 with filter tip attached. It is also possible to incubate at 55°C for a short time (1-2 min) to aid in resuspension.
10. Centrifuge at 20,000 ×g for 10 min at room temp to pellet any protein that is not in solution. Discard the pellet or return to step 9 and continue to pipette the pellet with smaller tip size to resuspend pellet further.
This is a critical step. If the protein is not in solution then alkylation of free cysteines will be impeded resulting in higher background.
Note: this centrifugation step is performed at room temp, aqueous solutions containing SDS in this protocol should not be chilled, as the SDS will precipitate.
11. Adjust the volume to 0.5 mg/mL (based on the input protein) using HEN buffer with 5% SDS (w/v) and 100 mM NEM.
12. Incubate at 55°C for 1h, vortexing every 15 min.
13. Following alkylation, precipitate the protein by pipetting the solution into clean tube containing 3× volume of chilled acetone.
Background levels are lowest by only precipitating the protein from the solution and discarding the bubble on top.
14. Incubate at −20°C for 30 min.
15. Centrifuge at 3000 ×g for 10 min at 4°C.
16. Discard the supernatant. And wash the pellet 3× with 5 mL of chilled acetone.
17. Invert the tube to allow residual acetone to dry.
Selective reduction of s-glutathionylated cysteine residues
18. When resuspending the precipitated protein pellet to prepare it for selective reduction, it is critical that the minimal essential amount of SDS is used, as SDS will inhibit the enzymatic activity of Grx1. To do this, breakup the pellet by transferring 1 mL of HEN without SDS onto the pellet and repeatedly pipetting until the larger pieces are broken up.
19. Transfer the suspension to a clean 1 mL low retention Eppendorf tube and pipette 8 μL of 25% (w/v) SDS in H2O. Vortex and let sit for several minutes.
20. The protein solution should turn from cloudy to transparent. If not, add another 8 μL of the 25% SDS.
21. To remove SDS that will interfere with Grx1 activity, precondition PD-10 desalting columns with PBS. Remove the top filter with forceps and fill the column with PBS, allowing the buffer to flow through the column by gravity. Repeat at least 3 times.
22. Fill the column a final time with PBS and then centrifuge at 1000 ×g for 2 min. Discard the flow through.
23. Transfer the PD-10 column to a new tube and pipette the resuspended protein solution into the middle of the resin.
24. Centrifuge at 1000 ×g for 2 min to elute the protein.
25. Add and equal volume of 2 mM NADPH and 4 mM GSH dissolved in PBS to the protein solution for a final concentration of 1 mM NADPH and 2 mM GSH. The final volume should be in the 2-3 mL range.
26. Add 13.5 μL of Grx1 to each of the samples.
Important: Omit this step for the negative controls.
27. Incubate at 37°C for 1 h.
28. Following incubation, precipitate the protein in 3× volume acetone for 30 min at −20°C.
Organomercury column binding
29. During the precipitation step, condition the organomercury columns according to Support Protocol 2.
It is important to not let the resin dry out. Once the columns are conditioned, cap the bottom of the column and pipette equilibration buffer over the resin until it is completely submerged. In the equilibration buffer, the protein should be loaded within 1-2 hrs.
30. Centrifuge the precipitated protein at 3000 ×g for 10 min at 4°C. Discard the supernatant.
31. Carefully wash the pellet with 5 mL chilled acetone. Repeat 3×.
32. Resuspend the protein pellet in 1 mL of loading buffer with 2.5% SDS.
33. Centrifuge the suspension at 10,000 ×g for 10 min at room temp to remove any insolubilized protein.
34. Remove the bottom cap to the column and let excess buffer flow through.
35. Pipette the protein directly onto the resin bed and allow buffer to flow through.
36. Replace the bottom cap of the column and add enough loading buffer to the top to completely cover the resin.
37. Continue with the reaction with organic mercury, Step 21 of the Basic Protocol 1, and proceed with Basic Protocol 2 or 3 for elution of proteins or peptides respectively.
