

Abstract
Cholesterol and cholesterol-derived oxysterols are critical for embryonic development, synapse formation and function, and myelination, among other biological functions. Indeed, alterations in levels of cholesterol, sterol precursors, and oxysterols results in a variety of developmental disorders, emphasizing the importance of cholesterol homeostasis. The ability of xenobiotics to reproduce similar phenotypes by altering cholesterol homeostasis has increasingly become of interest. Therefore, the ability to quantitatively assess alterations in cholesterol homeostasis resulting from exposure to xenobiotics is of value. This unit describes methods for the quantitative assessment of altered post-squalene cholesterol biosynthesis and subsequent oxysterol formation in various sample types using ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS). Understanding alterations in cholesterol homeostasis resulting from xenobiotic exposure can provide key insight into the toxicant’s mechanism of action and resulting phenotype.
Keywords: sterols, oxysterols, cholesterol, homeostasis, liquid chromatography-mass spectrometry, xenobiotics
INTRODUCTION
Cholesterol biosynthesis is a complex process involving many intermediates but can be broadly divided into two segments: pre-squalene and post-squalene syntheses. In pre-squalene synthesis, isoprenoids formed from the mevalonate pathway undergo a series of condensation reactions that leads first to squalene and then to squalene epoxide upon epoxidation. The 3-hydroxy-3-methyl-glutaryl-CoA reductase in the mevalonate pathway is the rate-determining step of the entire cholesterol synthesis pathway. In post-squalene synthesis, cyclization of squalene epoxide leads to the first sterol in the pathway, lanosterol, which is diverted into one of two pathways that both progress through a series of dehydrogenations, reductions, and demethylations (Figure 1) (Kelley & Herman, 2001). Once formed, cholesterol is further metabolized through both enzymatic (Pikuleva, 2006) and free radical (Yin, Xu, & Porter, 2011) processes to produce a variety of oxysterols (Figure 2) (Mutemberezi, Guillemot-Legris, & Muccioli, 2016).
Figure 1.
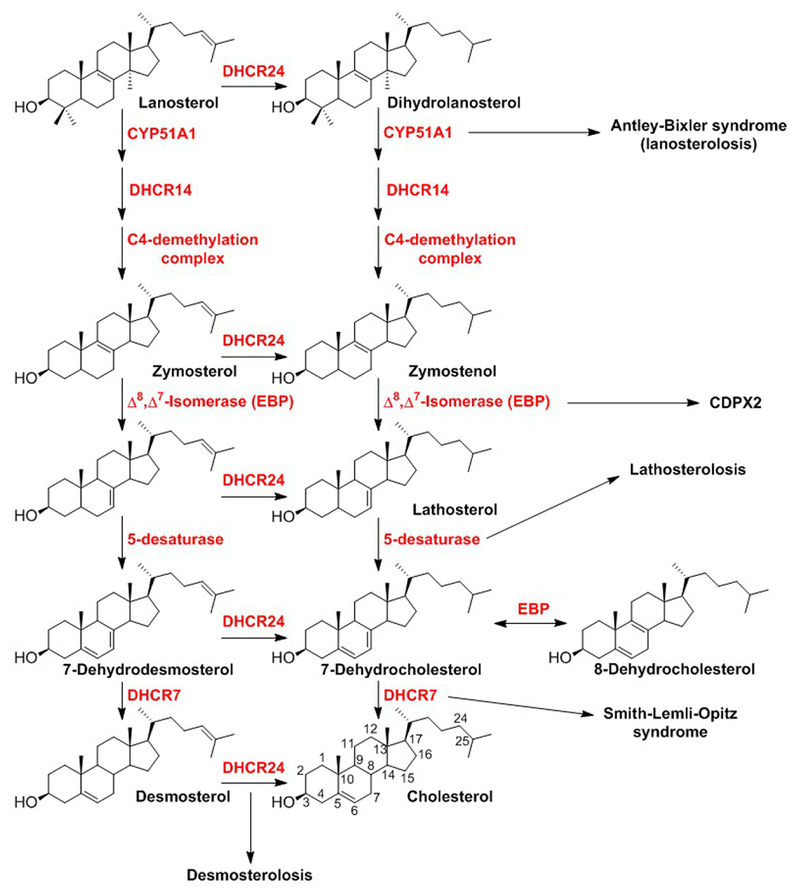
Post-squalene cholesterol biosynthetic pathway and examples of malformation disorders associated with defects at specific steps.
Figure 2.
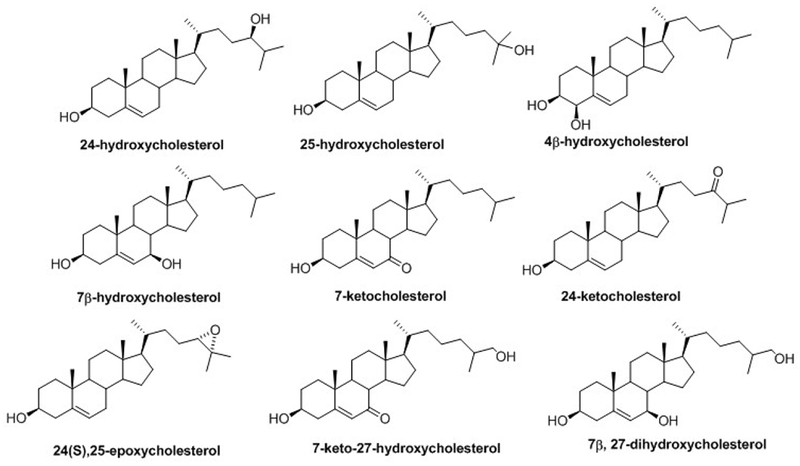
Structures of the main cholesterol-derived oxysterols.
Cholesterol plays important roles in embryonic development, synapse formation and function, and myelination. Alterations in cholesterol homeostasis, especially the deficiency of cholesterol and accumulation of its precursors, contribute to a variety of malformation disorders (F. D. Porter & Herman, 2010). The most common cholesterol biosynthesis disorder, Smith-Lemli-Opitz syndrome (SLOS), is characterized by multiple congenital malformations, developmental delay, cognitive impairment, and behavior problems. Cholesterol-derived oxysterols also have vast cellular roles as lipid mediators, targeting nuclear receptors, regulatory proteins, and cell membrane receptors (Mutemberezi et al., 2016).
Given the biological importance of the cholesterol biosynthetic pathway, the effect of xenobiotics on cholesterol homeostasis has become increasingly of interest. Alterations in cholesterol homeostasis have been shown to play a role in the developmental toxicity of ethanol and retinoic acid (Guizzetti, M. et al, 2011). Several pharmacological agents have been found to be inhibitors of various steps of cholesterol biosynthesis (Canfrán-Duque et al., 2013; de Medina et al., 2010; Hall et al., 2013; Kim et al., 2016; Korade et al., 2016). First trimester exposure to pharmacological modulators of DHCR7, the enzyme which converts 7-dehydrocholesterol (7-DHC) to cholesterol, is associated with adverse fetal outcomes (Boland & Tatonetti, 2016). Recently, we have demonstrated that a commonly used class of disinfectants, quaternary ammonium compounds, inhibit DHCR7 and alter cholesterol homeostasis in neuronal cells (Herron et al., 2016).
