

Abstract
Autism spectrum disorders (ASDs) are neurodevelopmental disorders that share behavioral features, the results of numerous studies have suggested that the underlying causes of ASDs are multifactorial. Behavioral and/or neurobiological analyses of ASDs have been performed extensively using a valid model of prenatal exposure to valproic acid (VPA). Abnormal synapse formation resulting from altered neurite outgrowth in neural progenitor cells (NPCs) during embryonic brain development has been observed in both the VPA model and ASD subjects. Although several mechanisms have been suggested, the actual mechanism underlying enhanced neurite outgrowth remains unclear. In this study, we found that VPA enhanced the expression of brain-derived neurotrophic factor (BDNF), particularly mature BDNF (mBDNF), through dual mechanisms. VPA increased the mRNA and protein expression of BDNF by suppressing the nuclear expression of methyl-CpG-binding protein 2 (MeCP2), which is a transcriptional repressor of BDNF. In addition, VPA promoted the expression and activity of the tissue plasminogen activator (tPA), which induces BDNF maturation through proteolytic cleavage. Trichostatin A and sodium butyrate also enhanced tPA activity, but tPA activity was not induced by valpromide, which is a VPA analog that does not induce histone acetylation, indicating that histone acetylation activity was required for tPA regulation. VPA-mediated regulation of BDNF, MeCP2, and tPA was not observed in astrocytes or neurons. Therefore, these results suggested that VPA-induced mBDNF upregulation was associated with the dysregulation of MeCP2 and tPA in developing cortical NPCs.
Keywords: Brain-derived neurotrophic factor, Methyl-CpG-binding protein 2, Tissue plasminogen activator, Valproic acid
INTRODUCTION
Autism spectrum disorders (ASDs) are neurodevelopmental disorders characterized by repetitive behaviors and impairments in social interactions and language [1,2]. The results of numerous studies have suggested that genetics and the environment, especially during pregnancy and the fetal period, play pivotal roles in the etiology of ASD.
Valproic acid (VPA), which is used to treat epilepsy and bipolar disorder, has a teratogenic effect on humans [3]. The prevalence of ASDs in the offspring of mothers treated with antiepileptic drugs during pregnancy has suggested that these medications are associated with ASD [4]. Prenatal exposure to VPA increases the risk of ASD phenotype characteristics, such as repetitive behavior, social impairment, neural tube developmental defects, and excitatory/inhibitory synapse imbalance [5,6,7,8]. Therefore, prenatal exposure to VPA has been extensively studied in rodents as a model of ASD to better understand the neurobiology of ASD and autistic behavior.
Several lines of evidence, including increased serum, plasma, and brain tissue levels of brain-derived neurotrophic factor (BDNF) in children with ASD, have suggested that BDNF is involved in ASD [9,10,11,12,13,14]. Similarly, exposure to VPA has been shown to increase BDNF expression in the immature cortical neuron [15] and fetal brain [16]. Because BDNF is widely expressed in the mammalian brain [17] and is critical for brain developmental mechanisms, such as the regulation of neuritogenesis and synapse formation [18], the aberrant hyperactivity of BDNF may play a key role in abnormal development in ASD [19].
Methyl-CpG-binding protein 2 (MeCP2) is a transcription factor that binds to methylated cytosines in CpG islands and that recruits other transcriptional repressors and histone deacetylases that silence target genes [20]. Numerous studies have reported that MeCP2 controls BDNF expression [20,21,22,23]. In addition, the similarities in the expression patterns of BDNF and MeCP2 during the prenatal and postnatal periods [24] and in vivo evidence of functional interactions of MeCP2 with BDNF [21] suggest that MeCP2 is crucial for the regulation of BDNF expression in normal brain development and that abnormalities in BDNF regulation contribute to physiological disorders, such as Rett syndrome.
Tissue plasminogen activator (tPA) is a serine protease that induces the mature and active form of BDNF (mBDNF) through the proteolytic cleavage of the precursor isoform of BDNF (pro-BDNF) [25]. tPA has been reported to increase neurite outgrowth through the degradation of the extracellular matrix or induction of mBDNF [26]. Although stimulatory effects of VPA on tPA have been reported in vascular endothelial cells [27], it is unclear whether VPA affects tPA levels in the developing embryonic brain.
Contradictory results of the regulation of BDNF by VPA have been reported. VPA enhances BDNF expression in primary cultured embryonic neural stem cells [28] and primary cultured neurons [29,30], whereas VPA-treated mice exhibited decreased BDNF mRNA levels in the somatosensory cortex [31]. Therefore, in this study, we first investigated the levels of BDNF expression in VPA-treated cortical neural progenitor cells (NPCs).
METHODS
Rat primary cortical neural stem cell culture
These animal studies were approved by the Institutional Animal Care and Use Committee of Chung-Ang University (Approval No. 2017-00093). Primary cortical neural stem cells were cultured from the cortices of embryonic-day-14-old Sprague–Dawley (SD) rats. A Pasteur pipette was used to separate the cortices into single cells, which were then incubated in Dulbecco's modified Eagle's medium/F12 (DMEM/F12) that was supplemented with 20 ng/ml of epidermal growth factor and 10 ng/ml of basic fibroblast growth factor in a 5% CO2, 90% N2, and 5% O2 incubator. The culture media were changed every 2 days until the single cells had grown into floating neurospheres. The neurospheres were dissociated into single cells with trypsin ethylenediaminetetraacetic acid and then regrown into neurospheres in epidermal growth factor- and basic fibroblast growth factor-supplemented media. The neurospheres were dissociated again into single cells and plated on poly-L-ornithine-coated plates with DMEM/F12 media containing 2% penicillin/streptomycin without growth factors or serum. The cells were incubated at 37℃ in a humidified atmosphere with 5% CO2, 90% N2, and 5% O2.
Rat primary neuron culture
Primary cortical neurons were prepared from embryonic-day-18-old SD rats. A Pasteur pipette was used to dissociate the cortices into single cells by pipetting the tissue several times. The cells were seeded onto poly-D-lysine-coated plates and incubated in neurobasal medium containing 1% penicillin/streptomycin, 20 µM glutamine, and B27. The cultures were maintained at 37℃ in a humidified 5% CO2 incubator.
