

Abstract
The toll-like receptor (TLR) signaling pathway plays an important role in the innate immune responses and antigen-specific acquired immunity. Aberrant activation of the TLR pathway has a significant impact on carcinogenesis or tumor progression. Therefore, we hypothesize that genetic variants in the TLR signaling pathway genes are associated with overall survival (OS) of patients with non-small cell lung cancer (NSCLC). To test this hypothesis, we first performed Cox proportional hazards regression analysis to evaluate associations between genetic variants of 165 TLR signaling pathway genes and NSCLC OS using the genome-wide association study (GWAS) dataset from the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (PLCO). The results were further validated by the Harvard Lung Cancer Susceptibility GWAS dataset. Specifically, we identified IRAK2 rs779901 C>T as a predictor of NSCLC OS, with a variant-allele (T) attributed hazards ratio (HR) of 0.78 [95% confidence interval (CI)=0.67-0.91, P=0.001] in the PLCO dataset, 0.84 (0.72-0.98, 0.031) in the Harvard dataset, and 0.81 (0.73-0.90, 1.08×10−4) in the meta-analysis of these two GWAS datasets. In addition, the T allele was significantly associated with an increased mRNA expression level of IRAK2. Our findings suggest that IRAK2 rs779901 C>T may be a promising prognostic biomarker for NSCLC OS.
Keywords: Non-small cell lung cancer (NSCLC), genome-wide association study (GWAS), single-nucleotide polymorphism (SNP), toll-like receptor (TLR), overall survival (OS)
Introduction
Lung cancer is the leading cause of cancer-related death worldwide1, with a 5-year survival rate of 17.7% in the United States, according to the data from the Surveillance, Epidemiology, and End Results (SEER) program between 2006 and 2012. Clinical characteristics, such as age, sex, smoking status, stage, histology and treatment options, are all recognized as the major factors to influence lung cancer survival2. Besides, genetic variants in critical genes can also play an important role in determining the prognosis of lung cancer.
Genome-wide association study (GWAS) is a powerful approach to identify novel single-nucleotide polymorphisms (SNPs) that are associated with risk and prognosis of many complex diseases, including lung cancer. For example, Sato and associates3 found that four SNPs (i.e. rs1656402, rs1209950, rs10074374 and rs2063681) were associated with prognosis of patients with advanced non-small cell lung cancer (NSCLC), who received carboplatin and paclitaxel as the first-line chemotherapy, in a GWAS study. Chang and coworkers identified that three SNPs (i.e. rs576732, rs476184 and rs1801260) at 4q12 were associated with progression-free survival among lung adenocarcinoma patients treated with EGFR-TKIs as the first-line therapy4. Cao and colleagues reported that SNPs at 21q22.3 led to the platinum-induced hepatotoxicity of NSCLC patients in a GWAS study5. However, most of the top SNPs identified by GWASs are lack of functional annotations. In addition, GWASs always focus on the top or most-significant SNPs/genes, paying little attention to the rest, which may have missed the SNPs that confer a true effect but not rank among the top significant ones.
Recently, the biological pathway-based approach, as a hypothesis-driven method, has been mostly used in the re-analysis of published GWAS datasets to test the cumulative effect of SNPs across multiple genes in the same pathway. This kind of pathway-based approach may improve the power to detect statistically significant associations, because much fewer SNPs in candidate genes of a significant biological pathway were included in the analysis. By using this approach, several novel and biologically functional variants have been reported to be associated with lung cancer survival6, 7. For example, Xu et al. found that five functional SNPs (ADAM12 rs10794069, DTX1 rs1732793, E2F3 rs3806116, TLE1 rs199731120 and rs35970494) in the Notch pathway were associated with NSCLC survival8 . Kong et al. reported that the SNP rs3782130 in the vitamin D pathway was associated with lung cancer survival by influencing the corresponding gene expression9. Tang et al. identified that SNPs in the PI3K/AKT pathway predicted severe radiation pneumonitis in lung cancer patients10.
Toll-like receptors (TLRs) are single, membrane-spanning and non-catalytic proteins that expressed on the surface of sentinel cells, such as macrophages and dendritic cells, which play an important role in the innate immune responses and antigen-specific acquired immunity. Specifically, the TLR signaling pathway consists of two sub-pathways: a MyD88-dependent pathway and a MyD88-independent pathway. Increasing evidence suggests that TLRs are important regulators of tumor biology11–13, and modulation of the TLR signaling has either anti- or pro-tumor effects on carcinogenesis or tumor progression, depending on TLR, cancer subtype and immune cells infiltrating the tumor12. For example, Grimmig et al. reported that the TLR signaling pathway promoted cell proliferation of pancreatic cancer, emphasizing a particular role of TLR2, TLR-4, and TLR-9 in the process14. Likewise, TLR3 was found to stimulate cancer cell survival, proliferation and progression in cancers of the pharynx15, breasts16 and head and neck17. For lung cancer, the TLR pathway may play a key role in tumorigenesis and progression, especially for NSCLC18–20. Therefore, some genetic variants in genes of this pathway may have a predictive value as a clinically potential biomarker for outcomes of NSCLC.
To date, there are no reported studies using large-scale GWAS datasets to investigate the role of genetic variants in the TLR signaling pathway genes in NSCLC survival. Therefore, we hypothesize that genetic variants in the TLR signaling pathway genes are associated with survival of NSCLC patients and tested this hypothesis by using the publically available GWAS dataset from the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (PLCO) as the discovery dataset and the Harvard GWAS dataset as the validation dataset.