This protocol describes methods for the quantitative assessment of altered post-squalene cholesterol homeostasis and subsequent oxysterol formation in various sample types using ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) and isotopically labeled internal standards.
BASIC PROTOCOL 1: LIPID EXTRACTION
To assess changes in cholesterol homeostasis, lipids must be isolated prior to analysis. The Folch extraction method is commonly used to extract both simple and complex lipids from virtually any type of biological sample (Folch, Lees, & Sloane Stanley, 1957). With this method, a chloroform-methanol extracting solvent is used in the presence of isotopically labelled sterol and oxysterol internal standards to disrupt lipoproteins and cell membranes and to solubilize lipids from either cells grown in culture or harvested tissues. The presence of internal standards accounts for any loss of endogenous sterols or oxysterols during the extraction process and enables quantitation following UHPLC-MS/MS analysis. Finally, the addition of an aqueous sodium chloride solution yields a layered mixture with a lower organic layer containing the lipids. This lower organic layer is collected, dried, reconstituted in methylene chloride, and stored at −80°C until UHPLC-MS/MS analysis.
Materials
Cultured cells rinsed with 1× PBS and centrifuged into a pellet or any harvested biological tissue stored at −80°C (See Extraction of lipids from cells on how to obtain cell pellet from either adherent or floating cell cultures).
Solutions & Reagents
1× phosphate buffered saline (PBS)
Deionized water (diH2O)
DC™ Protein Assay Kit II (Bio-Rad, 5000112)
Sterol internal standard (see recipe), 500/100 µg/mL and 50/10 µg /mL
Oxysterol internal standard (see recipe), 100 µg /mL and 10 µg /mL
Folch solution (chloroform:methanol = 2:1, with 1 mM butylated hydroxytoluene (BHT) and 1 mM triphenylphospine (PPh3)), chilled (see recipe)
Sodium chloride aqueous solution (0.9% w/v) (see recipe)
Methylene chloride (LC-MS grade)
Disposables
10 mL polystyrene, disposable serological pipets (Fisherbrand, 13-678-11E)10 mL disposable conical-bottom, Pyrex glass centrifuge tubes (Corning, 99502-10)
Flat bottomed, clear 96 well plate for protein assay (Fisherbrand, 12-565-501)
Disposable 9” glass Pasteur pipets (Fisherbrand, 13-678-20D)
Screw caps for 10 mL disposable glass tubes (Fisherbrand, 14-959-36A)
Glass screw thread vials (Thermo Scientific, C40001W)
Caps for screw thread vials (Thermo Scientific, C500053B)
Special Equipment
S1 Pipet Filler (Thermo Scientific)
Sorvall™ Legend™ X1 Centrifuge, chilled to 4°C (Thermo Scientific)
Ultrasonic bath (Fisher Scientific)
Absorbance microplate reader for protein assay
8-channel pipettor, 10-100 µL and 30-300 µL (Transferpette® S-8)
Mechanical pipet pump (Bel-Art Products)
Savant™ SpeedVac™ High Capacity Concentrator (Thermo Scientific)
Savant™ Refrigerated Vapor Trap (Thermo Scientific), chilled prior to extraction
Deep Vacuum Oil Pump for SpeedVac™ Concentrator (Thermo Scientific)
Polytron™ PT 1200 tissue homogenizer (Kinematica)
Extraction of lipids from cultured cells
- For adherent cells cultured in 100 mm dishes:
- Remove culture dishes from incubator, place on ice and allow to chill for 5 minutes before proceeding.
- Aspirate media from cells using pipette controller fitted with 10 mL serological pipette.
- Add 5 mL chilled 1× PBS, gently swirl dish without disrupting cells, and aspirate 1× PBS.
- Add another 5 mL chilled 1× PBS.
- Using cell scraper, lift adherent cells from dish by scraping back and forth across the dish.
- Using pipette controller fitted with 10 mL serological pipette, aspirate cells suspended in 5 mL 1× PBS and transfer to 10 mL disposable glass centrifuge tube.
- Centrifuge cells into pellet at 1693 × g (3000 rpm) for 5 minutes at 4°C.
- Aspirate the supernatant and either proceed to step 3 or store pellets at −80°C.
- For floating cells cultured in 150 mm dishes:
- Collect cell suspension with pipette controller fitted with 10 mL serological pipette and transfer to 10 mL disposable glass centrifuge tube.
- Centrifuge cells into pellet at 1693 × g (3000 rpm) for 5 minutes at 4°C.
- Aspirate the supernatant.
- Add 5 mL chilled 1× PBS and pipette up and down 5 times to dissociate.
- Centrifuge again at 1693 × g (3000 rpm) for 5 minutes at 4°C.
- Aspirate the supernatant and either proceed to step 3 or store pellets at −80°C.
-
Place freshly pelleted cells or cell pellets that have been stored at −80°C on ice.
If pellet is not already in a 10 mL glass tube, it needs to be transferred to one at this point, otherwise proceed to step 2. To transfer pellet, add 150 µL 1× PBS to the original tube, pipette up and down to dissociate, and transfer cell suspension to a 10 mL glass tube. Add another 150 µL to the original tube, pipette up and down, and transfer to the 10 mL glass tube, ensuring that all cells have been collected. Proceed to step 3.
Add 300 µL chilled 1× PBS to each sample and pipette up and down approximately 5 times, to dissociate cell pellet.
-
Sonicate cell suspension for 30 minutes to lyse cells, ensuring water in sonication bath remains ice cold.
The lysate may be stored at −80°C until extraction, otherwise continue with the extraction and keep samples on ice.
Sonication is the preferred method of lysing cells when lipid extracts will be used for additional analyses, such as untargeted lipidomics, which is often performed in our lab. Lysis buffers can introduce unwanted background signal in such assays, therefore sonication is used to circumvent this issue.
-
Conduct protein assay to determine the amount of protein in each cell lysate. A microplate reader is used at this step to measure absorbance and determine protein concentration.
Further details can be found in the DC Protein Assay Instruction Manual provided by Bio-Rad (LIT448).- Prepare the working reagent by adding 20 uL of reagent S to each mL of reagent A that will be needed for the run.
- From the 20 mg/mL BSA standard stock provided with the kit, prepare a BSA standard dilution series in 8 tubes labeled #1-8
- In tube #8, add 50 uL BSA stock to 150 uL diH2O and mix well to yield a 5 mg/mL solution
- Transfer 100 µL from tube #8 to 100 µL of diH2O in tube #7 and mix well to yield a 2.5 mg/mL solution
- Transfer 100 µL from tube #7 to 100 µL of diH2O in tube #6 and mix well to yield a 1.25 mg/mL solution
- Transfer 100 µL from tube #6 to 100 µL of diH2O in tube #5 and mix well to yield a 0.625 mg/mL solution
- Transfer 100 µL from tube #5 to 100 µL of diH2O in tube #4 and mix well to yield a 0.313 mg/mL solution
- Transfer 100 µL from tube #4 to 100 µL of diH2O in tube #3 and mix well to yield a 0.155 mg/mL solution
- Transfer 100 µL from tube #3 to 100 µL of diH2O in tube #2 and mix well to yield a 0.075 mg/mL solution
- Add only 100 µL of diH2O to tube #1 to yield a 0.0 mg/mL solution
- Pipette 5 µL of each BSA standard in triplicate wells of the flat bottomed, clear 96 well plate.