Alternate Protocol 2
Detection of S-acylation
For s-acylation, the same principle of selective reduction is utilized, however this alteration utilizes a chemical reduction process in the form of hydroxylamine, rather than an enzymatic reduction. Hydroxylamine selectively reduces thioester bonds (Drisdel, Alexander, Sayeed, & Green, 2006), which is the characteristic bond formed with the covalent adduction of fatty acids with cysteine residues. It should be noted that at times palmitoylation is used interchangeably to describe any lipidation of cysteine. This is a misnomer and may create confusion as to the relevant biological modifications under study. Although palmitate undoubtedly makes up a certain percentage of cysteine based lipidation, hydroxylamine-based reduction is not selective to differentiate between fatty acids with different length carbon chains. Thus, for accuracy we eschew the use of palmitoylation for the more precise S-acylation terminology.
Additional Materials
Hydroxylamine hydrochloride
N-ethylmaleimide (NEM)
PD-10 columns prepacked with Sephadex G-25 (GE Healthcare 17-0851-01)
Sample processing
- 1. Initial sample processing proceeds the same as for the analysis of S-glutathionylation above.
- Prepare tissue homogenate (Steps 1-3),
- Proceed with nitrosothiol photolysis (Steps 4-8)
- Blocking reduced cysteine residues (Steps 9-17)
Selective reduction of thioesters
2. When resuspending the precipitated protein pellet to prepare it for selective reduction, breakup the pellet by transferring 1 mL of HEN without SDS onto the pellet and repeatedly pipetting until the larger pieces are broken up.
3. Transfer the suspension to a clean 1 mL low retention Eppendorf tube and pipette 8 μL of 25% (w/v) SDS in H2O. Vortex and let sit for several minutes.
4. The protein solution should turn from cloudy to transparent. If not, add another 8 μL of the 25% SDS.
Although the SDS concentration does not interfere with a chemical reduction as much as with an enzymatic reduction, we have found that hydroxylamine and SDS are incompatible at high concentrations, thus it is advisable to limit SDS to the minimal necessary amount.
5. Make a stock concentration of 1M hydroxylamine in HEN buffer and adjust the pH to 7.7.
6. Add 1:1 hydroxylamine to protein solution for a final concentration of 500 mM.
Omit hydroxylamine and dilute with HEN buffer for the negative controls.
7. Incubate at 50°C for 1 h with occasional vortexing.
8. During the incubation period, prepare the PD-10 desalting columns.
Important: The removal of excess hydroxylamine is critical for precipitation. Failure to do so will result in an inefficient precipitation and protein loss.
9. Precondition PD-10 desalting columns with PBS. Remove the top filter with forceps and fill the column with HEN, allowing the buffer to flow through the column by gravity.
10. Repeat at least 3 times.
11. Fill the column a final time with HEN and then centrifuge at 1000 ×g for 2 min. Discard the flow through.
12. Transfer the PD-10 column to a new tube and pipette the hydroxylamine:protein solution into the middle of the resin.
13. Centrifuge at 1000 ×g for 2 min to elute the protein.
14. Precipitate the eluted protein by transferring the solution into a tube containing 3× volume of chilled acetone.
15. Incubate for 30 min at −20°C.
16. During the incubation, condition the organomercury columns as described in Support Protocol 2.
17. Centrifuge precipitate at 3000 ×g for 10 min at 4°C to pellet the precipitated protein.
Organomercury column binding
18. Resuspend the protein pellet in loading buffer with 2.5% SDS.
19. Centrifuge the suspension at 10,000 ×g for 10 min at room temp to remove any insolubilized protein.
20. Load onto mercury columns and incubate for 1 hour.
21. Continue with the reaction with organic mercury, Step 21 of the Basic Protocol 1, and proceed with Basic Protocol 2 or 3 for elution of proteins or peptides respectively.
Reagents and Solutions
All solutions should be made up using millipore water or equivalent ultrapure water unless otherwise noted and can be stored at room temperature for several months.
Organic mercury resin
100 mL Affi-gel 10 (Biorad)
2.10 g p-amino-phenylmercuric acetate
1 mL ethanolamine
Annhydrous isopropanol
Dimethylformamide
See synthesis procedure in methods. Store at 4°C shielded from light for up to 3 months. Equilibrate to room temperature before use.
