

Abstract
The geographical expansion of invasive species usually leads to temporary and/or permanent changes at multiple levels (genetics, epigenetics, gene expression, etc.) to acclimatize to abiotic and/or biotic stresses in novel environments. Epigenetic variation such as DNA methylation is often involved in response to diverse local environments, thus representing one crucial mechanism to promote invasion success. However, evidence is scant on the potential role of DNA methylation variation in rapid environmental response and invasion success during biological invasions. In particular, DNA methylation patterns and possible contributions of varied environmental factors to methylation differentiation have been largely unknown in many invaders, especially for invasive species in marine systems where extremely complex interactions exist between species and surrounding environments. Using the methylation‐sensitive amplification polymorphism (MSAP) technique, here we investigated population methylation structure at the genome level in two highly invasive model ascidians, Ciona robusta and C. intestinalis, collected from habitats with varied environmental factors such as temperature and salinity. We found high intrapopulation methylation diversity and significant population methylation differentiation in both species. Multiple analyses, such as variation partitioning analysis, showed that both genetic variation and environmental factors contributed to the observed DNA methylation variation. Further analyses found that 24 and 20 subepiloci were associated with temperature and/or salinity in C. robusta and C. intestinalis, respectively. All these results clearly showed significant methylation divergence among populations of both invasive ascidians, and varied local environmental factors, as well as genetic variation, were responsible for the observed DNA methylation patterns. The consistent findings in both species here suggest that DNA methylation, coupled with genetic variation, may facilitate local environmental adaptation during biological invasions, and DNA methylation variation molded by local environments may contribute to invasion success.
Keywords: biological invasion, DNA methylation, methylation divergence, methylation‐sensitive amplified polymorphism, tunicate
1. INTRODUCTION
Biological invasions provide promising “natural experiments” to advance our understanding of rapid environmental adaptation over contemporary time scales (Colautti & Lau, 2015; Lee, 2002; Prentis, Wilson, Dormontt, Richardson, & Lowe, 2008; Shine, 2012). Invasive species can rapidly spread over broad geographical scales by natural and/or human‐mediated dispersal and successfully colonize diverse habitats with varied local environmental conditions (Doney et al., 2012; Zhan, Briski, Bock, Ghabooli, & Macisaac, 2015). For marine invaders, introductions over long‐distance appear to be occurring with increased frequency due to intense human activities, such as shipping, aquaculture, and aquarium trades (Carlton, 1996; Lin, Gao, & Zhan, 2015; Zhan et al., 2017). In invaded habitats, invaders are often challenged by dramatically different abiotic and biotic conditions (Darling, Bagley, Roman, Tepolt, & Geller, 2008; Lee, 1999; Paul‐Pont, De Montaudouin, Gonzalez, Soudant, & Baudrimont, 2010; Sorte, Jones, & Miller, 2011; Zhan et al., 2015). Numerous studies suggest that rapid microevolution can occur in response to strong and persistent environmental challenges during biological invasions (Bock et al., 2015; Huang et al., 2017; Lande, 2015; Lin et al., 2017; Pu & Zhan, 2017; Sherman et al., 2016).
Epigenetic modification represent one important rapid mechanism for invasion success, as epigenetic modifications can influence phenotypes by regulating gene expression without changing the underlying DNA sequences (Bossdorf, Richards, & Pigliucci, 2008; Hawes et al., 2018; Rapp & Wendel, 2005). Although epigenetic variation can be predicted from its genotypic contexts or specific genetic polymorphisms elsewhere in the genome, still part of epigenetic variation can be independent from genetic control and is involved in response to environmental challenges as an autonomous system (Richards, 2006, 2008). Various types of epigenetic modifications, including modifications of DNA, histones and other chromosomal proteins, and the generation of extrachromosomal regulatory small RNAs and noncoding RNAs, have been found in many eukaryotes (Richards, 2008). Among these epigenetic modifications, DNA methylation, the addition of a methyl group to a specific base (mostly cytosine for eukaryotes), is one of the most intensively studied for its ubiquity in eukaryotes and essential biological functions (Jones, 2012; Suzuki & Bird, 2008). DNA methylation is involved in numerous biological processes, such as blocking transcription initiation, controlling transcription elongation, silencing transposon, inactivating X chromosome, and imprinting on genes (Duncan, Gluckman, & Dearden, 2014; Jones, 2012). In addition, studies of DNA methylation have been advanced by the development of new techniques, such as bisulfite conversion‐based methods (e.g., whole genome bisulfite sequencing—WGBS; reduced representation bisulfite sequencing—RRBS), immunoprecipitation‐based methods (e.g., methylated DNA immune‐precipitation sequencing—MeDIP‐seq; methylated DNA‐binding domain sequencing—MBD‐seq), and restriction enzyme‐based methods (e.g., methylation‐sensitive amplified polymorphism—MSAP; Methyl‐seq) (Suzuki & Bird, 2008; Wang et al., 2015). Although bisulfite conversion‐based sequencing techniques have been considered as promising tools for detecting methylation modifications, these methods are limited in studies at the population level (i.e., large‐scale population epigenomics studies), mainly owing to high experimental and computational costs as well as high‐quality reference genomes sequenced and assembled for genomewide analyses (Schulz, Eckstein, & Durka, 2013; Suzuki & Bird, 2008). Instead, the methylation‐sensitive amplification polymorphism (MSAP) technique, which can detect different methylation status at random restriction sites over a genome, is favorable as it allows to evaluate genomic methylation patterns of wild populations and identify associations between environmental conditions, phenotypic traits, and methylation status with low cost (Alonso, Pérez, Bazaga, Medrano, & Herrera, 2016). Thus, MSAP is the most widely used method in ecological epigenetics studies at the population level in a wide range taxa (Schulz et al., 2013).