Rat primary astrocyte culture
The prefrontal cortices 2-day-old SD rat pups were digested with trypsin for 10 min at 37℃. The triturated cells were cultured in DMEM/F12 containing 10% fetal bovine serum, 100 U/ml of penicillin, and 100 mg/ml of streptomycin for 7 days. The grown cells were washed twice with serum-free media and then detached using 0.25% trypsin with ethylenediaminetetraacetic acid. The cells were plated at low density (5,000 cells/cm2) on well plates, grown for 3 days, and then used for further analysis.
Casein zymography
tPA activity was measured using plasminogen-casein zymography. The culture supernatants were mixed with zymography buffer [200 mM Tris-HCl (pH 6.8), 8% w/v sodium dodecyl sulfate (SDS), 40% glycerol, 0.02% bromophenol blue, and without β-mercaptoethanol] and separated by electrophoresis on 8% polyacrylamide gels containing casein and plasminogen. After the electrophoresis, the gels were washed twice with 2.5% Triton X-100 for 30 min to eliminate the SDS. The gels were then incubated for 12–24 h at 37℃ in reaction buffer [20 mM Tris-HCl (pH 7.6) and 166 mM CaCl2,] for caseinolysis. To reveal the caseinolytic activity, the gels were stained with 0.1% Coomassie brilliant blue R-250 and destained using destaining solution (20% methanol, 10% acetic acid, and 70% deuterium-depleted water). tPA activity was visualized as the light bands that resulted from casein degradation. The caseinolysis band detected at 68 kDa matched the band of purified tPA standard that was present in the same gel. The gel was analyzed using a LAS 3000 detection system (Fujifilm, Minato-ku, Tokyo, Japan), and the band intensities were measured using ImageJ (https://imagej.nih.gov/ij/).
Western blotting
Twenty micrograms of protein were obtained from the cells and mixed with sample buffer [120 mM Tris-HCl (pH 6.8), 0.5% sodium deoxycholate, 0.1% SDS, and 1% Triton X-100]. The proteins were separated by 8% SDS-polyacrylamide gel electrophoresis and electrically transferred onto nitrocellulose membranes. The membranes were blocked with 5% skim milk at room temperature for 60 min and then incubated with the primary antibodies at 4℃ overnight. The membranes were washed three times with TBS-Tween (0.1%) for 10 min and subsequently incubated with horseradish peroxidase-conjugated secondary antibody for 90 min at room temperature. Each band of interest was detected with enhanced chemiluminescence solution, and the images were captured using LAS-3000. The band intensity was determined using ImageJ software.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
The levels of expression of tPA, MeCP2, and BDNF mRNA were determined by quantitative RT-PCR. The cells were washed twice with ice-cold phosphate-buffered saline (PBS), and total RNA was extracted using TRIzol reagent. First-strand cDNA synthesis was conducted using 2.5 µg of total RNA and reverse transcriptase. The extracted RNAs were incubated at 60℃ for 60 min and then heated at 94℃ for 5 min to terminate the reaction. A total of 5 ng of cDNA was used as a template for the PCR amplification of tPA, MeCP2, and BDNF. The primer sequences were as follows: tPA forward: AGT TGC AGC GAA CCA AGA TG, tPA reverse: TGC CAC GGT AAG TCA CAC CT; MeCP2 forward: CTG CTG CAG AGG CCA AAA AG, MeCP2 reverse: TGG TGG TGA TGA TGG TGC TC; and BDNF forward: GCT GCC TTG ATG TTT ACT TTG, BDNF reverse: ATG GGA TTA CAC TTG GTC TCG T. The PCR products were subjected to 1.0% agarose gel electrophoresis and stained with ethidium bromide. The intensity of each band was analyzed using ImageJ.
Immunofluorescent staining
The cells on the cover glass were fixed with 4% paraformaldehyde at room temperature for 20 min and then washed with PBS. The cells were permeabilized with 0.1% Triton in PBS for 15 min at room temperature and then washed twice with PBS. The cells were incubated with primary antibodies that were diluted with blocking solution (1% bovine serum albumin and 3% fetal bovine serum in PBS) at 4℃ overnight. After washing the cells three times with PBS, they were incubated with secondary antibodies conjugated with Alexa Fluor® 594 Dye and diluted in blocking buffer for 1 h at room temperature. After three washes, diamidino-2-phenylindole (DAPI) that was diluted in PBS was applied to the cells for 5 min. Cover glasses were mounted using Vectashield (Vector Laboratories, CA, US), and the cells were observed using confocal microscopy.
Statistical analysis
Data were displayed as mean±standard error of mean (S.E.M.) and were analyzed for statistical significance using one-way analysis of variance (ANOVA) followed by post hoc Tukey's comparisons test. Differences were considered statistically significant when the p value was less than 0.05 (p<0.05). All statistical analyses were conducted using GraphPad Prism software.
RESULTS
VPA increased BDNF expression in cultured cortical NPCs
To investigate whether VPA administration affected BDNF in developing brain, we first examined BDNF expression in VPA-treated embryonic cortical NPCs. Of the 9 exons, only exons 1, 4, and 6 of BDNF were detected in the fetal mice brains, and the levels of these exons were increased by VPA treatment [16]. However, only BDNF exon 4 expression was significantly increased by VPA treatment in the prefrontal cortex [32]. Thus, we then measured the expression levels of BDNF exon 4 after treatment with VPA (0.2 and 0.5 mM) in NPCs. VPA increased the expression of BDNF exon 4 in a concentration-dependent manner (Fig. 1A). To further investigate this, we investigated the levels of expression of pro-BDNF and mBDNF in VPA-treated NPCs. The expressions of mBDNF and acetylated histone 3 were increased, whereas pro-BDNF expression levels were suppressed by VPA (Fig. 1B). Therefore, these results suggested that VPA increased BDNF transcription in NPCs.
Fig. 1. Valproic acid (VPA)-induced upregulation of brain-derived neurotrophic factor (BDNF) in cultured neural progenitor cells (NPCs).