Equilibration buffer
50 mM NaCl
50 mM MES
1 mM DTPA, pH 6.0
Store at room temperature for up to 6 months.
Activation buffer
0.1M NaHCO3 pH 8.8
Store at room temperature for up to 1 year.
Homogenization buffer
250 mM Hepes,
1 mM DTPA,
0.1 mM neocuproine, pH 7.7,
1% TritonX-100
Add Triton X-100 fresh. Store the base buffer at room temperature for up to 1 year.
HEN buffer
250 mM Hepes,
1 mM DTPA, pH 7.7
0.1mM neocuproine.
Store at room temperature for up to 1 year.
Loading buffer
250 mM MES,
1 mM DTPA, pH 6.0
1% (v/v) SDS
SDS should be added fresh. Store the base buffer at room temperature for up to 1 year. Discard if yellow discoloration occurs.
Wash Buffer (containing 4 separate detergent conditions)
50 mM Tris-Cl
0.3M NaCl, pH 7.5
Plus:
0.5% (w/v) SDS or
0.05% (w/v) SDS or
1% (v/v) Triton X-100, 1M Urea or
0.1% (v/v) Triton X-100, 0.1M Urea
The base buffer can be made ahead of time and stored at room temperature. The SDS and Triton/Urea should be added fresh before use.
Performic Acid, 1% (v/v)
Combine 1% formic acid and 0.5% hydrogen peroxide for at least 60 minutes at room temperature in a glass vial shielded from light.
Commentary
Background Information
In recent years, as the investigation into the biological roles of protein s-nitrosylation expand, it is increasingly apparent that this signaling mechanism is involved in a number of fundamental processes and disease states (Foster, Hess, & Stamler, 2009; N. Gould, Doulias, Tenopoulou, Raju, & Ischiropoulos, 2013; Nakamura & Lipton, 2017). In fact, a number of thiol modifications have now been found to significantly contribute to biological signaling. In addition to glutathionylation and s-acylation (Alegre-Cebollada et al., 2014; Daniotti, Pedro, & Valdez Taubas, 2017; Zhang et al., 2018); the formation of protein sulfenic acids (Paulsen et al., 2012), persulfides (Albertolle, Phan, Pozzi, & Guengerich, 2018), and redox active disulfides (Groitl & Jakob, 2014) are thought to make up parts of the overall network of cellular redox signaling (Buettner, Wagner, & Rodgers, 2013; Thamsen & Jakob, 2011).
Due to the labile nature of most thiol modifications, particularly nitrosylation; along with the relative low abundance of most cysteine modifications, direct detection of the modifications is generally not feasible. Thus, the development of enrichment protocols that enable the indirect detection of these modifications on a wider scale. One of the most widely adopted methods that was also originally developed for nitrosylation is what is colloquially referred to as the biotin switch method (Jaffrey & Snyder, 2001) and commercially available in kits. This method utilizes Cu/Asc for selective nitrosothiol reduction followed by biotin derivitization and enrichment. Our organomercury method is essentially a technical refinement of the same methodological concept employed by the biotin switch method. The organomercury enables a more direct reactivity toward ntirosothiols (Saville, 1958) without relying on the Cu/Asc for nitrosothiol reduction. One of the primary drawbacks of our method is that as a consequence of the use of mercury containing compounds, appropriate waste handling and disposal needs to be employed. Based on the policies of the institution, this can be a significant hurdle to the implementation of these methods.
As the research into cysteine based modifications expands, additional resources and methods are being developed. One of these is the incorporation of MS compatible quantification strategies (Leichert et al., 2008; Lindemann & Leichert, 2012). With the advent of isobaric tags, whole proteome quantification can be incorporated prior to analysis or as part of the enrichment strategy. One consideration is downstream compatibility with derivitization agents. For instance, our base method for MS analysis elutes peptides through an oxidative reaction, this would effectively eliminates the use of a sulfhydryl reactive tag. Amine reactive tags are an option that we have had mixed success with. In general, the effectiveness of sample to sample quantification is wholly dependent on efficient and equal peptide derivitization across samples when performed following the enrichment.