During the process of local accommodation and adaptation, the most important feature of DNA methylation is its environmental effects (Verhoeven, VonHoldt, & Sork, 2016). Studies based on both common garden experiments and populations collected from the wild have detected correlations between DNA methylation variation and environmental stresses, indicating the environmental influence on DNA methylation divergence. For example, greenhouse experiments have shown that various stresses, such as low nutrients, salt stress, dietary components, and pathogen attack, can induce methylation variation throughout the whole genome (Dowen et al., 2012; Huang et al., 2017; Morán, Marco‐Rius, Megías, Covelo‐Soto, & Pérez‐Figueroa, 2013; Platt, Gugger, Pellegrini, & Sork, 2015; Verhoeven, Jansen, van Dijk, & Biere, 2010). In the wild, DNA methylation differentiation among populations has been frequently observed in different environments (Gugger, Fitz‐Gibbon, Pellegrini, & Sork, 2016; Paun et al., 2010; Wenzel & Piertney, 2014). In addition, maintenance and removal of DNA methylation are enzymatically mediated, thus the state of DNA methylation can change rapidly throughout the whole genome (Law & Jacobsen, 2010). Furthermore, changes in DNA methylation states can often cause important phenotypic consequences, leading to rapid accommodation and even adaptation to different local environments (Gao, Geng, Li, Chen, & Yang, 2010; Lea, Altmann, Alberts, & Tung, 2016; Pu & Zhan, 2017). Therefore, DNA methylation can integrate environmental signals into genomes rapidly and thereby modify gene expression and/or phenotypic variation to adapt to environmental changes/stresses.
DNA methylation variation is expected to be involved in the process of accommodation/adaptation to heterogeneous environments for invasive species (Hawes et al., 2018 and references therein; Pu & Zhan, 2017). Recent studies in invasive species showed that methylation diversity was much greater than genetic diversity in introduced populations of Japanese knotweed Fallopia species (Richards, Schrey, & Pigliucci, 2012) and Alligator weed Alternanthera philoxeroides (Gao et al., 2010). Liebl, Schrey, Richards, and Martin (2013) inferred that high methylation diversity could compensate for reduced genetic diversity during the invasion process of the house sparrow Passer domesticus. Interestingly, introduced populations of bluegrass Poa annua tended to have higher levels of methylation diversity than native populations (Chwedorzewska & Bednarek, 2012), and studies of the pygmy mussel (Xenostrobus securis) and tubeworm (Ficopomatus enigmaticus) found that recently introduced populations seemed to be less methylated in comparison with older introduced populations (Ardura, Zaiko, Morán, Planes, & Garcia‐Vazquez, 2017). Importantly, studies of invasive plants have also detected the association between DNA methylation variation and different habitats (Gao et al., 2010; Richards et al., 2012), and the authors further identified habitat‐related methylated loci (Richards et al., 2012). In order to further analyze possible contributions of local environments to methylation differentiation, studies assessed the degree of methylation differentiation among populations sampled from different environments. Interestingly, the level of methylation differentiation highly varied among invasive species, for example, strong methylation divergence was observed among populations of bluegrass (differentiation = 0.5; Chwedorzewska & Bednarek, 2012) and Japanese knotweed (differentiation = 0.5–0.8; Richards et al., 2012), while low methylation differentiation was found in introduced house sparrow populations in Kenya (differentiation = 0.004; Liebl et al., 2013). Collectively, it still remains largely unexplored on methylation patterns and possible contributions of varied environmental factors to methylation differentiation during range expansions in many invasive species.