Neurospheres were cultured from the cortical region of embryonic-day-14.5-old brains and dissociated into single NPCs. The NPCs were exposed to 0.2 or 0.5 mM VPA for 3 or 6 h and then analyzed. (A) The expression of BDNF mRNA was analyzed by reverse transcriptase-polymerase chain reaction (RT-PCR). Gapdh was used as a loading control. All data are expressed as mean±standard error of the mean (SEM, n=3). *p<0.05 vs. Vehicle 3 h, and ##p<0.01 vs. Vehicle 6 h [one-way analysis of variance (ANOVA) followed by post hoc Tukey's comparisons test]. (B) The expression levels of pro- and mature BDNF protein were analyzed by western blots. Acetylated histone 3 (AcH3) levels were increased by VPA treatment, and β-actin was used as a loading control. All data are expressed as mean±SEM (n=3). *p<0.05 vs. Vehicle 3 h, and #p<0.05 and ###p<0.001 vs. Vehicle 6 h (one-way ANOVA followed by post hoc Tukey's comparisons test). &&p<0.01 vs. VPA 0.2 mM 3 h, and †p<0.05 vs. VPA 0.5 mM 3 h (Student's t-test).
VPA decreased MeCP2 expression in NPC nuclei
Because BDNF exon 4 is the preferential target for epigenetic regulation as it contains the specific binding site for MeCP2, the epigenetic regulation of BDNF is thought to be involved in physiological conditions, such as psychiatric disorders [33]. MeCP2 suppresses BDNF gene transcription [34,35] by binding to methylated DNA in promoter 4 of Bdnf, which maintains the repressed state of the Bdnf gene [23]. Therefore, we investigated the effects of VPA on MeCP2 expression in VPA-treated NPCs. The levels of MeCP2 mRNA and protein were significantly decreased by treatment with 0.5 mM of VPA (Figs. 2A and B). In addition, the immunostaining results showed that MeCP2 expression was decreased by VPA treatment (Fig. 2C). Interestingly, the diffused pattern of MeCP2 expression was decreased by VPA treatment, which resulted in the dissociation of MeCP2 from the DAPI puncta region that contains enriched A/T bases.
Fig. 2. VPA reduced the levels of methyl-CpG-binding protein 2 (MeCP2) expression in NPCs.
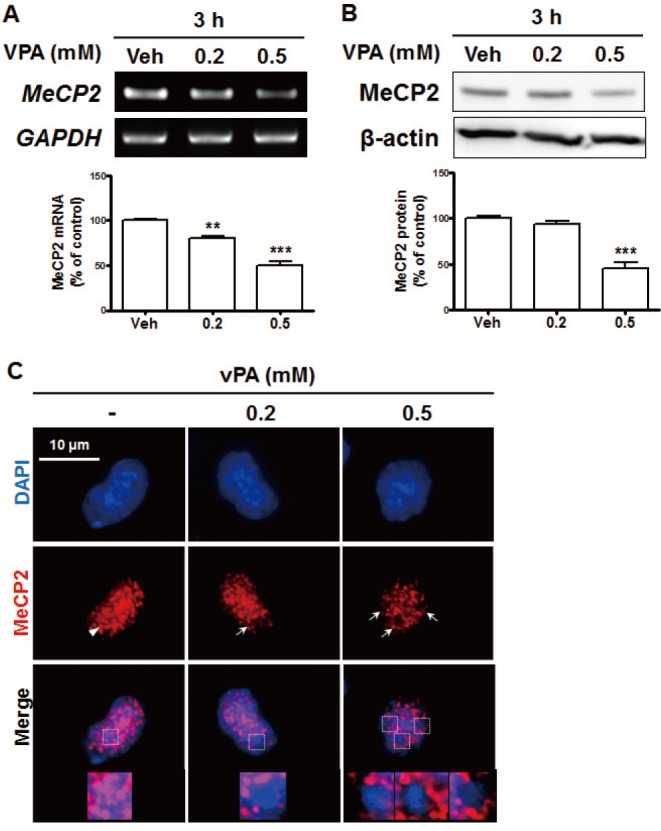
Cultured NPCs were treated with 0.2 or 0.5 mM VPA for 3 h. (A, B) MeCP2 mRNA levels were analyzed with RT-PCR (A), and protein levels were analyzed with western blots (B). All data are expressed as mean±SEM (n=3). **p<0.01 and ***p<0.001 vs. Vehicle 3 h (one-way ANOVA followed by post hoc Tukey's comparisons test). (C) Immunostaining of MeCP2 and diamidino-2-phenylindole (DAPI) in VPA-treated NPCs. The arrow indicates the coexpression of MeCP2 and DAPI, while the arrowhead indicates the region in which MeCP2 was expressed but DAPI staining cannot be observed. The magnified images are from the top row. All data are expressed as mean±SEM (n=3). **p<0.01 and ***p<0.001 vs. Vehicle 3 h (one-way ANOVA followed by post hoc Tukey's comparisons test). Scale bar=10 µm.
VPA increased the levels of tPA expression and activity in cultured NPCs
Our results showed that VPA increased mBDNF levels and suppressed pro-BDNF levels, which suggested that the processing of pro-BDNF into mBDNF was enhanced. Therefore, we investigated the levels of tPA expression in VPA-treated NPCs. The mRNA and protein levels of expression of tPA and acetylated histone 3 were increased in NPCs by VPA treatment (Figs. 3A and B). In addition, the caseinolytic assay showed that tPA activity was enhanced by VPA (Fig. 3C). To investigate whether VPA upregulated tPA through histone regulation, we treated the NPCs with other HDACis. TSA and SB also enhanced tPA activity, while valpromide, a VPA analog that lacks histone deacetylase inhibition activity [36,37], failed to enhance the activity (Fig. 3D). These results indicated that VPA increased tPA expression in NPCs through histone modification.
Fig. 3. VPA enhanced the levels of tissue plasminogen activator (tPA) expression in NPCs.

Cultured NPCs were treated with VPA (0.2 and 0.5 mM) for 3 h or 6 h. (A-C) The levels of tPA mRNA (A), protein (B), and activity (C) were analyzed with RT-PCR, western blot, and casein zymography, respectively. **p<0.01 and ***p<0.001 vs. Vehicle 3 h, and #p<0.05 and ##p<0.01 vs. Vehicle 6 h (one-way ANOVA followed by post hoc Tukey's comparisons test). (D) The NPCs were treated with histone acetylation inhibitors (HDAC) inhibitors or valpromide (VPM) for 6 h then tPA activity was analyzed; VPA (0.5 mM), trTSA (trichostatin A, 20 nM), SB (sodium butyrate, 0.1 mM), and VPM (0.5 mM). All data are expressed as mean±SEM (n=3). *p<0.05 and **p<0.01 vs. Vehicle (Student's t-test).