Critical Parameters
Blocking efficiency with MMTS (Step 6 of Basic Protocol 1) or NEM (Step 9 in Alternate Protocol 1)
This is a critical step that, if not done well, will lead to high background signal. The blocking agent should always be made fresh and never stored. Occasional vortexing helps the reaction as well.
Shielding samples from light
Nitrosothiols are incredibly labile and excessive exposure to fluorescent or UV lighting can cause a loss of signal. It is important that when analyzing nitrosylation, to keep the samples covered or otherwise protected from light until after they have been loaded onto the column. Beyond that point, it is not necessary to protect from light as the nitrosothiol is no longer present in the present due to the reaction with the phenylmercury.
Nitrosothiol photolysis
When conducting the alternate cysteine modifications protocols, or for the negative controls in the nitrosylation protocol, it is critical that the endogenous nitrosothiols are efficiently removed. Inadequate removal will contribute to high background levels and/or false positives. We have found that the nitrosothiol photolysis using UV is the least disruptive the other thiol modifications (See Fig 1 for a comparison of UV and copper catalyzed ascorbate). When using a UV method, it is important to add in the mannitol to prevent the reaction of the nitrosyl with other proteins in solution. Reductive agents that can break disulfide bonds are not compatible with glutathionylation. Cu/Ascorbate can be used as an alternate nitrosothiol reduction agent, although it needs to be validated that at the concentrations used, off target reduction of disulfide bonds is not occurring (Giustarini, Dalle-Donne, Colombo, Milzani, & Rossi, 2008)
Figure 1.
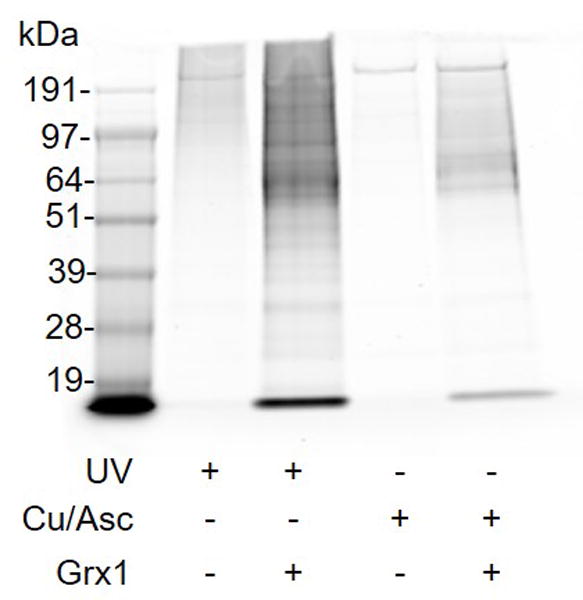
Colloidal blue stained gel comparing nitrosothiol photolysis. Lanes 1 and 2 use UV photolysis and lanes 3 and 4 use Cu/Asc. Lanes 1 and 3 are negative controls lanes 2 and 4 are treated with glutaredoxin 1 to reduce glutathionylated proteins prior to organic mercury enrichment.
Negative controls
Negative controls are also an important aspect of these methods. Invariably, some proteins or peptides will be present in the negative controls. Figure 2 shows the rough ratio of protein and peptide identifications in a positive sample and the negative controls. Since the organic mercury is reactive toward nitrosothiols, it is important to have a robust method of removal when using the Alternate Protocols for s-glutathionylation and s-acylation. Figure 1 shows a comparison of the signal using two methods of nitrosothiol reduction, UV photolysis and copper/ascorbate (Cu/Asc) catalyzed reduction, when processing for s-glutathionylation. Cu/Asc is more efficient at reducing nitrosothiols versus UV, but it also decreases the overall signal. Therefore, it is important to evaluate the collective signal from the negative controls along with the signal in the samples. Generally, any protein and peptide identifications that are found in the negative controls are removed from the sample lists irrespective of abundance.
Figure 2.
