

Abstract
Biotherapeutics are a promising class of molecules in drug discovery, but they are often limited to extracellular targets due to their poor cell penetration. High-throughput cell penetration assays are required for the optimization of biotherapeutics for enhanced cell penetration. We developed a HaloTag-based assay called the ChloroAlkane Penetration Assay (CAPA), which is quantitative, high-throughput, and compartment-specific. We demonstrate the ability of CAPA to profile extent of cytosolic penetration with respect to concentration, presence of serum, temperature, and time. We also used CAPA to investigate structure-penetration relationships for bioactive stapled peptides and peptides fused to cell-penetrating sequences. CAPA is not only limited to measuring cytosolic penetration. Using a cell line where HaloTag is localized to the nucleus, we show quantitative measurement of nuclear penetration. Going forward, CAPA will be a valuable method for measuring and optimizing the cell penetration of biotherapeutics.
Graphical Abstract
INTRODUCTION
In recent years the pharmaceutical industry has invested a great deal of resources in designing and developing biotherapeutics. Therapeutic peptides, proteins, gene-editing complexes, antibodies and nucleic acids are appealing due to their high affinity and specificity, which can lead to excellent potency and minimal off-target effects. The majority of biotherapeutics currently in use in humans are monoclonal antibodies, which target extracellular receptors.1 The slow progress in targeting intracellular processes emphasizes that a primary obstacle for biotherapeutic development is optimizing cytosolic penetration.
Many strategies have been applied to promote cell penetration for biomolecules.2–4 Perhaps the most widely applied method for biomolecule delivery is conjugation to polycationic cell-penetrating peptides (CPPs).5,6 For smaller biomolecules such as peptides, chemical modifications can be applied to improve cell penetration without the need for CPPs. For example, modifying helical peptides with all-hydrocarbon staples has yielded several cell-penetrant peptides with biological activity, and some are currently being tested in clinical trials.7,8 Despite decades of investigation and progress towards the clinic, we are only beginning to understand the underlying mechanisms of cell penetration for these and other candidate therapeutics.9,10
One of the largest obstacles to understanding and improving cell penetration is the difficulty of accurately measuring cytosolic localization. Several assays have been developed to measure the cell penetration of biomolecules, which we recently reviewed.11 Most commonly, molecules are tagged with a fluorescent dye and tracked using fluorescence or confocal fluorescence microscopy,12,13 but these methods cannot unambiguously quantitate localization to cytosolic and endosomal compartments. Alternatives have been developed that address some of these drawbacks.11,14–17 Still, there remains a need for compartment-specific, quantitative and high-throughput assays for measuring the cell penetration of biomolecules of interest.
Here, we describe the ChloroAlkane Penetration Assay (CAPA), a new assay for measuring cell penetration. We previously reported results from CAPA with a handful of autophagy-inducing peptides.18 In this work, we benchmark the assay to well-studied CPPs, hydrocarbon-stapled helices, cyclic peptides, and miniature proteins. We demonstrate how CAPA can generate data-rich profiles of cell penetration across many concentrations, time points and culture conditions. We also show how CAPA can help deconvolute phenotypic assay results by examining the structure-penetration relationships of autophagy-inducing stapled peptides and CPP-conjugated peptides.18,19 Finally, we use a nuclear-reporting cell line to reveal dose-dependent penetration profiles for penetration to the nucleus.
RESULTS
Assay design and methodology
CAPA relies on the HaloTag system. HaloTag, developed by Wood and co-workers in 2006, is a mutant bacterial haloalkane dehalogenase which catalyzes rapid, covalent bond formation to chloroalkane-containing molecules.20,21 The Halo-GFP-Mito cells used for CAPA are HeLa cells that stably express a fusion of HaloTag, GFP and a mitochondria-targeting peptide from the C-terminal domain of the ActA protein from Listeria monocytogenes. This fusion anchors HaloTag to the outer mitochondrial membrane, oriented in the cytosol.22 We selected this cell line to ensure that HaloTag was accessible from the cytosol, but not other cellular compartments such as the nucleus.
The general format for CAPA is a simple pulse-chase assay (Fig. 1a). First, cells are pulsed with a chloroalkane-tagged molecule (ct-molecule). If the molecule is cell-penetrant and accesses the cytosol, it will react with HaloTag and covalently block it. Cells are washed to remove ct-molecule that did not penetrate, then are chased with chloroalkane-tagged dye (ct-dye). The ct-dye rapidly penetrates cells and covalently binds unoccupied HaloTag. After washing the cells to remove excess ct-dye, fluorescence intensity of cells is quantitated using a benchtop flow cytometer. The fluorescence intensity is proportional to the amount of unreacted HaloTag, and is thus inversely related to the extent of penetration of the ct-molecule. Flow cytometry enabled high-throughput, quantitative measurements, allowing analysis of cells in 96-well plates with 10,000 cells per sample. Within each 96-well plate, cells not treated with any ct-molecule or ct-dye were used as a normalization standard for 0% fluorescence, and cells treated only with ct-dye were used as a normalization standard for 100% fluorescence. To reveal dose dependence, the mean fluorescence intensity of each histogram was plotted as a function of ct-molecule concentration, as shown in Figure 1a. Because the cell line produces HaloTag exclusively in the cytosolic compartment,22 CAPA signal is not affected by material trapped in endosomes, and thus reports on cytosolic penetration.
Figure 1. Chloroalkane penetration assay (CAPA) procedure and benchmarking to CPPs.
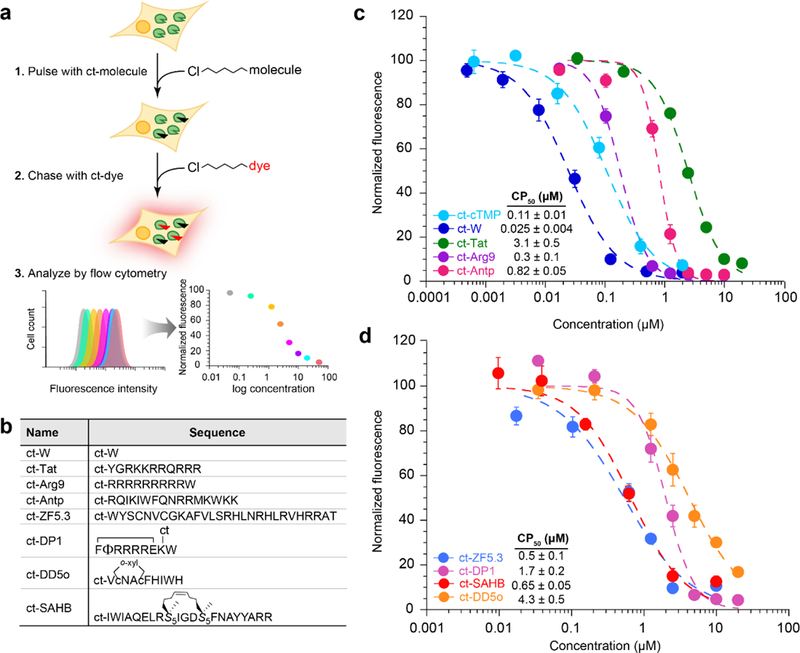
a) Schematic showing assay setup. Cells stably expressing HaloTag are pulsed with chloroalkane-tagged molecules, washed, chased with a chloroalkane-tagged dye, and finally analyzed by flow cytometry. b) Sequences of chloroalkane-tagged, cell-penetrant peptides and small molecules. ct represents the chloroalkane carboxylic acid (Fig. S1), which was attached via amide bond to the peptide N-terminus or, for DP1, a lysine side chain. Φ represents naphthylalanine, lowercase c represents d-cysteine, and S5 represents S-pentenylalanine. ct-DP1 was cyclized between the N-terminus and an internal glutamate side chain, DD5o was cyclized using ortho-dibromoxylene (o-xyl), and ct-SAHB was cyclized using ring-closing metathesis. c) CAPA results for ct-W, ct-cTMP (a caged trimethoprim used here as a second small-molecule control, see SI Fig. 1), ct-Tat, ct-Arg9 and ct-Antp. ct-molecules were incubated with cells in Opti-MEM for 4 h. Each point is the mean red fluorescence of 10,000 cells. The data were normalized using a no-pulse control as 100% signal, and a no-pulse/no-chase control as 0% signal. d) CAPA results for cell-penetrant peptides ct-ZF5.3, ct-DP1, ct-SAHB, and ct-DD5o. Error bars show standard error from three independent experiments. CP50 averages and standard errors are from three independent curve fits from three independent experiments.
