

Abstract
The misfolding, amyloid aggregation, and fibril formation of intrinsically disordered proteins/peptides (or amyloid proteins) have been shown to cause a number of disorders. The underlying mechanisms of amyloid fibrillation and structural properties of amyloidogenic precursors, intermediates, and amyloid fibrils have been elucidated in detail; however, in-depth examinations on physiologically relevant contributing factors that induce amyloidogenesis and lead to cell death remain challenging. A large number of studies have attempted to characterize the roles of biomembranes on protein aggregation and membrane-mediated cell death by designing various membrane components, such as gangliosides, cholesterol, and other lipid compositions, and by using various membrane mimetics, including liposomes, bicelles, and different types of lipid-nanodiscs.
We herein review the dynamic effects of membrane curvature on amyloid generation and the inhibition of amyloidogenic proteins and peptides, and also discuss how amyloid formation affects membrane curvature and integrity, which are key for understanding relationships with cell death. Small unilamellar vesicles with high curvature and large unilamellar vesicles with low curvature have been demonstrated to exhibit different capabilities to induce the nucleation, amyloid formation, and inhibition of amyloid-β peptides and α-synuclein. Polymorphic amyloidogenesis in small unilamellar vesicles was revealed and may be viewed as one of the generic properties of interprotein interaction-dominated amyloid formation. Several mechanical models and phase diagrams are comprehensively shown to better explain experimental findings. The negative membrane curvature-mediated mechanisms responsible for the toxicity of pancreatic β cells by the amyloid aggregation of human islet amyloid polypeptide (IAPP) and binding of the precursors of the semen-derived enhancer of viral infection (SEVI) are also described. The curvature-dependent binding modes of several types of islet amyloid polypeptides with high-resolution NMR structures are also discussed.
Keywords: Alpha-synuclein, amyloid-beta, hIAPP, SEVI, HIV, membrane interaction, protein misfolding disease
1. Introduction
1-1. Protein misfolding and aggregation and their relationships with various diseases
Most proteins fold spontaneously into three-dimensional native structures, which are unique and functional, below the solubility limit (Fig. 1) [1, 2]; however, intrinsically disordered proteins exist under physiological conditions and lack defined stereostructures [3]. The numerous biological functions of proteins are fundamentally achieved by the highly regulated intermolecular interactions of proteins with other molecules [4]. Nevertheless, endogenous and exogenous stresses decrease the stability of proteins in native states, which often causes the concurrent formation of insoluble aggregates (Fig. 1) [5]. Various machineries, such as quality control systems, contribute to the maintenance of diverse protein homeostasis in the body by helping to correct folding as well as the efficient and timely clearance of deleterious aggregates and misbehaving proteins [6, 7]. A failure in this control system due to aging or accidental and sporadic errors reinforces misregulated interprotein interactions. It consequently leads to the irreversible formation and accumulation of protein aggregates in various organelles, cells, and tissues, which, in turn, gives rise to a number of disorders including not only neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases, but also amyloidoses, including type II diabetes and dialysis-related amyloidosis, through the combined effects of the permanent loss of function and/or gain of toxic function [8–15].
Figure 1. General model of protein folding and misfolding-induced aggregation.

In the soluble region, unstructured proteins (light green) fold correctly to three-dimensional native conformations (dark green). Changes in the surrounding conditions decrease the solubility of unstable proteins and cause three types of self-assembled insoluble protein aggregates: amorphous aggregates (red), protofibrils (purple), and amyloid fibrils (blue). Thermodynamic solubility is shown with a vertical broken line. The apparently soluble region over solubility due to the metastability (M.S.) of supersaturation, which is a kinetically trapped state and produces the lag time, is represented. Increases in the lag time and metastability of supersaturation as well as in the structural order are shown using transverse and vertical triangles with differing levels of color gradation, respectively
Therefore, a large number of studies have targeted protein misfolding and aggregation and attempted to reveal their relationships to diseases based on whole protein behaviors together with protein folding and normal function. In the past two decades, the underlying and unique properties of the appearance (structure and morphology) and birth (mechanisms) of protein aggregates have been elucidated. Protein aggregates may be largely classified into three types: amyloid fibrils, protofibrils, and amorphous aggregates, depending on their structure and morphology, tinctorial properties, and aggregation mechanisms (Fig. 1). Amyloid fibrils are morphologically long in shape and commonly consist of one or multiple protofilaments that are rich in β-structures [16–18], whereas amorphous aggregates generally do not show ordered structures or a characteristic morphology [1]. Oligomers, which are preferably rounded in shape, exhibit highly variable structural properties, ranging from a well-structured β conformation [19–21] to a largely disordered structure [22–24]. Thioflavin T (ThT) and Congo red dyes detect the structures of amyloid fibrils, thereby allowing the various types of aggregates to be distinguished from each other [25]. Biophysical methods, such as circular dichroism [26] spectroscopy, Fourier transform-infrared spectroscopy (FT-IR), X-ray crystallography and fiber diffraction, and solution and solid-state NMR spectroscopy, have been effectively used in the detection and characterization of protein aggregates [25, 27–32]. Microscopy, such as atomic force microscopy (AFM) and transmission electron microscopy [33], visually identifies aggregate types [34, 35]. Although many elaborate and complex models, including Ostwald ripening [36] and two-step nucleation [37], have been proposed to explain amyloid fibrillation, amyloid fibrils are generally considered to self-assemble from soluble monomers through the two-step mechanism: slow nucleation, which generates the lag time, is followed by the rapid growth of fibrils (i.e., elongation). In contrast, amorphous aggregation often occurs without appreciable lag times [38]. Protofibrils share common structural, morphological, and kinetic properties between amyloid fibrils and amorphous aggregates.
Recent studies reported key macroscopic (i.e., thermodynamic) and kinetic concepts of solubility and supersaturation for protein aggregation [2, 38–40]. Protein aggregation must occur over thermodynamic solubility under given conditions and supersaturation kinetically controls aggregation because the metastability of supersaturation produces the lag time for productive nucleation (Fig. 1). The metastability of supersaturation may serve as an energy barrier (i.e., activation energy). Furthermore, it is generally accepted that amyloid fibrillation is a generic property of polypeptide chains [41]. All proteins, even disease-unrelated proteins, retain amyloidogenic regions [42], namely, the amylome [43], and are capable of forming amyloid fibrils under specific conditions [44, 45], indicating the continual risk of protein misfolding diseases [46]. However, a physical phenomenon of supersaturation counteracts the amylome because the high metastability of supersaturation maintains proteins over the solubility limit in a soluble state (Fig. 1) [2, 47–53]. This buffering function may prolong solubility and protect organisms against aggregation-induced impairments.
Protein aggregation exhibits strong susceptibility to environmental conditions. Thus, different types of protein aggregates form as a result of changing conditions. For example, polymorphic amyloid fibrillation appears in response to changes in conditions, such as pH, ionic strength, and temperature [29, 54–57]. The degree of cytotoxicity and damage to tissues depends on the types of aggregates and amyloid fibrils [46, 58]. Therefore, the characterization of protein aggregation under various conditions plays an essential role in a comprehensive understanding of the context-dependent mechanisms of amyloid formation and the structure of amyloid aggregates. Many factors affect amyloid formation. Not only changes in pH, salt types/concentrations, temperature, and pressure, but also the presence of additives, such as metal ions, have been shown to promote/inhibit amyloid formation and induce alternations in the aggregation pathway [59–66]. Membranes have been one of the most important biologically relevant contributors to amyloidogenesis because most proteins are exposed to the environments of various biological membranes. A number of roles have been demonstrated for biomembranes: representatively, membranes condense proteins, which increases the effective concentration (i.e., a decrease in the metastability of supersaturation and lag time (Fig. 1)), and reduce the diffusional dimension of proteins from three to two dimensions, which affects amyloidogenicity [26, 67–71]. Furthermore, the binding of amyloidogenic proteins to lipid membranes induces conformational changes in proteins for amyloid formation.
A deeper understanding of the mechanisms underlying amyloid fibrillation in the presence of membranes is one of the central goals in this field of research. The key question is “What are the properties of membranes that affect amyloidogenesis?”, and the general answers will be i) the components of membranes, ii) types of lipids, and iii) membrane curvature. Since several reviews have already described and summarized how lipid types and membrane compositions influence the structures and aggregation behaviors of many amyloid proteins, including Aβpeptides [13], hIAPP [72], and α-synuclein (αSN) [73, 74], we herein focus exclusively on the impact of membrane curvature on amyloid generation. We will review how the aggregation behaviors of Aβ peptides depend on curvature, and describe how αSN binds to the model membranes of synaptic vesicles with high curvature by changing its structure in the membrane-bound state. The effects of membranes on αSN amyloid formation and inhibition will be explained based on comprehensive mechanical models. We also show how IAPP amyloid formation influences membrane curvature as well as its mechanical and etiological links to islet cell toxicity. The membrane curvature-dependent binding modes of several types of IAPPs at the atomistic level, which are closely related to the amyloidogenicity of IAPP, will be addressed. A novel mechanism for the marked enhancement in infection by human immunodeficiency virus will be suggested based on negative curvature-induced membrane fusion by a highly amyloidogenic seminal peptide.
1-2. Size-dependent differences in physicochemical properties and the general usage of biomembrane-mimicking vesicles of distinct sizes
The biological membranes of organelles and cells have various shapes [75]. The cylindrical shapes of membrane tubes are observed under a dynamin coat, and the necks of membrane buds have the shape of a saddle. Synaptic vesicles are morphologically spherical in shape. These differences in shape engender an important physicochemical property of membranes, namely, curvature [76, 77]. Membrane curvature has been shown to affect the functions of proteins, which interact with membranes [76, 78]. Thus, in order to examine the effects of membrane curvature on protein functions in detail, several types of vesicles of different sizes were tailored for in vitro studies. Three types of vesicles are widely utilized: giant unilamellar vesicles (GUV), large unilamellar vesicles (LUV), and small unilamellar vesicles (SUV). The sizes of vesicles are in the following order: GUV > LUV > SUV (Fig. 2). Thus, the degree of curvature is in the order of SUV > LUV > GUV [79].
Figure 2. Model membrane size-dependent curvature.

Three types of vesicles of different sizes: GUV [95], LUV (yellow), and SUV (red), are shown with cartoons and colors. Changes in the vesicle size and curvature are indicated by the gradation in color. The relative sizes of vesicles are exaggerated for simple comparisons.
GUV are generally used instead of cell membranes because its diameter is ~5 μm and greater. The local membrane curvature of GUV was suggested to be almost flat relative to LUV and SUV [80]. GUV with the labeling of fluorescent lipids provide the advantage of visibility in fluorescence microscopy for observations of changes in membrane surfaces, such as membrane disruption, tubulation, and phase separation [81–83]. However, the preparation and quantification of GUV are not simple and, thus, difficulties are associated with obtaining key information, including detailed stoichiometry. In order to improve the usability of model membrane-based studies by overcoming quantitative issues, LUV and SUV have often been introduced. The diameter of LUV is ~100 – 500 nm, and may be regulated by the pore sizes of polycarbonate membranes. SUV of ~15 – 50 nm are prepared using ultrasonication [84]. LUV and SUV labeled with fluorescent lipids are not suitable for fluorescence microscopy because they are observed as spots and do not provide productive information [85]. However, it is markedly easier to control the concentrations of LUV and SUV than that of GUV, and, thus, they may be used in in vitro studies that require precise and accurate stoichiometric analyses and interpretation [79, 84]. Based on these findings, we focused on LUV and SUV in the stoichiometric discussion in this review.
The distinct sizes of LUV and SUV create a difference in the packing defect due to membrane bending and curvature [76]. Therefore, SUV show more packing defects than LUV. Packing defects are also affected by lipid compositions because the size and shape of lipid head groups and hydrophobic tails as well as the fluidity of membranes depend on the types of lipids [76]. The importance of packing defects in not only the functions of proteins, but also interactions between proteins and membranes has been demonstrated [33, 86, 87]. The sizes of viruses, mitochondria, and lysosomes are similar to that of LUV and the diameters of SUV and synaptic vesicles are similar. In order to investigate interactions between membranes and proteins in the exocytosis of synaptic vesicles, LUV and SUV are used to compare the effects of membrane curvature on protein functions [88, 89]. In the past two decades, LUV have also been utilized in studies on protein misfolding and aggregation with less focus on SUV [90, 91]. The following sections will be useful for understanding how SUV and LUV may be used in the study of protein aggregation.
2. Aggregation of amyloid β peptides in membrane environments
Amyloid β (Aβ) peptides consist of 39-42 residues and are produced by the sequential cleavage of the transmembrane amyloid precursor protein by β- and γ-secretases. Differences in the lengths of Aβ peptides depend on the cleavage site by γ-secretase [92–94]. Aβ(1-40) and Aβ(1-42) peptides are the most common types and have been implicated in the pathogenesis of Alzheimer’s disease. These two peptides both have nine positively- and negatively-charged residues and hydrophobic residues mainly in the C terminus. Aβ(1-40) and Aβ(1-42) peptides have been detected in senile plaques in the form of amyloid fibrils, and Aβ(1-42) peptides have been reported to exhibit higher cytotoxicity than Aβ(1-40) peptides. Thus, two additional hydrophobic residues in the C terminus make Aβ(1-42) more aggregation-prone and disease-related than Aβ(1-40) [95].
Previous studies suggested that nanomolar concentrations of Aβ peptides are present in vivo [93, 96–98], which is a lower concentration of Aβ needed for amyloid generation in vitro. A number of studies at the test tube level showed that more than a few micromolar concentrations of Aβ peptides are required for amyloidogenesis [99, 100]. Therefore, in order to obtain a plausible resolution for this issue, researchers have focused on the lipid membranes at which Aβ localizes from biological and pathological viewpoints. Yanagisawa and coworkers revealed the binding of Aβ peptides to ganglioside GM1, which accelerated the formation of amyloid fibrils [67]. A number of studies were subsequently performed in order to investigate the interaction between Aβ peptides and membranes in more detail. Matsuzaki and colleagues suggested that the binding of Aβ peptides to GM1-containing model membranes induced a structural transition in Aβ from a random coil to an α-helix at a low peptide/lipid ratio without aggregation (15 μM of Aβ(1-40) with 600 μM of GM1/cholesterol/SM (40:30:30) SUVs) [101]. However, membranes promoted the transition to β-sheet-rich conformations at high peptide/lipid ratios, which indicated that the formation of amyloid fibrils was expedited due to the acceleration of nucleation in membranes. Many other model membrane-based studies demonstrated that the propensity for and mechanisms underlying Aβ aggregation depended on the membrane components and physicochemical properties of constituent lipids. For example, a high level of cholesterol prevented fibrillation [102], while fibrillation was accelerated in low-polarity environments using a mixture of GM1, cholesterol, and SM and resulted in cytotoxicity [103]. Acidic lipids such as 1,2-dimyristoyl-sn-glycero-3-phosphor-rac-(1-glycerol) (DMPG) also accelerated fibrillation due to a direct interaction between the surfaces of membranes and Aβ [104].
One of the most critical physical features of membranes in living cells is curvature, which has attracted less attention in protein misfolding and aggregation in membrane environments. Pannuzzo and colleagues reported that the binding of Aβ to membranes bent membranes and increased curvature [105]. These findings also raised the question of whether differences in curvature affect the binding mode of Aβ to membranes and amyloid fibrillation. However, in spite of the importance of membrane curvature for protein aggregation, an insufficient number of detailed studies have been conducted. Therefore, we focused on the effects of membrane curvature on the amyloid formation of Aβ(1-40) and (1-42) peptides using lipid bilayers of different sizes [48]. We reviewed the key roles of membrane curvature in controlling the amyloidogenesis of Aβ(1-40) and (1-42) peptides.
2-1. Membrane curvature-dependent aggregation of Aβ peptide(1-40)
We initially examined the effects of the sizes of model membranes, i.e., curvature, on the amyloid fibrillation of Aβ(1-40) under physiological conditions (pH 7.4, 100 mM NaCl, and 37 °C) using the two types of vesicles, SUV with high curvature and LUV with low curvature [48]. We used 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) for the preparation of lipid bilayers because Aβ binding was previously shown to bend phosphocholine membranes, and, importantly, phosphocholine, a zwitterionic lipid, allowed us to focus on the effects of membrane curvature by eliminating the influence of electrostatic interactions between Aβ and membrane surfaces.
DOPC vesicles with diameters of 30, 50, 100, and 200 nm were added to solution containing Aβ(1-40) monomers, and aggregation kinetics were traced using the intensity of ThT fluorescence. The sizes of the DOPC vesicles had a dynamic impact on the amyloid fibrillation of Aβ(1-40) (Figs. 3 and 4A, B). Smaller vesicles (SUV of 30 and 50 nm) shortened the lag time and accelerated fibril growth (i.e., elongation) (Figs. 3A and 4B); however, the larger vesicles (LUV of 100 and 200 nm) did not significantly affect the lag time (Figs. 3D and 4B). Analyses of images obtained from TEM, AFM, and total internal reflection fluorescence microscopy (Fig. 3B and E) as well as the intensity of ThT fluorescence (Fig. 4A) revealed that i) SUV promoted the formation of amyloid fibrils and ii) LUV induced shorter amorphous-like amyloid fibrils. Similar amyloid-forming processes were observed when POPC SUV and LUV were incubated with Aβ(1-40).
Figure 3. Effects of model membranes of various sizes on Aβ(1-40) amyloid fibrillation and interactions between Aβ(1-40) and membranes.