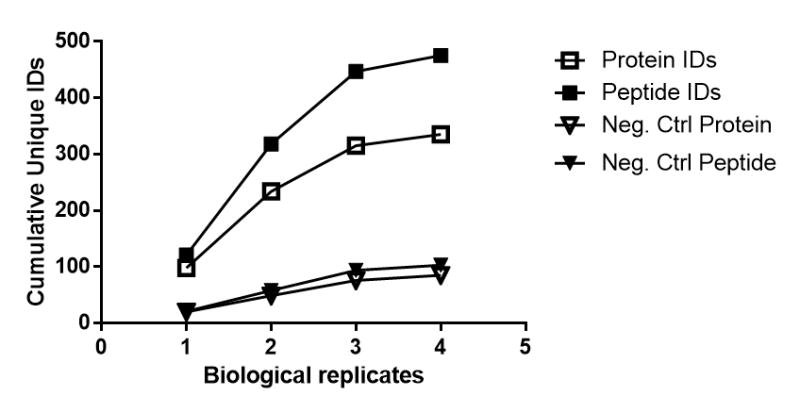
Counts of protein and peptide identification from a typical experiment. The cumulative number of unique protein (open symbols) or peptide (closed symbols) identifications added by each biological replicate. Samples were processed for s-glutathionylation in murine liver tissue using 6 mg of protein input with Alternative Protocol 1 followed by peptide enrichment following the steps in Basic Protocol 3 with matching negative controls. Samples analyzed using an Orbitrap Elite mass spectrometer.
Troubleshooting
High background/Low signal
Proper negative controls are necessary to have with each experimental run. At times, these negative controls may give a higher than typical background, this can be a consequence of several factors:
I. Incomplete nitrosothiol photolysis
Due to the reactivity of the organomercury, if the nitrosothiols are not removed prior to the selective reduction, they will bind with the column. Ensure that mannitol is added to the protein solution prior to photolysis, this increases the likelihood that the NO will not react with other proteins in solution. If you fail to do this, efficient photolysis will not occur. Another consideration is the UV setup. If the UV lamp is not sufficient for nitrosothiol removal, an alternative method using copper and ascorbate can be utilized. Cu/Asc selective reduction of nitrosothiol bonds is the theory behind another widely utilized method employing biotin based conjugation and enrichment. Of course, appropriate experimental conditions and concentrations will need to be determined for any modifications to the methodology presented herein.
II. Incomplete alkylation of reduced cysteine residues
The alkylation step is critical for background minimization. Always make the MMTS or NEM solution fresh, never use stored solutions. If the blocking step is insufficient, you will observe much higher levels of protein in the enriched samples and there will be roughly equivalent protein between the negative controls and samples. The first steps should be to ensure that the protein is completely in solution prior to the blocking step. Centrifuge and remove any insolubilized protein. Secondly, ensure that the samples are occasional vortexed throughout the incubation. Finally, if difficulty still persists, it may be advantageous to increase the concentration of alkylating agent or consider an alternative. In addition to the two presented in this protocol, iodoacetamide is also commonly used.
III. Insufficient resolubilization
Always ensure that following the precipitation steps the protein is completely in solution. Sometimes, even if the solution appears to be clear and all visible signs of protein appear to be in solution, brief centrifugation will ensure that there is no carryover of insolubilized protein.
IV. Ineffective reduction (for glutathionylation or acylation)
Positive controls are not entirely reliable for either glutathionylation or acylation. Using GSSG or Diamide and GSH to artificially induce the glutathionylation modification or palmitate to induce acylation has been met with mixed success in our hands. As such, it is not always feasible to incorporate a positive control. Near background levels in the samples could indicate that the enzyme is no longer viable, the cofactor concentration was too low, or the enzyme is not compatible with sample organism. We have had good results using the same enzyme solution up to 3 months, however if there is any doubt about the quality of the enzyme it is best to purchase new stock. Although Grx is fairly ubiquitous, this method has only been tested on mouse and human samples, the use of any samples originating from other organisms should be validated and modified as needed.
V. Sample carryover
Always ensure that you change tips between samples. For high sensitive MS analysis, use filter tips. Remain cognizant of the order of the samples, pipette the negative controls prior to the positive samples.
VI. Insufficient resin washing
There are times when the flow rates between columns will not be equivalent. Always ensure that the complete wash steps are followed for all columns. If problems persist, increase the wash volumes and occasionally mix the resin bed with a clean spatula or other device, ensuring no carryover between samples.