Benchmarking CAPA to widely-used CPPs
To assess the utility of this new assay, we decided to test some of the most widely used cell-penetrating peptides (Fig. 1b). These included Tat, which is derived from the HIV protein trans-activator of transcription;23 Antp (also known as penetratin), a 16-mer derived from the antennapedia homeodomain of Drosophila;24 and nona-arginine Arg9.25,26 As small molecule controls, we used a chloroalkane-tagged tryptophan (ct-W) and a trimethoprim analog (ct-cTMP) that had been previously tested with the HaloTag-expressing cell line.22 Figure 1c shows CAPA data for 4 h incubations of different concentrations of ct-Tat, ct-Antp, ct-Arg9, ct-W, and ct-cTMP. Individual data points showed high reproducibility in biological replicates, and the concentration-dependent data were reliably fit with sigmoidal curves. Given these data, we fit IC50 curves and determined midpoint values that we termed CP50, the concentration at which 50% cell penetration was observed for this cell line under the specific incubation conditions. We found CP50 values are highly reproducible and represent a convenient and quantitative means of comparing ct-molecules. The small molecule ct-W penetrated the cytosol best, with a CP50 of 0.025 μM, followed by ct-cTMP at 0.11 μM, ct-Arg9 at 0.3 μM, ct-Antp at 0.82 μM, and then ct-Tat at 3.1 μM (Fig. 1c). These values parallel similar relative values obtained by Jiao et al, who measured cell penetration of CPPs using quantitative MALDI-MS.27
Testing optimized cell-penetrant peptides
After benchmarking CAPA to commonly used CPPs, we sought to apply it to recently reported peptides that were optimized for cell penetration. We tested cell-penetrant mini protein ZF5.3 developed by Schepartz and co-workers,28 an all-hydrocarbon stapled peptide BIM-SAHBA1 developed by Walensky and co-workers,13 a cyclic oligoarginine peptide DP1 developed by Pei and co-workers,29 and also the autophagy-inducing stapled peptide DD5o developed in our lab (Fig. 1d).18 We found that ct-ZF5.3 and ct-SAHB accessed the cytosol best, with CP50 values of 0.5 and 0.65 μM respectively, followed by ct-DP1 at 1.7 μM and then ct-DD5o at 4.3 μM. While the CP50 value for ct-DP1 was higher than might be suggested by previous literature (peptide 1 in ref. 29), a DP1 derivative with a longer PEG linker had a CP50 of 0.16 μM (ct-PEG2-DP1, see below). Overall, CAPA data supported that ZF5.3, BIM-SAHBA1, and DP1 can access the cytosol more effectively than Tat, as suggested by previous literature.28–30
Effect of chloroalkane position and linker on CAPA
CAPA is not a tag-free assay, so it was important to evaluate the influence of the chloroalkane tag. The presence of the chloroalkane tag does not promote efficient cell penetration on its own, as observed by examples of ct-peptides with poor penetration as measured by CAPA (for instance, ct-DD5o-W9A, see below). To test effects of linker location, we synthesized two isomeric analogs of stapled peptide ct-DD5o, one of which incorporated a chloroalkane tag on the N-terminus and one with the tag on a lysine at the C-terminus (ct-DD5o-pa and pa-DD5o-ct, SI Fig. 1). These compounds were tested side-by-side in CAPA along with the original ct-DD5o, and we found no difference in their penetration profiles (SI Fig. 2). This suggests that the location of the chloroalkane does not affect the cell penetration, at least for DD5o.
Following work by Wood and others, we also tested the effects of linkers of varying lengths to determine whether they affected CAPA.21,31,32 Chloroalkane tags with PEG2, PEG3, PEG4, and hexane linkers were conjugated to peptide DD5o (SI Fig. 1). The CAPA data for all five of these DD5o analogs were very similar, and the CP50 values were the same within experimental error (SI Fig. 3a). We examined linker effects for several other peptides, comparing ct-peptide to ct-PEG2-peptide for ct-Tat, ct-Arg9, ct-Antp, ct-ZF5.3, ct-W, ct-DP1, ct-DD5o, and several alanine scan analogs of ct-DD5o (SI Fig. 3b-c). Most of the ct-peptide and ct-PEG2-peptide pairs showed similar CAPA profiles, but a few pairs had different CP50 values (SI Fig. 3d). Specifically, ct-DD5o-H7A, ct-DD5o-H10A, and ct-DP1 were each less cell penetrant than their ct-PEG2-tagged counterparts, and ct-ZF5.3 was more cell-penetrant than its ct-PEG2-tagged counterpart. Overall, the data suggest that CAPA is generally independent of the specific linker used to install the chloroalkane for most peptides, but that a small number of peptides are sensitive to the effects of the chloroalkane tag. Thus, sensitivity to the tag should be determined on a case-by-case basis.
We also wanted to rule out significant differences among ct-molecules with respect to intrinsic reactivity with HaloTag. Previous work from Wood and co-workers investigated the HaloTag reaction kinetics of different chloroalkane-tagged small molecules using a fluorescence gel-based assay.21 We designed a more direct gel-shift assay to detect the extent of HaloTag reaction for different ct-peptides. Recombinant, purified HaloTag was incubated with 1, 2, 4 or 8 equivalents of peptide, then separated by SDS-PAGE to analyze extent of reaction by observable band shift. Most peptides tested showed substantial band-shift at a 2:1 peptide:HaloTag ratio and near-complete reaction at 4:1 ratio (SI Fig. 4); some, like ct-DD5o, showed up to 2-fold less reactivity, but still had complete reaction at 8:1 ratio. We also applied the band-shift assay to compare ct-peptides that had drastically different results in CAPA. For instance, ct-DD5o and its W9A analog have strikingly different CAPA profiles, with CP50 values that differ by 50-fold (see below), so we tested them by gel-shift assay to ensure this was not due to poorer intrinsic reactivity of ct-DD5o-W9A analog with HaloTag. The gel shift assay demonstrated that the reactivities of these two peptides are similar (SI Fig. 4). We tested additional pairs of highly similar analogs that had drastically different CAPA profiles, including ct-DD5o-H7A and ct-PEG2-H7A, ct-H10A and ct-PEG2-H10A, and ct-DP1 and ct-PEG2-DP1. In each of these cases, the pair of peptides had similar reactivities with purified HaloTag protein (SI Fig. 4). Overall, the gel-shift assay verified that HaloTag reactivity is unlikely to be a predominant factor affecting CAPA readout, strengthening the interpretation that differences in CAPA data represent differences in extent of cell penetration.