Here, we used invasive model ascidians C. robusta (=C. intestinalis sp. A, Brunetti et al., 2015) and C. intestinalis (=C. intestinalis sp. B, Brunetti et al., 2015) to investigate population methylation structures and possible responsible environmental factors for observed patterns in diverse habitats. C. robusta and C. intestinalis, two morphologically similar but genetically distinct species of the C. intestinalis complex, have widely invaded temperate and warm‐temperate coastal zones over the past century (Carver, Mallet, & Vercaemer, 2006; Zhan, Macisaac, & Cristescu, 2010; Zhan et al., 2015). Due to ambiguous taxonomy within the genus Ciona, the native/invaded ranges and invasion histories of both C. robusta and C. intestinalis remain uncertain (Zhan et al., 2015 and references therein). C. robusta, which is generally considered as an East Asian native, has invaded coasts of Mediterranean Sea and many regions of Atlantic and Pacific coasts, while C. intestinalis, likely a native of Europe, has widely colonized northeast Atlantic coasts (Bouchemousse, Bishop, & Viard, 2016; Zhan et al., 2015 and references therein). Life history traits of Ciona, such as spawning time and life span, have a close relationship with its environmental conditions, particularly water temperature (Carver et al., 2006 and reference therein). For example, deep water populations in Scandinavia and sub‐Arctic populations can live 2–3 years and reproduce only once a year or less, while populations in the shallow regions of Scandinavian coast or in coastal regions of Atlantic Canada can survive for 12–18 months with two recruitment peaks per year (Carver et al., 2006 and reference therein). The broad geographical distribution, as well as varied environments in their native and/or invaded habitats, reflects the wide range environmental tolerance of both invasive species, especially for temperature and salinity (Dybern, 1967; Therriault & Herborg, 2008; Zhan et al., 2015). Both C. robusta and C. intestinalis have a short pelagic larval phase (1–5 days) and a sessile adult stage, suggesting that natural dispersal can only occur over relatively limited geographical ranges (Zhan et al., 2015). Therefore, the wide range expansion of both invasive species, particularly at the regional and continental scales, mainly is owing to human‐mediated pathways, which results in sudden shifts of habitats with varied environments. As such, these two ascidians provide good models to study mechanisms of rapid microevolution during biological invasions (Lin et al., 2017; Pu & Zhan, 2017; Zhan et al., 2015). Our previous study revealed significant genetic differentiation among C. robusta populations collected globally by using genomewide gene‐associated microsatellites, and genetic signatures and loci under selection were associated with varied local environmental conditions (Lin et al., 2017). Furthermore, when we analyzed the DNA methylation variation of salinity and temperature‐related genes using bisulfite sequencing at the population level, we found significant variation of DNA methylation among populations (Pu & Zhan, 2017). Interestingly, frequencies of several CpG loci were significantly correlated with local environmental factors (Pu & Zhan, 2017). However, Pu and Zhan's (2017) only focused on five genes which were putatively responsible for changes of temperature and salinity, and it remains largely unknown how environmental changes associated with habitat invasions shape methylation divergence among Ciona populations in different environments at the genome level.
In this study, we investigated the DNA methylation variation in C. robusta and C. intestinalis populations using the MSAP technique. We analyzed the DNA methylation patterns for populations of both species collected from different continents with varied local environments. In order to interpret the significant population methylation differentiation among populations, the possible contributions of genetic variation and two crucial environmental factors in marine ecosystems (i.e., temperature and salinity) were further tested.
2. MATERIALS AND METHODS
2.1. Sample collection
To cover the substantial environmental gradients in habitats of C. robusta and C. intestinalis, sampling sites were selected based on our former studies (Zhan et al., 2010, 2012) across four continents (Asia, Africa, Europe, and Oceania) for C. robusta and two continents (Europe and North America) for C. intestinalis (Table 1; Figure 1). To make it clear for further comparison between genetic and methylation data, populations of both C. robusta and C. intestinalis were named as our previous studies (Lin et al., 2017; Zhan et al., 2010, 2012). Temperature and salinity, two crucial environmental factors affecting numerous physiological processes in marine invertebrates, have a high degree of variation at the chosen sampling sites in this study (see Lin et al., 2017 for C. robusta; Table 2, for C. intestinalis). We chose a total of 10 populations, including five populations of C. robusta (N = 115) and five populations of C. intestinalis (N = 148; Table 1; Figure 1). For C. robusta, all of five collected populations were invasive populations, of which populations AM (Arenys de Mar) and BL (Blanes) established at the end of 19th, while populations SA (Cape Town) and NMF (Nelson) were introduced at mid‐20th (Bouchemousse et al., 2016 and references therein). Population GAP (Gampo) was reported on the coast of Korea at the late 1990s (Seo & Lee, 2009). For C. intestinalis, two European populations SL (Salzhaff) and SC (Schleimünde) were considered as putatively native populations, while the other three Canadian populations were invasive populations locally detected after 1997 (Bouchemousse et al., 2016 and references therein). All collected adult specimens were preserved in 100% ethanol at 4°C before analyses. All specimens were identified and confirmed to the species level by using one mitochondrial DNA fragment, cytochrome c oxidase subunit 3–NADH dehydrogenase subunit 1 (COX3‐ND1; Zhan et al., 2010).