VPA did not affect MeCP2 and tPA expression in neurons and astrocytes
VPA has previously been shown to increase BDNF expression in astrocytes [38], astrocytes from LEW/N rats [39], and human astrocytoma cells [18]. Moreover, a protective role of VPA was suggested by the increased expression of BDNF in neurons [15,40] and neuroblastomas [41]. To investigate whether VPA increased BDNF in other neuronal cells through the regulation of MeCP2 and tPA, we treated neurons and astrocytes with VPA. VPA treatment did not result in significant differences in the levels of BDNF, MeCP2, and tPA expression or tPA activity (Figs. 4A and B). Therefore, these results suggested that the VPA-induced stimulation of BDNF in cortical NPCs occurred through the regulation of MeCP2 and tPA expression.
Fig. 4. VPA did not affect the levels of MeCP2 or tPA expression in neurons or astrocytes.

Cortical neurons (A) or astrocytes (B) were treated with VPA for 6 h, then mRNA of BDNF, MeCP2, and tPA, and tPA activity were analyzed. GAPDH was used as a loading control.
DISCUSSION
In the present study, we found that VPA upregulated the levels of mBDNF in NPCs through dual mechanisms involving enhanced BDNF transcription and maturation. We found that the mRNA and protein expression levels of BDNF were increased, whereas those of MeCP2 were decreased, by VPA treatment. In addition, the levels of mRNA, protein, and activity of tPA were enhanced in VPA-treated NPCs. In addition, other histone acetylation inhibitors (HDACi) such as trichostatin A (TSA) and sodium butyrate (SB) enhanced tPA activity, while valpromide, which is an analog of VPA that lacks HDACi activity, failed to induce tPA activity. These results indicated that VPA stimulates BDNF expression in NPCs through the regulation of MeCP2 and tPA, which may contribute to aberrant cortical development in ASD.
Numerous studies have reported that stimulated BDNF is involved in the abnormal development of the developing brain. Because BDNF levels are normally low in the fetal brain and gradually increase during maturation [42], transient increases in BDNF levels might play a causal role in embryonic brain development defects, such as altered neuronal laminar fate and premature exit from the cell cycle in cortical development [43]. A rapid and uncorrected increase in BDNF levels by VPA treatment would leave an “architectural signature” during embryonic cortical development [44] and might lead to permanent defects in the postnatal period. Interestingly, VPA rapidly stimulated BDNF mRNA expression in 3 or 6 h, which was also observed in VPA-exposed embryonic-day-12.5-old fetal brain [16], whereas the VPA enhancement of BDNF expression took one [15] or a few days [28,29]. In addition, postmortem brain studies have shown increased immunoreactivity of BDNF in patients with ASD due to the suppression of BDNF cleavage, which was independent of transcriptional regulation [13].
In present study, VPA-induced upregulation of BDNF was observed only in NPCs not in astrocytes or neurons. In other studies, the upregulation of BDNF by VPA was investigated at relatively later time point after VPA exposure, from 24 h to a few days in neurons or astrocytes [30,32,45]. In addition, the concentration range of VPA was 0.6–1.2 mM [38,46], due to 0.3 and 0.4 mM of VPA did not have significant effect in astrocytes [46]. In present study, we investigated the effect of VPA on BDNF expression at relatively in earlier time, 3 and 6 h, and VPA concentration was lower (0.2 and 0.5 mM). Therefore, it assumes that early exposure and lower concentration of VPA would be a causal factor of no significant effect of VPA in BDNF expression in astrocytes or neurons. VPA would promote BDNF expression at higher concentration of VPA and further exposure time in astrocytes or neurons.
In addition, cell-type specific acetylation and methylation status would be a causal role of VPA-induced regulation of BDNF in NPCs. In recent study, cell-type specific ChIP-seq analyses displayed histone modification, such as acetylation and methylation of histone were variable in cell-types, which determine cell-type specific transcripts levels [47]. In addition, histone acetylation levels are gradually decline during differentiation of embryonic stem cells [48]. Considering VPA enhanced BDNF protein only in E12.5 fetal brain but not in matured brain [16], the developing brain seems to be more sensitive to histone acetylation than the matured brain. Although regional differences of histone acetylation have been observed during developing mouse embryonic brain and chick spinal cord [49], further studies would be required to clarify the cellular difference in acetylation and methylation status which determine cell-type specific transcripts levels in response to histone modification.
Because BDNF can cross blood brain barrier [50,51], highly correlation has been suggested between plasma and cortical BDNF levels [50,52]. It has been reported BDNF level in serum was significantly decreased in bipolar patients compared to healthy controls [53,54,55], and the enhanced BDNF was observed in patients treated with antidepressants [53,56,57]. Likewise, upregulated BDNF was monitored in bipolar patients treated with VPA and dextromethorphan [54], suggesting VPA have therapeutic effects on bipolar patients by promoting BDNF level. However, in other studies, serum BDNF level in bipolar patients was not significantly different compared to healthy control [58]. In addition, the BDNF levels in depressed patients treated with antidepressants were constitutively suppressed compared to healthy control [59], and serum BDNF was independent with antidepressant treatment [60], suggesting correlation between level of BDNF and VPA is required to be further investigated.
The results of this study showed that the VPA increase of BDNF mRNA expression accompanied the suppression of the expression of MeCP2, which is a transcriptional repressor of BDNF expression. The dysfunction of MeCP2 is related to Rett syndrome [61,62], and Rett-like features are induced by MeCP2 inactivation in adult brain [63]. A regulatory role of MeCP2 on BDNF expression has been suggested by three different models, including repression, activation, and dual operation [24]. Interestingly, the intracellular levels of BDNF are enhanced by both the knockdown as well as the overexpression of MeCP2 in hippocampal neurons [64], which suggests that adequate levels of MeCP2 are required for defining the optimal levels of BDNF. MeCP2 binds at the methylated CpG region that contains enriched A/T bases in Bdnf genes [65]. Our immunostaining results that showed that the nuclear expression of MeCP2 was mainly decreased in the DAPI puncta region suggested that VPA preferentially decreased the levels of MeCP2 expression in the A/T region.