(A, D) Aggregation kinetics monitored by ThT fluorescence in the absence (black) or presence of DOPC vesicles of 30 (pink) (A) and 100 nm [95] (D) at 37 °C without shaking. DOPC lipids in vesicles and Aβ concentrations were 10 μM. (B, E) AFM images of amyloid fibrils formed in the presence of DOPC vesicles of 30 (B) and 100 nm (E). (C, F) ITC measurements for DOPC vesicle:Aβ(1-40) interactions. ITC thermograms (upper panel) and binding isotherms (lower panel) for vesicles of 30 (C) or 100 nm (F). Figures were reproduced from reference [48].
Figure 4. Exposed hydrophobicity of liposomes and its effects on the fibrillation ofAβ.
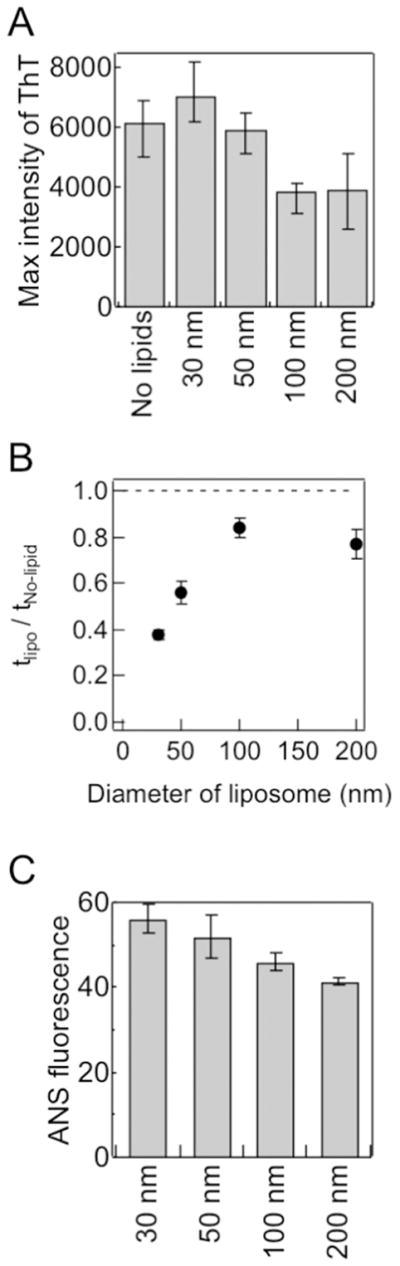
(A–C) Dependencies of the maximal ThT amplitude (A), relative lag time (B), and ANS fluorescence intensity (C) on vesicle size. Figures were reproduced from reference [48]. tlipo and tNo-lipid indicate the lag times of amyloid formation in the presence and absence of vesicles, respectively.
In an attempt to obtain mechanical insights into the aggregation mechanisms of Aβ(1-40), we characterized the binding of Aβ(1-40) with either SUV of 30 nm or LUV of 100 nm using isothermal titration calorimetry (ITC) (Fig. 3C and F). The ITC thermogram of the Aβ(1-40) titration to SUV of 30 nm showed a series of positive ITC peaks with gradual decreases in intensity, and endothermic reactions indicated intermolecular interactions between Aβ(1-40) and SUV (Fig. 3C). On the other hand, consecutive titrations of Aβ(1-40) to LUV of 100 nm showed gradual decreases in the amplitude of negative ITC peaks, which were indicative of exothermic binding and distinguishable from endothermic Aβ(1-40):SUV interactions (Fig. 3F). In addition, the extent of heat was larger and apparent affinity was stronger than those observed when Aβ(1-40) was titrated to SUV of 30 nm. Thus, these results revealed i) weak interactions between Aβ(1-40) and model membranes based on gradual changes in titration heat in the ITC thermogram, ii) the higher affinity of Aβ(1-40) for LUV than SUV, and iii) distinct binding modes of Aβ(1-40) depending on the difference in curvature in view of the reaction heat (endergonic vs exergonic). Similar endothermic and exothermic reactions were observed for the binding of magainin 2 amide and its analogs to LUVs and SUVs composed of POPC/POPG (3:1), respectively [106].
We then hypothesized that differences in packing defects between SUV and LUV [76] affected interactions between Aβ(1-40) and model membranes, thereby influencing the aggregation behavior and pathway of Aβ(1-40). In this context, packing defect-related hydrophobicity was tested using the fluorescence of 8-anilino- 1-naphthalenesulfonate (ANS). ANS is a dye that senses hydrophobic environments by enhancing its emission fluorescence, and is often used to detect the water-accessible hydrophobic regions of intermediates during protein folding [107]. The fluorescence intensity of ANS and vesicular size showed a linear relationship (Fig. 4C), which indicated that SUV with higher curvature and hydrophobicity has more packing defects than LUV, as suggested by the previous findings of other groups [108].
Collectively, we advocated models of the Aβ(1-40) aggregation pathway in membranes with distinct curvature based on the two types of interactions between Aβ(1-40) and model membranes (Fig. 5): one is the productive interaction leading to fibrillation-competent nucleation for typical mature amyloid fibrils and the other is the non-productive interaction that leads to amorphous-like amyloid formation in place of mature amyloid fibrils. The water-accessible hydrophobicity of model membranes probed by ANS binding and estimation of curvature suggested that SUV with higher surface curvature had more water-accessible hydrophobic regions and was effective for producing canonical amyloid nucleation. SUV with larger water-accessible hydrophobic regions weakly interacted with Aβ monomers, but adequately concentrated Aβ to initiate productive nucleation. Although the binding of Aβ(1-40) to LUV with lower curvature and smaller hydrophobic regions also occurred with higher affinity than SUV, it may not be fully productive for generating mature amyloid fibrils because the local concentration of Aβ(1-40) may not be sufficient to break the metastability of supersaturation. Alternatively, although the local concentration was sufficiently high, bound Aβ(1-40) molecules were tightly trapped on the surface of membranes and, thus, were unable to diffuse and transform to seed-competent conformations. These interactions of Aβ(1-40) with LUV may eventually result in the production of ThT-positive short amorphous-like amyloid fibrils.
Figure 5. Schematic models of amyloid nucleation and amorphous aggregation on the surface of liposome membranes.

(A, B) Schematic mechanism of Aβ(1-40) fibrillation in the presence of SUV (A) and LUV (B). Aβ(1-40) interacts weakly with the hydrophobic clefts of SUV, which promotes productive nucleation (A). Aβ(1-40) interacts with the surface of LUV more strongly than SUV, which may prevent the same nucleation observed in SUV and, thus, induce other types of nuclei that form amyloid fibrils with fast aggregation kinetics, such as rapid amorphous aggregation, i.e., the formation of amorphous-like amyloid fibrils (B) [48]. Amyloid fibrillation and amorphous aggregation were kinetically distinguishable based on productive nucleation, which generates the lag time [2, 38, 49, 51, 53, 149]. Amyloid fibrils typically form following the lag time with the sigmoidal pattern of the intensity of ThT fluorescence, while amorphous aggregation is very rapid without an appreciable lag time or marked ThT fluorescence emission intensity. We observed aggregation that was accompanied by relatively low ThT intensities with no or short lag times as well as the far-UV CD spectra of cross-β structures. These aggregates were small in the AFM image and appeared to be distant from mature amyloid fibrils, but adopted fibrillar structures in the TEM image. Furthermore, their morphologies differed from those of protofibrils. We referred to these aggregates as amorphous-like amyloid fibrils that share the properties of amyloid and amorphous aggregates in terms of kinetic, tinctorial, structural, and morphological features.
In this section, we described the curvature dependence of the binding mode and affinity, aggregation kinetics, and types of aggregates of Aβ(1-40). The effects of the curvature-dependent aggregation process and final aggregates on the membrane integrity of and toxicity against cells will provide more biologically and pathogenetically relevant implications. Not only the high-resolution structures of Aβ(1-40) in SUV and LUV, but also a molecular dynamics simulation will be a challenge for obtaining a more thorough understanding of the mechanical binding mode and nucleation at the atomistic and molecular levels.
2-2. Aggregation of Aβ peptides, including their peptide fragments
Although Aβ(1–40) is more prominent in physiological cellular environments, Aβ(1–42) is considered to be more neurotoxic possibly due to its higher propensity for aggregation [95]. However, the aggregation of Aβ(1-42) has been studied in less detail than that of Aβ(1-40) due to its strong propensity for aggregation, which has markedly hampered reproducible and accurate experiments and the efficient application of various biochemical and biophysical methods. In this section, we describe how different membrane curvatures impact on the aggregation behaviors of Aβ(1-42) with comprehensive models of the polymorphic and dilution-like inhibitory mechanisms. The stability of amyloid structures generated in model membranes and salt effects on Aβ amyloidogenesis will also be addressed.
2-2-1. Distinct self-assembly process of Aβ(1-42) in SUV and LUV
Aβ(1-42) aggregation depending on the sizes of the model membranes of POPC (SUV of ~30 nm and LUV of ~140 nm) was examined [53]. No significant changes in the secondary structures of Aβ(1-42) due to the addition of SUV by varying the concentration of POPC were observed before or after the incubation. Random-coil and β-sheet structures at any concentration of POPC were revealed before and after the reaction, respectively. However, the kinetic parameters of Aβ(1-42) amyloid formation (lag times, elongation rate constants, and the maximum intensities of ThT) largely depended on the concentration of POPC: lag times became longer and then shorter, elongation rates became higher, and the maximum intensities of ThT fluorescence decreased as the POPC concentration increased (Fig. 6A–D). Morphological analyses using AFM and TEM revealed the formation of long and short amyloid fibrils at low and high concentrations of POPC (high and low peptide/lipid ratios), respectively (Fig. 6I).
Figure 6. Effects of vesicle size on the amyloid formation of Aβ(1–42) at various POPC concentrations.

(A–H) The kinetic parameters and ThT maximum intensity of Aβ1-42 amyloid fibrillation in SUV (A–D) and LUV [229] are indicated. Representative fit curves (solid lines) of the raw kinetic data (markers) of Aβ1-42 amyloid formation (A, E) and lag times (B, F), elongation rates (C, G), and maximum ThT intensities of Aβ1-42 aggregation (D, H). The lag time and elongation rate were obtained using fitting analyses, and maximal ThT fluorescence intensities after an ~50-hr incubation are shown in A and E. Average values and error bars (B–D and F–H) were calculated using the fit results of three independent measurements that consisted of three data points. (I, J) AFM images of Aβ(1-42) solution after kinetic measurements at 10 μM (left) and 1 mM POPC [218] in SUV (I) and 10 μM (left) and 1 mM POPC [218] in LUV (J) are shown with scale bars indicating 1 mm. The inset in C is a TEM image of Aβ(1-42) solution after the kinetic measurement at 1 mM POPC lipid of SUV. Figures were reproduced from reference [53].
On the other hand, LUV affected the amyloidogenicity of Aβ(1-42) itself without a change in the initial (random coils) and final (β-rich) structural states. The increase observed in the POPC concentration (the lipid/peptide ratio) resulted in a decrease in the ThT fluorescence intensity at the stationary stage. Fitting analyses confirmed that amyloid fibrils formed without marked differences in their lag times or elongation rate constants at low and intermediate POPC concentrations (Fig. 6E–H). At high POPC concentrations, no amyloid fibrils or any other types of aggregates were detected (Fig. 6E–H and J). These results demonstrated that increases in the amounts of LUV prevented Aβ(1-42) amyloid formation, and indicated that the soluble molecular species of Aβ(1-42) remained in an unstructured state. Intermolecular affinity between Aβ(1-42) and two types of POPC vesicles was found to be weak in nature based on the lack of the appreciable binding heat of ITC thermograms.
Thus, the patterns of Aβ(1-42) amyloid formation varied with the curvature of model membranes, even at the same lipid concentration (the peptide/lipid ratio), and we suggested new mechanical models of curvature-dependent polymorphic amyloidogenesis as well as the inhibition of the amyloid generation of Aβ(1-42) (Fig. 7). SUV shows low packing efficiency among lipids (i.e., packing defects) [78, 88, 109–112], which may provide inhomogeneous hydrophobic clefts in the lipid bilayer [76, 78, 110]. In contrast, LUV shows fewer packing defects and lower hydrophobicity [48, 88, 111]. Aβ(1-42) monomers may bind to several types of hydrophobic clefts (binding sites) in SUV with different affinities through mainly hydrophobic interactions (Fig. 7A and B) [113, 114]. The accumulation of Aβ(1-42) at individual binding sites increased local concentrations of Aβ(1-42) and induced productive nucleation, which resulted in the formation of the distinct morphology of amyloid fibrils with different kinetics, i.e., polymorphs of amyloid fibrils (Fig. 7A). Different lengths of amyloid fibrils in equilibrium may be broadly referred to as polymorphs. Other experimental findings, such as TEM, CD, FT-IR, or NMR, will strengthen the suggestion of polymorphs. Caution is needed when the polymorphism of amyloid fibrils is addressed based on the lengths of amyloid fibrils. Similar polymorphic Aβ amyloid formation was observed when the physicochemical properties of surfaces were changed [115, 116].
Figure 7. Models of amyloid fibril formation of Aβ(1–42) in vesicles with distinct curvature.

(A–D) SUV (A and B) and a part of an LUV (C and D) are represented. The head groups and tails of POPC are colored in yellow and gray, respectively. Curves and rectangular figures indicate Aβ(1-42) monomers and amyloid fibrils, respectively. (A, B) Several clefts of a distinct size, shape, and hydrophobicity in SUV are illustrated. Aβ(1-42) monomers that interact weakly, moderately, and strongly with packing-defected clefts are colored in blue, orange, and red, respectively. Amyloid fibrils are represented by the same color of constituent monomers. (C, D) Blue curves and rectangles signify Aβ(1-42) monomers and amyloid fibrils, respectively [53].
Aβ(1-42) binds to the surfaces or shallow hydrophobic clefts of LUV, and increases in local concentrations at higher peptide/lipid ratios lead to amyloid formation (Fig. 7C). However, an increase in the amount of LUV inhibits Aβ(1-42) amyloid fibrillation due to insufficient local concentrations, i.e., dilution effects, for productive nucleation (Fig. 7D) [48], as observed in Aβ fibrillation with nanoparticles [117, 118]. Further decreases in the peptide/lipid ratio in SUV showed the same dilution-like inhibitory mechanism of Aβ(1-42) amyloid generation (Fig. 8A). The concept of the local concentration and peptide/lipid ratio may be a common and simple interpretation for the acceleration and blockade of amyloid formation in a membrane environment regardless of the sizes of biomimetic model membranes.
Figure 8. Stability of Aβ(1-42) amyloid fibrils formed in vesicles with distinct curvature and the inhibition of amyloidogenesis.