We also sought to demonstrate direct conjugation of ct-molecules to HaloTag in cells. For this purpose, we designed a probe containing chloroalkane and biotin (ct-PEG4-biotin) and demonstrated that it biotintylates HaloTag in vitro (SI Fig. 5a-b) To investigate reaction with HaloTag in cells, we first obtained dose-dependent CAPA data for ct-PEG4-biotin (CP50 = 0.7 μΜ, SI Fig. 5c). Cells from the same experiment, harvested prior to the chase with ct-dye, were lysed and biotin-conjugated proteins were pulled down using streptavidin-coated magnetic beads. Protein was eluted from the beads and blotted using anti-GFP antibody, in order to detect biotinylated HaloTag-GFP fusion protein. A band corresponding to the expected MW of the HaloTag fusion was observed after incubation with 5 μM ct-PEG4-biotin. The intensity of this band decreased at lower incubation concentrations, becoming undetectable at 0.1 μM (SI Fig. 5d). These results match the CAPA data, and support the interpretation that CAPA indeed reports on extent of HaloTag alkylated by ct-molecule during the first incubation step.
Cell penetration profiling
CAPA can easily be adapted to measure cell penetration quantitatively while varying incubation conditions. We designed a series of experiments that investigate variables that have been previously reported to affect cell penetration. For these “profiling” experiments, we employed a panel of five molecules that span different classes of cell-penetrant molecules. These included two widely used polycationic CPPs, Tat and Arg9, two different classes of stapled peptides, DD5o and BIM-SAHBA1, and the small molecule ct-W.
We investigated the temperature dependence of cell penetration by comparing CAPA data at 37 °C and 4 °C. These conditions have previously been used to investigate whether cell penetration of a molecule is energy-dependent.26,28,33–35 The results showed a stark decrease in penetration for ct-SAHB, ct-DD5o and ct-W, while ct-Tat and ct-Arg9 were less affected (Fig. 2a and SI Fig. 6). The temperature dependence of ct-W was unexpected, but temperature dependence of passively penetrant molecules has been observed for molecules that bind and diffuse through cell membranes.36,37 Another parameter we assessed was serum content in the media. Previous reports have shown that the cellular uptake of CPPs and certain all-hydrocarbon stapled peptides, such as BIM-SAHBA1, can be suppressed in the presence of fetal bovine serum (FBS).13,38 To test the effect of serum on cell penetration, we tested ct-peptides in the presence and absence of 10% FBS in DMEM. We measured slightly higher CP50 values for all molecules, suggesting lower penetration efficiency in the presence of serum (Fig. 2b and SI Fig. 7). The slight decrease could be attributed to peptide being bound by serum proteins, or serum might alter endocytic uptake mechanism or other internalization pathways.
Figure 2. Cell penetration profiling.
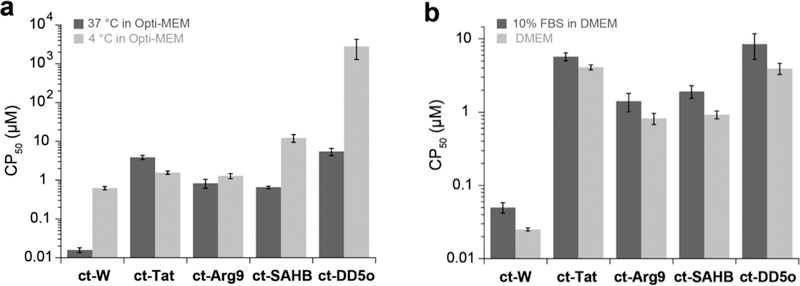
a) CAPA results for ct-W, ct-Tat, ct-Arg9, ct-SAHB, and ct-DD5o, tested at 37 °C and 4 °C. ct-molecules were incubated with cells for 4 h in Opti-MEM. Graph shows CP50 values on a log scale, and error bars show standard error from three independent experiments CP50 values are provided in SI Table 2. b) Effects of serum on cell penetration with CAPA. Peptides were incubated for 4 h at 37 °C in DMEM with or without 10% FBS. CP50 values are provided in SI Table 3. Graph shows CP50 values on a log scale, and error bars show standard error from three independent experiments.
Quantifying the time dependence of cell penetration has proven to be challenging using standard methods. We hypothesized that CAPA could provide the resolution necessary for comparing the penetration of different molecules at specific time points. We acquired full dose-response CAPA data after 0.5, 2, 4, 8 and 24 hours of incubation with ct-W, ct-Tat, ct-Antp, ct-SAHB, and ct-DD5o (Fig 3a). After 0.5 h, ct-W accessed the cytosol with a CP50 of 0.24 μM, while ct-Arg9 accessed the cytosol with a CP50 of 5.2 μM. The rest of the molecules did not penetrate the cytosol in that time frame. ct-W and all ct-peptides showed increasing penetration with time between 2h and 24h. Interestingly, ct-peptides appeared to have characteristic slopes to their dose-dependence curves, with some steeper than others regardless of time point. For instance, at the 4 h time point the CP50 values of ct-Arg9 and ct-SAHB are both roughly 0.5 μM, but the slopes of the dose-response curves are different (Fig. 3a). In fact, ct-Arg9 had a much steeper dose-response curve than the other compounds at all time points, while ct-SAHB had a shallower curve than the other compounds at all time points. In the presence of 10% serum, the dose-response curves for ct-Arg9 and ct-Tat are less steep, while serum had no effect on the slope of ct-SAHB (SI Fig. 7). Several mechanisms of penetration involve the accumulation of a critical local concentration of peptide above a certain threshold before translocation, endocytosis, and/or endosomal release can occur.9 The CAPA data imply that ct-Arg9 has a stronger threshold effect compared to the other peptides tested, and that ct-SAHB’s mechanism of entry may be less dependent on a critical concentration threshold.
Figure 3. Measurement of cell penetration over time using CAPA.