Accumulating evidences demonstrated that microRNA plays crucial role in neural development [66,67], and perturbation of microRNA is involved in neurodevelopmental disorders, such as ASD and fragile X-syndrome [68,69,70]. MiR-132 is one of the dominantly expressed microRNA in brain [71], and it represses MeCP2 levels by binding its miRNA recognition element (MRE) with MeCP2 3′UTR thus inhibits MeCP2 levels [72]. Interestingly, prenatal exposure to VPA displayed enhanced miR-132 whereas decrease of MeCP2 mRNA in embryonic brain [71]. Likewise, upregulated miR-132 in contrast to suppressed MeCP2 was observed in TSA treated NIH/3T3 cells [73], suggesting it would be possible VPA regulates MeCP2 expression through regulation of miR-132 levels.
In this study, we found that VPA enhanced the expression of tPA in NPCs. VPA also enhances tPA expression in cultured endothelial cells [27], and enhanced vascular tPA production decreases in vivo fibrin accumulation [74]. Reduced incidences of strokes and myocardial infarctions have been observed in patients with epilepsy who were treated with VPA [75,76], which also suggests that VPA enhances tPA activity. tPA expression is enhanced by other HDACis, such as TSA, SB, and MS-275, in cultured human endothelial cells [77,78,79], and tPA activity is not altered in the absence HDAC regulation; this suggests that histone acetylation is required for the upregulation of tPA [27]. In those study, chromatin immunoprecipitation assay displayed acetylated histone was enhanced in tPA transcription start site by VPA. The marginal enhancement of mRNA for PAI-1 or uPA, the other plasminogen activator [27], suggesting tPA is sensitive to histone acetylation. Indeed, tPA promoter contains binding sites for transcription factor Sp1, which is induced by HDAC inhibitors [80,81,82], and that is critical for consecutive expression of tPA [83,84]. Therefore, it assumes that HDACi promotes tPA transcription by inducing Sp1 expression in NPCs.
VPA has been reported to promote neurite outgrowth in NPCs [28]. Because tPA promotes neuritogenesis through the phosphorylation of LRP5/6 [85], tPA might play a causal role in the enhanced neurite outgrowth observed in VPA-treated NPCs. Although VPA did not directly stimulate tPA expression, VPA promoted tPA activity by suppressing the expression of the tPA inhibitor plasminogen activator inhibitor-1 (PAI-1) [86]. PAI-1 is highly expressed in astrocytes [87], and it terminates tPA activity by forming a complex with the catalytic domain of tPA [88]. We previously showed that tPA, but not PAI-1, is expressed in NPCs [85]. These findings suggest that the rapid induction of tPA by VPA resulted from the promotion of tPA expression by VPA, which was independent of PAI-1 activity.
As several studies have reported, a number of genes play a role in the abnormal brain and synapse development of subjects with ASD [89,90]. Likewise, our study showed that the exposure to VPA increased the risk of ASD through changes in the regulation of the expression of multiple genes in NPCs. In the future, studies should investigate the contribution of the rapid modulation of gene expression by histone remodeling towards abnormal gene expression in ASD development.
ACKNOWLEDGEMENTS
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2017R1D1A1B03031920).
Footnotes
Author contributions: H.M.K. cultured astrocytes and carried out molecular work including RT-PCR and zymography. Y.S.J. cultured neurons and carried out RT-PCR. H.H.P, supervised the experiments and J.H.L. analyzed data. S.H.J, carried out western blotting and S.Y.C. carried out immunocytochemistry. S.H.L. designed the experiments, cultured NPCs, and wrote the manuscript. C.Y.S. supervised the experiments and wrote the manuscript.
CONFLICTS OF INTEREST: The authors declare no conflicts of interest.
References
- 1.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29:349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Eadie MJ. Antiepileptic drugs as human teratogens. Expert Opin Drug Saf. 2008;7:195–209. doi: 10.1517/14740338.7.2.195. [DOI] [PubMed] [Google Scholar]
- 4.Rasalam AD, Hailey H, Williams JH, Moore SJ, Turnpenny PD, Lloyd DJ, Dean JC. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol. 2005;47:551–555. doi: 10.1017/s0012162205001076. [DOI] [PubMed] [Google Scholar]
- 5.Schneider T, Przewłocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30:80–89. doi: 10.1038/sj.npp.1300518. [DOI] [PubMed] [Google Scholar]
- 6.Schneider T, Turczak J, Przewłocki R. Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic acid: issues for a therapeutic approach in autism. Neuropsychopharmacology. 2006;31:36–46. doi: 10.1038/sj.npp.1300767. [DOI] [PubMed] [Google Scholar]
- 7.Roullet FI, Lai JK, Foster JA. In utero exposure to valproic acid and autism--a current review of clinical and animal studies. Neurotoxicol Teratol. 2013;36:47–56. doi: 10.1016/j.ntt.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Gao R, Penzes P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr Mol Med. 2015;15:146–167. doi: 10.2174/1566524015666150303003028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Correia CT, Coutinho AM, Sequeira AF, Sousa IG, Lourenco Venda L, Almeida JP, Abreu RL, Lobo C, Miguel TS, Conroy J, Cochrane L, Gallagher L, Gill M, Ennis S, Oliveira GG, Vicente AM. Increased BDNF levels and NTRK2 gene association suggest a disruption of BDNF/TrkB signaling in autism. Genes Brain Behav. 2010;9:841–848. doi: 10.1111/j.1601-183X.2010.00627.x. [DOI] [PubMed] [Google Scholar]
- 10.Connolly AM, Chez M, Streif EM, Keeling RM, Golumbek PT, Kwon JM, Riviello JJ, Robinson RG, Neuman RJ, Deuel RM. Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biol Psychiatry. 2006;59:354–363. doi: 10.1016/j.biopsych.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Nelson KB, Grether JK, Croen LA, Dambrosia JM, Dickens BF, Jelliffe LL, Hansen RL, Phillips TM. Neuropeptides and neurotrophins in neonatal blood of children with autism or mental retardation. Ann Neurol. 2001;49:597–606. [PubMed] [Google Scholar]