(A) Real-time monitoring of the ThT fluorescence intensity of the amyloid aggregation of Aβ(1-42) at 5 μM was performed at 1 [95], 5 (orange), and 10 mM POPC (red) in order to test the inhibition of amyloid formation through dilution effects, as detected in LUV at high concentrations of POPC lipids. (B, C) Far-UV CD spectra of amyloid fibrils of 5 μM Aβ(1-42) formed without vesicles (B) and with SUV (C) at 37 °C before (black) and after [95] heat scanning as well as at 110 °C (red). (D) The real-time monitoring of the ThT fluorescence of amyloid fibrils of Aβ(1-42) formed in the presence of SUV at POPC concentrations of 0 (black), 10 (cyan), and 1000 μM (red) was performed after the dilution of Aβ(1-42) amyloid solution. Figures were regenerated from reference [53].
2-2-2. Conformational stability of different curvature-induced Aβ amyloid fibrils
Studies on the stability of amyloid fibrils have greatly contributed to advances in our understanding of the physical and thermodynamic natures of proteins together with knowledge on globular proteins [39, 119]. They are also beneficial for elucidating the prion-like behaviors of amyloid fibrils via cell-to-cell propagation [120, 121] because the surrounding conditions for intercellular transmission may affect the structural stability of amyloid fibrils. In addition, amyloid stability and reversibility may be viewed as key factors to maintain proteostasis in terms of protein turnover and amyloid clearance [122–124] and are useful for examining polymorphic amyloidogenesis.
The hierarchical structures of amyloid fibrils elucidated using X-ray crystallography and solid-state NMR spectroscopy appear to be very stable [125–129]; however, amyloid fibrils showed nearly complete denaturation (i.e., depolymerization or dissociation) to monomers or other types of aggregates. External perturbations, including temperature [119, 122], chemical denaturants [130, 131], pressure [65, 66], mechanical perturbation [132], and the presence of additives, including polyphenols [133, 134], destabilized and denatured amyloid fibrils (several tens of kJ mol−1 of a change in Gibbs free energy) (Fig. 9). Previous studies reported that the stability of amyloid fibrils was similar to that of globular proteins [39, 119, 122, 135]. In contrast to the intramolecular interaction-steered folding of globular proteins, the stability of intermolecular interaction-driven amyloid formation depends on the critical concentration for aggregation, as described for micellization. Therefore, dilution also modulates amyloid stability [99, 124]. Increases in intra- and intermolecular contacts often stabilize globular proteins [136], and the binding of SDS or some types of salts enhanced the stability of amyloid fibrils against high temperatures (Fig. 9) [119].
Figure 9. Energy landscapes of the stability of amyloid fibrils.

Amyloid fibrils (black), which formed from monomers [95] under aggregating conditions (middle), are stable with a change in Gibbs free energy (ΔG)1 (ΔG1). Extrinsic stresses, such as temperature, pressure, and dilution, destabilize amyloid fibrils (red) with energetic loss by |ΔG1 + ΔG3|[218]. Other types of aggregates (orange), such as amorphous or fibrillar aggregates, often form. The presence of SDS or some types of salts, such as NaCl, stabilize amyloid fibrils (blue) by decreasing the level of G (ΔG2).
Based on these findings, the conformational stability of Aβ(1-42) amyloid fibrils, which made direct contact with membranes, and the effects of membrane curvature were examined using temperature and dilution as outer stresses. Increases in temperature from 37 to 110 °C decreased the CD signal intensity of amyloid fibrils formed in the absence and presence of either SUV with distinct concentrations of POPC or LUV (Fig. 8B and C). Far-UV CD spectra at 110 °C and after cooling to 37 °C were similar, and round aggregates without amyloid fibrils were observed in AFM images following heat scanning. These findings suggested that POPC membrane curvature and polymorphic properties did not markedly affect the stability of Aβ(1-42) amyloid fibrils, and the irreversibility of the heat denaturation of amyloids was attributed to amorphous aggregation. A decrease in the concentration of Aβ(1-42) to 1 μM by dilution, which is less than the critical concentration for amyloid formation, reduced ThT intensities with the incubation time (Fig. 8D), indicating the denaturation of amyloids. Although no marked differences were observed between the amyloids formed in the absence and presence of LUV, an increase in the stability of polymorphic amyloid fibrils formed in SUV at different concentrations of POPC against dilution was noted.
Therefore, it was concluded that the curvature of the POPC model membranes used here did not markedly affect the conformational stability of Aβ(1-42) amyloid fibrils for thermal stress; however, the stability of amyloids formed in the absence and presence of SUV was affected by dilution to some extent. More case studies based on various membrane compositions are required in order to obtain the general features of amyloid stability in a membrane environment with different curvatures, which will provide therapeutic perspectives for the better removal of amyloid fibrils.
2-2-3. Amyloidogenesis of two types of Aβ peptides
Packing defects in SUV with high curvature also induced the polymorphic amyloid formation of Aβ(1-40) in buffered solution without additional salts [48, 53, 137]; however, its amyloidogenicity was lower than Aβ(1-42). Aβ(1-40) did not form amyloid fibrils in LUV at the same POPC concentration range in SUV. The presence of additional salts (100 mM NaCl) increased the amyloidogenicity of Aβ(1-40) in SUV and still retained the polymorphism. The addition of salts markedly accelerated Aβ(1-40) amyloid formation in LUV. Thus, the importance of the salt concentration for the transition of non-productive nucleation to productive nucleation by condensing Aβ peptides in model membranes, particularly for LUV, was demonstrated.
Large differences in the amyloidogenicity of Aβ(1-40) and Aβ(1-42) depending on the curvature of vesicles are most likely attributed to the hydrophobic interaction between the C terminus of Aβ and membranes. Two additional C-terminal hydrophobic residues of Aβ(1-42) may promote nucleation more efficiently than Aβ(1-40) [138]. The more rigid C-terminal regions of Aβ(1-42) may also be related to membrane interactions and amyloidogenicity [139, 140]. Salt-enhanced hydrophobic interactions may be viewed as an accelerator of amyloid generation in LUV.
Overall, vesicle size-dependent differences in packing defects may be a key factor regulating the amyloid aggregation of Aβ peptides. Aβ amyloid fibrillation in membranes with large packing defects may be more deleterious to cells due to high amyloidogenicity than that on larger flat surfaces. Aβ sequestration by flat membranes at low Aβ/lipid ratios may help maintain Aβ solubility. Biological membranes play a role in elucidating the pathway of protein aggregation by adjusting the degree and metastability of supersaturation over the thermodynamic solubility limit [38, 49], which is essential for (non-)productive nucleation [48, 124]. Further examination of curvature-dependent Aβ aggregation in terms of the phase transition using charged lipids and other membrane components will provide valuable mechanical, thermodynamic, and kinetic insights into the molecular mechanisms underlying Aβ amyloid fibrillation and amorphous aggregation.
3. Parkinson’s disease and αSN aggregation in presynaptic vesicles
Parkinson’s disease is the second most neurodegenerative disorder and affects 6.2 million people globally, killing approximately 117,400 individuals in 2015 [141]. Parkinson’s disease is a long-term disorder that develops slowly; movement impairments in the early stage of the disease precede dementia in the advanced stage [142]. The aggregation of αSN and its deposition to the substantia nigra in the form of amyloid fibrils have been identified as the pathological hallmarks of Parkinson’s disease [11]. αSN and its aggregates are responsible for synucleinopathies, including Lewy body dementia [143]. Thus, studies on αSN aggregation and its structural aspects in monomeric and aggregated states as well as the resulting cytotoxicity have been extensively performed in vivo and in vitro for two decades [11, 12, 144, 145]. Interactions between αSN and other molecules have increasingly become research targets for amyloidogenesis and pathogenesis [146–148].
αSN is a 140-residue soluble protein that is intrinsically disordered under physiological conditions [147, 149]. It is largely divided into three regions based on the physicochemical properties of amino acid residues (Fig. 10A). The N-terminal region (residues 1 - ~60) is amphipathic in nature and contains many positive charges at neutral pH. The N-terminal region plays a central role in binding to membrane surfaces. The non-Aβ component [150] region (residues ~60 - 100) displays a high propensity for aggregation with high hydrophobicity and orchestrates core regions buried in the amyloid structure. The C-terminal region (residues ~100 - 140) is very negatively charged [151].
Figure 10. Amyloid aggregation kinetics of two types of αSNs in Mimic SUV with high curvature.

(A) Three major regions of αSNWT are schematically indicated with the residue number and color: the N-terminal region (NTR) (blue), non-amyloid β component [150] region [230], and C-terminal region (CTR) (red). (B, E) Kinetics of the amyloid aggregation of αSNWT (B) and αSN103 (E). Raw data and fit curves are shown using closed circles and solid lines, respectively. (C, D, F, G) Far-UV CD spectra of αSNWT (C, D) and αSN103 (F, G) were obtained immediately after sample preparation (~5 min) (C, F) and after a 24-h incubation (D, G). (H–M) Maximum ThT fluorescence intensities (H, K), lag times (I, L), and elongation rate constants (J, M) of the amyloid formation of αSNWT (H–J) and αSN103 (K–M). Average values were calculated from the data of three independent experiments. The color code in B–M represents the distinct concentrations of lipids (0 mM (black), 0.5 mM (light blue), 1 mM (blue), 2 mM (green), 3 mM (yellow), 4 mM (pink), and 5 mM (red)). Figures were reproduced from reference [149].
Although αSN may serve as a linker among small vesicles and function in the fusion of presynaptic vesicles, its biological functions remain unclear [152]. Historically, αSN was detected around the synaptic vesicle and nuclear membrane, and, thus, was named by the properties of its localization in cells [153, 154]. αSN also exists around other organelles, including mitochondria and endoplasmic reticula (refer to Section 4) [155–157]. Although a line of compelling evidence indicated the presence of αSN around synaptic vesicles, which may account for the molecular mechanism of αSN-mediated impairments in neuronal function and cell death [158], further studies are needed on the localization of αSN around organelles in order to clarify the onset and development of the disease. Therefore, the interactions between αSN and synaptic vesicles and the resulting effects on αSN aggregation have attracted increasing attention [159]. αSN has exhibited a binding capability for SUV mimicking the presynaptic vesicles of DOPC, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), and, subsequently, to change the conformational states from random coil to α-helical structures [114]. Electron paramagnetic resonance was successfully used to examine the structure of αSN bound to vesicles of anionic phospholipids [160]. The atomistic structural model based on simulated annealing molecular dynamics with restraints from electron paramagnetic resonance data showed an extended, curved α-helical conformation of αSN (residues of~10 - 90) in the membrane-bound state. Multiple lysine residues in the N-terminal region of αSN were suggested to be critical for membrane interactions. The lipid concentration-dependent β-structured amyloid formation of αSN in the SUV and LUV of 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine under artificial conditions, such as non-physiological ionic strength, has been suggested [161]. Intermediate states in the pathway of the structural transition of αSN from membrane-bound α-helical states to β-sheet fibrillar states have been characterized using solid-state NMR spectroscopy [162]. Although other model membrane systems have been used to investigate the effects of micelles or vesicles on the structures of αSN [159, 163] and aggregation behaviors of αSN [164, 165], the structural states of membrane-bound αSN and their relationship to aggregation in presynaptic vesicles have not yet been elucidated in detail due to the limited number of studies that have been conducted.
Thus, in this section, we provide a comprehensive description of a recent study on the amyloid aggregation of αSN using two types of αSNs and SUVs, including the presynaptic vesicle-mimicking model membrane. Comparisons of αSN structures in the absence and presence of each type of SUV as well as simple mechanical models for amyloid generation and inhibition will be described. Phase diagrams based on the non-two-state structural transition model will be shown, which is expected to be effective for understanding αSN aggregation from microscopic and macroscopic viewpoints.
3-1. αSN amyloid formation in SUV mimicking a presynaptic vesicle with high curvature
With the aim of obtaining insights into the relationship between the structural states of αSN and their amyloidogenesis in the presence of SUV at neutral pH and physiological ionic strength, we used unstructured wild-type αSN (αSNWT) and a C-terminally truncated mutant (αSN103) lacking negative charges [149]. The αSN103 mutant was previously reported to promote amyloid generation and aggregates containing a C-terminally truncated αSN were found in Lewy bodies implicating the pathological relevance of a C-terminally truncated form [166]. Two types of model membranes were utilized: SUV that mimics the presynaptic vesicle (Mimic SUV) using DOPC/DOPS/DOPE (2:3:5) [147, 149, 167] and another type of SUV that is composed of DOPC (DOPC SUV) as a control for the contribution of the charges of membranes.
CD analyses revealed that the presence of DOPC SUV did not significantly change the random coil structures of αSNWT and αSN103 in the initial membrane-bound states. However, the addition of Mimic SUV markedly altered the initial structures of the two types of αSNs from random coil structures to α-helical structures in a lipid concentration-dependent manner. The effects of these initial states on the aggregation of αSNWT and αSN103 were investigated using the ThT assay as well as CD and AFM analyses. Random coil-like αSNWT in DOPC SUV did not show significant changes in amyloidogenicity. However, Mimic SUV markedly affected the amyloid aggregation of helical αSNWT in a dose-dependent manner. αSNWT fibrillation was promoted at the lower concentration of Mimic lipids, and increases in the lipid concentration blocked amyloid formation (Fig. 10B, C, and D). The amyloid formation of αSN103 in the absence and presence of the two types of SUVs was generally faster than that of αSNWT. Although no marked influence of DOPC SUVs on the amyloid generation of largely unstructured αSN103 was detected, similar to αSNWT fibrillation, the amyloid formation of helical αSN103 was prominent in the presence of Mimic SUV. At all of the Mimic lipid concentrations used, αSN103 in helical structures formed amyloid fibrils with distinct kinetic properties. The lag time did not depend on the concentration of Mimic lipids: however, the elongation rate was enhanced by increases in the concentrations of lipids (Fig. 10E, F, and G).
We attributed the prominent effects of Mimic SUV on the amyloid aggregation of αSNWT and αSN103 using two mechanical models with the feature of the initial structural state of αSNs: a two-state binding model (Fig. 11) and a non-two-state structural transition model (Fig. 12) (refer to the next section). The two-state binding model assumes a transition between unstructured αSN in membrane-unbound states and helical αSN in membrane-bound states. In this model, the population of membrane-bound αSN plays a pivotal role in amyloid formation. Low and moderate populations of helical αSNWT (i.e., high and moderate αSN/lipid ratios) in Mimic SUV accelerate amyloidogenesis in a lipid concentration-dependent manner. Further increases in the population of membrane-bound helical αSNWT (i.e., the low αSN/lipid ratio) result in the inhibition of amyloid formation (Fig. 11A).
Figure 11. αSN amyloid formation based on the two-state binding model.
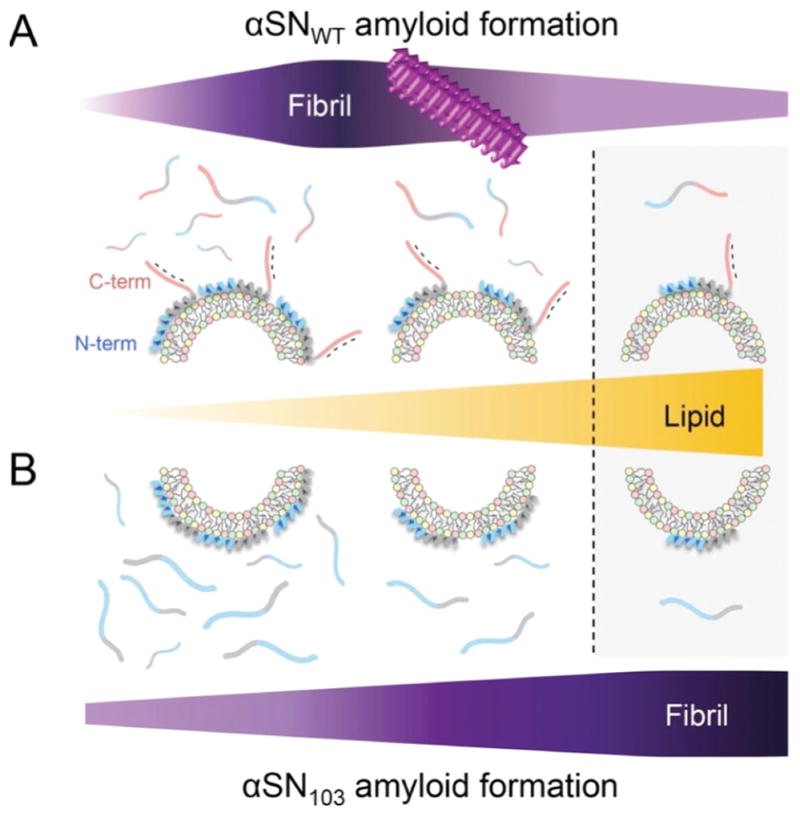
(A, B) The two-state binding model between unstructured αSNs in membrane-unbound states and helical αSNs in membrane-bound states as well as the amyloid formation of αSNWT (A) and αSN103 (B) with Mimic SUV are schematically represented. Increases in the concentration of lipids (orange triangle, middle) and amyloidogenicity (purple figures, top and bottom) are highlighted with darker colors in A and B. The vertical broken line indicates the highest concentration of Mimic lipids used in this study (5 mM), with the population of membrane-bound αSNs being ~ 65 - 70%. Three major regions of αSN are represented using different colors, as described in Figure 10A. Negative charges in the C-terminal region are indicated with “-”. The figure is reproduced from reference [149].
Figure 12. Initial structure-dependent αSN amyloidogenesis based on the non-two-state binding model.