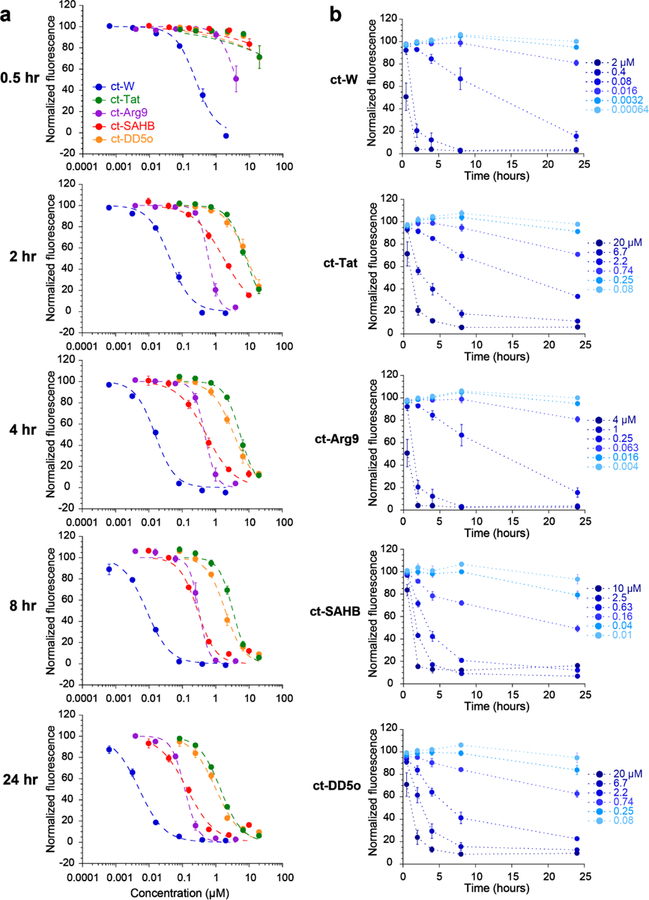
a) Data from time-course experiments. Each graph shows dose-dependent cell penetration at a different time point. ct-molecules were incubated with cells in Opti-MEM for 0.5 h, 2 h, 4 h, 8 h or 24 h. Error bars show standard error from three independent experiments. CP50 values are provided in SI Table 4. b) Data from time-course experiments, plotted as a function of time. Each graph shows time-dependent cell penetration for a different ct-molecule at six different concentrations. Error bars show standard error from three independent experiments.
The same data can also be presented as CAPA signal versus time for each concentration of each ct-molecule (Fig. 3b). These data provide valuable insight on the extent of penetration at specific concentrations and specific time points. For example, there are specific concentrations at which cell penetration scales roughly linearly with time for each ct-molecule (ct-W at 0.08 μM, ct-Tat at 2.2 μM, ct-Arg9 at 0.25 μM, ct-SAHB at 0.63 μM, and ct-DD5o at 2.2 μM; Fig. 3b). The time-dependent CAPA data also reveal concentrations below which cytosolic penetration is barely achieved even with a 24 h incubation, such as ct-DD5o below 0.25 μM. Because the phenotypic effects of CPPs and stapled helices have been observed to be time-dependent, having an independent means of measuring cytosolic penetration at different time points will be very valuable for choosing relevant experimental conditions.
Investigating structure-penetration relationships using CAPA
In our previous work on stapled, autophagy-inducing peptides, we produced structure-activity relationships (SAR) using an alanine scan of peptide DD5o.18 Activity of autophagy-inducing peptides is evaluated in vitro by measuring dose-dependent degradation of autophagy adaptor protein p62, as well as dose-dependent lipidation of LC3.18,19 These are rigorous standards for judging autophagy-inducing activity,39 but these are also phenotypic assays which cannot differentiate the extent of cell penetration from other properties. For this reason, we decided to use CAPA to investigate structure-penetration relationships more directly. We first applied CAPA to an alanine scan of ct-DD5o (Fig. 4a and SI Fig. 8). The data showed that when Val1, Asn3 and Phe6 were substituted to Ala, the cytosolic penetration was similar or slightly better (Fig. 4a). The Ile8 substitution yielded a slightly less cell-penetrant peptide. Substituting His7 or His10 with Ala, however, increased the CP50 values to above 30 μM, which is more than 4-fold worse than ct-DD5o, and the substitution of Trp9 to Ala abolished cell penetration altogether. The observation that Trp9 contributes to cell penetration was novel, but it matched what was assumed about the role of Trp9 from structure-activity data. Specifically, solving the 3D structure of DD5o revealed that Trp9 was part of a large, continuous hydrophobic surface.18 Having a continuous hydrophobic surface has been shown to be crucial for the cell penetration of hydrocarbon-stapled helices,13 a trait which DD5o likely shares. From the CAPA data, it is now possible to attribute the decreased activity of the W9A analog in autophagy-induction assays (ref. 18 and SI Fig. 9) to abolished cell penetration.
Figure 4. Investigation of structure-penetration relationships using CAPA.
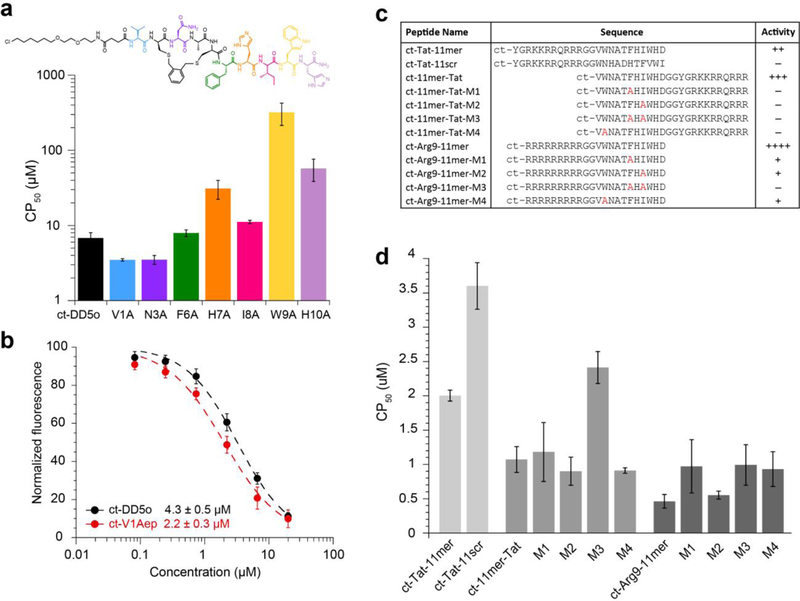
a) CAPA results for alanine-scanning analogs of ct-DD5o. The CP50 values are shown on a log scale for each alanine-substituted analog. ct-molecules were incubated with cells in Opti-MEM for 4 h. CP50 values are provided in SI Table 5. b) Dose dependence of cell penetration for ct-DD5o and ct-V1Aep. c) Table showing the sequences and relative autophagy-inducing activity of CPP-linked autophagy-inducing peptides, including ct-Tat-11mer, scrambled analog ct-Tat-11scr, ct-11mer-Tat and its mutants, and ct-Arg9–11mer and its mutants. Estimates of relative activity are based on LC3 and p62 immunoblots (SI Fig. 12) and are presented as increasing activity from + (minimal activity) to ++++ (very strong activity), or – for no activity. d) CP50 values of CPP-linked autophagy-inducing peptides. CP50 values are provided in SI Table 6. All ct-peptides were incubated with cells in Opti-MEM for 4 h. All error bars show standard error from three independent experiments.
In the context of the original DD5o structure, which was capped with pentynoic acid instead of the chloroalkane, the residue Val1 was shown to be necessary for autophagy-inducing activity despite being nonessential for the activity of the CPP-linked peptide Tat-11mer.18 At the time, our hypothesis was that Val1 was important for cell penetration of DD5o. Testing this hypothesis using CAPA, we found that ct-DD5o and ct-DD5o-V1A had similar CP50 values (Fig. 4a), but also that these two ct-peptides had similar activity with respect to autophagy induction (SI Fig. 9). These data suggest that Val1 is either not required for cell penetration of DD5o, or it is required for cell penetration of DD5o but not for chloroalkane-tagged analogs. Next, we designed and tested additional analogs of DD5o with substitutions at the Val1 position. We observed that substituting Val1 with 2-amino-3-ethyl-pentanoic acid (Aep) led to a peptide with 2-fold better autophagy-inducing activity (pa-V1Aep, SI Fig. 10), but as with DD5o-V1A, the chloroalkane-tagged version (ct-V1Aep) had comparable autophagy-inducing activity to ct-DD5o (SI Fig. 10). Testing ct-V1Aep in CAPA (Fig. 4b), we obtained a CP50 value of 2.2 μM, which is roughly two-fold lower than the CP50 of ct-DD5o (4.3 μM). Thus, CAPA guided the design of a more cell-penetrant stapled peptide, though cooperative effects of the chloroalkane tag remain to be deconvoluted.