- Pipette 5 µL of each cell lysate in duplicate wells of the flat bottomed, clear 96 well plate.
- Add 25 µL per well of the working reagent using the 8-channel 10-100 µL pipettor.
- Add 200 µL per well of the Reagent B, provided in the assay kit, using the 8-channel 30-300 µL pipettor in the same order the working reagent was added.
- Place plate into microplate reader and mix for 5 seconds if the microplate reader has a mixing function, otherwise gently agitate the plate by hand to mix the reagents.
-
After 15 minutes, read absorbances using the plate reader at 750 nm.Absorbances stable for 1 hour.
- To calculate protein concentration, subtract the average optical density (OD of the 0.0 mg/mL standard from the average OD of each BSA standard.
- Plot these data and determine the slope and y-intercept of the line.
- Subtract the average OD of the 0.0 mg/mL standard from the average OD of each sample to obtain a corrected OD value.
- Calculate the concentration of protein in each sample by subtracting the y-intercept from each corrected OD value and then dividing by the slope.
- Multiply by the total volume of lysate (300 µL) to obtain the absolute µg amount of protein in each sample. This value will be used as a normalization factor in step 6 under Quantitation of sterols and oxysterols in Basic Protocol 2.
- Add 50/10 µg/mL sterol and 10 µg/mL oxysterol internal standard solutions to each sample and the extraction blank.
- For sterols, add 0.5 µg d7-cholesterol and d7-7-DHC and 0.1 µg 13C3-lanosterol and 13C3-desmosterol per 100 µg protein.
- For oxysterols, add 0.1 µg oxysterol internal standard per 100 µg protein.
Add 1 mL of aqueous sodium chloride solution to each sample.
Vortex each sample for 30 seconds.
Add 4 mL of chilled Folch solution to each sample.
Vortex each sample for 30 seconds.
-
Centrifuge samples for 5 minutes at 1693 × g (3000 rpm) and 4°C.
Following centrifugation, the samples should separate into 2 layers, a lower organic phase consisting primarily of chloroform and lipids, and an upper aqueous phase consisting of methanol, water, and non-lipid contaminants. Note that a layer of protein may be visible between these layers, depending on the amount of starting material (Figure 3).
-
Recover the lower phase using a pipet pump fitted with a 9” glass Pasteur pipet and transfer to a glass centrifuge tube.
Collect a small amount of air into the pipet. Lower the glass pipet into the lower phase by gently pushing aside the protein layer. Discharge the air in the pipet to gently disperse any liquid that may have entered the pipet tip as it passed through the aqueous phase. Collect the organic phase with the pipet and discharge into glass centrifuge tube. This may require two full pipet volumes. Leave a small amount of organic phase at the bottom of the tube to avoid collecting the protein pellet.If part of the protein pellet or upper phase is collect, re-disperse the sample back into the tube and repeat steps 11-13.
Use of a mechanical pipet pump versus a pipet bulb is recommended here for better control when removing liquid near the aqueous-pellet-organic interface.
-
To dry samples in the SpeedVac, turn on the Savant™ Refrigerated Vapor Trap and allow to cool for 1 hour until the ready indicator light turns green.
It is recommended that the Refrigerated Vapor Trap is chilled prior to initiating the extraction.
Uncap samples and place in the Savant™ SpeedVac™ High Capacity Concentrator, ensuring that the samples in the centrifuge are balanced.
-
Next, turn on the Deep Vacuum Oil Pump and adjust gas ballast control knob to position “I”.
Adjustment of the gas ballast allows any volatile organic solvent that has not be collected in the cold trap to escape the vacuum pump. This will prolong the life of the oil in the vacuum pump.
Finally, close the lid on the Savant™ SpeedVac™ High Capacity Concentrator and turn the Concentrator switch to “On” and set the Drying Rate switch to “Low”.
Drying should take approximately 2 hours.
-
Once dry, reconstitute extracts by adding 300 µL methylene chloride, capping samples, and vortexing for 3 seconds.
Given the small volume of the reconstituted extract, it may be beneficial to centrifuge the samples briefly to collect any liquid that is on the wall of the tube.
Transfer reconstituted extracts to a labelled screw top vial, cap, and store at −80°C until UHPLC-MS/MS analysis.
Figure 3.

Representative image of aqueous (top), protein (thin, middle layer), and lipid (bottom) layers from a cell sample.
Extraction of lipids from biological tissues
-
Remove tissues from −80°C and place on ice.
If tissue is not already in a 10 mL glass tube, it needs to be transferred to one at this point. Otherwise proceed to step 2.
-
Record the weight of each tissue.
Ideally, 10-100 mg of tissue yields is sufficient for assessing changes in sterol homeostasis. Analysis of tissues larger than 100 mg may require additional dilution of the lipid extract prior to UHPLC-MS/MS analysis. Analysis of tissues less than 10 mg may result in low peak areas for less abundant analytes.
- Add 500/100 µg/mL sterol and 100 µg/mL oxysterol internal standard solution separately to each sample.
- For sterols, add 5 µg d7-cholesterol and d7-7-dehydrocholesterol and 1 µg 13C3-lanosterol and 13C3-desmosterol per 100 mg tissue.
- For oxysterols, add 1 µg oxysterol internal standard per 100 mg tissue.
Add 4 mL of chilled Folch solution to each sample.
Vortex each sample for 30 seconds.
Rinse the probe of the tissue homogenizer in a separate fresh, chilled Folch solution.
-
Homogenize each tissue sample thoroughly.
Continue to rinse the blade with fresh, chilled Folch solution in between samples to prevent cross contamination.
Add 1 mL of aqueous sodium chloride solution to each sample.
Follow steps 9-18 under “Extraction of lipids from cells”, except for step 15. Tissue samples below 100 mg should be reconstituted in 300 µL methylene chloride and samples at or above 100 mg should be reconstituted in 1 mL methylene chloride.
BASIC PROTOCOL 2: UHPLC-MS/MS ANALYSIS OF CHOLESTEROL HOMEOSTASIS
This section describes the analysis of sterols or cholesterol-derived oxysterols in the lipid extracts using UHPLC-MS/MS. It should be noted that the concentration of endogenous oxysterols in biological samples is typically much lower (1/100 or less) than sterols. Thus, separate sample preparation and UHPLC-MS/MS analysis is conducted for sterols and oxysterols. In this protocol, lipid extracts, prepared in Basic Protocol 1, are dried and re-constituted in mobile phase and then a 10-µL aliquot is injected into the UHPLC-MS/MS system. Reverse phase chromatography is used to elute the sample through a C18 column with an isocratic 90% methanol mobile phase, separating the oxysterols and sterols. Selective reaction monitoring is then employed to monitor the dehydration process of the ion [M+H]+ or [M+H-H2O]+ (Finno et al., 2016; Fliesler et al., 2018). The total time for analysis is 15 minutes per run for sterols and oxysterols, with oxysterols eluting between 0.5 and 6.0 minutes and sterols eluting between 6 and 12.5 minutes. Subsequent quantitation is based on relative response factors (RRFs) obtained from the standard sample, which is prepared with a mix of standard sterols, oxysterols and corresponding isotopically labelled internal standards. Using the RRF for each sterol or oxysterol analyte allows the calculation of each endogenous sterol and oxysterol concentration based on the peak area ratio and internal standard concentration in the sample. Finally, statistical analyses are used to determine significant changes in sterol and/or cholesterol-derived oxysterol levels, which indicates changes in cholesterol homeostasis.