Table 1.
Sampling sites and environmental parameters for the two highly invasive ascidians Ciona robusta and C. intestinalis
| Population ID | Region/state and country | Colonized time | Coordinates | n | AveT (°C) | MaxT (°C) | MinT (°C) | AveS (‰) | MaxS (‰) | MinS (‰) |
|---|---|---|---|---|---|---|---|---|---|---|
| C. robusta | ||||||||||
| AM | Arenys de Mar, Spain | End‐19th | 41°33′41″N, 2°34′37″E | 29 | 17.98 | 25.28 | 13.29 | 37.67 | 38.21 | 36.87 |
| BL | Blanes, Spain | End‐19th | 41°41′12″N, 2°53′22″E | 12 | 17.44 | 24.38 | 12.98 | 37.95 | 38.32 | 37.48 |
| SA | Cape town, South Africa | Mid‐20th | 33°54′33″S, 18°25′59″E | 32 | 16.03 | 16.92 | 15.16 | 35.18 | 35.30 | 34.99 |
| GAP | Gampo, Korea | End‐20th | 35°48′26″N, 129°30′13″E | 30 | 17.72 | 24.31 | 12.18 | 33.72 | 34.48 | 32.17 |
| NMF | Nelson, New Zealand | Mid‐20th | 41°15′29″S, 173°16′42″E | 12 | 13.55 | 16.37 | 11.22 | 34.78 | 34.92 | 34.62 |
| C. intestinalis | ||||||||||
| HF | Halifax, Canada | End‐20th | 44°38′48″N, 63°34′8″W | 30 | 8.51 | 17.98 | 1.09 | 31.03 | 31.54 | 30.68 |
| MR | Murray River, Canada | Early‐21st | 46°0′53″N, 62°36′30″W | 30 | 7.77 | 18.37 | −1.25 | 29.28 | 30.64 | 28.20 |
| YM | Yarmouth, Canada | End‐20th | 43°50′06″N, 66°07′22″W | 30 | 7.08 | 11.48 | 2.16 | 31.81 | 32.20 | 31.50 |
| SL | Salzhaff, Germany | Putatively native | 54°2′22″N, 11°31′36″E | 30 | 9.25 | 17.44 | 1.82 | 15.67 | 18.52 | 13.36 |
| SC | Schleimünde, Germany | Putatively native | 54°41′25″N, 10°7′15″E | 28 | 9.45 | 17.94 | 1.90 | 14.84 | 17.67 | 12.71 |
S: salinity; T: temperature.
Figure 1.
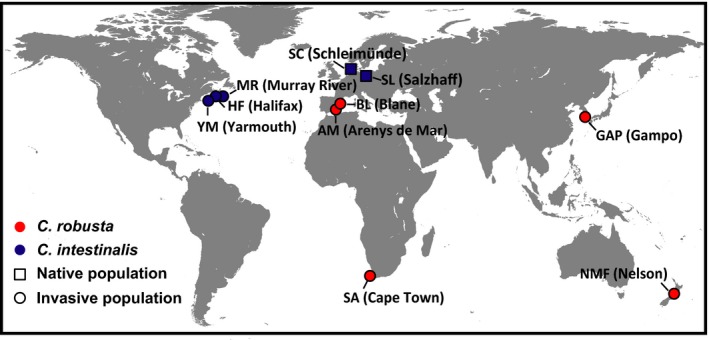
Sampling sites of Ciona robusta (red) and C. intestinalis (blue)
Table 2.
p‐Values for the exact test for the difference of the temperature (above diagonal) and salinity (below diagonal) among sample sites of Ciona intestinalis based on a nonparametric test, Mann–Whitney U test
p < 0.01; —, not significant.