Materials and methods
Study populations
The discovery dataset includes 1,185 NSCLC patients from the PLCO Trial, which is a randomized controlled study conducted by the National Cancer Institute (NCI)21. The PLCO trial enrolled 154,901 participants aged 55-74 from ten centers across the United States between 1993 and 200122, and all the participants were randomized to either the screening arm which consisted of initial chest x-ray followed by annual chest x-rays or the control arm with the standard care, with a followed-up for at least 13 years after enrollment23, 24. The information in the PLCO database included demographics, family history of cancer, smoking status, medical history and demographic characteristics of each person. Genomic DNA extracted from the blood samples was genotyped with Illumina HumanHap240Sv1.0, HumanHap300v1.1 and HumanHap550v3.0 (dbGaP accession: phs000093.v2.p2 and phs000336.v1.p1)25, 26. To expand the genotyping data, imputation was performed with IMPUTE2 according to the CEU data from the 1000 Genomes Project (phase 1 release V3). Among all the participants, 1,185 Caucasian NSCLC patients with complete individual information about age, sex, smoking status, clinical stage, histology, treatment options, follow-up information and genotype data were available for survival analysis. The NSCLC overall survival (OS) was considered as the major endpoint in the analysis. The follow-up time was defined from the diagnosis of NSCLC to the last follow-up or the time of death. The study protocol was reviewed and approved by the institutional review board of NCI and a written informed consent was obtained from each participant.
The validation dataset includes 984 NSCLC patients from the GWAS dataset of the Harvard Lung Cancer Susceptibility Study. The blood sample of each patient was obtained within 1-4 weeks of the diagnosis. Genomic DNA extracted from the blood samples was genotyped with Illumina Humanhap610-Quad arrays, and imputation was performed by using MaCH1.0 based on the 1000 Genomes Project. Details of the patients from the Harvard study have also been described elsewhere27.
Gene and SNP selection
The TLR signaling pathway genes were identified from the Molecular Signatures Database (MsigDB), which is a collection of annotated gene sets that can be analyzed by the gene set enrichment analysis (GSEA) software (http://software.broadinstitute.org/gsea/). SNPs within these genes and their ± 2 kb flanking regions were selected as the following quality control criteria: (1) genotyping rate ≥ 95%, (2) minor allelic frequency (MAF) ≥ 0.05, and (3) Hardy-Weinberg equilibrium (HWE) P value ≥ 1×10−5. Function prediction online tools including SNPinfo and RegulomeDB were applied to identify potentially representative functional SNPs. Specifically, SNPinfo28 was used for functional prediction of protein structure, gene regulation, splicing, and microRNA binding, and RegulomeDB29 was used to identify the correlation between a SNP and expression of the corresponding gene.
Statistical analysis
In the PLCO dataset, Cox proportional hazards regression analysis (under an additive genetic model) was performed with adjustments for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, and surgery as well as the first four principal components of the population structures obtained from the GWAS dataset. We estimated associations between SNPs in the TLR signaling pathway genes and NSCLC OS by calculating hazards ratio (HR) and its 95% confidence interval (CI) with the GenABEL package of R software. For multiple testing correction, the false-positive report probability (FPRP) approach with a cut-off value of 0.20 was used to reduce the probability of false positive findings30, which was used under the consideration that the majority of the SNPs were derived from the imputation and thus have a high degree of linkage disequilibrium (LD); therefore, these SNPs are not independent from each other, an assumption used in the Bonferroni correction31. We assigned a prior probability of 0.01 to detect an HR of 1.5 for an association with variant genotypes or minor alleles of the SNPs with P ≤ 0.05. In the Harvard dataset, Cox regression analysis with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery and the first three principal components was performed to validate the findings from the PLCO dataset. Finally, a meta-analysis was performed to combine the results of PLCO and Harvard studies by using PLINK 1.07, for which a fixed-effects model was applied, when no heterogeneity was found between two studies (Q-test P-value > 0.10 and I2 < 50.0%); otherwise, a random-effects model was used. In the stratified analysis, the heterogeneity test of associations between subgroups of each clinical characteristic was performed by using the Chi-square-based Q-test, and P < 0.05 was considered statistically significant for differences between the subgroups of each clinical characteristic. Besides, a pairwise LD was constructed by using the data from the 1000 Genomes Project of 373 European individuals. The expression quantitative trait loci (eQTL) analysis32 was performed to evaluate correlations between SNPs and mRNA expression levels of their genes by using sequencing data from lymphoblastoid cells derived from the same 373 individuals of European descent in the 1000 Genomes Project. Linear regression analysis was performed to analyze these correlations by using PLINK 1.07. Haploview v4.233 was used to construct the Manhattan plot, and LocusZoom34 was employed to produce the regional association plots. All statistical analyses were performed with SAS software (version 9.4; SAS Institute, Cary, NC, USA), if not specified otherwise.
Results
Gene and SNP extraction
We selected 165 TLR signaling pathway genes from the MsigDB website (http://software.broadinstitute.org/gsea/) (Supporting Information Table 1) and performed imputation for SNPs within 500 kb up- and down-streams of these genes with IMPUTE2 according to the 1000 Genomes Project CEU data (phase 1 release V3). After imputation, we extracted the data of SNPs within 2 kb up- and down-streams of each gene for further analysis. As a result, 1,384 genotyped SNPs and 13,047 imputed SNPs were extracted after quality control of information matric > 0.9, MAF ≥ 0.05, and HWE P value ≥ 1×10−5. In total, 14,431 SNPs were included in further analysis.
Associations between SNPs in the TLR signaling pathway genes and NSCLC OS in the PLCO dataset
As shown in the work flowchart (Figure 1), we first used Cox regression analysis to evaluate associations between 14,431 SNPs of TLR signaling pathway genes and NSCLC OS. The Cox models were performed with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery and first four principal components that were imbalance between cases and controls (Supporting Information Table 2). As a result, a total of 1,296 SNPs were significantly associated with NSCLC OS with P ≤ 0.05 in an additive genetic model (Supporting Information Figure 1). Among these SNPs, only the top 68 with FPRP ≤ 0.200 were selected for further validation.