Materials
Solutions and Reagents
Methanol (LC-MS grade)
10 µg/mL sterol standard (see recipe)
10 µg/mL oxysterol standard (see recipe)
50/10 µg/mL sterol internal standard (see recipe)
10 µg/mL oxysterol internal standard (see recipe)
Mobile phase: 90% methanol with 0.1% formic acid (see recipe)
Disposables
Glass crimp/snap top vials (11 mm) with fused 500 µL insert (Thermo Scientific, C40001-LV1)
Pre-slit snap caps with PTFE lining for 11mm glass vials (Thermo Scientific, C4011-55)
Special Equipment
Reacti-Vap™ Evaporator (Thermo Scientific) with Argon gas supply
C18 column (Kinetex®, 100 mm × 2.1 mm, 1.7 µm particle diameter; Phenomenex)
Triple-quadrupole mass spectrometer equipped with an atmospheric-pressure chemical ionization source (Sciex QTRAP® 6500™ system)
ACQUITY UPLC® liquid chromatography system with binary solvent, sample, and column manager (Waters®)
Computer interface for mass spectrometer with Windows operating system
Software
Analyst® 5.1 instrument control and data processing software (2013 SCIEX Analyst® 5.1, version 1.6.2)
Waters® ACQUITY Console instrument component software (2013 Waters®, version 1.60)
Microsoft® Excel® (Microsoft Corp.)
Sample preparation for sterols
On day of analysis, prepare standard sample by adding 2 µL each of the 50/10 µg/mL sterol internal standard and 10 µg/mL sterol standard to an autosampler vial.
Wash needles of Reacti-Vap™ Evaporator with methanol, let air-dry for 1 minute.
-
Dry standard sample under a gentle flow of Argon using the Reacti-Vap™ Evaporator and reconstitute in 100 µL mobile phase.
This yields a standard sample containing 1 µg/mL of d7-7-DHC and d7-cholesterol, 0.2 µg/mL each of 13C3-desmosterol and 13C3-lanosterol, and 0.2 µg/mL all sterol standards. These amounts are similar to the amount of internal standard used in the samples. This is important as quantitation relies on single point calibration.
Cap with pre-slit snap cap and vortex 3 seconds.
-
For each sample, prepare a 10× dilution of reconstituted extract (from Basic Protocol 1 step 13) by transferring 10 µL to a glass autosampler vial, drying under Argon, and reconstituting in 100 µL mobile phase.
Further dilution or concentration of samples may be needed based on instrument sensitivity and nature of the sample.
Cap with pre-slit snap cap and vortex each sample 3 seconds.
Sample preparation for oxysterols
On day of analysis, prepare standard sample by adding 2 µL each of the 10 µg/mL oxysterol internal standard and 10 µg/mL oxysterol standard to an autosampler vial.
Wash needles of Reacti-Vap™ Evaporator with methanol, let air-dry.
-
Dry under Argon using the Reacti-Vap™ Evaporator and reconstitute in 100 µL mobile phase.
This yields a standard sample containing 0.2 µg/mL of oxysterol standards and internal standard, which is similar to the amount of internal standard used in the samples. This is important as quantitation relies on single point calibration.
Cap with pre-slit snap cap and vortex 3 seconds.
-
Prepare a 1:1 dilution of the reconstituted extract from Basic Protocol 1 step 13 by transferring 40 µL to a glass autosampler vial, drying, and reconstituting in 40 µL mobile phase.
Further dilution or concentration of samples may be needed based on instrument sensitivity and nature of the sample.
A minimum volume of 40 µL is needed in the autosampler vial to provide adequate depth for the injection needle to draw sample.
Cap with pre-slit snap cap and vortex each sample 3 seconds.
Analysis of sterols and oxysterols using UHPLC-MS/MS
This protocol is designed for the following instrumentation: Sciex QTRAP® 6500™ system with an atmospheric-pressure chemical ionization source and the ACQUITY UPLC® liquid chromatography system, which consists of a binary solvent manager, sample manager, and column manager. The instrumentation is controlled by the Analyst® software and the ACQUITY UPLC® Console instrument component software.
-
Build an acquisition method in the instrumentation software following the parameters outlined in Table 1, including the mass transitions outlined for sterols, oxysterols and corresponding internal standards in Tables 2–3.
As noted previously, one acquisition method may be used for both sterols and oxysterols, but often the amount of oxysterols is much lower than sterols in biological samples. Therefore, two separate acquisition methods are recommended, as the samples will need to be prepared much more concentrated for oxysterols than for sterols. Steps 2-5 in this section describe a general protocol for either type of analysis.
Equilibrate column and mass spectrometer to the parameters outlined in Table 1.
Once equilibrated, inject a blank sample to verify the column is in good condition and the instrument is running properly.
-
Inject the extraction blank, which contains internal standard, and evaluate for unwanted analyte that may have been introduced in the extraction process or from the internal standard.
In the case that analyte has been introduced into the extraction blank, see step 4 under Quantitation of sterols and oxysterols.
-
Inject 10 µL of the standard sample and check that the chromatograms appear similar to those shown in Figures 4 and 5.
See Troubleshooting section for guidance if unexpected changes in peak intensity or retention time occur.
Build the sample batch so that biological samples are randomized and there is an injection of the standard sample at the beginning, middle, and end of the run.
Submit the sample batch to the run queue.
Table 1.
UHPLC-MS/MS parameters.
| UHPLC Conditions | |
| Column | Kinetex® C18, 100 mm × 2.1 mm, 1.7µm particle diameter |
| Injection volume | 10 µL |
| Flow rate | 0.4 mL/min |
| Mobile phase | 90% methanol with 0.1% formic acid |
| MS/MS Conditions | |
| Declustering potential | 80 V |
| Entrance potential | 10 V |
| Collision energy | 25 V |
| Collision cell exit potential | 20 V |
| APCI Conditions | |
| Nebulizer current | 3 mA |
| Temperature | 300°C |
| Curtain gas | 20 psi |
| Ion source gas | 55 psi |
Table 2.