2.2. Methylation‐sensitive amplification polymorphism assay
Total genomic DNA was isolated from approximately 50 mg of siphon tissues following the proteinase K method (Waters, Dijkstra, & Wallis, 2000). The quality and quantity of DNA were measured using Nanodrop 2000 spectrophotometer (Thermo Scientific). Total genomic DNA (300 ng) was digested at 37°C for 3 hr in two parallel reactions using 5 U of EcoRI‐HF and 5 U of either MspI or HpaII (New England Biolabs) in a final volume of 10 μl. MspI and HpaII can recognize and cleave the same sequence 5′‐CCGG‐3′, but their susceptibility is different depending on the methylation state of cytosines at restriction sites (Schulz et al., 2013). After digestion, enzymes were heat‐deactivated at 80°C for 10 min. EcoRI_ADAPTER (F) 5′‐CTCGTAGACTGCGTACC‐3′/EcoRI_ADAPTER (R) 5′‐AATTGGTACGCAGTCTAC‐3′ and HpaII/MspI_ADAPTER (F) 5′‐GACGATGAGTCTAGAA‐3′/HpaII/MspI_ADAPTER (R) 5′‐CGTTCTAGACTCATC‐3′ (Wenzel & Piertney, 2014) were used to prepare EcoRI and HpaII/MspI adapters by mixing an equal amount of complementary oligonucleotides. A ligation mixture in a volume of 5 μl contained 5 pmol of EcoRI adaptor and 50 pmol of HpaII/MspI adaptor, and 30 U of T4 DNA ligase (New England Biolabs) was added to digestion products and incubated at 37°C for 3 hr, followed by 16°C for 9 hr, and then 65°C for 20 min.
Preselective polymerase chain reaction (PCR) was carried out in a total volume of 20 μl containing 2 μl of the ligation product, 0.4 μM each of EcoRI_ADAPTER (F)+A and HpaII/MspI_ADAPTER (F)+T preselective primers, 0.5 U of Taq DNA polymerase (Takara), 2 mM of MgCl2, and 0.2 mM of each nucleotide. The PCR profile was as follows: 72°C for 2 min followed by 30 cycles of 95°C for 20 s, 56°C for 30 s and 72°C for 1 min, and a final elongation step at 60°C for 2 min.
Based on the number of amplified fragments, five primer pairs (Table 3) were chosen for selective PCRs. Selective PCRs were performed in a total volume of 30 μl using 0.5 μl of preselective PCR product, 0.2 μM of fluorescently labeled (6‐FAM) forward primer, 0.2 μM of reverse primer, 0.5 U of Taq DNA polymerase (Takara), 1.25 mM of MgCl2 and 0.125 mM of each nucleotide. The PCR profile was as follows: 94°C for 3 min, 13 touchdown cycles of 94°C for 30 s, 65°C for 30 s reduced by 0.7°C per cycle and 72°C for 2 min, 24 cycles of 94°C for 30 s, 56°C for 30 s and 72°C for 2 min, and a final elongation step at 72°C for 2 min. Selective amplification products were separated on an ABI 3730 DNA analyzer (Applied Biosystems) along with GeneScan LIZ 500 size standard (Applied Biosystems). GeneMarker v.2.2.0. (SoftGenetics) was used for fragment scrutiny. GeneMarker panels were created for each primer pair with the fragment range from 150 bp to 500 bp, as fragment size <150 bp may increase the potential impact of size homoplasy (Caballero, Quesada, & Rolán‐Alvarez, 2008). Monomorphic epiloci and singletons (i.e., when only one sample had a deviating status) were excluded from further analyses. Replicated samples (6% of the total) starting from DNA extraction were included in all steps to test the reproducibility of MSAP assay (Bonin et al., 2004). Epiloci with more than two mismatches across the replicated samples were discarded from our datasets. The error rate based on replicated samples was estimated as 5.71% and 5.66% for C. robusta and C. intestinalis, respectively.
Table 3.
Selective primer combinations used in methylation‐sensitive amplification polymorphism (MSAP)
| EcoRI | HpaII/MspI |
|---|---|
| GACTGCGTACCAATTCACA | GATGAGTCTAGAACGGTTA |
| GACTGCGTACCAATTCAGA | GATGAGTCTAGAACGGTGTT |
| GACTGCGTACCAATTCAGA | GATGAGTCTAGAACGGCT |
| GACTGCGTACCAATTCAGA | GATGAGTCTAGAACGGAT |
| GACTGCGTACCAATTCATC | GATGAGTCTAGAACGGCA |
2.3. Data analyses
Individuals were scored based on the presence (as “1”) or absence (as “0”) of EcoRI‐HpaII and EcoRI‐MspI fragments, and four methylation conditions of restriction sites are distinguished as: Type I) fragments detected in both MspI and HpaII profiles are unmethylated loci; Type II) fragments only detected in HpaII profile are methylation of the external cytosine on only one strand (HMeCCG); Type III) fragments only detected in the MspI profile are methylation of the inner cytosine on one or both strands (HMeCG or MeCG); and Type IV) fragments absent from both HpaII and MspI profiles are methylation of both inner and outer cytosines on one or both strands (HMeCHMeCG, HMeCMeCG, MeCHMeCG, or MeCMeCG), methylation of the both outer cytosine (MeCCG) and absence of restriction sites (Figure 2; Schulz et al., 2013). Multistate data matrices were generated by combining scores of the two parallel digestion profiles. To separate the contribution of unmethylated and methylated states of the same epiloci, the multistate epiloci were transformed into two separate binary subepiloci, u‐subepiloci (Figure 2; only type I scored as “1,” others scored as “0,” i.e., unmethylated epiloci) and m‐subepiloci (Figure 2; type II and type III scored as “1,” others scored as “0,” i.e., methylated epiloci). Thus, the multistate raw data matrices were recorded into two binary data sets: dataset U and dataset M following the “Mixed Scoring 1” approach (Figure 2; Schulz et al., 2013). This approach can temper the confusion of data interpretation between types II and III, which may be caused by the methylation variation at two closely spaced CCGG sites (Fulneček & Kovařík, 2014).