Figure 1.
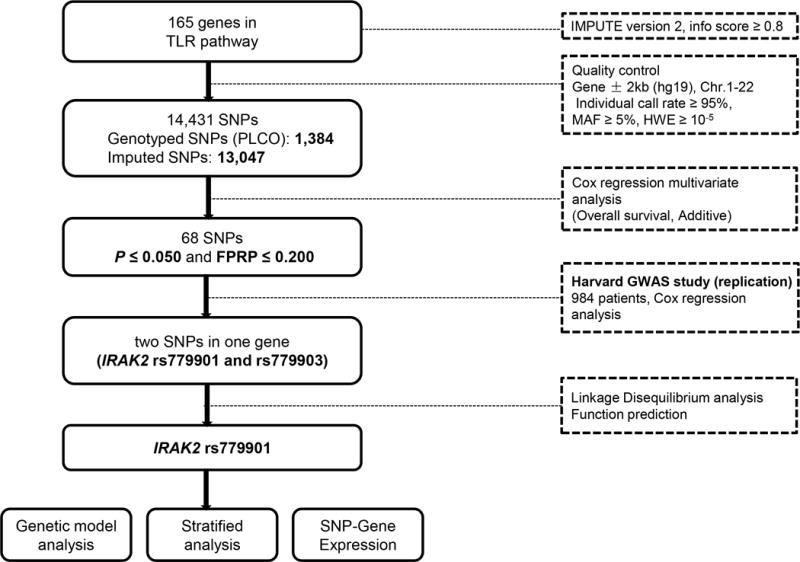
Research workflow chart. SNP, single nucleotide polymorphisms; FPRP, false-positive report probability; MAF, minor allelic frequency; HWE, Hardy-Weinberg equilibrium.
Validation analysis with the Harvard dataset
The top 68 SNPs initially identified from the PLCO trial were further validated by the Harvard GWAS dataset. Two SNPs (i.e., rs779901 and rs779903) in the intron region of IRAK2 remained significantly associated with NSCLC OS with P ≤ 0.05 in an additive genetic model after Cox regression analysis with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery and principal components. The details of associations between 68 SNPs and NSCLC OS in the two independent studies are described in Supporting Information Table 3.
Potentially functional and representative SNP selection
The information of the two validated SNPs is described in Table 1 and Supporting Information Table 4. We performed the LD analysis of these two SNPs and found that they were in a complete LD (r2=1.0) with each other (Supporting Information Figure 2). We then used two function prediction online tools (i.e., SNPinfo and RegulomeDB) to search for their potential functional relevance but found nothing available for rs779903. We only found that rs779901 had a score of 4 in the RegulomeDB (Table 1) and thus further explored its potential function by using the ENCODE project data. As shown in Supporting Information Figure 3, rs779901 is located in the intron region of IRAK2, which shows some considerable H3K4Me1 enrichment. To visualize the locations of the SNPs in IRAK2, we showed all genotyped and imputed SNPs in a regional association plot with an expansion of ±500 kb in the flanks of the gene region, in which rs779901 was marked in purple, shown on the top of the plot (Supporting Information Figure 4). As a result, we selected rs779901 as the representative SNP for further analysis. In addition, we also performed an independent test for the SNP identified in the present study and previously published studies that used the PLCO dataset, and we found that SNP rs779901 remained as an independent predictor for OS in this dataset, not influenced by other SNPs previously published (Supporting Information Table 5).
Table 1.
Summary of the two validated SNPsin the toll-like receptor signaling pathway geneIRAK2
| SNP | Allelea | Gene | PLCO (n=1185) | Harvard (n=984) | Meta-analysis | Regulo meDB | SNP info | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| EAF | HR (95%CI)b | Pb | EAF | HR (95%CI)c | Pc | Phetd | I2 | HR (95%CI)e | Pe | |||||
| rs779901 | C/T | IRAK2 | 0.14 | 0.78 (0.67-0.91) | 0.001 | 0.15 | 0.84 (0.72-0.98) | 0.031 | 0.47 | 0 | 0.81 (0.73-0.90) | 1.08×10−4 | 4 | no |
| rs779903 | G/A | IRAK2 | 0.14 | 0.78 (0.67-0.91) | 0.001 | 0.15 | 0.84 (0.72-0.98) | 0.032 | 0.47 | 0 | 0.81 (0.73-0.90) | 1.09×10−4 | no data | no |
Abbreviations: SNP, single-nucleotide polymorphism; TLR, toll-like receptor; PLCO, Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial; EAF, effect allele frequency; HR, hazards ratio; CI, confidence interval.
Reference allele/effect allele;
Adjusted for age, sex, stage, histology, smoking status, chemotherapy, radiotherapy, surgery, PC1, PC2, PC3,PC4 in the PLCO dataset;
Adjusted for age, sex, stage, histology, smoking status, chemotherapy, radiotherapy, surgery, PC1, PC2, PC3 in the Harvard dataset;
Phet: P value for heterogeneity by Cochrane’s Q test;
Meta-analysis in the fix-effects model.
All the tests of the proportional hazards assumption for the validated SNPs in PLCO and Harvard studies were not statistically significant (P> 0.05).