We further wanted to show the utility of CAPA by investigating the type and location of CPP that would improve cell penetration for a CPP-conjugated peptide. The peptide Tat-11mer has previously been tested in vitro and in vivo and shown to induce autophagy at 5 μM.18,19 We hypothesized that we could improve the activity of Tat-11mer by altering its CPP to improve cell penetration. We tested ct-Tat-11mer in CAPA, along with an analog with a C-terminal Tat (ct-11mer-Tat) and an analog with an N-terminal nona-arginine (ct-Arg9–11mer; sequences in Fig. 4c). Full dose-response curves were obtained for all peptides, and CP50 values were calculated (Fig. 4d and SI Fig. 11). The CP50 value for ct-11mer-Tat was two-fold less than ct-Tat-11mer, and the CP50 value of ct-Arg9–11mer was four-fold less than ct-Tat-11mer (Fig. 4d). Autophagy-inducing activity of these CPP fusions matched these trends, with ct-11mer-Tat active at 2-fold lower concentration than the original ct-Tat-11mer, and ct-Arg9–11mer active at 4-fold lower concentration than ct-Tat-11mer. We note that ct-Arg9–11mer is the most potent autophagy-inducing peptide to date, at least in vitro (SI Fig. 12).
Since CPP fusions can have unpredictable effects, it is common to use scrambled sequences or point mutants as negative controls. An ideal negative control would enter cells with identical efficiency but would not be active in phenotypic assays. However, the effects of sequence changes on cell penetration are seldom measured for CPP-peptide fusions. For instance, we designed a scrambled analog of Tat-11mer, as well as single and double Ala mutants of 11mer-Tat and Arg9–11mer, as potential negative controls.18,19 Each of these analogs showed either greatly reduced autophagy induction or no induction at all (Fig. 4c and SI Fig. 12), but it was unclear whether some of this effect might be due to reduced cell penetration. To directly measure cell penetration for these negative controls, we performed CAPA on chloroalkane-tagged analogs (Fig. 4d and SI Fig. 11). The sequence-scrambled analog of ct-Tat-11mer, ct-Tat-11scr, had a 2-fold decrease in penetration compared to ct-Tat-11mer. Single Ala substitutions in ct-11mer-Tat and ct-Arg9–11mer resulted in ct-peptides with similar cell penetration. However, a mutant of ct-Arg9–11mer with two Ala substitutions had a 2-fold decrease in cell penetration compared to ct-Arg9–11mer. Overall, these data demonstrated that cell penetration of CPP-conjugated peptides is not cargo-independent, a phenomenon that has been observed in other systems.40 These data also allowed us to select optimal CPP-conjugated peptides and negative controls for ongoing development efforts.
Investigating nuclear penetration using CAPA
CAPA is not limited to the quantitation of cytosolic penetration. If performed with cell lines that express HaloTag fusions that localize to specific organelles, the assay will report on penetration to those subcellular compartments. To demonstrate this versatility, we used a HeLa cell line that stably expresses HaloTag and GFP fused to the C-terminus of histone 3. This cell line was previously used for nuclear localization studies.22 As shown in Figure 5a, the GFP-HaloTag fusion localizes to nuclei, and the nuclei exhibit red fluorescence when treated with ct-TAMRA. Pulsing for 4 h with 2 μM ct-W prior to chasing with ct-TAMRA prevented nearly all this red signal (Fig. 5a), and signal from nuclear ct-dye was readily quantified using flow cytometry.
Figure 5. Measurement of nuclear penetration using CAPA.
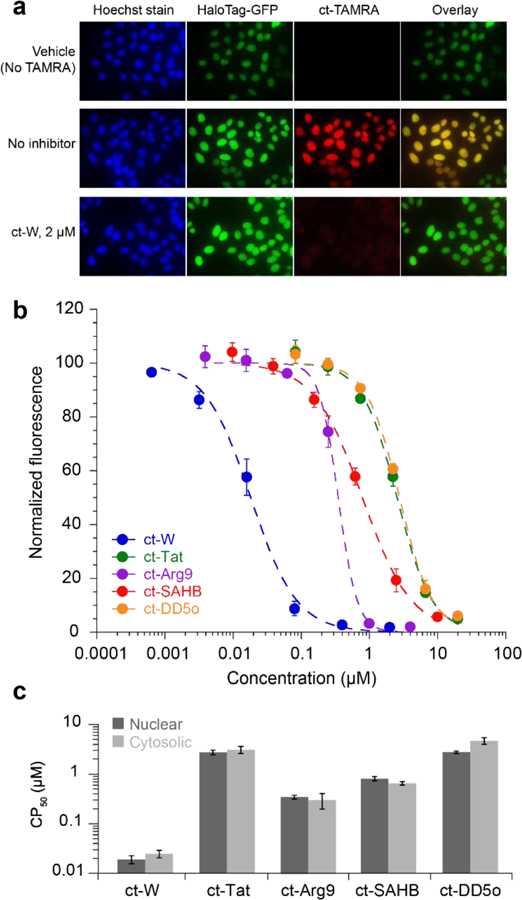
a) Images showing HeLa stably expressing H3-GFP-HaloTag. The first column shows fluorescence from Hoechst DNA stain, the second column shows fluorescence from GFP, and the third column shows red fluorescence due to captured ct-TAMRA. The fourth column is an overlay of the GFP and TAMRA signals. b) Dose dependence of nuclear penetration for indicated peptides. ct-peptides were incubated with cells in Opti-MEM for 4 h as described for the cytosolic cell line. Error bars show standard error from three independent experiments. c) Comparison of CP50 values between cytosolic and nuclear cell lines. Error bars show standard error from three independent experiments. CP50 values are provided in SI Table 7. CP50 averages and standard errors are from three independent curve fits from three independent experiments.
Dose dependence of nuclear penetration was quantitated using CAPA for ct-W, ct-Tat, ct-Arg9, ct-SAHB, and ct-DD5o (Fig. 5b). The concentration curves were very similar to those measured in the cytosolic HaloTag cell line (Fig. 1c,d). Even the relative slopes of the concentration dependence matched the profile that was observed for the cytosolic HaloTag cell line, reinforcing the hypothesis that the slope reflects the mechanism of penetration. The CP50 values from curve fits to nuclear CAPA data were very similar to those from the cytosolic cell line (Fig. 5c). Molecules smaller than 40,000 Da can typically diffuse freely through the nuclear pore,41 so it makes sense that the CP50 values of all five molecules were similar for both nuclear and cytosolic penetration.
DISCUSSION
Measuring and optimizing cell penetration remains a challenge in drug development. CAPA is a novel, quantitative assay for measuring cell penetration in a high-throughput and compartment-specific manner. In this work, we showed that CAPA can be used to quantitate the dose-dependent penetration of different types of molecules. Relative extents of cytosolic penetration are readily and quantitatively compared using CAPA-derived CP50 values. CP50 values obtained for small molecules, known CPPs, and recently developed bioactive peptides are all consistent with previous results from other cell penetration assays and phenotypic assays.9,13,18,27,29,42 Importantly, CAPA provides a much richer data set than most existing methods, but requires low culture volumes and allows parallel analysis of up to 96 compounds or conditions at a time.