Figure 2.
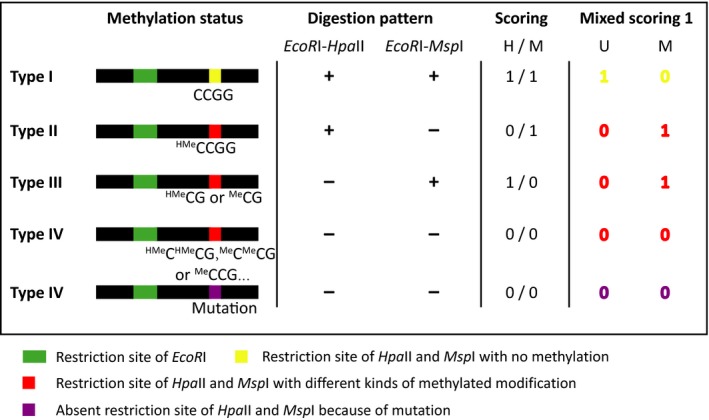
Scoring scheme based on the “Mixed Scoring 1” method
Intrapopulation methylation diversity was characterized by the percentage of private loci, percentage of polymorphic loci, and Shannon's information index for both u‐ and m‐subepiloci. Using GENALEX version 6.5 (Peakall & Smouse, 2006), population methylation differentiation (ΦPT) was determined based on both u‐ and m‐subepiloci for all intraspecific population pairs in both species by pairwise analysis of molecular variance (AMOVA) with 999 randomizations (Excoffier, Smouse, & Quattro, 1992). Pairwise distances between intraspecific individuals were computed by Euclidean measure for both u‐ and m‐subepiloci with GENALEX version 6.5. To visualize the methylation differentiation among populations of C. robusta and C. intestinalis, principal coordinates analyses (PCoA) of u‐ and m‐subepiloci were conducted separately with the individual‐by‐individual methylation distance matrices using GENALEX version 6.5.
As genetic analyses based on genomewide microsatellites have been conducted for C. robusta (Lin et al., 2017), we used C. robusta as an example to evaluate the relationship between genetic and methylation differentiation for populations using Genodive (Meirmans & Van Tienderen, 2004) with 99,999 permutations. The genetic differentiation matrix was obtained based on pairwise ΦPT values calculated by 152 genomewide microsatellites from Lin et al. (2017), where the individuals used were exactly the same as these in this study. To investigate the relative contributions of genetic variation and environmental factors to the observed methylation variation, partial redundancy analysis (pRDA) was conducted for C. robusta using vegan package (function varpart; Oksanen et al., 2017) in the R (version 3.3.1.; R Core Team, 2016). The total methylation variation matrix, which combined U and M datasets, was set as the response matrix. The environmental factors and genetic variation matrices were set as explanatory variable matrices. Comparing with the annual mean temperature and salinity, recent studies emphasized that changes of maximum and minimum values may have greater effects on ascidians (Stachowicz, Terwin, Whitlatch, & Osman, 2002). Studies on both plants and animals suggest that the environmental stresses can be associated with long‐term epigenetic modifications, which can be transmitted through generations (Jablonka, 2012; Weyrich et al., 2016). Thus, the matrix of environmental factors was represented by the raw data of six environmental factors including annual water temperature, maximum water temperature, minimum water temperature, annual water salinity, maximum water salinity, and minimum water salinity. For the ambiguous invasion histories of both C. robusta and C. intestinalis, here we used the average values (among 1955–2012) of the six environmental factors collected from NOAA with the resolution as quarter‐degree (http://www.nodc.noaa.gov/OC5/SELECT/woaselect/woaselect.html). Although the environmental data from NOAA mainly focus on large‐scale variation in the open ocean, the difference of environmental factors among sample sites across continents still can be extracted from the NOAA datasets and is sufficient for our continent‐level study here. The genetic variation matrix was transformed from the 152 genomewide microsatellites data from Lin et al. (2017) according to the number of mutation steps among individuals (Leung, Breton, & Angers, 2016). To reduce the possible strong linear dependencies among the explanatory variables in pRDA model, forward selection method was applied on six environmental factors and microsatellites separately using R package packfor (function forward.sel; Dray, Legendre, & Blanchet, 2013), and the resulted variables were further used in pRDA. The total methylation variation explained by genetics or/and environmental factors was represented by the adjusted R 2 (R adj 2), and permutation tests were performed to test the significance of each fraction with 999 randomizations (function anova.cca in package vegan; Oksanen et al., 2017).