- 12.Miyazaki K, Narita N, Sakuta R, Miyahara T, Naruse H, Okado N, Narita M. Serum neurotrophin concentrations in autism and mental retardation: a pilot study. Brain Dev. 2004;26:292–295. doi: 10.1016/S0387-7604(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 13.Garcia KL, Yu G, Nicolini C, Michalski B, Garzon DJ, Chiu VS, Tongiorgi E, Szatmari P, Fahnestock M. Altered balance of proteolytic isoforms of pro-brain-derived neurotrophic factor in autism. J Neuropathol Exp Neurol. 2012;71:289–297. doi: 10.1097/NEN.0b013e31824b27e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Z, Zhang L, Zhu T, Huang J, Qu Y, Mu D. Peripheral brain-derived neurotrophic factor in autism spectrum disorder: a systematic review and meta-analysis. Sci Rep. 2016;6:31241. doi: 10.1038/srep31241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasan MR, Kim JH, Kim YJ, Kwon KJ, Shin CY, Kim HY, Han SH, Choi DH, Lee J. Effect of HDAC inhibitors on neuroprotection and neurite outgrowth in primary rat cortical neurons following ischemic insult. Neurochem Res. 2013;38:1921–1934. doi: 10.1007/s11064-013-1098-9. [DOI] [PubMed] [Google Scholar]
- 16.Almeida LE, Roby CD, Krueger BK. Increased BDNF expression in fetal brain in the valproic acid model of autism. Mol Cell Neurosci. 2014;59:57–62. doi: 10.1016/j.mcn.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 18.Nishino S, Ohtomo K, Numata Y, Sato T, Nakahata N, Kurita M. Divergent effects of lithium and sodium valproate on brain-derived neurotrophic factor (BDNF) production in human astrocytoma cells at therapeutic concentrations. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39:17–22. doi: 10.1016/j.pnpbp.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Tsai SJ. Is autism caused by early hyperactivity of brain-derived neurotrophic factor. Med Hypotheses. 2005;65:79–82. doi: 10.1016/j.mehy.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 20.Murgatroyd C, Wu Y, Bockmuhl Y, Spengler D. Genes learn from stress: how infantile trauma programs us for depression. Epigenetics. 2010;5:194–199. doi: 10.4161/epi.5.3.11375. [DOI] [PubMed] [Google Scholar]
- 21.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 22.Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 24.Li W, Pozzo-Miller L. BDNF deregulation in Rett syndrome. Neuropharmacology. 2014;76 Pt C:737–746. doi: 10.1016/j.neuropharm.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandel AL, Ozdener H, Utermohlen V. Identification of pro- and mature brain-derived neurotrophic factor in human saliva. Arch Oral Biol. 2009;54:689–695. doi: 10.1016/j.archoralbio.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- 27.Larsson P, Ulfhammer E, Magnusson M, Bergh N, Lunke S, El-Osta A, Medcalf RL, Svensson PA, Karlsson L, Jern S. Role of histone acetylation in the stimulatory effect of valproic acid on vascular endothelial tissue-type plasminogen activator expression. PLoS One. 2012;7:e31573. doi: 10.1371/journal.pone.0031573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu L, Zhou H, Pan B, Li X, Fu Z, Liu J, Shi Z, Chu T, Wei Z, Ning G, Feng S. c-Jun amino-terminal kinase is involved in valproic acid-mediated neuronal differentiation of mouse embryonic NSCs and neurite outgrowth of NSC-derived neurons. Neurochem Res. 2017;42:1254–1266. doi: 10.1007/s11064-016-2167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oikawa H, Goh WW, Lim VK, Wong L, Sng JC. Valproic acid mediates miR-124 to down-regulate a novel protein target, GNAI1. Neurochem Int. 2015;91:62–71. doi: 10.1016/j.neuint.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 30.Park SW, Lee JG, Seo MK, Cho HY, Lee CH, Lee JH, Lee BJ, Baek JH, Seol W, Kim YH. Effects of mood-stabilizing drugs on dendritic outgrowth and synaptic protein levels in primary hippocampal neurons. Bipolar Disord. 2015;17:278–290. doi: 10.1111/bdi.12262. [DOI] [PubMed] [Google Scholar]
- 31.Roullet FI, Wollaston L, Decatanzaro D, Foster JA. Behavioral and molecular changes in the mouse in response to prenatal exposure to the anti-epileptic drug valproic acid. Neuroscience. 2010;170:514–522. doi: 10.1016/j.neuroscience.2010.06.069. [DOI] [PubMed] [Google Scholar]
- 32.Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boulle F, van den Hove DL, Jakob SB, Rutten BP, Hamon M, van Os J, Lesch KP, Lanfumey L, Steinbusch HW, Kenis G. Epigenetic regulation of the BDNF gene: implications for psychiatric disorders. Mol Psychiatry. 2012;17:584–596. doi: 10.1038/mp.2011.107. [DOI] [PubMed] [Google Scholar]
- 34.Klose R, Bird A. Molecular biology. MeCP2 repression goes nonglobal. Science. 2003;302:793–795. doi: 10.1126/science.1091762. [DOI] [PubMed] [Google Scholar]
- 35.Im HI, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci. 2010;13:1120–1127. doi: 10.1038/nn.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 37.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 38.Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei R, Lin CM, Tu YY. Strain-specific BDNF expression of rat primary astrocytes. J Neuroimmunol. 2010;220:90–98. doi: 10.1016/j.jneuroim.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Harrison IF, Crum WR, Vernon AC, Dexter DT. Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson's is associated with histone acetylation and upregulation of neurotrophic factors. Br J Pharmacol. 2015;172:4200–4215. doi: 10.1111/bph.13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croce N, Mathe AA, Gelfo F, Caltagirone C, Bernardini S, Angelucci F. Effects of lithium and valproic acid on BDNF protein and gene expression in an in vitro human neuron-like model of degeneration. J Psychopharmacol. 2014;28:964–972. doi: 10.1177/0269881114529379. [DOI] [PubMed] [Google Scholar]