Survival analysis of the representative SNP and NSCLC OS
As shown in Table 1, the rs779901 T variant allele was associated with a decreased death risk of NSCLC with a variant-allele attributed HR of 0.78 (95% CI = 0.67-0.91, P = 0.001) in the PLCO trial, 0.84 (95% CI = 0.72-0.98, P = 0.031) in the Harvard study, and 0.81 (95% CI = 0.73-0.90, P = 1.08×10−4) for the meta-analysis of the two studies. The results of univariate and multivariate Cox regression analyses with other genetic models (codominant/dominant/recessive) for the representative SNP rs779901 in the PLCO trial were presented in Table 2, which shows that CT+TT genotypes were associated with a decreased death risk of NSCLC (HR = 0.79, 95% CI= 0.67-0.93, and P= 0.005), compared with the CC genotype. We then conducted stratified analysis to identify any modification by clinical characteristics of age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy and surgery in the PLCO dataset. We found that there was heterogeneity between patients with age >71 and ≤ 71 years (P = 0.001). The protective T variant genotypes (CT+TT) were significantly associated with a decreased death risk only among those patients with age >71 years. However, such survival advantage in older populations of cancer patients is also likely due to survival bias in these patients. Results of interaction analysis showed that the rs779901 CT+TT genotypes interacted with age and radiotherapy (Pint = 0.008 and 0.048, respectively) (Table 3).
Table 2.
Associations between the representative SNP in IRAK2 and overall survival of NSCLC patients in PLCO database
| Genotype | Frequency | Univariate analysis | Multivariate analysisa | |||
|---|---|---|---|---|---|---|
|
| ||||||
| All | Death (%) | HR (95%CI) | P | HR (95%CI) | P | |
| IRAK2 | ||||||
| rs779901 C>T | ||||||
| CC | 873 | 595 (68.2) | 1.00 | 1.00 | ||
| CT | 286 | 188 (65.7) | 0.93 (0.79-1.10) | 0.379 | 0.82 (0.70-0.97) | 0.023 |
| TT | 24 | 14 (58.3) | 0.76 (0.45-1.30) | 0.315 | 0.46 (0.26-0.82) | 0.009 |
| CT+TT | 310 | 202 (65.2) | 0.92 (0.78-1.07) | 0.276 | 0.79 (0.67-0.93) | 0.005 |
| Trend | 0.91 (0.79-1.05) | 0.214 | 0.78 (0.68-0.91) | 0.001 | ||
| CC+CT | 1159 | 783 (67.6) | 1.00 | 1.00 | ||
| TT | 24 | 14 (58.3) | 0.78 (0.46-1.32) | 0.347 | 0.49 (0.27-0.87) | 0.015 |
Abbreviations: SNP, single-nucleotide polymorphisms; TLR, toll-like receptor; NSCLC, non-small cell lung cancer; PLCO, Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial; HR, hazards ratio; CI, confidence interval.
Multivariate Cox regression analyses with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery, PC1, PC2, PC3, and PC4.
Table 3.
Stratified analysis of the protective genotypes and NSCLC OS in the PLCO dataset
| Characteristics | CC | CT+TT | Univariate analysis | Multivariate analysisa | Phetb | Interactionc | ||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| All | Death (%) | All | Death (%) | HR (95%CI) | P | HR (95%CI) | P | |||
| Age (years) | 0.001 | 0.008 | ||||||||
| ≤ 71 | 481 | 303 (63.0) | 153 | 96 (62.8) | 1.02 (0.81-1.28) | 0.873 | 1.10 (0.87-1.40) | 0.415 | ||
| > 71 | 392 | 292 (74.5) | 157 | 106 (67.5) | 0.74 (0.59-0.93) | 0.008 | 0.63 (0.50-0.79) | 9.05×10−5 | ||
| Sex | 0.074 | 0.078 | ||||||||
| Male | 509 | 381 (74.9) | 188 | 126 (67.0) | 0.79 (0.64-0.96) | 0.019 | 0.71 (0.58-0.88) | 0.001 | ||
| Female | 364 | 214 (58.8) | 122 | 76 (62.3) | 1.13 (0.87-1.46) | 0.374 | 0.98 (0.74-1.31) | 0.907 | ||
| Smoking status | 0.413 | 0.085 | ||||||||
| Never | 95 | 50 (52.6) | 20 | 13 (65.0) | 1.38 (0.75-2.55) | 0.306 | 1.24 (0.61-2.51) | 0.551 | ||
| Former | 461 | 338 (73.3) | 184 | 124 (67.4) | 0.87 (0.71-1.07) | 0.182 | 0.76 (0.62-0.94) | 0.013 | ||
| Current | 317 | 207 (65.3) | 106 | 65 (61.3) | 0.88 (0.67-1.17) | 0.379 | 0.83 (0.62-1.12) | 0.223 | ||
| Histology | 0.068 | 0.548 | ||||||||
| Adenocarcinoma | 428 | 256 (59.8) | 148 | 91 (61.5) | 1.08 (0.85-1.37) | 0.553 | 0.91 (0.71-1.17) | 0.454 | ||
| Squamous cell carcinoma | 206 | 145 (70.4) | 79 | 47 (59.5) | 0.70 (0.50-0.97) | 0.034 | 0.56 (0.39-0.78) | 0.001 | ||
| Others | 239 | 194 (81.2) | 83 | 64 (77.1) | 0.87 (0.65-1.15) | 0.318 | 0.87 (0.64-1.18) | 0.358 | ||
| Tumor stage | 0.514 | 0.991 | ||||||||
| I-IIIA | 472 | 232 (49.2) | 181 | 82 (45.3) | 0.90 (0.70-1.15) | 0.397 | 0.75 (0.58-0.98) | 0.032 | ||
| IIIB-IV | 400 | 363 (90.8) | 128 | 119 (93.0) | 1.04 (0.85-1.28) | 0.689 | 0.84 (0.68-1.05) | 0.123 | ||
| Chemotherapy | 0.283 | 0.209 | ||||||||
| No | 460 | 268 (58.3) | 177 | 98 (55.4) | 0.97 (0.77-1.22) | 0.766 | 0.68 (0.53-0.87) | 0.002 | ||
| Yes | 405 | 319 (78.8) | 133 | 104 (78.2) | 0.88 (0.70-1.10) | 0.246 | 0.82 (0.65-1.04) | 0.099 | ||
| Radiotherapy | 0.276 | 0.048 | ||||||||
| No | 546 | 322 (59.0) | 214 | 127 (59.4) | 1.00 (0.81-1.23) | 1.000 | 0.86 (0.70-1.07) | 0.179 | ||
| Yes | 319 | 265 (83.1) | 96 | 75 (78.1) | 0.87 (0.67-1.13) | 0.292 | 0.71 (0.54-0.93) | 0.013 | ||
| Surgery | 0.565 | 0.220 | ||||||||
| No | 470 | 419 (89.2) | 167 | 147 (88.0) | 0.91 (0.76-1.10) | 0.343 | 0.77 (0.63-0.93) | 0.008 | ||
| Yes | 395 | 168 (42.5) | 143 | 55 (38.5) | 0.88 (0.65-1.19) | 0.395 | 0.86 (0.62-1.18) | 0.335 | ||
Abbreviations: NSCLC, non-small cell lung cancer; OS, overall survival; PLCO, Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial; HR, hazards ratio; CI, confidence interval;
Adjusted for age, sex, stage, histology, smoking status, chemotherapy, radiotherapy, surgery, PC1, PC2, PC3,PC4;
Phet: P value for heterogeneity by Cochrane’s Q test;
The interaction between SNP rs779901 and each of other covariates.