Sterols and internal standards transitions and retention times.
| Analyte Name | Quadrupole 1 (m/z) | Quadrupole 3 (m/z) | Retention Time (min) | Peak ID |
|---|---|---|---|---|
| 7-Dehydrodesmosterol | 365.3 | 365.3 | 5.88 | A |
| Zymosterol | 367.3 | 367.3 | 6.84 | B |
| Desmosterol | 367.3 | 367.3 | 7.32 | C |
| 13C3-Desmosterol | 370.3 | 370.3 | 7.30 | D |
| 8-Dehydrocholesterol | 367.3 | 367.3 | 7.81 | E |
| d7-7-Dehydrocholesterol | 374.3 | 374.3 | 7.93 | F |
| 7-Dehydrocholesterol | 367.3 | 367.3 | 8.02 | G |
| Lathosterol | 369.3 | 369.3 | 9.57 | H |
| d7-Cholesterol | 376.3 | 376.3 | 9.96 | I |
| Cholesterol | 369.3 | 369.3 | 10.09 | J |
| 13C3-Lanosterol | 412.3 | 412.3 | 11.53 | K |
| Lanosterol | 409.3 | 409.3 | 11.54 | L |
Table 3.
Oxysterols and internal standards transitions and retention times.
| Analyte Name | Quadrupole 1 (m/z) | Quadrupole 3 (m/z) | Retention Time (min) | Peak ID |
|---|---|---|---|---|
| 7β,27-dihydroxycholesterol | 401.3 | 383.3 | 0.93 | M |
| 7-keto-27-hydroxycholesterol | 417.3 | 399.3 | 0.97 | N |
| 24-hydroxycholesterol* | 385.3 | 367.3 | 1.69 | O |
| 25-hydroxycholesterol* | 385.3 | 367.3 | 1.69 | O |
| 24-ketocholesterol | 383.3 | 365.3 | 1.84 | P |
| 24(S),25-epoxycholesterol | 383.3 | 365.3 | 1.97 | Q |
| 7β-hydroxycholesterol | 385.3 | 367.3 | 2.85 | R |
| d7-7-ketocholesterol | 408.3 | 390.3 | 3.12 | S |
| 7-ketocholesterol | 401.3 | 383.3 | 3.17 | T |
| 4β-hydroxycholesterol | 385.3 | 367.3 | 5.60 | U |
24- and 25-hydroxycholesterol peaks cannot be resolved under these chromatographic conditions.
Figure 4.
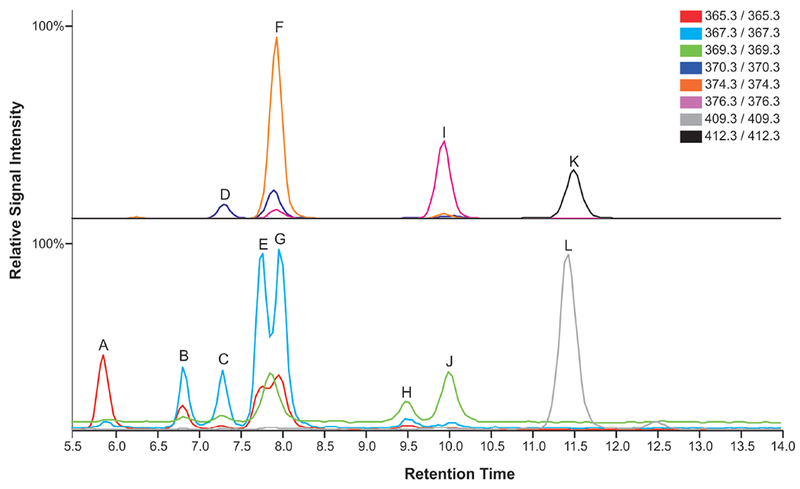
LC-MS chromatogram of the oxysterol standard sample, including the internal standard d7-7-ketocholesterol (peak S) and all oxysterol standards. Mass transitions are denoted in color legend.
Figure 5.
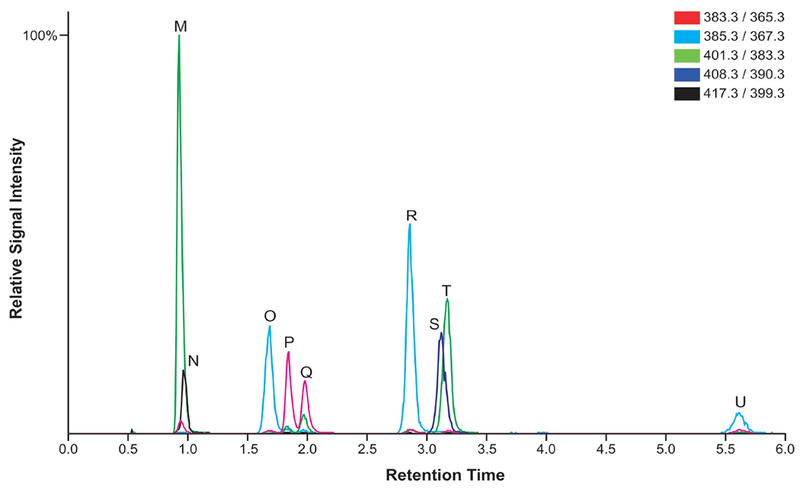
LC-MS chromatogram of the sterol standard sample, including the internal standards 13C3-desmosterol, d7-7-DHC, d7-cholesterol, and 13C3-lanosterol (top panel) and all sterol standards (bottom panel). Mass transitions are denoted in color legend.
Quantitation of sterols and oxysterols
- Within the Analyst® software, set up the quantitation method to allow for automated peak integration.
- In quantitation mode, select a standard sample as the representative sample to use.
- In the internal standards table, enter each internal standard along with its mass transition.
- For each analyte listed in the table, select the internal standard that will be used to calculate the area ratio for that specific analyte.
- Select the integration tab and ensure that the correct peak is selected for each analyte and internal standard. Adjust the retention time if needed.
- Using the quantitation method, conduct automated peak integration.
- Integrate the peaks of each analyte and internal standard for all extraction blanks, samples and standard samples injected.
- Review individual peaks for proper integration and consistency of peak shape, retention time, and area.
-
Calculate the peak area ratio of the analyte to internal standard.Note that isotopically-labeled internal standards are not available for all analytes. To overcome this problem, an alternative with similar chemical composition to that of the analyte can be substituted. For calculating the peak area ratio of zymosterol, 7-DHC, and 8-DHC use d7-7-DHC; for 7-dehydrodesmosterol (7-DHD) and desmosterol use 13C3-desmosterol; for cholesterol use d7-cholesterol; and for lanosterol and lathosterol use 13C3-lanosterol. Use d7-7-ketocholesterol for all cholesterol-derived oxysterols. However, users can add additional internal standards if they are available, such as d6-24(S),25-epoxycholesterol, d7-24-hydroxycholesterol, and d7-4β-hydroxycholesterol.
-
2.Using an Excel® spreadsheet, generate the relative response factor (RRF) of each analyte from each injection of the standard sample.
- Calculate the response factor (RF) for each sterol and oxysterol by dividing the standard analyte peak area by its concentration in the standard sample.
- Calculate the RF of each internal standard in the standard sample by dividing the internal standard peak area by its concentration in the standard sample.
- Calculate the RRF by dividing the RF of the analyte by the RF of its corresponding internal standard.
As noted previously, this quantitation method relies on single point calibration. For the best results, the standard and internal standard concentrations should be in the range of the estimated concentration in the sample. This may require additional fine-tuning to get the correct levels.
-
3.