(A, B) Membrane-bound structures and the amyloid formation of αSNWT (A, upper) and αSN103 (B, lower) are schematically represented. Increases in the content of helical structures (blue triangle, left), concentration of lipids (orange triangle, middle), and amyloidogenicity (purple figure, right) are highlighted with darker colors in A and B. Lipid concentration-dependent structural changes in αSNs on Mimic and DOPC membranes are guided using arrows with yellow and light grey, respectively. Three major regions of αSN are represented using different colors, as described in Figure 10A. Negative charges in the C-terminal region are indicated with “-”. N and C termini are labeled as N-term and C-term, respectively. The random coil-like, partially-helical, and largely-helical structures of αSNs are indicated by RCS, PHS, and LHS, respectively. Amyloid fibrils on model membranes are also schematically shown on the right. This figure is reproduced from reference [149].
On the other hand, helical αSN103 aggregates to amyloid fibrils at all concentrations of Mimic lipids without showing amyloid inhibition, even at the same concentration range of lipids and with a similar population of bound αSN to αSNWT (Fig. 11B). Thus, the initial structural states of αSN as well as the charged states responsible for electrostatic repulsion between αSN and negatively charged membranes were concluded to be critical for modulating amyloidogenicity. Low populations of largely disordered αSNWT or αSN103 in DOPC membrane-bound states showed low amyloidogenicity. These findings also suggested that the population of membrane-bound αSNs is essential for amyloid-competent nucleation (i.e., productive nucleation).
The biological and pathological importance of the relative ratio of αSN strains to synaptic vesicles in neurons was also indicated. When the ratio of αSNWT/synaptic vesicles is low, the suppression of amyloidogenesis will be favored due to the thermodynamic stabilization of highly helical αSNWT, which is nucleation-incompetent, or the low effective local concentration, which is insufficient for nucleation. In this ratio, αSN may function as a regulator of neurotransmission and synaptic vesicle fusion [145, 146]. High and intermediate ratios may be risky with the loss of biological function due to the kinetic and thermodynamic promotion of pathogenic amyloid formation. In addition, it is important to note that interactions between synaptic vesicles and C-terminally truncated αSN variants may be more deleterious to neurons than those with αSNWT due to the higher amyloidogenicity of a C-terminally truncated mutant. Thus, mutations, post-translational modifications, and environmental conditions, including factors affecting the expression levels of αSNs, lipid metabolism, and metal homeostasis, will have an impact on the αSN/lipid ratio that limits amyloidogenicity.
3-2. A non-two-state structural transition model and phase diagram
The amyloid formation of helical αSN103 in Mimic SUV was not inhibited at lipid concentrations at which the amyloid aggregation of helical αSNWT was blocked in spite of a similar helical content and membrane-bound population. In addition, the Mimic lipid concentration-dependent amyloid formation of helical αSN103 with distinct kinetics was also revealed, and may be ascribed to polymorphic amyloid formation. These aggregation behaviors of helical αSN103 in Mimic SUV were not completely explained by the two-state binding model. Thus, a non-two-state structural transition model that postulates the formation of multiple helical conformations of membrane-bound αSNs depending on the concentrations of Mimic lipids has been suggested (Fig. 12) [149]. Detailed CD analyses revealed the lipid concentration-dependent initial structures of αSNs in membrane-bound states. Increases in the concentration of Mimic lipids generally resulted in higher α-helical contents in αSNs and induced different helical structures.
Specific structures of αSNWT with partially helical folds at moderate concentrations of Mimic lipids were suggested to be the most amyloidogenic, although largely disordered structures with less helical regions formed amyloid fibrils with lower amyloidogenicity at low concentrations of Mimic lipids (Fig. 12A). The largely helical structures of αSNWT at higher concentrations of Mimic lipids may be non-amyloidogenic due to physical properties, such as high solubility and phase stability. Multiple specific structures of αSN103 with different helical folds at various concentrations of Mimic lipids were all suggested to be amyloidogenic, which induced different nucleation for the polymorphism of amyloids (Fig. 12B). The lack of disordered regions and the charges of the C-terminal region of αSN103 relative to αSNWT were also considered to produce differences in amyloid inhibition.
Amyloidogenic intermediates of αSN have also been observed with changes in pH, temperature, and the concentration of alcohol [148]. Different binding modes of αSN on model membranes have been detected [88, 147], and multiple distinct phospholipid-binding modes of αSN were indicated based on the findings of solution NMR spectroscopy [164]. Furthermore, recent studies showed that a native-like mutant of the SH3 domain, a folding intermediate of β2-microglobulin, and partially unfolded lysozyme and insulin were amyloidogenic, which supported the importance of the specific structural states of aggregation-prone precursors for amyloid generation [168, 169]. The distinct helical conformations of unfolded proteins and/or peptides, such as lysozyme, insulin, cytochrome c, Aβ peptide, glucagon, and β2-microglobulin, may be explained by a non-two-state structural transition model [9, 38, 49, 51, 56]. Collectively, these findings suggest that the non-two-state structural transition model will also be useful for understanding amyloidogenesis with the two-state binding model.
Phase diagrams are effective for understanding the aggregation of several proteins in terms of thermodynamics and kinetics because they show phase and structural transitions based on the solubility and metastability of supersaturation (Figs. 13 and 14) [8, 12, 151, 170]. The apparently soluble states of αSNWT and αSN103, which are kinetically trapped by high metastability for nucleation are shown in DOPC and Mimic SUVs (Fig. 13). Thus, reaching an equilibrium between amyloid fibrils and residual monomers takes time (i.e., the lag time). The mechanical stimulation of various agitations and other factors, such as the presence of membranes, disrupt the metastability of supersaturation and promote equilibration. The phase states of αSNWT and αSN103 in real equilibrium are shown in Figure 14. Phase diagrams will be also useful for any phase transition and separation of substances.
Figure 13. Phase diagrams of αSN aggregation on model membranes in initial states.

(A–D) The phase states of αSNWT (A and C) and αSN103 (B and D) in the presence of DOPC (A and B) and Mimic SUV (C and D) in the initial states before the incubation. Regions of unstructured and helically-folded αSN are shown in blue and light green, respectively. Denser colors represent greater possibilities for the existence of corresponding molecular species depending on the concentrations of αSN and lipids. The yellow broken lines of the conceptual solubility curves in each phase diagram are represented for comparing solubility before (Figure 13) and after equilibrium (Figure 14) (i.e., before and after the disruption of the metastability of supersaturation). Broken black lines are also shown to represent the conceptual concentration of αSNs used in the present study for a better understanding of molecular species. Molecular species that are present under each condition are schematically shown. Three major regions of αSN are represented using different colors, as described in Figure 10A. Increases in the content of α-helix in αSNs with higher concentrations of lipids are shown above individual phase diagrams. RCS, PHS, and LHS indicate random coil-like structures with less helicity, partially-helical structures, and largely-helical structures, respectively. X and Y axes indicate the concentrations of lipids and αSNs, respectively. This figure is reproduced from reference [149].
Figure 14. Phase diagrams of αSN aggregation on model membranes in equilibrium.

(A–D) The phase states of αSNWT (A and C) and αSN103 (B and D) in the presence of DOPC (A and B) and Mimic SUV (C and D) in the two-state equilibrium between residual αSN monomers and αSN amyloid fibrils after incubations. The regions of unstructured and helically-folded αSN are shown in blue and light green, respectively. Insoluble αSN amyloid fibrils are also represented in red. Denser colors represent greater possibilities for the existence of corresponding molecular species depending on the concentrations of αSN and lipids. The yellow solid lines in each phase diagram indicate conceptual solubility curves. The conceptual solubility curve represented in Figures 13 and 14 is experimentally obtained using the concentrations of monomers [38, 39, 49, 51]. We did not construct actual solubility curves for Figures 13 and 14 because we did not measure the concentrations of residual monomers in αSN, which must be in equilibrium with amyloid fibrils under each condition in Figure 14. Thus, we attempted to show conceptual solubility curves in Figures 13 and 14 based on other findings in reference [149] and our previous studies [38, 39, 49, 51], which may account for a propensity for changes in solubility. In the initial states (i.e., before the equilibrium state) (Figure 13), αSN maintained kinetically thermodynamic solubility by taking advantage of the metastability of supersaturation. The amyloidogenicity of αSN may increase at higher concentrations of αSN, as revealed in our previous study [141]; for example, broadening of the red region in C. Broken black lines are also shown to represent the conceptual concentration of αSNs used in the present study in order to obtain a better understanding of molecular species. The molecular species that are present under each condition are schematically shown. Three major regions of αSN are represented using different colors, as described in Figure 10A. The increase in the α-helix content in αSNs in the initial states after sample preparation with higher concentrations of lipids is shown above individual phase diagrams. RCS, PHS, and LHS indicate random coil-like structures with less helicity, partially-helical structures, and largely-helical structures, respectively. X and Y axes indicate the concentrations of lipids and αSNs, respectively. This figure is reproduced from reference [149].
4. Localization of amyloidogenic precursors in subcellular organelles may open new avenues
A large body of evidence has suggested the localization of amyloidogenic proteins and peptides in subcellular organelles. αSN is localized to several intracellular organelles, including the nucleus [171], cytosol [172], mitochondrion [173], and endoplasmic reticulum (ER) [174]. In addition, αSN has been detected in mitochondria-associated ER membranes (MAM) [175–179], which is a subregion of the ER connected to mitochondria. Aβ peptides, which have predominantly been reported to form extracellular aggregates, were found to localize and bind to mitochondria, leading to cytochrome c-dependent apoptosis [180]. In this context, a number of the physicochemical properties of the membranes of these organelles, including curvature, may affect aggregation behaviors, thereby causing dysfunctions in organelles and subsequent cytotoxicity. Of particular interest will be the effects of MAM on the amyloid generation of αSN and Aβ peptides and the resulting deleterious effects. The membranes of MAM are composed of cholesterol, negatively charged phospholipids, and intracellular lipid rafts [175], which are expected to facilitate the binding of amyloidogenic precursors. Thus, it is important to note that αSN and Aβ peptides as well as their pathogenic mutants and fragment peptides may interact with MAM membranes by showing α-helical conformational transitions, which modulate populations of membrane-bound species and the nucleation of amyloidogenesis. A change in the curvature of model membranes mimicking MAM may provide further insights into the effects of membrane curvature.
Of note, pathogenic mutations in αSN have been shown to disrupt calcium homeostasis in MAM [157], which, in turn, results in alterations in respiration and adenosine triphosphate production and enhances oxidative stress. Mitochondria communicate with ER via MAM to regulate several important physiological phenomena [181], including phospholipid and cholesterol metabolism, cellular calcium homeostasis, ER stress, the unfolded protein response, autophagy, and mitochondrial biogenesis by using calcium as a second messenger. Therefore, these important biological roles of MAM may contribute to clarifying the mechanisms of cytotoxicity by the aggregation of αSN and possibly Aβ peptides. Twenty members of the PDI family, which controls protein homeostasis mainly in the ER [182], in the mammalian ER [183, 184] and some PDI family members in MAM, such as PDI, ERp46, and ERp57 [185], have recently been identified, suggesting interactions between PDI family members and amyloidogenic precursors. Thus, examinations of the interactions of PDIs with αSN and Aβ peptides in membrane environments and their effects on amyloid formation and cell toxicity are warranted and will be useful for revealing not only the underlying mechani sms of the formation and inhibition of amyloid fibrils, but also the physiological function and trafficking of soluble αSN, which currently remain unclear.
5. Effects of membrane curvature on IAPP islet cell toxicity
Lipid membranes have been shown to play very important roles in the aggregation and fibril formation of many amyloid peptides and proteins. While the amyloid aggregation process is influenced by lipid membranes and depends on the membrane composition, the process of aggregation has been shown to induce cell toxicity. A two-step process for membrane disruption and cell toxicity has been demonstrated for amyloid-beta peptide and Islet Amyloid Polypeptide (IAPP, also known as amylin) [186]. This two-step process is broadly characterized as pore formation and the fibril formation-induced detergent-like mechanism of membrane disruption. These mechanisms of membrane disruption have similar characteristics to those reported for antimicrobial peptides [184, 187, 188]. A number of biophysical studies elucidated the two-step mechanism for amyloid peptides and demonstrated that pore formation was altered by pH, metal ions, and small molecule compounds, whereas the fibril formation mechanism was influenced by small molecule inhibitors, metals, and other proteins [13, 189, 190]. For example, the presence of calcium and an acidic pH suppressed pore formation, whereas the presence of zinc and insulin suppressed fiber formation by IAPP [64, 191–193]. In addition to these factors, the membrane disruption process also depends on the lipid composition. The presence of anionic lipids enhancing the binding of IAPP with membranes, two different roles of cholesterol, and membrane curvature have also been investigated in detail for IAPP [194, 195].
Previous studies reported that the growth of amyloid fibers on the surface of the lipid membrane cause curvature strain, while the membrane relieves this strain by changing its shape. In addition, curvature strain induced by fibril growth has also been shown to fragment the lipid membrane. These distorted membranes contribute to cell toxicity by allowing the permeation of small solutes such as ions. In order to fully investigate the role of membrane curvature on the pathology of IAPP, IAPP aggregation-induced membrane curvature was measured by differential scanning calorimetry (DSC) and NMR experiments. Four different IAPP peptides were examined: full-length 37-residue human-IAPP (hIAPP1-37: KCNTATCATQRLANFLVHSSNNFGAI LSSTNVGSNTY), full-length 37-residue rat peptide (rIAPP1-37: KCNTATCATQRLANFLVRSSNNLGPVLPPTNVGSNTY), and the 1-19 N-terminal segments of human (hIAPP1-19: KCNTATCATQRLANFLVHS) and rat (rIAPP1-19: KCNTATCATQRLANFLVRS) IAPP peptides. Previous studies demonstrated that hIAPP1-37 aggregates to form fibers and kill islet cells, neither rIAPP1-37 nor rIAPP1-19 forms fibers or are toxic to cells, and hIAPP1-19 is not fiber-competent, but is toxic to cells [196, 197]. NMR structural studies revealed very similar helical structures for the hIAPP1-19 and rIAPP1-19 peptides in a membrane environment; however, their membrane orientations differed. The rIAPP1-19 peptide prefers to associate with the membrane surface, while the hIAPP1-19 peptide aggregates in lipid bilayers under non-acidic pH conditions. The hIAPP1-37 peptide forms a helix-turn-helix-helix in membranes, whereas the rIAPP1-37 peptide has a helical structure in the N terminus and the presence of three Pro residues results in a C terminus that is randomly structured and, thus, differs from that of hIAPP1-37.
DSC experiments have been used to measure the peptide-induced curvature strain on 1,2-dipalmitoleoyl-phosphatidylethanolamine (DiPoPE) lipid bilayers. The effects of four different IAPP peptides on the liquid crystalline (Lα) to the inverted hexagonal [198] phase transition temperature of DiPoPE lipid bilayers were examined (Fig. 15). The full-length human peptide, which exhibits toxicity to islet cells, induced negative curvature in the lipid membrane by decreasing the Lα to HII phase transition temperature. On the other hand, non-amyloidogenic rIAPP1-37 exerted negligible effects on the phase transition of DiPoPE. The non-fiber-forming human and rat 1-19 fragments both exhibited weaker effects on phase transition than hIAPP1-37. Overall, toxic IAPP peptides were found to induce greater negative curvature strain than non-toxic IAPP peptides.
Figure 15. IAPP-induced membrane curvature measured by DSC experiments.
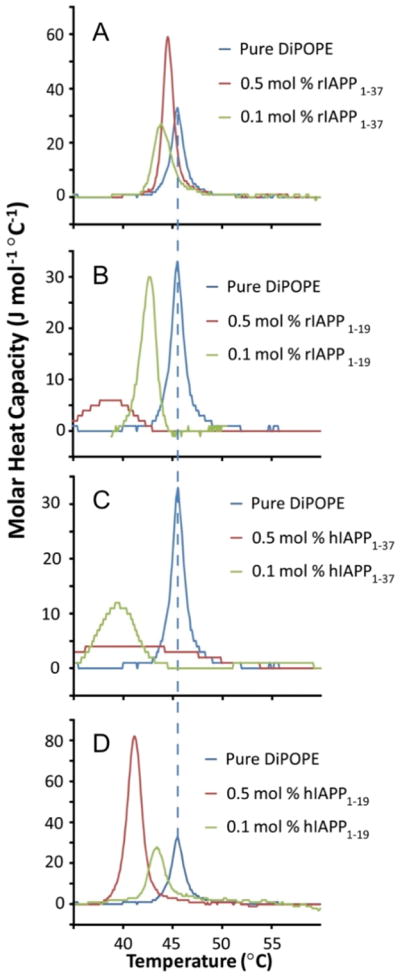
Effects of IAPP peptides on the Lα to HII phase transition of multilamellar vesicles (MLV) of DiPoPE. This figure is reproduced from reference [231].
In order to further evaluate the role of membrane curvature on IAPP peptides, solid-state NMR experiments were performed on magnetically-aligned bicelles constituting short (1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC)) and long (1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC) and DMPG) acyl chain phospholipids with a 9:9:4 molar ratio of DMPC: DMPG: DHPC (Fig. 16). Previous studies demonstrated that large bicelles are composed of a planar bilayer containing DMPC and DMPG lipids, while short chain DHPC are located in toroidal pores. Therefore, toroidal perforations possess positive and negative curvatures: positive curvature (convex) perpendicular to the bilayer surface and negative curvature (concave) in the plane of the lipid membrane. One-dimensional (1D) phosphorus-31 (spin = 1/2) and nitrogen-14 (spin = 1) NMR spectra of the magnetically-aligned 9:9:4 molar ratio of DMPC: DMPG: DHPC bicelles (q ratio of 4.5) were used to investigate peptide-induced effects on lipid head groups (Fig. 17). In addition, peptide-induced changes in the hydrophobic core of lipid bilayers were measured using two-dimensional (2D) PDLF solid-state NMR experiments. These experiments revealed differences in the membrane interactions of both rat peptides (rIAPP1-37 and rIAPP1-19). In contrast to the full-length rat peptide, the toxic rIAPP1-19 peptide binds to the short-chain DHPC in the pores and also affects the order of long chain lipids (Fig. 17).
Figure 16. Solid-state NMR experiments on bicelles.