Anticipated Results
The use of the organic mercury resin has been repeatedly validated (Gould et al., 2015; Raju et al., 2015, Chen et al. 2017). An average of roughly 1.5 sites of modification per protein is typically observed. An example of protein and peptide data from a typical experiment is shown in Figure 2. With each biological replicate, fewer unique protein identifications and peptide identifications will be returned. Figure 2 shows the beginning of the plateau of proteome coverage, which is typically based on the sensitivity of the mass spectrometry instrument and run conditions. This data shows that adequate proteome coverage can be achieved with as little as 4 biological replicates, but for full proteome coverage 6 biological replicates is more useful.
To date, over a thousand sites of endogenous protein s-nitrosylation have been found across different organ systems including brain, lung, liver, thymus, and kidney (Doulias et al., 2013). In addition, using the sample processing alterations, over 883 endogenous sites of glutathionylation and 585 sites of s-acylation have been identified in mouse liver (Gould et al., 2015). Provided that proper sample handling techniques are conducted and familiarity with the methods, the various thiol modifications should be readily detectable under normal physiological conditions.
Time Considerations
In general the time that it will take to complete this protocol will vary based on the analysis to be conducted as well as the experience with the experimental protocol. This protocol, by all measures, is long even for experienced investigators. Conservatively, it is advisable to plan to be performing the protocol for the majority of a normal working day.
Most buffers can be premade and stored on the benchtop. For buffers that contain critical reagents such as SDS, Triton X-100, Urea, NEM, or MMTS, the base buffer can be made ahead of time and then the additional reagents added when needed. Assuming advanced preparations are made, generating the tissue homogenate, centrifugation, and generating the negative controls/nitrosothiol photolysis will take roughly 30-45 min. Followed by a precipitation step which takes 40 min (30 min incubation + 10 min centrifugation). The blocking step will take another 30 min to 1 hour followed by an additional precipitation step. The alternative protocols for glutathionylation and S-acylation will add an additional 1-1.5 h of time to the sample processing. If begun in the morning, the samples can be loaded onto the columns usually by midday or slightly after if performing the glutathionylation or S-acylation protocol. The incubation time on the column can be lengthened if needed, but should not exceed several hours. The column wash times are typically variable and require more hands on attention to keep the columns filled. The time for completion will be dependent on the resin bed volume and flow rate, but typical time to completion is 1 hour. If processing the samples for protein capture, the method can be complete by mid to late afternoon. Once the protein is eluted from the columns, it can be stored frozen until ready to concentrate. If preparing peptides for site analysis via MS, an additional overnight incubation plus 1-2 hours of additional wash steps is needed.
With experience an investigator can usually processes six samples at once. It is inadvisable to attempt more, as the multiple resuspension steps can take a significant amount of time if difficulty is encountered getting the protein back into suspension. Whenever conducting this method, it is always advisable to run at least one, but preferably matched, negative controls in parallel with each batch of samples processed. Overall, a successful analysis will largely depend on the familiarity with the protocol and should increase with experience conducting the protocol.
Significance Statement
Arising as a result of the wide reactivity of thiols, an array of covalent modifications with biological significance are formed on cysteine residues. However, the general instability and low abundance, at normal physiological levels, of most cysteine modifications makes direct detection of the modifications difficult. Therefore, enrichment strategies are employed that capture or selectively reduce the modification of interest. When coupled with downstream analysis tools such as mass spectrometry or western blotting, accurate characterization of the endogenous modifications is possible at the site and protein level in a targeted or proteome wide scale.
Acknowledgments
The authors would like to thank Protein Core at The Children's Hospital of Philadelphia Research Institute, particularly Dr. Steve Seeholzer, Lynn Spruce and Hua Ding for help with sample analysis. The authors would also like to thank Harry Ischiropoulos for support during the development of these methods. This work was supported in part by NIH grant HL54926.