Average the RRFs calculated from each injection of the standard sample and use this number to account for any instrumental drift.
-
4.
Calculate the concentration of each endogenous sterol and oxysterol in the sample, as well as the extraction blank, by multiplying the peak area ratio by the internal standard concentration in the sample and then dividing by the average RRF.
If quantifiable levels of any analyte were introduced in the extraction blank, subtract that amount from each sample, to correct for analyte that did not originate from the cell or tissue.
-
5.
The absolute amount of each endogenous sterol and oxysterol can by calculated by multiplying the concentration by the dilution factor and volume of mobile phase used to reconstitute extract.
-
6.
The absolute amount of sterol or oxysterol should then be normalized to the amount of protein or tissue used in the extraction, as differences in protein content or tissue weight could yield different sterol or oxysterol levels. Alternatively, normalization of sterol precursors and/or oxysterols to cholesterol content can also be used to assess relative changes in cholesterol synthesis and/or metabolism.
Statistical Analyses
This data is amenable to a variety of statistical analyses to determine significant differences in sterol and oxysterol levels between treatment groups. Student’s t-test to compare two means or analysis of variance (ANOVA) for more than two means are two commonly used analyses to determine if xenobiotic treatment alters sterol or oxysterol levels. Regression analyses could also be useful for determining a dose-response relationship between doses of xenobiotic and resulting sterol or oxysterol levels.
Reagents and Solutions
500/100 µg/mL sterol internal standard
Prepare a solution containing 500 µg/mL d7-cholesterol (Avanti Polar Lipids, Inc., 700041) and d7-7-DHC (Avanti Polar Lipids, Inc., 700116P) and 100 µg/mL 13C3-lanosterol (Kerafast®, EVU135) and 13C3-desmosterol (Kerafast®, EVU127).
Prepare a stock solution of each internal standard by dissolving solid in benzene.
Aliquot an amount of each internal standard into the same screw top vial, depending on starting stock concentration, to achieve the desired final concentration.
Dry this mixture under Argon using the Reacti-Vap™ Evaporator and reconstitute in benzene (LC-MS grade) to achieve the desired final concentration of each sterol internal standard.
Store at −80°C up to 6 months.
50/10 µg/mL sterol internal standard
Prepare a 10× dilution of the 500/100 µg/mL sterol internal standard solution in a new screw top vial.
Store at −80°C up to 6 months.
100 µg/mL oxysterol internal standard
Prepare a 100 µg/mL d7-7-ketocholesterol solution in benzene (LC-MS grade).
Prepare a stock solution by dissolving 1 mg d7-7-ketocholesterol (Avanti Polar Lipids, Inc., 700046P) in 1 mL benzene in a screw top vial.
Transfer 100 µL of the 1 mg/mL mixture to a new screw top vial and dry under Argon using the Reacti-Vap™ Evaporator.
Reconstitute in 1 mL benzene.
Store at −80°C up to 6 months.
10 µg/mL oxysterol internal standard
Prepare a 10× dilution of the 100 µg/mL oxysterol internal standard solution in a new screw top vial.
Store at −80°C up to 6 months.
Sodium chloride aqueous solution (0.9% w/v)
Weight out 0.900 g of sodium chloride (certified ACS crystalline) and transfer to a 100 mL volumetric flask.
Adjust volume to 100 mL with water (LC-MS grade).
Transfer to glass storage bottle.
Store at room temperature up to 1 month.
Folch solution (chloroform:methanol = 2:1, with 1 mM BHT and 1 mM PPh3)
Weigh out 0.131 g of PPh3 and 0.11 g of BHT.
Measure 167 mL methanol (LC-MS grade) in graduated cylinder and add to 500 mL volumetric flask.
Transfer PPh3 and BHT to the volumetric flask containing methanol.
Fill the volumetric flask to 500 mL with chloroform (LC-MS grade).
Transfer solution to glass storage bottle.
Make fresh and chill prior to extraction.
This recipe makes enough for approximately 125 samples.
10 µg/mL sterol standard
Dissolve 1 mg of each sterol (listed below) in 1 mL of benzene (LC-MS grade).
Prepare a 10× dilution of each sterol standard to yield a 100 µg/mL stock solution in benzene.
Add 100 µL of each sterol standard to the same screw top vial.
Add 200 µL of benzene to yield a 10 µg/mL sterol standard solution.
Cap and store at −80°C up to 6 months.
7-dehydrodesmosterol (Avanti Polar Lipids, Inc., 700138P)
Zymosterol (Avanti Polar Lipids, Inc., 700068P)
Desmosterol (Avanti Polar Lipids, Inc., 700060P)
8-dehyrocholesterol (Avanti Polar Lipids, Inc., 700075P)
7-dehydrocholesterol (Avanti Polar Lipids, Inc., 700066P)
Lathosterol (Avanti Polar Lipids, Inc., 700069P)
Cholesterol (Sigma-Aldrich®, C8667)
Lanosterol (Avanti Polar Lipids, Inc., 700063P)
10 µg/mL oxysterol standard
Dissolve 1 mg of each oxysterol (listed below) in 1 mL of benzene (LC-MS grade).
Prepare a 10× dilution of each oxysterol standard to yield a 100 µg/mL stock solution of each in benzene.
Add 100 µL of each oxysterol standard from 100 µg/mL stock to the same screw top vial.
Add 200 µL of benzene to yield a 10 µg/mL oxysterol standard solution.
Cap and store at −80°C up to 6 months.
7β,27-dihydroxycholesterol (Avanti Polar Lipids, Inc., 700025P)
7-keto-27-hydroxycholesterol (Avanti Polar Lipids, Inc., 700033P)
24-hydroxycholesterol (Avanti Polar Lipids, Inc., 700061P)
25-hydroxycholesterol (Avanti Polar Lipids, Inc., 700019P)
24-ketocholesterol (Steraloids, Inc., C7030-000)
24(S),25-epoxycholesterol (Avanti Polar Lipids, Inc., 700039P)
4β-hydroxycholesterol (Avanti Polar Lipids, Inc., 700036P)
7β-hydroxycholesterol (Avanti Polar Lipids, Inc., 700035P)
7-ketocholesterol (Avanti Polar Lipids, Inc., 700015P)
Mobile phase: 90% methanol with 0.1% formic acid
Measure out 100 mL of water (LC-MS grade) using a graduated cylinder and add to 1 L volumetric flask. Fill the volumetric flask half-way with methanol.
Add 1 mL formic acid (LC-MS grade) to volumetric flask with 1000 µL pipet.
Adjust volume to 1L with methanol.
Prepare the mobile phase fresh prior to each analysis.
COMMENTARY
Background Information
Mass spectrometry is the modern technology for sterols and oxysterols analysis, including both gas chromatography-mass spectrometry (GC-MS) and LC-MS, as well as matrix assisted laser desorption/ionization (MALDI)-MS for imaging studies (Griffiths et al., 2017). Conventionally, sterols are analyzed by GC or GC-MS after derivatization, which is still often used, but LC-MS has become the technique of choice over the past decade (Gerst, Ruan, Pang, Wilson, & Schroepfer, 1997). In LC-MS analysis, sterols and oxysterols can be analyzed either underivatized or with derivatization to facilitate ionization (Griffiths & Wang, 2009; Honda et al., 2008; McDonald, Thompson, McCrum, & Russell, 2007).