In drug development, when a compound is potent in biochemical assays but not in cell-based assays, this failure is frequently attributed to poor cytosolic penetration. This may not always be the case, since there are many other possible explanations for this outcome such as off-target binding. CAPA represents a straightforward method to interrogate cytosolic penetration directly, and to derive optimal incubation times and conditions for cell-based assays. For example, the bioactivity of some hydrocarbon-stapled peptides is dependent on incubation time and the absence of serum in the culture media.7,43 Our results with BIM-SAHBA1 reflect that its cytosolic penetration is reduced somewhat (up to 50%, Fig. 2b) in the presence of serum.13 Dose- and time-dependence data (Fig. 3a,b) suggest very specific conditions for maximizing cell penetration for BIM-SAHBA1: 10 μM incubation for 2 h, 2.5 μM for 3–4 h, or 0.6 μM for 8 h. These values match results from phenotypic assays.13,30 Similarly, concentration and time dependence of activity for autophagy-inducing peptides, including Tat-linked peptides and the stapled peptide DD5o, match the concentration and time dependence reported by CAPA.18,19 Overall, these results suggest that CAPA will be useful as a predictive tool to choose conditions for cell-based assays in which the molecule of interest achieves efficient cytosolic penetration.
Though CAPA has several advantages, it is not without limitations. The assay is not tag-free, as the compounds being tested must contain the chloroalkane tag. The chloroalkane is generally non-perturbing and bio-orthogonal, as observed by us and others using the HaloTag system.18,20–22 Importantly, we did not observe any significant decrease in solubility due to the chloroalkane tag for any of the peptides tested, even for the hydrophobic stapled peptides BIM-SAHBA1 and DD5o. We did find CAPA results to be sensitive to linker length for a subset of peptides tested, including the cyclic CPP DP1 and the miniprotein ZF5.3.28,29 Also, the DD5o analogs ct-DD5o-H7A, ct-DD5o-W9A, and ct-DD5o-H10A were particularly sensitive to linker length, which is notable because these were the same analogs that showed decreased cell penetration compared to ct-DD5o. We also observed that ct-DD5o, ct-V1A and ct-V1Aep were more potent inducers of autophagy than their non-chloroalkane-tagged analogs. All together, these data suggest that there may be some degree of cooperativity between specific side chains and the chloroalkane tag, and that the chloroalkane tag can improve cell penetration for some peptides. The assay’s sensitivity is currently limited by the level of HaloTag produced by the stable cell lines, but this could be controlled by careful cell line construction. Another potential concern is peptide degradation, either extracellularly or in the endolysomal compartment, which could lead to release of the chloroalkane tag. This is a potential artifact for any tag-based assay, including those dependent on dye-conjugated molecules. Effects of degradation can be controlled for by evaluating the stability of the molecules of interest in cell culture. We note that, despite their different susceptibilities to proteolytic degradation, we measured CP50 values for small molecules, CPPs, and stapled peptides that match previous results from other cell-penetration assays and phenotypic assays.9,13,18,27,29,42 Our control experiments using ct-PEG2-biotin also correlated CAPA signal to direct reaction of ct-molecule with HaloTag. Finally, recent work by Lim and co-workers demonstrated the cellular uptake of cyclic peptoids using CAPA, and their CAPA results matched results from other cell penetration assays.36 Since peptoids are extremely resistant to proteolytic degradation,36,44 these results also support the interpretation that CAPA reports on cytosolic penetration of intact ct-molecules at shorter time points.
A valuable application of CAPA is the analysis of structure-penetration relationships. In this work, we demonstrated how CAPA can be used to optimize type and location of CPP for CPP-conjugated peptides. We also demonstrated how scrambled mutants and point mutants of CPP conjugates can be screened by CAPA to find negative controls with similar cell penetration as the bioactive peptide. Using CAPA, we interrogated the effects of single amino acid substitutions on the cell penetration of stapled peptide DD5o. Because the molecular targets of Tat-11mer and DD5o are not yet known, CAPA answered an otherwise difficult question: why were Val1 and Trp9 required for cellular activity of pa-DD5o even though the corresponding residues were not required for activity of Tat11-mer?18,19 Our prior assumption was that these residues were required for cell penetration of the stapled peptide. This was proven true for Trp9 but not for Val1. This observation led to the design of a more cell-penetrant stapled peptide, V1Aep, which is substituted with 2-amino-3-ethyl-pentanoic acid (Aep) at Val1. These results illustrate how CAPA can inform and enrich medicinal chemistry and compound optimization.
We anticipate that obtaining cell penetration profiles will be greatly beneficial for the development of peptide therapeutics. While it has limitations, CAPA avoids many pitfalls of other assays, particularly by reporting exclusively on cytoplasmic localization. For specific lead compounds, CAPA can help optimize cell penetration efficiency without requiring large amounts of material or highly specialized equipment. CAPA could be readily applied to compound libraries and high-throughput screening for cell penetration, provided the libraries incorporate the chloroalkane tag. CAPA is also well-suited for future studies to broadly define the cargo dependence of CPP-mediated cell penetration. Our data comparing cytosolic and nuclear penetration imply that CAPA will also be a useful tool for designing drugs targeted to specific subcellular compartments or organelles. Finally, we note that CAPA is readily applicable beyond peptide therapeutics. We are currently pursuing applications in nucleic acid and protein therapeutics, including high-throughput evaluation of different chemical modifications and delivery methods that are intended to improve penetration to the cytosol or other subcellular compartments.
METHODS
Peptide synthesis and purification.
Peptides were synthesized on Rink Amide resin (0.58 mmol/g) using standard Fmoc chemistry. For each coupling, 5 eq. of benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), 5 eq. of hydroxybenzotriazole (HOBt) and 13 eq. of diisopropylethylamine (DIPEA) were dissolved in N,N-dimethylformamide (DMF), added and shaken with the resin for 30 min at rt. To deprotect Fmoc, a 20% piperidine/DMF solution was used 2 × 10 min. To cap the N-terminus with the chloroalkane, 3 eq. of chloroalkane carboxylate, 3 eq. of PyBOP, and 10 eq. of DIPEA were dissolved in DMF and added to the resin for 1 hr at room temperature. The coupling efficiency was checked using a Kaiser test, and a second coupling was performed if reaction was incomplete. Peptides were cleaved from the resin using cleavage cocktail (94% TFA, 2.5% EDT, 2.5% H2O, 1% TiPS) and precipitated in diethyl ether. Peptide ct-DD5o was cyclized using thiol bisalkylation chemistry as previously described in detail.45 The linear peptide DP1 (FΦRRRREKW) was synthesized as described above with Fmoc-Glu-OAll and Fmoc-Lys(Mtt)-OH. To cyclize the peptide, first the Glu was deprotected using 0.1 eq. Pd(PPh3)4, 10 eq. phenylsilane, in CHCl3 for 3h in the dark, then washed alternating 5% sodium diethyldithiocarbamate in DMF and 5% DIPEA in DMF. Next, the N-terminal Fmoc was deprotected as shown above. Then the peptide was cyclized using 10 eq. PyBOP, 10 eq. HOBt, and 20 eq. of DIPEA in DMF, and incubated overnight. The Mtt group was then deprotected using 1% TFA/DCM 2×10 mins. The chloroalkane carboxylate was then reacted with the deprotected Lys as described above. Peptides were purified by RP-HPLC and lyophilized. Peptide stocks for the experiments were made in DMSO and the concentration was determined by measuring UV absorption at 280 nm. We incorporated a tryptophan on the C-terminus of Arg9, to be able to determine the concentration by UV spectroscopy.