- 42.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 43.Fukumitsu H, Ohtsuka M, Murai R, Nakamura H, Itoh K, Furukawa S. Brain-derived neurotrophic factor participates in determination of neuronal laminar fate in the developing mouse cerebral cortex. J Neurosci. 2006;26:13218–13230. doi: 10.1523/JNEUROSCI.4251-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ben-Ari Y. Neuro-archaeology: pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci. 2008;31:626–636. doi: 10.1016/j.tins.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 45.Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51–59. doi: 10.1038/sj.mp.4002099. [DOI] [PubMed] [Google Scholar]
- 46.Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, Wilson B, Lu RB, Gean PW, Chuang DM, Hong JS. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- 47.Girdhar K, Hoffman GE, Jiang Y, Brown L, Kundakovic M, Hauberg ME, Francoeur NJ, Wang YC, Shah H, Kavanagh DH, Zharovsky E, Jacobov R, Wiseman JR, Park R, Johnson JS, Kassim BS, Sloofman L, Mattei E, Weng Z, Sieberts SK, et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat Neurosci. 2018;21:1126–1136. doi: 10.1038/s41593-018-0187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JH, Hart SR, Skalnik DG. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis. 2004;38:32–38. doi: 10.1002/gene.10250. [DOI] [PubMed] [Google Scholar]
- 49.Cho B, Kim HJ, Kim H, Sun W. Changes in the histone acetylation patterns during the development of the nervous system. Exp Neurobiol. 2011;20:81–84. doi: 10.5607/en.2011.20.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karege F, Schwald M, Cisse M. Postnatal developmental profile of brain-derived neurotrophic factor in rat brain and platelets. Neurosci Lett. 2002;328:261–264. doi: 10.1016/s0304-3940(02)00529-3. [DOI] [PubMed] [Google Scholar]
- 51.Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology. 1998;37:1553–1561. doi: 10.1016/s0028-3908(98)00141-5. [DOI] [PubMed] [Google Scholar]
- 52.Klein AB, Williamson R, Santini MA, Clemmensen C, Ettrup A, Rios M, Knudsen GM, Aznar S. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int J Neuropsychopharmacol. 2011;14:347–353. doi: 10.1017/S1461145710000738. [DOI] [PubMed] [Google Scholar]
- 53.Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry. 2008;64:527–532. doi: 10.1016/j.biopsych.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen SL, Lee SY, Chang YH, Chen PS, Lee IH, Wang TY, Chen KC, Yang YK, Hong JS, Lu RB. Therapeutic effects of add-on low-dose dextromethorphan plus valproic acid in bipolar disorder. Eur Neuropsychopharmacol. 2014;24:1753–1759. doi: 10.1016/j.euroneuro.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 55.Cunha AB, Frey BN, Andreazza AC, Goi JD, Rosa AR, Goncalves CA, Santin A, Kapczinski F. Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neurosci Lett. 2006;398:215–219. doi: 10.1016/j.neulet.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 56.Yoshimura R, Mitoma M, Sugita A, Hori H, Okamoto T, Umene W, Ueda N, Nakamura J. Effects of paroxetine or milnacipran on serum brain-derived neurotrophic factor in depressed patients. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1034–1037. doi: 10.1016/j.pnpbp.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 57.Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, Nakazato M, Watanabe H, Shinoda N, Okada S, Iyo M. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. 2003;54:70–75. doi: 10.1016/s0006-3223(03)00181-1. [DOI] [PubMed] [Google Scholar]
- 58.Dias VV, Brissos S, Frey BN, Andreazza AC, Cardoso C, Kapczinski F. Cognitive function and serum levels of brain-derived neurotrophic factor in patients with bipolar disorder. Bipolar Disord. 2009;11:663–671. doi: 10.1111/j.1399-5618.2009.00733.x. [DOI] [PubMed] [Google Scholar]
- 59.Piccinni A, Marazziti D, Catena M, Domenici L, Del Debbio A, Bianchi C, Mannari C, Martini C, Da Pozzo E, Schiavi E, Mariotti A, Roncaglia I, Palla A, Consoli G, Giovannini L, Massimetti G, Dell'Osso L. Plasma and serum brain-derived neurotrophic factor (BDNF) in depressed patients during 1 year of antidepressant treatments. J Affect Disord. 2008;105:279–283. doi: 10.1016/j.jad.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 60.Hellweg R, Ziegenhorn A, Heuser I, Deuschle M. Serum concentrations of nerve growth factor and brain-derived neurotrophic factor in depressed patients before and after antidepressant treatment. Pharmacopsychiatry. 2008;41:66–71. doi: 10.1055/s-2007-1004594. [DOI] [PubMed] [Google Scholar]
- 61.Calfa G, Percy AK, Pozzo-Miller L. Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp Biol Med (Maywood) 2011;236:3–19. doi: 10.1258/ebm.2010.010261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li W, Pozzo-Miller L. Beyond widespread Mecp2 deletions to model rett syndrome: conditional spatio-temporal knockout, single-point mutations and transgenic rescue mice. Autism Open Access. 2012;2012:5. doi: 10.4172/2165-7890.S1-005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen MV, Du F, Felice CA, Shan X, Nigam A, Mandel G, Robinson JK, Ballas N. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J Neurosci. 2012;32:10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larimore JL, Chapleau CA, Kudo S, Theibert A, Percy AK, Pozzo-Miller L. Bdnf overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobiol Dis. 2009;34:199–211. doi: 10.1016/j.nbd.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. 2005;19:667–678. doi: 10.1016/j.molcel.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 66.Sun AX, Crabtree GR, Yoo AS. MicroRNAs: regulators of neuronal fate. Curr Opin Cell Biol. 2013;25:215–221. doi: 10.1016/j.ceb.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Volvert ML, Rogister F, Moonen G, Malgrange B, Nguyen L. MicroRNAs tune cerebral cortical neurogenesis. Cell Death Differ. 