eQTL analysis
We performed the eQTL analysis to identify the correlation between the representative SNP rs779901and IRAK2 mRNA expression by using data from the 1000 Genomes Project of 373 European individuals. As shown in Figure 2, we found that the rs779901 T allele was associated with an increased expression level of IRAK2 mRNA (P = 0.004), compared with the C allele in the additive model. We also observed that CT+TT genotypes were significantly associated with an increased expression level of IRAK2 mRNA, compared with the CC genotype in the dominant model (P = 0.007).
Figure 2.
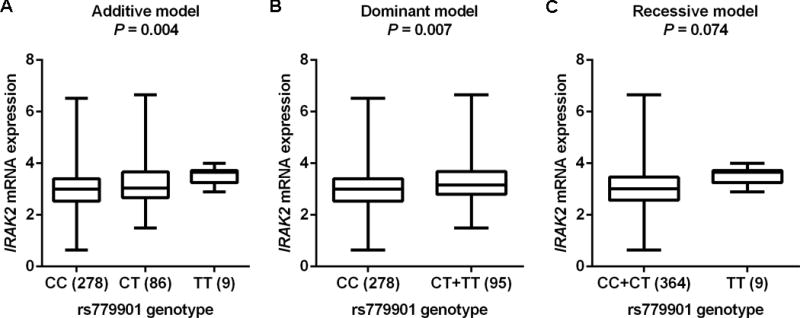
eQTL analysis of the representative SNP rs779901 and mRNA expression levels of IRAK2. All the data were from 373 individuals of European descent from the 1000 Genomes Project. A. Additive model; B. Dominant model; C. Recessive model.
Comparison of IRAK2 mRNA expression between normal and NSCLC tissues
Finally, we looked for the evidence whether IRAK2 would possibly be an oncogene or a suppressor gene by assessing its expression levels in the target tissues. The data used for the comparison of IRAK2 mRNA expression between normal and NSCLC tissues were extracted from the Oncomine Platform (https://www.oncomine.org/resource/login.html). The dataset of Hou Lung Statistics demonstrated that the expression level of IRAK2 mRNA was lower in cancer tissues than that in normal lung tissues for both large cell lung carcinoma and lung adenocarcinoma (Supporting Information Figure 5).
Discussion
In the present study, we investigated associations between 14,431 genetic variants of 165 genes in the TLR signaling pathway and NSCLC OS by a two-phase analysis of previously published GWAS datasets. We identified IRAK2 rs779901 as a predictor of NSCLC OS. Specifically, the rs779901 T allele, as a protective allele, was associated with a favorite OS in NSCLC patients. In addition, the rs779901 T variant genotypes were associated with an increased expression level of IRAK2 mRNA, which provides further support for the biological plausibility of our findings.
TLRs, as trans-membrane proteins, are expressed on the surface of immune cells and play a protective role against pathogens. Previous studies have reported that aberrant expression of TLRs and activation of the TLR signaling pathway was significantly associated with carcinogenesis or tumor progression12, 35. The TLR signaling pathway consists of various molecules, such as MyD88, IRAKs, TIRAP, TRIF and TRAM, which play an important role in infectious and non-infectious diseases and may be promising therapeutic targets for cancers as well35. IRAKs, i.e., interleukin-1 receptor-associated kinases, are the key mediators of the MyD88-dependent TLR signaling pathway. They first bind to the adaptor molecules, such as MyD88, TIRAP, TRIF and TRAM, and then promote the activation of downstream molecules, such as TRAF6. In humans, there are four members of the IRAK genes, including IRAK1, IRAK2, IRAK3 and IRAK4, which encode proteins that have different biological functions36. All of the IRAK proteins share a similar N-terminal domain important for the MyD88 interaction. However, only IRAK1, IRAK2 and IRAK3 contain a C-terminal domain for the activation of TRAF6. IRAK4 is the most upstream kinase of the IRAK family members, and it recruits either IRAK1 or IRAK2 by trans-autophosphoryIation of serine and threonine residues. IRAK1 or IRAK2, in turn, recruits TRAF6 and leads to the activation of the nuclear factor κB (NF-κB) and mitogen-activated protein kinases37. Among the four IRAK family members, IRAK1 and IRAK4 exhibit kinase activities, while IRAK2 and IRAK3 contain an inactive pseudokinase domain. IRAK1 and IRAK4 have been reported to be associated with cancer development38–40. Especially for IRAK4, the loss of its kinase function is associated with an increased susceptibility to various pathogens, while its over-activation leads to autoimmune diseases, such as cancers37. Therefore, the development of IRAK4 inhibitors will be promising for target therapies that improve the prognosis of cancer patients. To the best of our knowledge, little is known about the role of IRAK2.