Chloroalkane tag.
In this work, we prepared a chloroalkane carboxylate (SI Fig. 1) which has been previously used with the HaloTag system.22 The chloroalkane carboxylic acid was synthesized as previously described, with some modifications.46,47 The complete synthesis scheme in shown in SI Fig. 11, and the complete synthesis description in given in Supporting Methods. Peptides tested in this work were conjugated to the chloroalkane carboxylic acid on their N-terminus, unless otherwise specified.
Chloroalkane-TAMRA synthesis.
Ten mg of 5/6-TAMRA-succinimidyl ester (Anaspec) was dissolved in 200 μL of DMF. To the solution of TAMRA, 4.5 mg of the chloroalkane amine 3 in SI Figure 13 and 32 μL of N,N-diisopropylethyamine were added. The reaction was stirred for 2h at room temperature. The reaction mixture was diluted with 200 μL of water and the product was purified by RP-HPLC.
Mammalian cell culture.
HeLa cells stably expressing the HaloTag-GFP-Mito construct and H3-GFP-HaloTag construct were provided by the Chenoweth lab.22 Cells were cultured using DMEM + 10% FBS + 1% Pen/Strep + 1 μg/mL puromycin and kept at 37 °C with 5% CO2. To select for HaloTag-expressing populations, cells were seeded in T75 flasks and incubated with culture media plus 20 μg/mL puromycin.
HaloTag expression and purification.
The pH6HTN plasmid (Promega) was mutated using site-directed mutagenesis to introduce a stop codon after the HaloTag sequence. The plasmid was transformed into competent BL21 E. coli cells. One liter of LB was inoculated using a 10 mL overnight from a single colony. The cells were grown at 37 °C until OD600 of 0.6 and induced using 1 mM isopropyl β-D-1-thiogalactopyranoside, then shaken overnight at room temperature. Cells were lysed by sonication (10 min, 30 sec on / 30 sec off), and purified in batch format using a Ni-NTA column (Thermo Fisher). The eluted fraction was further purified by gel filtration (Superdex 75 10/300 GL, GE Life Sciences) on an ÄKTA pure FPLC system. Purity and identity of the protein was confirmed using SDS-PAGE gel electrophoresis and MALDI-TOF mass spectrometry. The concentration was determined by measuring absorbance at 280 nm and using a calculated extinction coefficient of 58,400 M−1 cm−1.
Chloroalkane penetration assay.
The cell lines used for CAPA were HeLa cell lines, generated by Chenoweth and co-workers, that stably expresses HaloTag exclusively in the cytosol or nucleus.22 Cells were seeded in a 96-well plate the day before the experiment at density of 4 × 104 cells per well. The day of the experiment the media was aspirated, and 100 μL of Opti-MEM was added to the cells. Peptide stocks in DMSO were diluted in filtered water and serial dilutions of the peptides were performed in a separate 96-well plate, ensuring the final DMSO concentration was kept consistent and below 1%. 25 μL of peptide solution was added to each well and the plate was incubated for 4 hr at 37 °C with 5% CO2. The contents of the wells were aspirated off and wells were washed using fresh Opti-MEM for 15 mins. The wash was aspirated off and the cells were chased using 5 μM ct-TAMRA for 15 mins, except for the No-ct-TAMRA control wells which were incubated with Opti-MEM alone. The contents of the wells were aspirated and washed with fresh Opti-MEM for 30 mins. After aspiration, cells were rinsed once with PBS. The cells were then trypsinized, resuspended in PBS and analyzed using a benchtop flow cytometer (Guava EasyCyte, EMD Millipore). For the experiment that varied temperature, two identical 96-well plates were prepared, and one was incubated at 37 °C with 5% CO2 for 4 h, the other at 4 °C for 4 h. For the experiments that varied the presence of serum, two identical plates were prepared, one with DMEM and the other with DMEM + 10% FBS. For the time-course experiments, five identical plates were prepared and incubated at 37 °C with 5% CO2. After 30m, 2h, 4 h, 8h and 2 4 h of incubation, one plate was removed from the incubator and analyzed as described above.
Microscopy.
Cells were seeded at 4 × 105 cells in a glass-bottom, black-wall 24-well plate and grown overnight. Cells were treated in Opti-MEM with or without 2 μM ct-W, and incubated for 4 h as described above. Cells were washed with Opti-MEM for 15 min, then incubated with 5 μΜ ct-TMR for 15 mins. Cells were washed with fresh Opti-MEM for 30 mins, then rinsed with PBS. PBS was added to the wells before imaging. Images were taken on inverted fluorescence microscope (IX71, Olympus).
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank D. Chenoweth and C. Aonbangkhen for cell lines, and L. Walensky for reagents and helpful discussions. This work was supported by NIH U19AI109725 (B.L. and J.A.K.) and NIH R01GM127585 (J.A.K.).
B.L. is a scientific founder of Casma Therapeutics, Inc.