In the current protocol, UHPLC-MS/MS with APCI is used to quantify the major sterols in the cholesterol biosynthetic pathway as well as the cholesterol-derived oxysterols without chemical derivatization (Finno et al., 2016; Fliesler et al., 2018; Herron et al., 2016). APCI is advantageous over GC-MS and LC-MS paired with electrospray ionization (LC-ESI-MS/MS), as sterols and oxysterols are nonvolatile and poorly ionizable. The use of APCI circumvents the need to derivatize samples while still affording sufficient sensitivity. The elimination of derivatization procedures can reduce undesired ex vivo autoxidation of cholesterol and its precursors, which could interfere with the analysis of endogenous oxysterols (Lamberson et al., 2017; Xu & Porter, 2015; Yin et al., 2011). Additionally, the UHPLC-MS/MS method in the current protocol has a run time of 15 minutes for each sample, which is quite efficient compared to previously published methods (McDonald et al., 2007).
Critical Parameters and Troubleshooting
Autoxidation of cholesterol and its precursors.
Cholesterol and cholesterol precursors are highly vulnerable to autoxidation, where oxysterols can be formed from sterols in air (Lamberson et al., 2017)(Xu & Porter, 2015; Yin et al., 2011). Artefactual formation of cholesterol-derived oxysterols during freezing and thawing of samples or during extraction could hamper accurate measurements. It is important to use only freshly collected or flash frozen samples to prevent oxidation prior to homogenization in the Folch solution. In addition, antioxidants, such as BHT and PPh3, are added to the Folch solution to prevent undesired autoxidation.
UHPLC maintenance and troubleshooting.
The UHPLC system, including tubing, column, injection port, and needle can become clogged by dirty samples, precipitated salts, solvents, and even autosampler caps. Therefore, it is important to use only high-quality, HPLC-grade solvents and pre-slit autosampler caps and to syringe filter samples prior to UHPLC-MS/MS analysis if extracts look cloudy or contain particulates. System pressure can also be an indicator of alterations in the column, mobile phase, flow rate, and temperature. It is important to record system pressure to establish a pressure reference point. If changes in pressure occur, longer column equilibration might help in stabilizing the pressure to the reference point. The mobile phase composition may change over time so one should make it fresh prior to sample analysis. If pressure problem persists, consider replacing the column as it has a finite lifetime. Additionally, altered retention times most often indicate poor equilibration of the system and mobile phase, as well as column contamination and flow rate. Overloading the sample or improper column equilibration might also yield distorted peak shapes that could result in inaccurate quantitation. Finally, random baseline noise spikes and disrupted flow rates might indicate gas bubbles in the UHPLC pumps, which would require mobile phase degassing or replacement of pump seals. Further information on these issues and how to avoid them can be found in the manufacturers user guide.
Mass spectrometer maintenance.
The mass spectrometer should be properly and routinely maintained for optimal performance. Loss of sensitivity, increased background noise, and additional peaks not part of the sample could be observed in the absence of proper and timely instrument maintenance, indicative of system contamination. To avoid these issues as well as damage to the mass spectrometer, follow the manufacturer’s guidance on maintenance tasks and schedule.
Anticipated Results
A chromatogram for each sterol and oxysterol standard sample, each containing a mixture of sterols and oxysterols and corresponding internal standards, is shown in Figures 4 and 5. With this method, oxysterols are eluted through the column between 1 and 5 minutes and sterols are eluted through the column between 5 and 14 minutes.
The utility of this protocol is demonstrated in Table 4. In this case, we investigated the effect of a known cholesterol biosynthesis inhibitor, AY9944, on cholesterol homeostasis in neurospheres, which are a three-dimensional in vitro model used for developmental neurotoxicity testing. We hypothesized that AY9944, which inhibits cholesterol biosynthesis at the step of DHCR7, would lead to an accumulation of sterol precursors and a decrease in cholesterol and cholesterol-derived oxysterols in treated neurospheres. Following a 72-hour treatment with AY9944 or vehicle control, neurospheres were collected into a cell pellet and a protein mass assay was used to determine sample protein content. Lipid extraction and UHPLC-MS/MS analysis for sterols and oxysterols was conducted. The endogenous sterol and oxysterol content was then normalized to the protein content for each sample. The Student’s t-test assuming unequal variance was used to determine statistically significant differences in sterol and oxysterol levels, using the significance value of p ≤ 0.05.
Table 4.
Treatment of neurospheres with AY9944 induced significant changes in levels of sterols and cholesterol-derived oxysterols.
| Sterols | Control | AY9944 | Units |
|---|---|---|---|
| Cholesterol | 15.01 ± 4.39 | 2.82 ± 1.3 * | µg/mg |
| 7-DHC | 0.14 ± 0.05 | 11 ± 3.02 * | ng/mg |
| Desmosterol | 6.4 ± 1.2 | 0.19 ± 0.15 ** | µg/mg |
| 7-DHD | 0.81 ± 0.39 | 10.52 ± 2.57 ** | ng/mg |
| Lanosterol | 0.38 ± 0.13 | 0.25 ± 0.05 | µg/mg |
| 8-DHC | 0.36 ± 0.02 | 2.52 ± 0.19 | ng/mg |
| Zymosterol | 1.01 ± 0.39 | 1.23 ± 0.41 | ng/mg |
| Lathosterol | 0.65 ± 0.60 | 0.43 ± 0.48 | µg/mg |
| Cholesterol-derived Oxysterols | |||
| 24(S),25-epoxycholesterol | 352.04 ± 176.27 | 76.03 ± 54.61 * | ng/mg |
| 24-ketocholesterol | 22.3 ± 7.68 | 38.31 ± 2.7 * | ng/mg |
| 7-hydroxycholesterol | 2.17 ± 0.52 | 5.55 ± 1.41 * | ng/mg |
| 24/25-hydroxycholesterol | 9.25 ± 2.45 | 1.32 ± 1.75 ** | ng/mg |
| 7-ketocholesterol | 13.67 ± 8.32 | 10.81 ± 5.27 | ng/mg |
denotes p≤0.05;
denotes p≤0.005;
t-test assuming unequal variances was used.
The results of this experiment show that AY9944 is a potent inhibitor of cholesterol biosynthesis in neurospheres. This is indicated by the significant accumulation of the cholesterol precursors 7-DHC and 7-DHD, which rely on DHCR7 to be converted to cholesterol and desmosterol, respectively. Further alteration of cholesterol homeostasis was observed in the almost 5-fold decrease in 24(S),25-epoxycholesterol and 7-fold decrease in 24/25-hydroxycholesterol, two of the main cholesterol-derived oxysterols. Significant decreases in these cholesterol-derived oxysterols provide further evidence that cholesterol biosynthesis is downregulated in the AY9944 treated samples.