The present study demonstrated a role of IRAK2 rs779901 in the prognosis of NSCLC for the first time, suggesting a potential role of IRAK2 in the progression of NSCLC. It seems that IRAK2 may play a critical role in the TLR signaling pathway through a multimeric helical MyD88-IRAK4-IRAK2 complex, which induces TRAF6 ubiquitination and leads to the activation of downstream signaling38–40. As mentioned above, there is no C-terminal domain in IRAK4, while IRAK2 provides C-terminal domain by combining with IRAK4 and then promotes the downstream of the TLR signaling pathway38–40. According to the data from the Oncomine Platform, expression levels of IRAK2 mRNA were decreased in NSCLC tissues, in both large cell carcinoma and lung adenocarcinoma, although the specific mechanism remains unknown.
In the present study, we found that the rs779901 T allele was significantly associated with a favorite OS in NSCLC patients. SNP rs779901 is located in the intron region of IRAK2, where considerable levels of H3K4Me1 enrichment are accessible to transcription factors to enhance transcriptional activity. More importantly, the rs779901 T allele is correlated with an increased IRAK2 mRNA expression in a variant allele dose-response manner. Therefore, we propose that rs779901 may influence IRAK2 mRNA expression by affecting the transcriptional activity, a possible mechanism underlying the observed association with the prognosis of NSCLC, which needs to be further validated by mechanistic studies.
There are some limitations in the present study. Firstly, both of the two available GWAS datasets we used were from Caucasian populations; therefore, our findings may not be generalized to the general population. Secondly, the present study was a pathway-based analysis. We obtained the genes of the TLR signaling pathway from the canonical pathway, GO biological process and GO molecular function datasets, the three major publicly recognized datasets of GSEA/MSigDB website. It is likely that some important or unknown genes of this pathway might be excluded. Thirdly, only a few clinical characteristics were included in the present study, and other information, such as nutrition status, performance, somatic mutations and details of treatment, was not available for further analysis. Finally, we were unable to explore the biological mechanisms by which the SNPs of TLR signaling pathway genes may influence NSCLC OS, because the target NSCLC tissues from the study participants were unavailable.
In conclusion, we performed a two-phase analysis for associations of genetic variants in 165 genes in the TLR signaling pathway with NSCLC OS by using two previously published GWAS datasets. We found that IRAK2 rs779901 was a prognostic factor for OS in NSCLC patients. Additional population replications from other ethnic groups and functional validation by mechanistic studies are needed to further substantiate our findings. Once validated, our findings may add a promising prognostic biomarker to personalized management and treatment of NSCLC patients.
Supplementary Material
Supporting Information Figure 1. Manhattan plot of 14,431 SNPs of toll-like receptor signaling pathway genes in the PLCO study. The statistical values across the autosomes for associations between 14,431 SNPs and overall survival are plotted as −log10 P values. The red horizontal line indicates P = 0.05, the two validated SNPs in IRAK2 were marked red.
Supporting Information Figure 2. The linkage disequilibrium plot for the two validated SNPs in IRAK2.
Supporting Information Figure 3. ENCODE project data of the representative SNP rs779901 from the University of California Santa Cruz (UCSC, http://genome.ucsc.edu/).
Supporting Information Figure 4. Regional association plot of the representative SNP rs779901. The left-hand Y-axis shows the -log10 transformation of P-value of individual SNPs, which is plotted against the chromosomal base-pair position with an expansion of 500 kb in the flanks of the gene region. The right-hand Y-axis shows the recombination rate estimated for European populations from HapMap Data Rel 22/phase II.
Supporting Information Figure 5. The comparison of IRAK2 mRNA expression levels between normal and non-small cell lung cancer tissues from the Oncomine Platform (https://www.oncomine.org/resource/login.html).
Brief description.
The toll-like receptor signaling pathway plays an important role in the innate immune responses and antigen-specific acquired immunity. Aberrant activation of the pathway has a significant impact on carcinogenesis or tumor progression. In the present study of re-analyzing published genome-wide association study datasets, we found that IRAK2 rs779901 C>T in the TLR pathway predicted overall survival of patients with non-small cell lung cancer. This genetic variant may be a promising prognostic biomarker for these patients.
Acknowledgments
We thank all the participants of the PLCO Cancer Screening Trial. We also thank the National Cancer Institute (NCI) for providing the access to the PLCO data. The statements contained herein are solely those of the authors and do not represent or imply concurrence or endorsement by NCI. The authors would also like to acknowledge dbGaP repository for providing the cancer genotyping datasets. The accession numbers for the datasets of lung cancer are phs000336.v1.p1 and phs000093.v2.p2. A list of contributing investigators and funding agencies for those studies can be found in the Supplemental Data.
Grant support
Qingyi Wei was supported by start-up funds from Duke Cancer Institute, Duke University Medical Center, and Qingyi Wei is partly supported by the Duke Cancer Institute as part of the P30 Cancer Center Support Grant (Grant ID: NIH/NCI CA014236). Yinghui Xu was supported by Youth Foundation of The First Hospital of Jilin University (Grant ID: JDYY82017020). Yinghui Xu was also supported by Xisike Clinical Oncology Research Foundation (CSCO-Haosen) (Grant ID: Y-HS2017-062). The Harvard Lung Cancer Susceptibility Study was supported by NIH grants 5U01CA209414, CA092824, CA074386, and CA090578 to David C. Christiani.