Footnotes
COMPETING INTEREST
ASSOCIATED CONTENT
Supporting Information. Chemical structures and characterization, additional data figures and tables, additional methods. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Walsh G Nat. Biotechnol 2014, 32 (10), 992–1000. [DOI] [PubMed] [Google Scholar]
- (2).Stewart MP; Sharei A; Ding X; Sahay G; Langer R; Jensen KF Nature 2016, 538 (7624), 183–192. [DOI] [PubMed] [Google Scholar]
- (3).D’Astolfo DS; Pagliero RJ; Pras A; Karthaus WR; Clevers H; Prasad V; Lebbink RJ; Rehmann H; Geijsen N Cell 2015, 161 (3), 674–690. [DOI] [PubMed] [Google Scholar]
- (4).Bruce VJ; McNaughton BR Cell Chem. Biol 2017, 24 (8), 924–934. [DOI] [PubMed] [Google Scholar]
- (5).Bechara C; Sagan S FEBS Lett 2013, 587 (12), 1693–1702. [DOI] [PubMed] [Google Scholar]
- (6).Stanzl EG; Trantow BM; Vargas JR; Wender PA Acc. Chem. Res 2013, 46 (12), 2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Walensky LD; Bird GH J. Med. Chem 2014, 57 (15), 6275–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Meric-Bernstam F; Saleh MN; Infante JR; Goel S; Falchook GS; Shapiro G; Chung KY; Conry RM; Hong DS; Wang JS-Z; Steidl U; Walensky LD; Guerlavais V; Payton M; Annis DA; Aivado M; Patel MR J. Clin. Oncol 2017, 35 (15_suppl), 2505. [Google Scholar]
- (9).Kauffman WB; Fuselier T; He J; Wimley WC Trends Biochem. Sci 2015, 40 (12), 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Brock R Bioconjug. Chem 2014, 25, 863–868. [DOI] [PubMed] [Google Scholar]
- (11).Peraro L; Kritzer JA Getting in: Emerging Methods and Design Principles for Cell-Penetrant Peptides. Angew. Chem. Int. Ed 2018, 10.1002/anie.201801361. [DOI] [PMC free article] [PubMed]
- (12).Chu Q; Moellering RE; Hilinski GJ; Kim Y-W; Grossmann TN; Yeh JT-H; Verdine GL Medchemcomm 2015, 6 (1), 111–119. [Google Scholar]
- (13).Bird GH; Mazzola E; Opoku-Nsiah K; Lammert MA; Godes M; Neuberg DS; Walensky LD Nat. Chem. Biol 2016, 12 (10), 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yu P; Liu B; Kodadek T Nat. Biotechnol 2005, 23 (6), 746–751. [DOI] [PubMed] [Google Scholar]
- (15).Michnick SW; Ear PH; Manderson EN; Remy I; Stefan E Nat. Rev. Drug Discov 2007, 6 (7), 569–582. [DOI] [PubMed] [Google Scholar]
- (16).Larochelle JR; Cobb GB; Steinauer A; Rhoades E; Schepartz AJ Am. Chem. Soc 2015, 137 (7), 2536–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Burlina F; Sagan S; Bolbach G; Chassaing G Nat. Protoc 2006, 1 (1), 200–205. [DOI] [PubMed] [Google Scholar]
- (18).Peraro L; Zou Z; Makwana KM; Cummings AE; Ball HL; Yu H; Lin YS; Levine B; Kritzer JA J. Am. Chem. Soc 2017, 139 (23), 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shoji-Kawata S; Sumpter R; Leveno M; Campbell GR; Zou Z; Kinch L; Wilkins AD; Sun Q; Pallauf K; MacDuff D; Huerta C; Virgin HW; Helms JB; Eerland R; Tooze SA; Xavier R; Lenschow DJ; Yamamoto A; King D; Lichtarge O; Grishin NV; Spector SA; Kaloyanova DV; Levine B Nature 2013, 494 (7436), 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Los GV; Encell LP; McDougall MG; Hartzell DD; Karassina N; Zimprich C; Wood MG; Learish R; Ohana RF; Urh M; Simpson D; Mendez J; Zimmerman K; Otto P; Vidugiris G; Zhu j.; Darzins A; Klaubert DH; Bulleit RF; Wood KV ACS Chem. Biol 2008, 3 (6), 373–382. [DOI] [PubMed] [Google Scholar]
- (21).Friedman Ohana R; Kirkland TA; Woodroofe CC; Levin S; Uyeda HT; Otto P; Hurst R; Robers MB; Zimmerman K; Encell LP; Wood KV ACS Chem. Biol 2015, 10 (10), 2316–2324. [DOI] [PubMed] [Google Scholar]
- (22).Ballister ER; Aonbangkhen C; Mayo AM; Lampson MA; Chenoweth DM Nat. Commun 2014, 5, 5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Frankel AD; Pabo CO. Cell 1988, 55 (6), 1189–1193. [DOI] [PubMed] [Google Scholar]
- (24).Derossi D; Joliot AH; Chassaing G; Prochiantz AJ Biol. Chem 1994, 269 (14), 10444–10450. [PubMed] [Google Scholar]
- (25).Wender PA; Mitchell DJ; Pattabiraman K; Pelkey ET; Steinman L; Rothbard JB Proc. Natl. Acad. Sci 2000, 97 (24), 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Futaki S; Suzuki T; Ohashi W; Yagami T; Tanaka S; Ueda K; Sugiura YJ Biol. Chem 2001, 276 (8), 5836–5840. [DOI] [PubMed] [Google Scholar]
- (27).Jiao CY; Delaroche D; Burlina F; Alves ID; Chassaing G; Sagan SJ Biol. Chem 2009, 284 (49), 33957–33965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Appelbaum JS; Larochelle JR; Smith BA; Balkin DM; Holub JM; Schepartz A Chem. Biol 2012, 19 (7), 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D Biochemistry 2016, 55 (18), 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Edwards AL; Wachter F; Lammert M; Huhn AJ; Luccarelli J; Bird GH; Walensky LD ACS Chem. Biol 2015, 10 (9), 2149–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Neklesa TK; Tae HS; Schneekloth AR; Stulberg MJ; Corson TW; Sundberg TB; Raina K; Holley SA; Crews CM Nat. Chem. Biol 2011, 7 (8), 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Friedman Ohana R; Levin S; Wood MG; Zimmerman K; Dart ML; Schwinn MK; Kirkland TA; Hurst R; Uyeda HT; Encell LP; Wood KV ACS Chem. Biol 2016, 11 (9), 2608–2617. [DOI] [PubMed] [Google Scholar]
- (33).Console S; Marty C; García-Echeverría C; Schwendener R; Ballmer-Hofer KJ Biol. Chem 2003, 278 (37), 35109–35114. [DOI] [PubMed] [Google Scholar]
- (34).Kim HY; Yum SY; Jang G; Ahn D-R Sci. Rep 2015, 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Drin G; Cottin S; Blanc E; Rees AR; Temsamani JJ Biol. Chem 2003, 278 (33), 31192–31201. [DOI] [PubMed] [Google Scholar]
- (36).Shin M-K; Hyun Y-J; Lee JH; Lim H-S ACS Comb. Sci 2018, 20, 237–242. [DOI] [PubMed] [Google Scholar]
- (37).Augustijns PF; Brown SC; Willard DH; Consler TG; Annaert PP; Hendren RW; Bradshaw TP Biochemistry 2000, 39 (25), 7621–7630. [DOI] [PubMed] [Google Scholar]
- (38).Kosuge M; Takeuchi T; Nakase I; Jones AT; Futaki S Bioconjug. Chem 2008, 19 (3), 656–664. [DOI] [PubMed] [Google Scholar]
- (39).Klionsky DJ; Abdelmohsen K; Abe A; Abedin MJ; Abeliovich H; Arozena AA; Adachi H; Adams CM; Adams PD; Adeli K; et al. Autophagy 2016, 12 (1), 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Maiolo JR; Ferrer M; Ottinger EA Biochim. Biophys. Acta - Biomembr 2005, 1712 (2), 161–172. [DOI] [PubMed] [Google Scholar]
- (41).D’Angelo MA; Hetzer MW Trends Cell Biol 2008, 18 (10), 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Holub JM; Larochelle JR; Appelbaum JS; Schepartz A Biochemistry 2013, 52 (50), 9036–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Chang YS; Graves B; Guerlavais V; Tovar C; Packman K; To K-H; Olson KA; Kesavan K; Gangurde P; Mukherjee A; Baker T; Darlak K; Elkin C; Filipovic Z; Qureshi FZ; Cai H; Berry P; Feyfant E; Shi XE; Horstick J; Annis DA; Manning AM; Fotouhi N; Nash H; Vassilev LT; Sawyer TK Proc. Natl. Acad. Sci 2013, 110 (36), E3445–E3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mándity IM; Fülöp F Expert Opin. Drug Discov 2015, 10 (11), 1163–1177. [DOI] [PubMed] [Google Scholar]
- (45).Peraro L; Siegert TR; Kritzer JA Methods Enzymol 2016, 580, 303–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Vistain LF; Rotz MW; Rathore R; Preslar AT; Meade TJ Chem. Commun 2016, 52 (1), 160–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).So M. kyung; Yao H; Rao J Biochem. Biophys. Res. Commun 2008, 374 (3), 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.