To identify adaptive epiloci that likely are influenced by local environmental conditions, spatial analysis method (SAM) was performed for both C. robusta and C. intestinalis to test the relevance between allelic frequencies of epiloci and corresponding environmental parameters at sampling locations by computing multiple univariate logistic regression (Joost, Kalbermatten, & Bonin, 2008). Correlations between six environmental factors included in pRDA and each epiloci were conducted using SAM v.1 with the initial confidence level of 0.05. The input matrix included raw environmental data and methylated information for each individual. To ensure the robustness of SAM, two statistical tests including likelihood ratio G and Wald, which assess the significance of coefficients, were employed by the logistic regression function. A model was considered to be significant only if both tests reject the corresponding null hypothesis.
To verify whether these environment‐related epiloci were potentially involved in local environmental response, BAYESCAN (Foll & Gaggiotti, 2008) was used for both C. robusta and C. intestinalis to detect outliers by separating adaptive effects from neutral effects through the locus‐specific population differentiation coefficient. For each locus, BAYESCAN v2.1 directly calculates q‐values and an outlier is detected when its q‐value is lower than 5%. The input files (U and M datasets) were transformed as the format of dominant binary markers and tested following 10 pilot runs of 5,000 iterations with a 50,000 burn‐in and a thinning interval of 20. The prior odds for the neutral model were set as 10.
3. RESULTS
3.1. Intrapopulation methylation diversity
We generated 332 and 456 epiloci for C. robusta and C. intestinalis, respectively, by combining parallel results of EcoRI‐HpaII and EcoRI‐MspI reactions. Using the Mixed Scoring approach, 604 subepiloci including 275 u‐subepiloci and 329 m‐subepiloci for C. robusta, and 841 subepiloci including 395 u‐subepiloci and 446 m‐subepiloci for C. intestinalis were obtained by splitting the epiloci with more than one methylation type. The intrapopulation methylation diversity was measured by both u‐subepiloci and m‐subepiloci for C. robusta and C. intestinalis (Table 4). For both species, the percentages of private u‐subepiloci in each population were much higher than their corresponding values of private m‐subepiloci (Table 4). However, the polymorphism of u‐subepiloci, which ranged from 60.36% to 81.09% in C. robusta populations and varied from 67.59% to 73.92% in C. intestinalis populations (Table 4), were lower than corresponding estimators of m‐subepiloci in both C. robusta (79.03% to 92.40%; Table 4) and C. intestinalis (81.39% to 94.39%; Table 4). The values of Shannon index based on u‐subepiloci were similar among populations of C. robusta, ranging from 0.245 to 0.299 except for an European population BL (Blanes) where we detected a much higher value of 0.416 (Table 4). Interestingly, Shannon index of m‐subepiloci was much higher than that of u‐subepiloci in both C. robusta and C. intestinalis (Table 4). For both species, no obvious positive or negative correlations were detected between invasion time and methylation diversity at u‐ and m‐subepiloci. For C. intestinalis, one of native populations SC (Schleimünde) showed the highest values of polymorphism and Shannon index in both U and M profiles, while methylation diversity of the other native population SL (Salzhaff) was similar to that in invasive populations (Table 4).
Table 4.