Abbreviations
- TLR
toll-like receptor
- NSCLC
non-small cell lung cancer
- OS
overall survival
- GWAS
genome-wide association studies
- SNP
single-nucleotide polymorphisms
- HR
hazards ratio
- CI
confidence interval
- SEER
Surveillance, Epidemiology, and End Results
- PLCO
Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial
- NCI
National Cancer Institute
- MsigDB
Molecular Signatures Database
- GSEA
gene set enrichment analysis
- MAF
minor allelic frequency
- HWE
Hardy-Weinberg equilibrium
- FPRP
false-positive report probability
- LD
linkage disequilibrium
- eQTL
expression quantitative trait loci
- IRAK
interleukin-1 receptor-associated kinase
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Liu H, Ready NE, Su L, Wei Y, Christiani DC, Wei Q. Genetic variants in ABCG1 are associated with survival of nonsmall-cell lung cancer patients. Int J Cancer. 2016;138:2592–2601. doi: 10.1002/ijc.29991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sato Y, Yamamoto N, Kunitoh H, Ohe Y, Katori N, Sawada J, Sakamoto H, Saijo N, Yoshida T, Tamura T. Genome-wide association scan detected candidate polymorphisms associated with overall survival (OS) in advanced non-small cell lung cancer (NSCLC) treated with carboplatin (CBDCA) and paclitaxel (PTX) J Clin Oncol. 2009;27:8031. [Google Scholar]
- 4.Chang IS, Jiang SS, Yang JC, Su WC, Chien LH, Hsiao CF, Lee JH, Chen CY, Chen CH, Chang GC, Wang Z, Lo FY, et al. Genetic Modifiers of Progression-free Survival in Never-smoking Lung Adenocarcinoma Patients Treated with First-line TKIs. Am J Respir Crit Care Med. 2017;195:663–673. doi: 10.1164/rccm.201602-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao S, Wang C, Ma H, Yin R, Zhu M, Shen W, Dai J, Shu Y, Xu L, Hu Z, Shen H. Genome-wide Association Study on Platinum-induced Hepatotoxicity in Non-Small Cell Lung Cancer Patients. Sci Rep. 2015;5:11556. doi: 10.1038/srep11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian DC, Han Y, Byun J, Shin HR, Hung RJ, McLaughlin JR, Landi MT, Seminara D, Amos CI. A Novel Pathway-Based Approach Improves Lung Cancer Risk Prediction Using Germline Genetic Variations. Cancer Epidemiol Biomarkers Prev. 2016;25:1208–1215. doi: 10.1158/1055-9965.EPI-15-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y, Wang Y, Liu H, Kang X, Li W, Wei Q. Genetic variants of genes in the Notch signaling pathway predict overall survival of non-small cell lung cancer patients in the PLCO study. Oncotarget. 2016;7:61716–61727. doi: 10.18632/oncotarget.11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong J, Xu F, Qu J, Wang Y, Gao M, Yu H, Qian B. Genetic polymorphisms in the vitamin D pathway in relation to lung cancer risk and survival. Oncotarget. 2015;6:2573–2582. doi: 10.18632/oncotarget.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang Y, Liu B, Li J, Wu H, Yang J, Zhou X, Yi M, Li Q, Yu S, Yuan X. Genetic variants in PI3K/AKT pathway are associated with severe radiation pneumonitis in lung cancer patients treated with radiation therapy. Cancer Med. 2016;5:24–32. doi: 10.1002/cam4.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto A, Morello S, Sorrentino R. Lung cancer and Toll-like receptors. Cancer Immunol Immunother. 2011;60:1211–1220. doi: 10.1007/s00262-011-1057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dajon M, Iribarren K, Cremer I. Toll-like receptor stimulation in cancer: A pro- and anti-tumor double-edged sword. Immunobiology. 2017;222:89–100. doi: 10.1016/j.imbio.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Khan AA, Khan Z, Warnakulasuriya S. Cancer-associated toll-like receptor modulation and insinuation in infection susceptibility: association or coincidence? Ann Oncol. 2016;27:984–997. doi: 10.1093/annonc/mdw053. [DOI] [PubMed] [Google Scholar]
- 14.Grimmig T, Moench R, Kreckel J, Haack S, Rueckert F, Rehder R, Tripathi S, Ribas C, Chandraker A, Germer CT, Gasser M, Waaga-Gasser AM. Toll Like Receptor 2, 4, and 9 Signaling Promotes Autoregulative Tumor Cell Growth and VEGF/PDGF Expression in Human Pancreatic Cancer. Int J Mol Sci. 2016:17. doi: 10.3390/ijms17122060. pii: E2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matijevic Glavan T, Cipak Gasparovic A, Verillaud B, Busson P, Pavelic J. Toll-like receptor 3 stimulation triggers metabolic reprogramming in pharyngeal cancer cell line through Myc, MAPK, and HIF. Mol Carcinog. 2017;56:1214–1226. doi: 10.1002/mc.22584. [DOI] [PubMed] [Google Scholar]
- 16.Jia D, Wang L. The other face of TLR3: A driving force of breast cancer stem cells. Mol Cell Oncol. 2015;2:e981443. doi: 10.4161/23723556.2014.981443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veyrat M, Durand S, Classe M, Glavan TM, Oker N, Kapetanakis NI, Jiang X, Gelin A, Herman P, Casiraghi O, Zagzag D, Enot D, et al. Stimulation of the toll-like receptor 3 promotes metabolic reprogramming in head and neck carcinoma cells. Oncotarget. 2016;7:82580–82593. doi: 10.18632/oncotarget.12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei F, Yang F, Li J, Zheng Y, Yu W, Yang L, Ren X. Soluble Toll-like receptor 4 is a potential serum biomarker in non-small cell lung cancer. Oncotarget. 2016;7:40106–40114. doi: 10.18632/oncotarget.9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dajon M, Iribarren K, Cremer I. Dual roles of TLR7 in the lung cancer microenvironment. Oncoimmunology. 2015;4:e991615. doi: 10.4161/2162402X.2014.991615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ke X, Wu M, Lou J, Zhang S, Huang P, Sun R, Huang L, Xie E, Wang F, Gu B. Activation of Toll-like receptors signaling in non-small cell lung cancer cell line induced by tumor-associated macrophages. Chin J Cancer Res. 2015;27:181–189. doi: 10.3978/j.issn.1000-9604.2015.03.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mabie J, Riley T, Marcus PM, Black A, Rozjabek H, Yu K, Young M, Austin J, Rathmell J, Williams C, Prorok PC. Data Processing and Analytic Support in the PLCO Cancer Screening Trial. Rev Recent Clin Trials. 2015;10:233–237. doi: 10.2174/1574887110666150730122723. [DOI] [PubMed] [Google Scholar]