(A) Representation of magnetically-aligned bicelles. The lipid bilayer normal was aligned perpendicular to the direction of the magnetic field of the NMR spectrometer (B0). (B) Schematics showing the curvature strain of bicelles. Phosphorus-31 NMR spectra of magnetically-aligned 9:9:4 DMPC:DMPG:DHPC bicelles without (C) and with (D and E) the rat peptides, rIAPP1-19 (D) and rIAPP1-37 (E). This figure is reproduced from reference [231].
Figure 17. 2D PDLF solid-state NMR experiments on magnetically-aligned bicelles.

(A) Molecular structures of long-chain lipids DMPG and DMPC. (B and C) 2D PDLF solid-state NMR spectra correlating the anisotropic chemical shifts of 13C and 1H-13C dipolar couplings of bicelles with the lipid bilayer normal aligned perpendicular to the direction of the magnetic field of the NMR spectrometer. 2D PDLF spectra without (black, B and C) and with the rIAPP peptides, rIAPP1-37 (B, blue) and rIAPP1-19 (C, red). (D) Selected 1D 1H-13C dipolar coupling spectral slices extracted from 2D PDLF spectra (B and C) and zoomed in the region of the spectrum in C (E) showing the effects of rIAPP1-19 on the terminal methyl groups of lipids. This figure is reproduced from reference [231].
6. Negative curvature membrane promotes fiber-dependent membrane disruption by IAPP
A biophysical investigation of the role of phosphatidylethanolamine (PE) on the aggregation and membrane interactions of hIAPP showed the promotion of fiber formation-dependent detergent-like membrane disruption [195]. Fluorescence dye-leakage experiments demonstrated that the presence of PE lipids decreased membrane leakage induced by non-fibrillar hIAPP, whereas increased dye leakage was observed for the fibrils of hIAPP (Fig. 18). CD experiments monitoring the coil-to-helix conformational transition revealed a higher binding affinity of fibrillary IAPP species than the non-fibrillar species. NMR experiments showed a greater amount of PE lipids present in the fragments of membranes induced by IAPP. These findings confirmed the role of lipids that have intrinsic negative curvature strain on IAPP-induced membrane disruption and, thus, in the loss of insulin-producing islet cells in type 2 diabetes.
Figure 18. Human-IAPP induced dye leakage.

(A) Release of 6-carboxyfluorescein from 250 μM POPC/POPS (7:3), POPE/POPC/POPS (3:4:3), or POPC/POPS/LysoPC (6.8:3:0.2) LUVs induced by hIAPP of 2.5 μM IAPP. The initial phase of membrane disruption was fit with a double-exponential curve (dotted lines). The presence of POPE decreases the amount of dye released during the first phase and markedly increases the efficiency of dye release during the second phase. The dotted line shows leakage only due to pore formation and not by amyloid fibrils. (B) Release of 6-carboxyfluorescein from seeded fibril growth. This figure is reproduced from reference [195].
7. Negative curvature is key in the marked increase in infection by HIV induced by SEVI
The presence of seminal amyloid fibrils of a proteolytic fragment of prostatic acid phosphatase (PAP), a protein that is abundant in human semen, has been shown to cause an approximately 400,000-fold increase in the rate of HIV infection. An in vitro biophysical study investigated the lipid membrane interactions of the amyloid aggregates of PAP (called SEVI). Turbidity measurements showed the fusion of lipid vesicles induced by a fragment of PAP (PAP248-286), suggesting the ability of the PAP248-286 peptide to induce membrane fusion. A FRET-based lipid assay further confirmed membrane fusion by PAP248-286 (Fig. 19A and B). DSC experiments on DiPoPE MLV revealed the effects of PAP248-286 on the Lα to HII phase transition, suggesting that the peptide facilitates the formation of a negatively curved membrane (Fig. 19C). PAP248-286 is an intrinsically disordered peptide and its amyloid aggregation has been shown to depend on a number of factors as well as the ability of the bacterial curli amyloid protein to cross-seed the amyloid aggregation and formation of SEVI amyloid. NMR structural studies revealed a highly disordered, but partially helical structure for the SEVI precursor of PAP248-286 in a membrane environment [199]. These studies collectively proposed a molecular mechanism by which membrane-associated SEVI promotes the fusion of viral and host cell membranes for the easy transfer of RNA from HIV. The SEVI-like mechanism has recently been used to design new compounds for gene therapeutic approaches [200]. Thus, the marked enhancement in infection by HIV induced by SEVI was explained based on the ability of the membrane-bound PAP peptide to promote membrane fusion via the induction of negative curvature strain.
Figure 19. SEVI promotes membrane fusion and the induction of a negative curvature strain by PAP peptides.