2012;19:1573–1581. doi: 10.1038/cdd.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 69.Im HI, Kenny PJ. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012;35:325–334. doi: 10.1016/j.tins.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Geaghan M, Cairns MJ. MicroRNA and posttranscriptional dysregulation in psychiatry. Biol Psychiatry. 2015;78:231–239. doi: 10.1016/j.biopsych.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 71.Hara Y, Ago Y, Takano E, Hasebe S, Nakazawa T, Hashimoto H, Matsuda T, Takuma K. Prenatal exposure to valproic acid increases miR-132 levels in the mouse embryonic brain. Mol Autism. 2017;8:33. doi: 10.1186/s13229-017-0149-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci. 2007;10:1513–1514. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- 73.Good KV, Martinez de, Tyagi M, Cheema MS, Thambirajah AA, Gretzinger TL, Stefanelli G, Chow RL, Krupke O, Hendzel M, Missiaen K, Underhill A, Landsberger N, Ausio J. Trichostatin A decreases the levels of MeCP2 expression and phosphorylation and increases its chromatin binding affinity. Epigenetics. 2017;12:934–944. doi: 10.1080/15592294.2017.1380760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Larsson P, Alwis I, Niego B, Sashindranath M, Fogelstrand P, Wu MC, Glise L, Magnusson M, Daglas M, Bergh N, Jackson SP, Medcalf RL, Jern S. Valproic acid selectively increases vascular endothelial tissue-type plasminogen activator production and reduces thrombus formation in the mouse. J Thromb Haemost. 2016;14:2496–2508. doi: 10.1111/jth.13527. [DOI] [PubMed] [Google Scholar]
- 75.Olesen JB, Hansen PR, Abildstrøm SZ, Andersson C, Weeke P, Schmiegelow M, Erdal J, Torp-Pedersen C, Gislason GH. Valproate attenuates the risk of myocardial infarction in patients with epilepsy: a nationwide cohort study. Pharmacoepidemiol Drug Saf. 2011;20:146–153. doi: 10.1002/pds.2073. [DOI] [PubMed] [Google Scholar]
- 76.Dregan A, Charlton J, Wolfe CD, Gulliford MC, Markus HS. Is sodium valproate, an HDAC inhibitor, associated with reduced risk of stroke and myocardial infarction? A nested case-control study. Pharmacoepidemiol Drug Saf. 2014;23:759–767. doi: 10.1002/pds.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kooistra T, van den, Tons A, Platenburg G, Rijken DC, van den Berg J. Butyrate stimulates tissue-type plasminogen-activator synthesis in cultured human endothelial cells. Biochem J. 1987;247:605–612. doi: 10.1042/bj2470605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arts J, Lansink M, Grimbergen J, Toet KH, Kooistra T. Stimulation of tissue-type plasminogen activator gene expression by sodium butyrate and trichostatin A in human endothelial cells involves histone acetylation. Biochem J. 1995;310:171–176. doi: 10.1042/bj3100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dunoyer-Geindre S, Kruithof EK. Epigenetic control of tissue-type plasminogen activator synthesis in human endothelial cells. Cardiovasc Res. 2011;90:457–463. doi: 10.1093/cvr/cvr028. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Dufau ML. Silencing of transcription of the human luteinizing hormone receptor gene by histone deacetylase-mSin3A complex. J Biol Chem. 2002;277:33431–33438. doi: 10.1074/jbc.M204417200. [DOI] [PubMed] [Google Scholar]
- 81.Kim S, Kang JK, Kim YK, Seo DW, Ahn SH, Lee JC, Lee CH, You JS, Cho EJ, Lee HW, Han JW. Histone deacetylase inhibitor apicidin induces cyclin E expression through Sp1 sites. Biochem Biophys Res Commun. 2006;342:1168–1173. doi: 10.1016/j.bbrc.2006.02.081. [DOI] [PubMed] [Google Scholar]
- 82.Sowa Y, Orita T, Minamikawa S, Nakano K, Mizuno T, Nomura H, Sakai T. Histone deacetylase inhibitor activates the WAF1/Cip1 gene promoter through the Sp1 sites. Biochem Biophys Res Commun. 1997;241:142–150. doi: 10.1006/bbrc.1997.7786. [DOI] [PubMed] [Google Scholar]
- 83.Arts J, Herr I, Lansink M, Angel P, Kooistra T. Cell-type specific DNA-protein interactions at the tissue-type plasminogen activator promoter in human endothelial and HeLa cells in vivo and in vitro. Nucleic Acids Res. 1997;25:311–317. doi: 10.1093/nar/25.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Medcalf RL, Ruegg M, Schleuning WD. A DNA motif related to the cAMP-responsive element and an exon-located activator protein-2 binding site in the human tissue-type plasminogen activator gene promoter cooperate in basal expression and convey activation by phorbol ester and cAMP. J Biol Chem. 1990;265:14618–14626. [PubMed] [Google Scholar]
- 85.Lee SH, Ko HM, Kwon KJ, Lee J, Han SH, Han DW, Cheong JH, Ryu JH, Shin CY. tPA regulates neurite outgrowth by phosphorylation of LRP5/6 in neural progenitor cells. Mol Neurobiol. 2014;49:199–215. doi: 10.1007/s12035-013-8511-x. [DOI] [PubMed] [Google Scholar]
- 86.Cho KS, Kwon KJ, Choi CS, Jeon SJ, Kim KC, Park JH, Ko HM, Lee SH, Cheong JH, Ryu JH, Han SH, Shin CY. Valproic acid induces astrocyte-dependent neurite outgrowth from cultured rat primary cortical neuron via modulation of tPA/PAI-1 activity. Glia. 2013;61:694–709. doi: 10.1002/glia.22463. [DOI] [PubMed] [Google Scholar]
- 87.Gravanis I, Tsirka SE. Tissue plasminogen activator and glial function. Glia. 2005;49:177–183. doi: 10.1002/glia.20115. [DOI] [PubMed] [Google Scholar]
- 88.Hastings GA, Coleman TA, Haudenschild CC, Stefansson S, Smith EP, Barthlow R, Cherry S, Sandkvist M, Lawrence DA. Neuroserpin, a brain-associated inhibitor of tissue plasminogen activator is localized primarily in neurons. Implications for the regulation of motor learning and neuronal survival. J Biol Chem. 1997;272:33062–33067. doi: 10.1074/jbc.272.52.33062. [DOI] [PubMed] [Google Scholar]
- 89.Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biol. 2012;13:247. doi: 10.1186/gb-2012-13-7-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci. 2011;14:1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]