- 22.Gohagan JK, Prorok PC, Greenwald P, Kramer BS. The PLCO Cancer Screening Trial: Background, Goals, Organization, Operations, Results. Rev Recent Clin Trials. 2015;10:173–180. doi: 10.2174/1574887110666150730123004. [DOI] [PubMed] [Google Scholar]
- 23.Ten Haaf K, van Rosmalen J, de Koning HJ. Lung cancer detectability by test, histology, stage, and gender: estimates from the NLST and the PLCO trials. Cancer Epidemiol Biomarkers Prev. 2015;24:154–161. doi: 10.1158/1055-9965.EPI-14-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oken MM, Marcus PM, Hu P, Beck TM, Hocking W, Kvale PA, Cordes J, Riley TL, Winslow SD, Peace S, Levin DL, Prorok PC, et al. Baseline chest radiograph for lung cancer detection in the randomized Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. J Natl Cancer Inst. 2005;97:1832–1839. doi: 10.1093/jnci/dji430. [DOI] [PubMed] [Google Scholar]
- 25.Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L, Lee M, Popova N, Sharopova N, Kimura M, Feolo M. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014;42:D975–D979. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhai R, Yu X, Wei Y, Su L, Christiani DC. Smoking and smoking cessation in relation to the development of co-existing non-small cell lung cancer with chronic obstructive pulmonary disease. Int J Cancer. 2014;134:961–970. doi: 10.1002/ijc.28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009;37:W600–W605. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96:434–442. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conneely KN, Boehnke M. So many correlated tests, so little time! Rapid adjustment of P values for multiple correlated tests. Am J Hum Genet. 2007;81:1158–1168. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120362. doi: 10.1098/rstb.2012.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 34.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu J, Mohan C. Toll-like receptor signaling pathways–therapeutic opportunities. Mediators Inflamm. 2010;2010:781235. doi: 10.1155/2010/781235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhyasen GW, Starczynowski DT. IRAK signalling in cancer. Br J Cancer. 2015;112:232–237. doi: 10.1038/bjc.2014.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patra MC, Choi S. Recent Progress in the Molecular Recognition and Therapeutic Importance of Interleukin-1 Receptor-Associated Kinase 4. Molecules. 2016;21 doi: 10.3390/molecules21111529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srivastava R, Geng D, Liu Y, Zheng L, Li Z, Joseph MA, McKenna C, Bansal N, Ochoa A, Davila E. Augmentation of therapeutic responses in melanoma by inhibition of IRAK-1,-4. Cancer Res. 2012;72:6209–6216. doi: 10.1158/0008-5472.CAN-12-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Z, Younger K, Gartenhaus R, Joseph AM, Hu F, Baer MR, Brown P, Davila E. Inhibition of IRAK1/4 sensitizes T cell acute lymphoblastic leukemia to chemotherapies. J Clin Invest. 2015;125:1081–1097. doi: 10.1172/JCI75821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang D, Chen W, Xiong J, Sherrod CJ, Henry DH, Dittmer DP. Interleukin 1 receptor-associated kinase 1 (IRAK1) mutation is a common, essential driver for Kaposi sarcoma herpesvirus lymphoma. Proc Natl Acad Sci U S A. 2014;111:E4762–E4768. doi: 10.1073/pnas.1405423111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1. Manhattan plot of 14,431 SNPs of toll-like receptor signaling pathway genes in the PLCO study. The statistical values across the autosomes for associations between 14,431 SNPs and overall survival are plotted as −log10 P values. The red horizontal line indicates P = 0.05, the two validated SNPs in IRAK2 were marked red.
Supporting Information Figure 2. The linkage disequilibrium plot for the two validated SNPs in IRAK2.
Supporting Information Figure 3. ENCODE project data of the representative SNP rs779901 from the University of California Santa Cruz (UCSC, http://genome.ucsc.edu/).
Supporting Information Figure 4. Regional association plot of the representative SNP rs779901. The left-hand Y-axis shows the -log10 transformation of P-value of individual SNPs, which is plotted against the chromosomal base-pair position with an expansion of 500 kb in the flanks of the gene region. The right-hand Y-axis shows the recombination rate estimated for European populations from HapMap Data Rel 22/phase II.
Supporting Information Figure 5. The comparison of IRAK2 mRNA expression levels between normal and non-small cell lung cancer tissues from the Oncomine Platform (https://www.oncomine.org/resource/login.html).