FRET-based lipid mixing measurements of membrane fusion at (A) pH 7.3 and (B) pH 4 by PAP248-286 in POPC:POPG (7:3) vesicles. The amino acid sequence of PAP248-286 is shown. (C) DSC heating scans of MLV of DiPoPE in the absence and presence of the PAP248-286 peptide. Figures were reproduced from reference [199].
8. Use of novel membrane mimetics to obtain high-resolution structural insights into amyloid aggregation and the roles of membrane curvature
Live cells are too complex to be used in high-resolution structural studies using techniques such as NMR. However, the development of aligned lipid bilayers enabled the application of a number of solution and solid-state NMR experiments. Membrane mimetics, such as mechanically-aligned lipid bilayers [201–204] and bicelles [205–209], enabled the in-depth probing of amyloid-lipid interactions. The recent development of membrane-curvature free lipid bilayer nanodiscs [210–216] has been valuable for trapping amyloid intermediates that are not affected by curvature [21, 217]. Recently developed polymer-based macro-nanodiscs have been shown to spontaneously align in an external magnetic field [214, 215], and this is a very valuable property that has been utilized in the molecular imaging of membrane proteins and, thus, may be useful for elucidating the atomistic-resolution structures of amyloid oligomers bound to lipids bilayers [150, 218–223]. High-resolution studies performed with and without the roles of membrane curvature being employed will provide an in-depth understanding of the roles of membrane curvature in the pathological progression of amyloid diseases.
9. Perspective
Amyloid aggregation is known to play very important roles in more than 50 severe diseases. Indeed, recent findings indicated that metabolite amyloids induce more than 50 diseases. The direct involvement of SEVI amyloid fibrillary aggregates in the marked enhancement in infection by HIV is unexpected. While amyloid generation is extremely context-dependent, the majority of amyloidogenic proteins and peptides are exposed to the cell membrane environment. Many physicochemical factors originating from biomembranes have been shown to affect amyloidogenesis. Not only the important roles of the cell membrane environment in protein aggregation, including amyloid fibrillation and the formation of other types of aggregates, but also the influence of the aggregation process and aggregates on membrane integrity and cell toxicity have been extensively covered in this review.
Many recent studies have demonstrated the many important roles of lipids constituting the cell membrane on the nucleation, kinetics, oligomer formation, and protofiber and fiber formation of amyloid aggregation. However, although membrane curvature has been extensively investigated in order to obtain a clearer understanding of the mechanisms underlying membrane disruption by antimicrobial peptides [77, 224–228], its significance for protein aggregation, the binding of amyloidogenic precursors, and cytotoxicity is relatively new and not examined in as much detail in spite of its obvious importance, as described herein. More case studies are expected to advance and generalize our understanding of the indispensable impact of membrane curvature. Other undiscovered aspects of membranes that affect protein aggregation and membrane interactions as well as the resulting damage to organelles and cells are expected to be characterized. Furthermore, more membrane-based studies are needed on phase separation and the transition from soluble to insoluble molecular species, droplet formation, two-step nucleation, and liquid-liquid phase separation from a macroscopic point of view in combination with kinetics. In-depth analyses of interactions between membranes and either precursor proteins or aggregates using calorimetry, which offers a plethora of thermodynamic and kinetic information, are promising.
Structural biology using the high-resolution structures of soluble precursors, prefibrillar aggregates, and amyloid fibrils is highly beneficial for obtaining microscopic insights. Macroscopic and microscopic studies together with thermodynamics and kinetics will provide a more complete understanding of protein aggregation in membranes. While the transient nature of toxic intermediates and amyloid aggregates and lipid components have been the major obstacles to obtaining high-resolution structures, recent studies have overcome these challenges by using lipid nanodiscs [21]. These approaches are key for obtaining structures that will be used in the screening of small molecule compounds that may potentially treat aging-related amyloid diseases.
Highlights.
The relationships between membrane curvature and amyloid aggregation were surveyed.
SUV promoted amyloidogenesis and LUV induced amorphous-like amyloids of Aβ(1-40).
SUV caused polymorphic Aβ(1-42) amyloid formation and LUV blocked amyloid generation.
The initial structures of αSN in synaptic mimic SUV are key to its amyloid fibrillation.
IAPP aggregation and the SEVI precursor affected curvature and resulting cytotoxicity.
Acknowledgments
We thank Prof. Yasushi Kawata (Tottori University, Japan) for his valuable contribution to the αSN studies. This study was supported by Grants-in-Aid for Young Scientists (B) (15K18518 and 25870407) and for Scientific Research (C) (15K07038) for Y.-H.L, funds from the National Institutes of Health (AG048934) to A.R., and a Grant-in-Aid from the Japan Society for the Promotion of Science Fellows (253790) for M.S.T.
Abbreviations
- Aβ
amyloid-beta
- AFM
atomic force microscopy
- ANS
8-anilino-1-naphthalenesulfonate
- αSNWT
wild-type α-synuclein
- αSN103
C-terminally truncated α-synuclein
- CD
circular dichroism
- CTR
C-terminal region
- DHPC
1,2-dihexanoyl-sn-glycero-3-phosphocholine
- DiPoPE
1,2-dipalmitoleoyl-phosphatidylethanolamine
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine
- DMPG
1,2-dimyristoyl-sn-glycero-3-phosphor-rac-(1-glycerol)
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPC SUV
SUV composed of DOPC
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DOPS
1,2-dioleoyl-sn-glycero-3-phospho-L-serine
- DSC
differential scanning calorimetry
- ER
endoplasmic reticulum
- FT-IR
Fourier transform-infrared spectrometer
- GUV
giant unilamellar vesicle
- HII
inverted hexagonal
- HIAPP
human-IAPP
- IAPP
islet amyloid polypeptide
- ITC
isothermal titration calorimetry
- Lα
liquid crystalline
- LUV
large unilamellar vesicle
- MAM
mitochondria-associated ER membranes Mimic
- SUV
SUV that mimics the presynaptic vesicle
- MLV
multilamellar vesicles
- NAC
non-amyloid β component
- NTR
N-terminal region
- RIAPP
rat-IAPP
- PAP
prostatic acid phosphatase
- PD
Parkinson’s disease
- PDI
protein disulfide isomerase
- PE
phosphatidylethanolamine
- POPC
1-palmitoyl-2-oleoyl-phosphatidylcholine
- POPE
1-palmitoyl-2-oleoyl-phosphatidylethanolamine
- POPG
1-palmitoyl-2-oleoyl-phosphatidylglycerol
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine
- SEVI
semen-derived enhanced viral infection
- SUV
small unilamellar vesicle
- TEM
transmission electron microscopy
- ThT
thioflavin T
- 1D
one-dimensional
- 2D
two-dimensional
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Yoshimura Y, Lin Y, Yagi H, Lee YH, Kitayama H, Sakurai K, So M, Ogi H, Naiki H, Goto Y. Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc Natl Acad Sci USA. 2012;109:14446–14451. doi: 10.1073/pnas.1208228109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 4.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 5.Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 6.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 7.Deleidi M, Maetzler W. Protein clearance mechanisms of alpha-synuclein and amyloid-Beta in lewy body disorders. Int J Alzheimers Dis. 2012;2012:391438. doi: 10.1155/2012/391438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 11.Dauer W, Przedborski S. Parkinson’s disease: Mechanisms and Models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 12.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 13.Kotler SA, Walsh P, Brender JR, Ramamoorthy A. Differences between amyloid-beta aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem Soc Rev. 2014;43:6692–6700. doi: 10.1039/c3cs60431d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeToma AS, Salamekh S, Ramamoorthy A, Lim MH. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem Soc Rev. 2012;41:608–621. doi: 10.1039/c1cs15112f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oikawa T, Nonaka T, Terada M, Tamaoka A, Hisanaga S, Hasegawa M. alpha-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly. J Biol Chem. 2016;291:15046–15056. doi: 10.1074/jbc.M116.736355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenberg DS, Sawaya MR. Structural Studies of Amyloid Proteins at the Molecular Level. Annu Rev Biochem. 2017;86:69–95. doi: 10.1146/annurev-biochem-061516-045104. [DOI] [PubMed] [Google Scholar]
- 17.Fandrich M, Schmidt M, Grigorieff N. Recent progress in understanding Alzheimer’s beta-amyloid structures. Trends Biochem Sci. 2011;36:338–345. doi: 10.1016/j.tibs.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 19.Lorenzen N, Nielsen SB, Buell AK, Kaspersen JD, Arosio P, Vad BS, Paslawski W, Christiansen G, Valnickova-Hansen Z, Andreasen M, Enghild JJ, Pedersen JS, Dobson CM, Knowles TP, Otzen DE. The role of stable alpha-synuclein oligomers in the molecular events underlying amyloid formation. J Am Chem Soc. 2014;136:3859–3868. doi: 10.1021/ja411577t. [DOI] [PubMed] [Google Scholar]
- 20.Haupt C, Leppert J, Ronicke R, Meinhardt J, Yadav JK, Ramachandran R, Ohlenschlager O, Reymann KG, Gorlach M, Fandrich M. Structural basis of beta-amyloid-dependent synaptic dysfunctions. Angew Chem Int Ed Engl. 2012;51:1576–1579. doi: 10.1002/anie.201105638. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez Camargo DC, Korshavn KJ, Jussupow A, Raltchev K, Goricanec D, Fleisch M, Sarkar R, Xue K, Aichler M, Mettenleiter G, Walch AK, Camilloni C, Hagn F, Reif B, Ramamoorthy A. Stabilization and structural analysis of a membrane-associated hIAPP aggregation intermediate. Elife. 2017;6 doi: 10.7554/eLife.31226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 23.Fu Z, Aucoin D, Davis J, Van Nostrand WE, Smith SO. Mechanism of Nucleated Conformational Conversion of Abeta42. Biochemistry. 2015;54:4197–4207. doi: 10.1021/acs.biochem.5b00467. [DOI] [PubMed] [Google Scholar]
- 24.Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, Cecchi C, Vendruscolo M, Chiti F, Cremades N, Ying L, Dobson CM, De Simone A. Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science. 2017;358:1440–1443. doi: 10.1126/science.aan6160. [DOI] [PubMed] [Google Scholar]
- 25.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric Determination of Amyloid Fibrils in Vitro Using the Fluorescent Dye, Thioflavine T. Anal Biochem. 1989;177:224–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 26.Soong R, Brender JR, Macdonald PM, Ramammorthy A. Association of highly compact type II diabetes related islet amyloid polypeptide intermediate species at physiological temperature revealed by diffusion NMR spectroscopy. J Am Chem Soc. 2009;131:7079–7085. doi: 10.1021/ja900285z. [DOI] [PubMed] [Google Scholar]
- 27.Eanes ED, Glenner GG. J Histichem Cytochem. 1968;16:673–677. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- 28.Halverson K, Fraser PE, Kirschner DA, Lansbury PTJ. Molecular Determinants of Amyloid Deposition in Alzheimer’s Disease: Conformational Studies of Synthetic β-Protein Fragments. Biochemistry. 1990;29:2639–2644. doi: 10.1021/bi00463a003. [DOI] [PubMed] [Google Scholar]
- 29.Tycko R. Amyloid Polymorphism: Structural Basis and Neurobiological Relevance. Neuron. 2015;86:632–645. doi: 10.1016/j.neuron.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 32.Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem Biophys Res Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Attard GS, Templer RH, Smith WS, Hunt AN, Jackowski S. Modulation of CTP:phosphocholine cytidylyltransferase by membrane curvature elastic stress. Proc Natl Acad Sci USA. 2000;97:9032–9036. doi: 10.1073/pnas.160260697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ban T, Hamada D, Hasegawa K, Naiki H, Goto Y. Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J Biol Chem. 2003;278:16462–16465. doi: 10.1074/jbc.C300049200. [DOI] [PubMed] [Google Scholar]
- 35.Adamcik J, Mezzenga R. Study of amyloid fibrils via atomic force microscopy. Curr Opin Colloid Interface Sci. 2012;17:369–376. [Google Scholar]
- 36.Levin A, Mason TO, Adler-Abramovich L, Buell AK, Meisl G, Galvagnion C, Bram Y, Stratford SA, Dobson CM, Knowles TP, Gazit E. Ostwald’s rule of stages governs structural transitions and morphology of dipeptide supramolecular polymers. Nat Commun. 2014;5:5219. doi: 10.1038/ncomms6219. [DOI] [PubMed] [Google Scholar]
- 37.Auer S, Ricchiuto P, Kashchiev D. Two-step nucleation of amyloid fibrils: omnipresent or not? J Mol Biol. 2012;422:723–730. doi: 10.1016/j.jmb.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 38.Lin Y, Lee YH, Yoshimura Y, Yagi H, Goto Y. Solubility and supersaturation-dependent protein misfolding revealed by ultrasonication. Langmuir. 2014;30:1845–1854. doi: 10.1021/la403100h. [DOI] [PubMed] [Google Scholar]
- 39.Ikenoue T, Lee YH, Kardos J, Yagi H, Ikegami T, Naiki H, Goto Y. Heat of supersaturation-limited amyloid burst directly monitored by isothermal titration calorimetry. Proc Natl Acad Sci USA. 2014;111:6654–6659. doi: 10.1073/pnas.1322602111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adachi M, So M, Sakurai K, Kardos J, Goto Y. Supersaturation-limited and Unlimited Phase Transitions Compete to Produce the Pathway Complexity in Amyloid Fibrillation. J Biol Chem. 2015;290:18134–18145. doi: 10.1074/jbc.M115.648139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 42.Fandrich M, Fletcher MA, Dobson CM. Amyloid fibrils from muscle myoglobin. Nature. 2001;410:165–166. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- 43.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jahn TR, Radford SE. Folding versus aggregation: polypeptide conformations on competing pathways. Arch Biochem Biophys. 2008;469:100–117. doi: 10.1016/j.abb.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frare E, Polverino De Laureto P, Zurdo J, Dobson CM, Fontana A. A highly amyloidogenic region of hen lysozyme. J Mol Biol. 2004;340:1153–1165. doi: 10.1016/j.jmb.2004.05.056. [DOI] [PubMed] [Google Scholar]
- 46.Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 47.Ciryam P, Tartaglia GG, Morimoto RI, Dobson CM, Vendruscolo M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013;5:781–790. doi: 10.1016/j.celrep.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terakawa MS, Yagi H, Adachi M, Lee YH, Goto Y. Small liposomes accelerate the fibrillation of amyloid beta (1-40) J Biol Chem. 2015;290:815–826. doi: 10.1074/jbc.M114.592527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin Y, Kardos J, Imai M, Ikenoue T, Kinoshita M, Sugiki T, Ishimori K, Goto Y, Lee YH. Amorphous Aggregation of Cytochrome c with Inherently Low Amyloidogenicity Is Characterized by the Metastability of Supersaturation and the Phase Diagram. Langmuir. 2016;32:2010–2022. doi: 10.1021/acs.langmuir.5b03810. [DOI] [PubMed] [Google Scholar]
- 50.Ling X, Fang W, Lee YH, Araujo PT, Zhang X, Rodriguez-Nieva JF, Lin Y, Zhang J, Kong J, Dresselhaus MS. Raman enhancement effect on two-dimensional layered materials: graphene, h-BN and MoS2. Nano Lett. 2014;14:3033–3040. doi: 10.1021/nl404610c. [DOI] [PubMed] [Google Scholar]
- 51.Muta H, Lee YH, Kardos J, Lin Y, Yagi H, Goto Y. Supersaturation-limited amyloid fibrillation of insulin revealed by ultrasonication. J Biol Chem. 2014;289:18228–18238. doi: 10.1074/jbc.M114.566950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kundra R, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M. Protein homeostasis of a metastable subproteome associated with Alzheimer’s disease. Proc Natl Acad Sci USA. 2017;114:E5703–E5711. doi: 10.1073/pnas.1618417114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kinoshita M, Kakimoto E, Terakawa MS, Lin Y, Ikenoue T, So M, Sugiki T, Ramamoorthy A, Goto Y, Lee YH. Model membrane size-dependent amyloidogenesis of Alzheimer’s amyloid-beta peptides. Phys Chem Chem Phys. 2017;19:16257–16266. doi: 10.1039/c6cp07774a. [DOI] [PubMed] [Google Scholar]
- 54.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 56.Chatani E, Yagi H, Naiki H, Goto Y. Polymorphism of beta2-microglobulin amyloid fibrils manifested by ultrasonication-enhanced fibril formation in trifluoroethanol. J Biol Chem. 2012;287:22827–22837. doi: 10.1074/jbc.M111.333310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pellarin R, Schuetz P, GE, Caflisch A. Amyloid fibril polymorphism is under kinetic control. J Am Chem Soc. 2010;132:14960–14970. doi: 10.1021/ja106044u. [DOI] [PubMed] [Google Scholar]
- 58.Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA. 2004;101:2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yagi H, Mizuno A, So M, Hirano M, Adachi M, Akazawa-Ogawa Y, Hagihara Y, Ikenoue T, Lee YH, Kawata Y, Goto Y. Ultrasonication-dependent formation and degradation of alpha-synuclein amyloid fibrils. Biochim Biophys Acta. 2015;1854:209–217. doi: 10.1016/j.bbapap.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 60.Gursky O, Aleshkov S. Temperature-dependent β-sheet formation in β-amyloid Aβ1-40 peptide inwater: uncoupling β-structure folding from aggregation. Biochim Biophys Acta. 2000;1476:93–102. doi: 10.1016/s0167-4838(99)00228-9. [DOI] [PubMed] [Google Scholar]
- 61.Klement K, Wieligmann K, Meinhardt J, Hortschansky P, Richter W, Fandrich M. Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer’s abeta(1-40) amyloid fibrils. J Mol Biol. 2007;373:1321–1333. doi: 10.1016/j.jmb.2007.08.068. [DOI] [PubMed] [Google Scholar]