Time Considerations
For approximately 20 cell samples, the protein assay takes 2 hours to complete. The lipid extraction for 20 cell samples takes approximately 5 hours, including drying in the SpeedVac, reconstituting in methylene chloride, and transferring to a screw top storage vial. For sterols and oxysterols separately, UHPLC-MS/MS analysis takes approximately 16 minutes per sample for a total run time of approximately 6 hours for injections of 20 cell samples, 3 standard samples (at minimum), and 1 extraction blank. For the same number of tissues samples, the lipid extraction and UHPLC-MS/MS analyses take approximately same amount of time.
Significance Statement.
Many xenobiotics, including pharmaceuticals and environmental toxicants, have been shown to alter cholesterol homeostasis, which is critical for normal growth and development. Ultra-high performance liquid chromatography-tandem mass spectrometry analysis can be used to assess alterations in cholesterol homeostasis by quantitatively measuring levels of cholesterol, sterol precursors, and cholesterol-derived oxysterols in exposed cells or tissues. Determining the effect of a xenobiotic’s against cholesterol homeostasis can yield valuable mechanistic information and provide insight on whether altered cholesterol homeostasis could contribute to the exposure phenotype.
ACKNOWLEDGEMENT
This work was supported by grants from the National Institutes of Health (R00HD073270 and R01HD092659). J.H. was also supported by a National Institute of Environmental Health Sciences training grant (T32ES007032).
LITERATURE CITED
- Boland MR, & Tatonetti NP. (2016). Investigation of 7-dehydrocholesterol reductase pathway to elucidate off-target prenatal effects of pharmaceuticals: a systematic review. The pharmacogenomics journal, 16(5), 411–429. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfrán-Duque A, Casado ME, Pastor O, Sánchez-Wandelmer J, la Peña, de G, Lerma M, Mariscal P, et al. (2013). Atypical antipsychotics alter cholesterol and fatty acid metabolism in vitro. Journal of Lipid Research, 54(2), 310–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Medina P, Paillasse MR, Segala G, Poirot M, Silvente-Poirot S, & Mangelsdorf DJ (2010). Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proceedings of the National Academy of Sciences of the United States of America, 107(30), 13520–13525. National Academy of Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finno CJ, Bordbari MH, Valberg SJ, Lee D, Herron J, Hines K, Monsour T, et al. (2016). Transcriptome profiling of equine vitamin E deficient neuroaxonal dystrophy identifies upregulation of liver X receptor target genes. Free Radical Biology and Medicine, 101, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliesler SJ, Peachey NS, Herron J, Hines KM, Weinstock NI, Ramachandra Rao S, & Xu L (2018). Prevention of Retinal Degeneration in a Rat Model of Smith-Lemli-Opitz Syndrome. Scientific reports, 8(1), 1286 Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, & Sloane Stanley GH (1957). A simple method for the isolation and purification of total lipides from animal tissues. Journal of Biological Chemistry, 226(1), 497–509. [PubMed] [Google Scholar]
- Gerst N, Ruan B, Pang J, Wilson WK, & Schroepfer GJ (1997). An updated look at the analysis of unsaturated C27 sterols by gas chromatography and mass spectrometry. The Journal of Lipid Research, 38(8), 1685–1701. [PubMed] [Google Scholar]
- Griffiths WJ, & Wang Y (2009). Analysis of neurosterols by GC-MS and LC-MS/MS. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences, 877(26), 2778–2805. [DOI] [PubMed] [Google Scholar]
- Griffiths WJ, Abdel-Khalik J, Yutuc E, Morgan AH, Gilmore I, Hearn T, & Wang Y (2017). Cholesterolomics: An update. Analytical biochemistry, 524, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizzetti M, Chen J, & Costa LG (2011) Chapter 65: Disruption of cholesterol homeostasis in developmental neurotoxicity In Gupta RC (Ed.), Reproductive and Developmental Toxicology (pp. 855–862). Academic Press. [Google Scholar]
- Hall P, Michels V, Gavrilov D, Matern D, Oglesbee D, Raymond K, Rinaldo P, et al. (2013). Aripiprazole and trazodone cause elevations of 7-dehydrocholesterol in the absence of Smith-Lemli-Opitz Syndrome. Molecular genetics and metabolism, 110(1–2), 176–178. [DOI] [PubMed] [Google Scholar]
- Herron J, Reese RC, Tallman KA, Narayanaswamy R, Porter NA, & Xu L (2016). Identification of Environmental Quaternary Ammonium Compounds as Direct Inhibitors of Cholesterol Biosynthesis. Toxicological Sciences : an official journal of the Society of Toxicology, 151(2), 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda A, Yamashita K, Miyazaki H, Shirai M, Ikegami T, Xu G, Numazawa M, et al. (2008). Highly sensitive analysis of sterol profiles in human serum by LC-ESI-MS/MS. The Journal of Lipid Research, 49(9), 2063–2073. [DOI] [PubMed] [Google Scholar]
- Kelley RI, & Herman GE (2001). Inborn errors of sterol biosynthesis. Annual review of genomics and human genetics, 2(1), 299–341. [DOI] [PubMed] [Google Scholar]
- Kim H-YH, Korade Z, Tallman KA, Liu W, Weaver CD, Mirnics K, & Porter NA (2016). Inhibitors of 7-Dehydrocholesterol Reductase: Screening of a Collection of Pharmacologically Active Compounds in Neuro2a Cells. Chemical research in toxicology, 29(5), 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korade Z, Kim H-YH, Tallman KA, Liu W, Koczok K, Balogh I, Xu L, et al. (2016). The Effect of Small Molecules on Sterol Homeostasis: Measuring 7-Dehydrocholesterol in Dhcr7-Deficient Neuro2a Cells and Human Fibroblasts. Journal of medicinal chemistry, 59(3), 1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberson CR, Muchalski H, McDuffee KB, Tallman KA, Xu L, & Porter NA (2017). Propagation rate constants for the peroxidation of sterols on the biosynthetic pathway to cholesterol. Chemistry and physics of lipids, 207(Pt B), 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JG, Thompson BM, McCrum EC, & Russell DW (2007). Extraction and analysis of sterols in biological matrices by high performance liquid chromatography electrospray ionization mass spectrometry. Methods in enzymology, 432, 145–170. Elsevier. [DOI] [PubMed] [Google Scholar]
- Mutemberezi V, Guillemot-Legris O, & Muccioli GG (2016). Oxysterols: From cholesterol metabolites to key mediators. Progress in lipid research, 64, 152–169. [DOI] [PubMed] [Google Scholar]
- Pikuleva IA (2006). Cholesterol-metabolizing cytochromes P450. Drug metabolism and disposition: the biological fate of chemicals, 34(4), 513–520. American Society for Pharmacology and Experimental Therapeutics. [DOI] [PubMed] [Google Scholar]
- Porter FD, & Herman GE (2010). Malformation syndromes caused by disorders of cholesterol synthesis. The Journal of Lipid Research, 52(1), 6–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, & Porter NA (2015). Free radical oxidation of cholesterol and its precursors: Implications in cholesterol biosynthesis disorders. Free radical research, 49(7), 835–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Xu L, & Porter NA (2011). Free radical lipid peroxidation: mechanisms and analysis. Chemical reviews, 111(10), 5944–5972. [DOI] [PubMed] [Google Scholar]