References
- Albertolle M, Phan TTN, Pozzi A, Guengerich FP. Sulfenylation of Human Liver and Kidney Microsomal Cytochromes P450 and Other Drug Metabolizing Enzymes as a Response to Redox Alteration. Molecular and Cellular Proteomics. 2018 doi: 10.1074/mcp.RA117.000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Fernández JM. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell. 2014;156(6):1235–1246. doi: 10.1016/j.cell.2014.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner GR, Wagner BA, Rodgers VG. Quantitative redox biology: an approach to understand the role of reactive species in defining the cellular redox environment. Cell Biochemistry and Biophysics. 2013;67(2):477–483. doi: 10.1007/s12013-011-9320-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Wang Y, Rafikov R, Haigh S, Zhi WB, Kumar S, Fulton DJR. RhoA S-nitrosylation as a regulatory mechanism influencing endothelial barrier function in response to G+-bacterial toxins. Biochemical Pharmacology. 2017;127:34–45. doi: 10.1016/j.bcp.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniotti JL, Pedro MP, Valdez Taubas J. The role of S-acylation in protein trafficking. Traffic. 2017;18(11):699–710. doi: 10.1111/tra.12510. [DOI] [PubMed] [Google Scholar]
- Doulias P-T, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(39):16958–16963. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulias PT, Raju K, Greene JL, Tenopoulou M, Ischiropoulos H. Mass spectrometry-based identification of S-nitrosocysteine in vivo using organic mercury assisted enrichment. Methods. 2013;62(2):165–170. doi: 10.1016/j.ymeth.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drisdel RC, Alexander JK, Sayeed A, Green WN. Assays of protein palmitoylation. Methods. 2006;40(2):127–134. doi: 10.1016/j.ymeth.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends in Molecular Medicine. 2009;15(9):391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustarini D, Dalle-Donne I, Colombo R, Milzani A, Rossi R. Is ascorbate able to reduce disulfide bridges? A cautionary note. Nitric Oxide. 2008;19(3):252–258. doi: 10.1016/j.niox.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. Journal of Biological Chemistry. 2013;288(37):26473–26479. doi: 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould NS, Evans P, Martínez-Acedo P, Marino SM, Gladyshev VN, Carroll KS, Ischiropoulos H. Site-Specific Proteomic Mapping Identifies Selectively Modified Regulatory Cysteine Residues in Functionally Distinct Protein Networks. Chemistry & Biology. 2015;22(7):965–975. doi: 10.1016/j.chembiol.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groitl B, Jakob U. Thiol-based redox switches. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2014;1844(8):1335–1343. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamnell-Pamment Y, Lind C, Palmberg C, Bergman T, Cotgreave IA. Determination of site-specificity of S-glutathionylated cellular proteins. Biochemical and Biophysical Research Communications. 2005;332(2):362–369. doi: 10.1016/j.bbrc.2005.04.130. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Science’s STKE. 2001;2001(86):pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(24):8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann C, Leichert LI. Quantitative redox proteomics: the NOxICAT method. Methods in Molecular Biology. 2012;893:387–403. doi: 10.1007/978-1-61779-885-6_24. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA. ‘SNO’-Storms Compromise Protein Activity and Mitochondrial Metabolism in Neurodegenerative Disorders. Trends in Endocrinology and Metabolism. 2017;28(12):879–892. doi: 10.1016/j.tem.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nature Chemical Biology. 2012;8(1):57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju K, Doulias PT, Evans P, Krizman EN, Jackson JG, Horyn O, Ischiropoulos H. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci Signal. 2015;8(384):ra68. doi: 10.1126/scisignal.aaa4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saville B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst. 1958;83(993):670–672. doi: 10.1039/AN9588300670. [DOI] [Google Scholar]
- Thamsen M, Jakob U. The redoxome: Proteomic analysis of cellular redox networks. Current Opinion in Chemical Biology. 2011;15(1):113–119. doi: 10.1016/j.cbpa.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ye ZW, Chen W, Manevich Y, Mehrotra S, Ball LE, Townsend DM. S-Glutathionylation of estrogen receptor alpha affects dendritic cell function. Journal of Biological Chemistry. 2018 doi: 10.1074/jbc.M117.814327. [DOI] [PMC free article] [PubMed] [Google Scholar]