- 62.Miura T, Suzuki K, Kohata N, Takeuchi H. Metal binding modes of Alzheimer’s amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry. 2000;39:7024–7031. doi: 10.1021/bi0002479. [DOI] [PubMed] [Google Scholar]
- 63.Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. Solid State NMR Reveals a pH-dependent Antiparallel β-Sheet Registry in Fibrils Formed by a β-Amyloid Peptide. J Mol Biol. 2004;335:247–260. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 64.Salamekh S, Brender JR, Hyung SJ, Nanga RP, Vivekanandan S, Ruotolo BT, Ramamoorthy A. A two-site mechanism for the inhibition of IAPP amyloidogenesis by zinc. J Mol Biol. 2011;410:294–306. doi: 10.1016/j.jmb.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rezaei-Ghaleh N, Amininasab M, Kumar S, Walter J, Zweckstetter M. Phosphorylation modifies the molecular stability of beta-amyloid deposits. Nat Commun. 2016;7:11359. doi: 10.1038/ncomms11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foguel D, Suarez MC, Ferrao-Gonzales AD, Porto TC, Palmieri L, Einsiedler CM, Andrade LR, Lashuel HA, Lansbury PT, Kelly JW, Silva JL. Dissociation of amyloid fibrils of alpha-synuclein and transthyretin by pressure reveals their reversible nature and the formation of water-excluded cavities. Proc Natl Acad Sci USA. 2003;100:9831–9836. doi: 10.1073/pnas.1734009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid β-protein (Aβ): A possible form of preamyloid in Alzheimer’s disease. Nat Med. 1995;1:1062–1066. doi: 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]
- 68.Aisenbrey C, Borowik T, Bystrom R, Bokvist M, Lindstrom F, Misiak H, Sani MA, Grobner G. How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur Biophys J. 2008;37:247–255. doi: 10.1007/s00249-007-0237-0. [DOI] [PubMed] [Google Scholar]
- 69.Bokvist M, Grobner G. Misfolding of Amyloidogenic Proteins at Membrane Surfaces: The Impact of Macromolecular Crowding. J Am Chem Soc. 2007;129:14848–14849. doi: 10.1021/ja076059o. [DOI] [PubMed] [Google Scholar]
- 70.Michalek M, Salnikov ES, Werten S, Bechinger B. Membrane interactions of the amphipathic amino terminus of huntingtin. Biochemistry. 2013;52:847–858. doi: 10.1021/bi301325q. [DOI] [PubMed] [Google Scholar]
- 71.Michalek M, Aisenbrey C, Bechinger B. Investigation of membrane penetration depth and interactions of the amino-terminal domain of huntingtin: refined analysis by tryptophan fluorescence measurement. Eur Biophys J. 2014;43:347–360. doi: 10.1007/s00249-014-0966-9. [DOI] [PubMed] [Google Scholar]
- 72.Jayasinghe SA, Langen R. Membrane interaction of islet amyloid polypeptide. Biochim Biophys Acta. 2007;1768:2002–2009. doi: 10.1016/j.bbamem.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 73.Pfefferkorn CM, Jiang Z, Lee JC. Biophysics of alpha-synuclein membrane interactions. Biochim Biophys Acta. 2012;1818:162–171. doi: 10.1016/j.bbamem.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Butterfield SM, Lashuel HA. Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew Chem Int Ed Engl. 2010;49:5628–5654. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 75.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bigay J, Antonny B. Curvature, lipid packing, and electrostatics of membrane organelles: defining cellular territories in determining specificity. Dev Cell. 2012;23:886–895. doi: 10.1016/j.devcel.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 77.Hallock KJ, Lee D-K, Ramammorthy A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys J. 2003;84:3052–3060. doi: 10.1016/S0006-3495(03)70031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vanni S, Hirose H, Barelli H, Antonny B, Gautier R. A sub-nanometre view of how membrane curvature and composition modulate lipid packing and protein recruitment. Nat Commun. 2014;5:4916. doi: 10.1038/ncomms5916. [DOI] [PubMed] [Google Scholar]
- 79.Lasic DD, Needham D. The “stealth” liposome: A prototypical biomaterial. Chem Rev. 1995;95:2601–2628. [Google Scholar]
- 80.Kozlov MM. Joint effort bends membrane. Nature. 2010;463:439–440. doi: 10.1038/463439a. [DOI] [PubMed] [Google Scholar]
- 81.Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA, Webb WW. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc Natl Acad Sci USA. 2007;104:3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roux A, Cappello G, Cartaud J, Prost J, Goud B, Bassereau P. A minimal system allowing tubulation with molecular motors pulling on giant liposomes. Proc Natl Acad Sci USA. 2002;99:5394–5399. doi: 10.1073/pnas.082107299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van Rooijen BD, Claessens MM, Subramaniam V. Membrane binding of oligomeric alpha-synuclein depends on bilayer charge and packing. FEBS Lett. 2008;582:3788–3792. doi: 10.1016/j.febslet.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 84.Szoka FJ. Comparative properties and methods of preparation of lipid vesicles (liposomes) Annu Rev Biophys Bioeng. 1980;9:467–508. doi: 10.1146/annurev.bb.09.060180.002343. [DOI] [PubMed] [Google Scholar]
- 85.Hoekstra D, de Boer T, Klappe K, Wilschut J. Fluorescence method for measuring the kinetics of fusion between biological membranes. Biochemistry. 1984;23:5675–5681. doi: 10.1021/bi00319a002. [DOI] [PubMed] [Google Scholar]
- 86.Antonny B. Mechanisms of membrane curvature sensing. Annu Rev Biochem. 2011;80:101–123. doi: 10.1146/annurev-biochem-052809-155121. [DOI] [PubMed] [Google Scholar]
- 87.Antonny B, Huber I, Paris S, Chabre M, Cassel D. Activation of ADP-ribosylation factor 1 GTPase-activating protein by phosphatidylcholine-derived diacylglycerols. J Biol Chem. 1997;272:30848–30851. doi: 10.1074/jbc.272.49.30848. [DOI] [PubMed] [Google Scholar]
- 88.Snead D, Eliezer D. Alpha-synuclein function and dysfunction on cellular membranes. Exp Neurobiol. 2014;23:292–313. doi: 10.5607/en.2014.23.4.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, Langen R. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–32493. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dominguez L, Meredith SC, Straub JE, Thirumalai D. Transmembrane Fragment Structures of Amyloid Precursor Protein Depend on Membrane Surface Curvature. J Am Chem Soc. 2014;136:854–857. doi: 10.1021/ja410958j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mizuno N, Varkey J, Kegulian NC, Hegde BG, Cheng N, Langen R, Steven AC. Remodeling of lipid vesicles into cylindrical micelles by alpha-synuclein in an extended alpha-helical conformation. J Biol Chem. 2012;287:29301–29311. doi: 10.1074/jbc.M112.365817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 93.Seubert P, Oltersdorf T, Lee MG, Barbour R, Blomquist C, Davis DL, Bryant K, Fritz LC, Galasko D, Thal LJ, Lieberburg I, Schenk D. Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nature. 1993;361 doi: 10.1038/361260a0. [DOI] [PubMed] [Google Scholar]
- 94.Itkin A, Salnikov ES, Aisenbrey C, Raya J, Glattard E, Raussens V, Ruysschaert JM, Bechinger B. Structural Characterization of the Amyloid Precursor Protein Transmembrane Domain and Its gamma-Cleavage Site. ACS Omega. 2017;2:6525–6534. doi: 10.1021/acsomega.7b00619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 96.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 97.Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci USA. 2009;106:20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski ML, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid b-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 99.Yagi H, Hasegawa K, Yoshimura Y, Goto Y. Acceleration of the depolymerization of amyloid beta fibrils by ultrasonication. Biochim Biophys Acta. 2013;1834:2480–2485. doi: 10.1016/j.bbapap.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 100.Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TPJ. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Matsuzaki Katsumi, Horikiri C. Interactions of Amyloid β-Peptide (1-40) with Ganglioside-Containing Membranes. Biochemistry. 1999;38:4137–4142. doi: 10.1021/bi982345o. [DOI] [PubMed] [Google Scholar]
- 102.Ji SR, Wu Y, Sui SF. Cholesterol is an important factor affecting the membrane insertion of beta-amyloid peptide (A beta 1-40), which may potentially inhibit the fibril formation. J Biol Chem. 2002;277:6273–6279. doi: 10.1074/jbc.M104146200. [DOI] [PubMed] [Google Scholar]
- 103.Fukunaga S, Ueno H, Yamaguchi T, Yano Y, Hoshino M, Matsuzaki K. GM1 cluster mediates formation of toxic Abeta fibrils by providing hydrophobic environments. Biochemistry. 2012;51:8125–8131. doi: 10.1021/bi300839u. [DOI] [PubMed] [Google Scholar]
- 104.Bokvist M, Lindström F, Watts A, Gröbner G. Two Types of Alzheimer’s β-Amyloid (1–40) Peptide Membrane Interactions: Aggregation Preventing Transmembrane Anchoring Versus Accelerated Surface Fibril Formation. J Mol Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- 105.Pannuzzo M, Milardi D, Raudino A, Karttunen M, La Rosa C. Analytical model and multiscale simulations of Aβ peptide aggregation in lipid membranes: towards a unifying description of conformational transitions, oligomerization and membrane damage. Phys Chem Chem Phys. 2013;15:8940–8952. doi: 10.1039/c3cp44539a. [DOI] [PubMed] [Google Scholar]
- 106.Wieprecht T, Beyermann M, Seelig J. Thermodynamics of the coil-alpha-helix transition of amphipathic peptides in a membrane environment: the role of vesicle curvature. Biophys Chem. 2002;96:191–201. doi: 10.1016/s0301-4622(02)00025-x. [DOI] [PubMed] [Google Scholar]
- 107.Goto Y, Fink AL. Conformational States of β-Lactamase: Molten-Globule States at Acidic and Alkaline pH with High Salt. Biochemistry. 1989;28:949–952. doi: 10.1021/bi00429a004. [DOI] [PubMed] [Google Scholar]
- 108.Sugiura Y, Ikeda K, Nakano M. High Membrane Curvature Enhances Binding, Conformational Changes, and Fibrillation of Amyloid-beta on Lipid Bilayer Surfaces. Langmuir. 2015;31:11549–11557. doi: 10.1021/acs.langmuir.5b03332. [DOI] [PubMed] [Google Scholar]
- 109.Shintani M, Yoshida K, Sakuraba S, Nakahara M, Matubayasi N. NMR-NOE and MD simulation study on phospholipid membranes: dependence on membrane diameter and multiple time scale dynamics. J Phys Chem B. 2011;115:9106–9115. doi: 10.1021/jp204051f. [DOI] [PubMed] [Google Scholar]
- 110.Zhernenkov M, Bolmatov D, Soloviov D, Zhernenkov K, Toperverg BP, Cunsolo A, Bosak A, Cai YQ. Revealing the mechanism of passive transport in lipid bilayers via phonon-mediated nanometre-scale density fluctuations. Nat Commun. 2016;7:11575. doi: 10.1038/ncomms11575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nuscher B, Kamp F, Mehnert T, Odoy S, Haass C, Kahle PJ, Beyer K. Alpha-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J Biol Chem. 2004;279:21966–21975. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- 112.Dikiy I, Eliezer D. N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound alpha-synuclein and increases its affinity for physiological membranes. J Biol Chem. 2014;289:3652–3665. doi: 10.1074/jbc.M113.512459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Giacomelli CE, Norde W. Influence of hydrophobic teflon particles on the structure of amyloid β-peptide. Biomacromolecules. 2003;4:1719–1726. doi: 10.1021/bm034151g. [DOI] [PubMed] [Google Scholar]
- 114.Lee SJ, Lee JW, Choi TS, Jin KS, Lee S, Ban C, Kim HI. Probing conformational change of intrinsically disordered alpha-synuclein to helical structures by distinctive regional interactions with lipid membranes. Anal Chem. 2014;86:1909–1916. doi: 10.1021/ac404132g. [DOI] [PubMed] [Google Scholar]
- 115.Kowalewski T, Holtzman DM. In situ atomic force mictoscopy study of Alzheimer’s β-amyloid peptide on different substrates: New insights into mechanism of β-sheet formation. Proc Natl Acad Sci USA. 1999;96:3688–3693. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ban T, Goto Y. Direct Observation of Amyloid Growth Monitored by Total Internal Reflection Fluorescence Microscopy. Methods Enzymol. 2006;413:91–102. doi: 10.1016/S0076-6879(06)13005-0. [DOI] [PubMed] [Google Scholar]
- 117.Vacha R, Linse S, Lund M. Surface effects on aggregation kinetics of amyloidogenic peptides. J Am Chem Soc. 2014;136:11776–11782. doi: 10.1021/ja505502e. [DOI] [PubMed] [Google Scholar]
- 118.Cabaleiro-Lago C, Quinlan-Pluck F, Lynch I, Dawson KA, Linse S. Dual effect of amino modified polystyrene nanoparticles on amyloid beta protein fibrillation. ACS Chem Neurosci. 2010;1:279–287. doi: 10.1021/cn900027u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kardos J, Micsonai A, Pal-Gabor H, Petrik E, Graf L, Kovacs J, Lee YH, Naiki H, Goto Y. Reversible heat-induced dissociation of beta2-microglobulin amyloid fibrils. Biochemistry. 2011;50:3211–3220. doi: 10.1021/bi2000017. [DOI] [PubMed] [Google Scholar]
- 120.Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med. 2014;20:130–138. doi: 10.1038/nm.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ikenoue T, Lee YH, Kardos J, Saiki M, Yagi H, Kawata Y, Goto Y. Cold denaturation of alpha-synuclein amyloid fibrils. Angew Chem Int Ed Engl. 2014;53:7799–7804. doi: 10.1002/anie.201403815. [DOI] [PubMed] [Google Scholar]
- 123.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 124.Wetzel R. Kinetics and Thermodynamics of Amyloid FIbril Assembly. Acc Chem Res. 2006;49:671–679. doi: 10.1021/ar050069h. [DOI] [PubMed] [Google Scholar]
- 125.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VM, George JM, Rienstra CM. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 2016;23:409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jimenez JL, Nettleton EJ, Bouchard M, Robinson CV, Dobson CM, Saibil HR. The protofilament structure of insulin amyloid fibrils. Proc Natl Acad Sci USA. 2002;99:9196–9201. doi: 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fitzpatrick AW, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang L, Ladizhansky V, Muller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TP, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, Dobson CM. Atomic structure and hierarchical assembly of a cross-beta amyloid fibril. Proc Natl Acad Sci USA. 2013;110:5468–5473. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, Tycko R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell. 2017;171:615–627. e616. doi: 10.1016/j.cell.2017.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Peng Z, Peralta MDR, Toney MD. Extraordinarily Stable Amyloid Fibrils Engineered from Structurally Defined beta-Solenoid Proteins. Biochemistry. 2017;56:6041–6050. doi: 10.1021/acs.biochem.7b00364. [DOI] [PubMed] [Google Scholar]
- 131.Hirota-Nakaoka N, Hasegawa K, Naiki H, Goto Y. Dissolution of beta2-microglobulin amyloid fibrils by dimethylsulfoxide. J Biochem. 2003;134:159–164. doi: 10.1093/jb/mvg124. [DOI] [PubMed] [Google Scholar]
- 132.Karsai A, Slack TJ, Malekan H, Khoury F, Lin WF, Tran V, Cox D, Toney M, Chen X, Liu GY. Local Mechanical Perturbation Provides an Effective Means to Regulate the Growth and Assembly of Functional Peptide Fibrils. Small. 2016;12:6407–6415. doi: 10.1002/smll.201601657. [DOI] [PubMed] [Google Scholar]
- 133.Palhano FL, Lee J, Grimster NP, Kelly JW. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J Am Chem Soc. 2013;135:7503–7510. doi: 10.1021/ja3115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ahmad B, Borana MS, Chaudhary AP. Understanding curcumin-induced modulation of protein aggregation. Int J Biol Macromol. 2017;100:89–96. doi: 10.1016/j.ijbiomac.2016.06.053. [DOI] [PubMed] [Google Scholar]
- 135.Baldwin AJ, Knowles TP, Tartaglia GG, Fitzpatrick AW, Devlin GL, Shammas SL, Waudby CA, Mossuto MF, Meehan S, Gras SL, Christodoulou J, Anthony-Cahill SJ, Barker PD, Vendruscolo M, Dobson CM. Metastability of native proteins and the phenomenon of amyloid formation. J Am Chem Soc. 2011;133:14160–14163. doi: 10.1021/ja2017703. [DOI] [PubMed] [Google Scholar]
- 136.Francesco RD, Pastore A, Vecchio G, Cortese R. Circular dichroism study on the conformational stability of the dimerization domain of transcription factor LFB1. Biochemistry. 1991;30:143–147. doi: 10.1021/bi00215a021. [DOI] [PubMed] [Google Scholar]
- 137.Schmidt M, Sachse C, Richter W, Xu C, Fandrich M, Grigorieff N. Comparison of Alzheimer Abeta(1-40) and Abeta(1-42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci USA. 2009;106:19813–19818. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shabestari MH, Meeuwenoord NJ, Filippov DV, Huber M. Interaction of the amyloid beta peptide with sodium dodecyl sulfate as a membrane-mimicking detergent. J Biol Phys. 2016;42:299–315. doi: 10.1007/s10867-016-9408-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. The Alzheimer’s peptides Abeta40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J Mol Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yan Y, Liu J, McCallum SA, Yang D, Wang C. Methyl dynamics of the amyloid-beta peptides Abeta40 and Abeta42. Biochem Biophys Res Commun. 2007;362:410–414. doi: 10.1016/j.bbrc.2007.07.198. [DOI] [PubMed] [Google Scholar]
- 141.G.D.a.I.I.a.P. Collaborators. Global, regional, and national incidence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1545–1602. doi: 10.1016/S0140-6736(16)31678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sveinbjornsdottir S. The clinical symptoms of Parkinson’s disease. J Neurochem. 2016;139(Suppl 1):318–324. doi: 10.1111/jnc.13691. [DOI] [PubMed] [Google Scholar]
- 143.Spillantini MG, Crowther RA, Jales R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TP, Linse S, Dobson CM. Solution conditions determine the relative importance of nucleation and growth processes in alpha-synuclein aggregation. Proc Natl Acad Sci USA. 2014;111:7671–7676. doi: 10.1073/pnas.1315346111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lundblad M, Decressac M, Mattsson B, Bjorklund A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci USA. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Auluck PK, Caraveo G, Lindquist S. alpha-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- 147.Fusco G, De Simone A, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM, Veglia G. Direct observation of the three regions in alpha-synuclein that determine its membrane-bound behaviour. Nat Commun. 2014;5:3827. doi: 10.1038/ncomms4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Anderson VL, Ramlall TF, Rospigliosi CC, Webb WW, Eliezer D. Identification of a helical intermediate in trifluoroethanol-induced alpha-synuclein aggregation. Proc Natl Acad Sci USA. 2010;107:18850–18855. doi: 10.1073/pnas.1012336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Terakawa MS, Lee YH, Kinoshita M, Lin Y, Sugiki T, Fukui N, Ikenoue T, Kawata Y, Goto Y. Membrane-induced initial structure of alpha-synuclein control its amyloidogenesis on model membranes. Biochim Biophys Acta. 2018;1860:757–766. doi: 10.1016/j.bbamem.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 150.Salnikov ES, Sarrouj H, Reiter C, Aisenbrey C, Purea A, Aussenac F, Ouari O, Tordo P, Fedotenko I, Engelke F, Bechinger B. Solid-State NMR/Dynamic Nuclear Polarization of Polypeptides in Planar Supported Lipid Bilayers. J Phys Chem B. 2015;119:14574–14583. doi: 10.1021/acs.jpcb.5b07341. [DOI] [PubMed] [Google Scholar]
- 151.Bisaglia M, Mammi S, Bubacco L. Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J. 2009;23:329–340. doi: 10.1096/fj.08-119784. [DOI] [PubMed] [Google Scholar]
- 152.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yu S, Li X, Liu G, Han J, Zhang C, Li Y, Xu S, Liu C, Gao Y, Yang H, Ueda K, Chan P. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience. 2007;145:539–555. doi: 10.1016/j.neuroscience.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 155.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J Neurosci. 2012;32:3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cali T, Ottolini D, Negro A, Brini M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012;287:17914–17929. doi: 10.1074/jbc.M111.302794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cookson MR. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci USA. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci USA. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Galvagnion C, Buell AK, Meisl G, Michaels TC, Vendruscolo M, Knowles TP, Dobson CM. Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol. 2015;11:229–234. doi: 10.1038/nchembio.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Comellas G, Lemkau LR, Zhou DH, George JM, Rienstra CM. Structural intermediates during alpha-synuclein fibrillogenesis on phospholipid vesicles. J Am Chem Soc. 2012;134:5090–5099. doi: 10.1021/ja209019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rao JN, Jao CC, Hegde BG, Langen R, Ulmer TS. A combinatorial NMR and EPR approach for evaluating the structural ensemble of partially folded proteins. J Am Chem Soc. 2010;132:8657–8668. doi: 10.1021/ja100646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bodner CR, Dobson CM, Bax A. Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J Mol Biol. 2009;390:775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jensen PH, Nielsen MS, Jales R, Dotti CG, Goedert M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 166.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 168.Neudecker P, Robustelli P, Cavalli A, WP, Lundstrom P, Zarrine-Afsar A, Shaepe S, Vendruscolo M, KLE Strunture of an Intermediate State in Protein Folding and Aggregation. Science. 2012;336:362–366. doi: 10.1126/science.1214203. [DOI] [PubMed] [Google Scholar]
- 169.Karamanos TK, Pashley CL, Kalverda AP, Thompson GS, Mayzel M, Orekhov VY, Radford SE. A Population Shift between Sparsely Populated Folding Intermediates Determines Amyloidogenicity. J Am Chem Soc. 2016;138:6271–6280. doi: 10.1021/jacs.6b02464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lomakin L, Chung DS, Bendek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid β-protein fibrils: Detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci USA. 1996;93:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL. Nuclear localization of α-synuclein and its interaction with histones. Biochemistry. 2003;42:8465–8471. doi: 10.1021/bi0341152. [DOI] [PubMed] [Google Scholar]
- 172.Specht CG, Tigaret CM, Rast GF, Thalhammer A, Rudhard Y, Schoepfer R. Subcellular localisation of recombinant α- and γ-synuclein. Mol Cell Neurosci. 2005;28:326–334. doi: 10.1016/j.mcn.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 173.Shavali S, Brown-Borg HM, Ebadi M, Porter J. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett. 2008;439:125–128. doi: 10.1016/j.neulet.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhang L, Zhang C, Zhu Y, Cai Q, Chan P, Ueda K, Yu S, Yang H. Semi-quantitative analysis of α-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 2008;1244:40–52. doi: 10.1016/j.brainres.2008.08.067. [DOI] [PubMed] [Google Scholar]
- 175.Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, Voos W, Schon EA, Przedborski S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci. 2014;34:249–259. doi: 10.1523/JNEUROSCI.2507-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sanz-Blasco S, Valero RA, Rodriguez-Crespo I, Villalobos C, Nunez L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PloS one. 2008;3:e2718. doi: 10.1371/journal.pone.0002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 178.Kim HS, Lee JH, Lee JP, Kim EM, Chang KA, Park CH, Jeong SJ, Wittendorp MC, Seo JH, Choi SH, Suh YH. Amyloid β peptide induces cytochrome c release from isolated mitochondria. Neuroreport. 2002;13:1989–1893. doi: 10.1097/00001756-200210280-00032. [DOI] [PubMed] [Google Scholar]
- 179.Camilleri A, Zarb C, Caruana M, Ostermeier U, Ghio S, Hogen T, Schmidt F, Giese A, Vassallo N. Mitochondrial membrane permeabilisation by amyloid aggregates and protection by polyphenols. Biochim Biophys Acta. 2013;1828:2532–2543. doi: 10.1016/j.bbamem.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 180.Kim J, Yang Y, Song SS, Na JH, Oh KJ, Jeong C, Yu YG, Shin YK. Beta-amyloid oligomers activate apoptotic BAK pore for cytochrome c release. Biophys J. 2014;107:1601–1608. doi: 10.1016/j.bpj.2014.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–625. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cheng H, Wang L, Wang CC. Domain a’ of protein disulfide isomerase plays key role in inhibiting alpha-synuclein fibril formation. Cell Stress Chaperones. 2010;15:415–421. doi: 10.1007/s12192-009-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Okumura M, Kadokura H, Inaba K. Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic Biol Med. 2015;83:314–322. doi: 10.1016/j.freeradbiomed.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 184.Bechinger B. The SMART model: Soft Membranes Adapt and Respond, also Transiently, in the presence of antimicrobial peptides. J Pept Sci. 2015;21:346–355. doi: 10.1002/psc.2729. [DOI] [PubMed] [Google Scholar]
- 185.Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, Simmen T. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM) Cell Stress Chaperones. 2010;15:619–629. doi: 10.1007/s12192-010-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sciacca MF, Kotler SA, Brender JR, Chen J, Lee DK, Ramamoorthy A. Two-step mechanism of membrane disruption by Abeta through membrane fragmentation and pore formation. Biophys J. 2012;103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bechinger B, Lohner K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim Biophys Acta. 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 188.Ramamoorthy A. Beyond NMR spectra of antimicrobial peptides: dynamical images at atomic resolution and functional insights. Solid State Nucl Magn Reson. 2009;35:201–207. doi: 10.1016/j.ssnmr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Patel HR, Pithadia AS, Brender JR, Fierke CA, Ramamoorthy A. In Search of Aggregation Pathways of IAPP and Other Amyloidogenic Proteins: Finding Answers through NMR Spectroscopy. J Phys Chem Lett. 2014;5:1864–1870. doi: 10.1021/jz5001775. [DOI] [PubMed] [Google Scholar]
- 190.Brender JR, Salamekh S, Ramamoorthy A. Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc Chem Res. 2012;45:454–462. doi: 10.1021/ar200189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sciacca MF, Milardi D, Messina GM, Marletta G, Brender JR, Ramamoorthy A, La Rosa C. Cations as switches of amyloid-mediated membrane disruption mechanisms: calcium and IAPP. Biophys J. 2013;104:173–184. doi: 10.1016/j.bpj.2012.11.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Brender JR, Hartman K, Nanga RP, Popovych N, de la Salud Bea R, Vivekanandan S, Marsh EN, Ramamoorthy A. Role of zinc in human islet amyloid polypeptide aggregation. J Am Chem Soc. 2010;132:8973–8983. doi: 10.1021/ja1007867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Brender JR, Lee EL, Hartman K, Wong PT, Ramamoorthy A, Steel DG, Gafni A. Biphasic effects of insulin on islet amyloid polypeptide membrane disruption. Biophys J. 2011;100:685–692. doi: 10.1016/j.bpj.2010.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sciacca MF, Lolicato F, Di Mauro G, Milardi D, D’Urso L, Satriano C, Ramamoorthy A, La Rosa C. The Role of Cholesterol in Driving IAPP-Membrane Interactions. Biophys J. 2016;111:140–151. doi: 10.1016/j.bpj.2016.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sciacca MF, Brender JR, Lee DK, Ramamoorthy A. Phosphatidylethanolamine enhances amyloid fiber-dependent membrane fragmentation. Biochemistry. 2012;51:7676–7684. doi: 10.1021/bi3009888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J Am Chem Soc. 2008;130:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shimanouchi T, Tasaki M, Vu HT, Ishii H, Yoshimoto N, Umakoshi H, Kuboi R. Abeta/Cu-catalyzed oxidation of cholesterol in 1,2-dipalmitoyl phosphatidylcholine liposome membrane. J Biosci Bioeng. 2010;109:145–148. doi: 10.1016/j.jbiosc.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 199.Nanga RP, Brender JR, Vivekanandan S, Popovych N, Ramamoorthy A. NMR structure in a membrane environment reveals putative amyloidogenic regions of the SEVI precursor peptide PAP(248–286) J Am Chem Soc. 2009;131:17972–17979. doi: 10.1021/ja908170s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Vermeer LS, Hamon L, Schirer A, Schoup M, Cosette J, Majdoul S, Pastre D, Stockholm D, Holic N, Hellwig P, Galy A, Fenard D, Bechinger B. Vectofusin-1, a potent peptidic enhancer of viral gene transfer forms pH-dependent alpha-helical nanofibrils, concentrating viral particles. Acta Biomater. 2017;64:259–268. doi: 10.1016/j.actbio.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 201.Hallock KJ, Wildman KH, Lee D-K, Ramammorthy A. An innovation precedure using a sublimable solid to align lipid bilayers for solid-state NMR studies. Biophys J. 2002;82:2499–2503. doi: 10.1016/S0006-3495(02)75592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Separovic F, Killian JA, Cotten M, Busath DD, Cross TA. Modeling the membrane environment for membrane proteins. Biophys J. 2011;100:2073–2074. doi: 10.1016/j.bpj.2011.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sani MA, Separovic F. Antimicrobial Peptide Structures: From Model Membranes to Live Cells. Chemistry. 2018;24:286–291. doi: 10.1002/chem.201704362. [DOI] [PubMed] [Google Scholar]
- 204.Salnikov ES, Aisenbrey C, Aussenac F, Ouari O, Sarrouj H, Reiter C, Tordo P, Engelke F, Bechinger B. Membrane topologies of the PGLa antimicrobial peptide and a transmembrane anchor sequence by Dynamic Nuclear Polarization/solid-state NMR spectroscopy. Sci Rep. 2016;6:20895. doi: 10.1038/srep20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sanders CR, Prosser RS. Biocelles: a model membrane system for all seasons? Structure. 1998;6:1228–1234. doi: 10.1016/s0969-2126(98)00123-3. [DOI] [PubMed] [Google Scholar]
- 206.Durr UH, Soong R, Ramamoorthy A. When detergent meets bilayer: birth and coming of age of lipid bicelles. Prog Nucl Magn Reson Spectrosc. 2013;69:1–22. doi: 10.1016/j.pnmrs.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Durr UH, Gildenberg M, Ramamoorthy A. The magic of bicelles lights up membrane protein structure. Chem Rev. 2012;112:6054–6074. doi: 10.1021/cr300061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yamamoto K, Pearcy P, Lee DK, Yu C, Im SC, Waskell L, Ramamoorthy A. Temperature-resistant bicelles for structural studies by solid-state NMR spectroscopy. Langmuir. 2015;31:1496–1504. doi: 10.1021/la5043876. [DOI] [PubMed] [Google Scholar]
- 209.Yamamoto K, Pearcy P, Ramamoorthy A. Bicelles exhibiting magnetic alignment for a broader range of temperatures: a solid-state NMR study. Langmuir. 2014;30:1622–1629. doi: 10.1021/la404331t. [DOI] [PubMed] [Google Scholar]
- 210.Denisov IG, Sligar SG. Nanodiscs in Membrane Biochemistry and Biophysics. Chem Rev. 2017;117:4669–4713. doi: 10.1021/acs.chemrev.6b00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dorr JM, van Coevorden-Hameete MH, Hoogenraad CC, Killian JA. Solubilization of human cells by the styrene-maleic acid copolymer: Insights from fluorescence microscopy. Biochim Biophys Acta. 2017;1859:2155–2160. doi: 10.1016/j.bbamem.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 212.Ravula T, Hardin NZ, Ramadugu SK, Cox SJ, Ramamoorthy A. Formation of pH-Resistant Monodispersed Polymer-Lipid Nanodiscs. Angew Chem Int Ed Engl. 2018;57:1342–1345. doi: 10.1002/anie.201712017. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 213.Yasuhara K, Arakida J, Ravula T, Ramadugu SK, Sahoo B, Kikuchi JI, Ramamoorthy A. Spontaneous Lipid Nanodisc Fomation by Amphiphilic Polymethacrylate Copolymers. J Am Chem Soc. 2017;139:18657–18663. doi: 10.1021/jacs.7b10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ramadugu VSK, Di Mauro GM, Ravula T, Ramamoorthy A. Polymer nanodiscs and macro-nanodiscs of a varying lipid composition. Chem Commun. 2017;53:10824–10826. doi: 10.1039/c7cc06409h. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 215.Ravula T, Ramadugu SK, Di Mauro G, Ramamoorthy A. Bioinspired, Size-Tunable Self-Assembly of Polymer-Lipid Bilayer Nanodiscs. Angew Chem Int Ed Engl. 2017;56:11466–11470. doi: 10.1002/anie.201705569. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 216.Ravula T, Hardin NZ, Ramadugu SK, Ramamoorthy A. pH Tunable and Divalent Metal Ion Tolerant Polymer Lipid Nanodiscs. Langmuir. 2017;33:10655–10662. doi: 10.1021/acs.langmuir.7b02887. [DOI] [PubMed] [Google Scholar]
- 217.Nath A, Miranker AD, Rhoades E. A membrane-bound antiparallel dimer of rat islet amyloid polypeptide. Angew Chem Int Ed Engl. 2011;50:10859–10862. doi: 10.1002/anie.201102887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Cross TA, Ekanayake V, Paulino J, Wright A. Solid state NMR: The essential technology for helical membrane protein structural characterization. J Magn Reson. 2014;239:100–109. doi: 10.1016/j.jmr.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Das N, Murray DT, Cross TA. Lipid bilayer preparations of membrane proteins for oriented and magic-angle spinning solid-state NMR samples. Nat Protoc. 2013;8:2256–2270. doi: 10.1038/nprot.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ramamoorthy A, Wei Y, Lee D-K. PISEMA Solid-State NMR Spectroscopy. Ann Rep NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- 221.Bechinger B. Lipid multilayers: Domains stack up. Nat Mater. 2012;11:1005–1006. doi: 10.1038/nmat3507. [DOI] [PubMed] [Google Scholar]
- 222.Aisenbrey C, Michalek M, Salnikov ES, Bechinger B. Solid-state NMR approaches to study protein structure and protein-lipid interactions. Methods Mol Biol. 2013;974 doi: 10.1007/978-1-62703-275-9_16. [DOI] [PubMed] [Google Scholar]
- 223.Sani MA, Separovic F. How Membrane-Active Peptides Get into Lipid Membranes. Acc Chem Res. 2016;49:1130–1138. doi: 10.1021/acs.accounts.6b00074. [DOI] [PubMed] [Google Scholar]
- 224.Thennarasu S, Huang R, Lee DK, Yang P, Maloy L, Chen Z, Ramamoorthy A. Limiting an antimicrobial peptide to the lipid-water interface enhances its bacterial membrane selectivity: a case study of MSI-367. Biochemistry. 2010;49:10595–10605. doi: 10.1021/bi101394r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ramamoorthy A, Thennarasu S, Lee DK, Tan A, Maloy L. Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys J. 2006;91:206–216. doi: 10.1529/biophysj.105.073890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Powers JP, Tan A, Ramamoorthy A, Hancock RE. Solution structure and interaction of the antimicrobial polyphemusins with lipid membranes. Biochemistry. 2005;44:15504–15513. doi: 10.1021/bi051302m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Thennarasu S, Lee D-K, Tan A, Kari UP, Ramammorthy A. Antimicrobial activity and membrane selective interactions of a synthetic lipopeptide MSI-843. Biochim Biophys Acta. 2005;1711:49–58. doi: 10.1016/j.bbamem.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Henzler Wildman KA, Lee D-K, Ramamoorthy A. Mechanism of lipid bilayer disruption by the human antimicrobial pepride, LL-37. Biochemistry. 2003;42:6545–6558. doi: 10.1021/bi0273563. [DOI] [PubMed] [Google Scholar]
- 229.Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. alpha-Synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 230.Grey M, Linse S, Nilsson H, Brundin P, Sparr E. Membrane interaction of alpha-synuclein in different aggregation states. J Parkinsons Dis. 2011;1:359–371. doi: 10.3233/JPD-2011-11067. [DOI] [PubMed] [Google Scholar]
- 231.Smith PE, Brender JR, Ramamoorthy A. Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: the case of islet amyloid polypeptide. J Am Chem Soc. 2009;131:4470–4478. doi: 10.1021/ja809002a. [DOI] [PMC free article] [PubMed] [Google Scholar]