Methylation diversity for the analyzed populations of Ciona robusta and C. intestinalis based on u‐subepiloci (unmethylated loci) and m‐subepiloci (methylated loci)
| Population ID | u‐subepiloci | m‐subepiloci | ||||
|---|---|---|---|---|---|---|
| Private loci % | Polymorphic loci % | Shannon index | Private loci % | Polymorphic loci % | Shannon index | |
| C. robusta | ||||||
| AM (Arenys de Mar) | 0.70 | 60.72 | 0.245 | 0 | 82.37 | 0.409 |
| BL (Blanes) | 3.27 | 81.09 | 0.416 | 0.30 | 82.67 | 0.433 |
| SA (Cape town) | 0.36 | 74.55 | 0.280 | 0 | 80.85 | 0.411 |
| GAP (Gampo) | 4.36 | 61.45 | 0.261 | 3.04 | 92.40 | 0.502 |
| NMF (Nelson) | 2.18 | 60.36 | 0.295 | 0 | 79.03 | 0.425 |
| Average | 2.17 | 67.63 | 0.299 | 0.67 | 83.46 | 0.436 |
| C. intestinalis | ||||||
| HF (Halifax) | 0.25 | 73.67 | 0.290 | 0 | 91.70 | 0.385 |
| MR (Murray River) | 0.76 | 69.87 | 0.254 | 0 | 88.57 | 0.336 |
| YM (Yarmouth) | 0.25 | 70.38 | 0.279 | 0 | 90.36 | 0.356 |
| SL (Salzhaff) | 2.78 | 67.59 | 0.271 | 0 | 81.39 | 0.339 |
| SC (Schleimünde) | 2.53 | 73.92 | 0.295 | 0.2 | 94.39 | 0.496 |
| Average | 1.31 | 71.09 | 0.278 | 0.04 | 89.28 | 0.382 |
3.2. Interpopulation methylation differentiation
For C. intestinalis, higher ΦPT values in U profile than those in M profile were observed in all population pairs (Table 5B). Putative native populations from Europe (SL and SC) were highly and significantly differentiated from invasive populations collected from North America (HF, MR and YM) in both U (ΦPT = 0.133–0.216, p < 0.05; Table 5B) and M profiles (ΦPT = 0.053–0.083, p < 0.05; Table 5B). The differentiation among three Canadian invasive populations (HF, MR & YM) was relatively low but significant in both U (ΦPT = 0.018–0.069, p < 0.05; Table 5B) and M profiles (ΦPT = 0.010–0.023, p < 0.05; Table 5B). The highest methylation differentiation was detected between two adjacent European native populations SC and SL in the U profile (ΦPT = 0.224, p < 0.05; Table 5B), while in M profile, the highest value was found between an European native population SL and a Canadian invasive population HF (ΦPT = 0.083, p < 0.05; Table 5B).
Table 5.
Estimates of population methylation differentiation in two highly invasive ascidians Ciona robusta (A) and C. intestinalis (B). Above diagonal: pairwise ΦPT based on m‐subepiloci, below diagonal: pairwise ΦPT based on u‐subepiloci
| (A) | AM | BL | SA | GAP | NMF |
|---|---|---|---|---|---|
| AM (Arenys de Mar) | 0.058a | 0.018a | 0.132a | 0.095a | |
| BL (Blanes) | 0.187a | 0.067a | 0.073a | 0.037a | |
| SA (Cape town) | 0.047a | 0.163a | 0.124a | 0.078a | |
| GAP (Gampo) | 0.274a | 0.262a | 0.220a | 0.085a | |
| NMF (Nelson) | 0.262a | 0.154a | 0.184a | 0.194a |
| (B) | HF | MR | YM | SL | SC |
|---|---|---|---|---|---|
| HF (Halifax) | 0.023a | 0.010a | 0.083a | 0.058a | |
| MR (Murray River) | 0.054a | 0.014a | 0.077a | 0.056a | |
| YM (Yarmouth) | 0.018a | 0.069a | 0.076a | 0.053a | |
| SL (Salzhaff) | 0.210a | 0.216a | 0.215a | 0.068a | |
| SC (Schleimünde) | 0.136a | 0.156a | 0.133a | 0.224a |
p < 0.01.
When interpopulation methylation differentiation was assessed by pairwise AMOVA, significant methylation differentiation in C. robusta was observed between all population pairs in both U and M profiles (p < 0.05). Interestingly, overall pairwise ΦPT values in U profiles were higher than their corresponding values in M profiles (Table 5A). The highest methylation differentiation for both U (ΦPT = 0.274; Table 5A) and M profiles (ΦPT = 0.132; Table 5A) was consistently observed between the population pair of AM (Arenys de Mar, Europe) and GAP (Gampo, Asia). The more recently introduced population GAP (Gampo) was also highly differentiated from the other three earlier introduced populations in both U (ΦPT = 0.194–0.274; Table 5A) and M profiles (ΦPT = 0.073–0.132; Table 5A). Intriguingly, the lowest ΦPT values in both U (ΦPT = 0.047; Table 5A) and M profiles (ΦPT = 0.018; Table 5A) were detected between a European population AM (Arenys de Mar) and an African population SA (Cape Town), two populations with a distant geographical distance.
Principal coordinates analysis (PCoA) of both U and M profiles showed largely consistent results with pairwise AMOVA for C. robusta and C. intestinalis (Figure 3). In the U profile of C. robusta, the Asian population (GAP) was completely separated from the other populations to form a unique cluster. Individuals of European population BL (Blanes) and Oceanian population NMF (Nelson) were scattered widely and slightly separated from the intermingled European (AM) and African (SA) populations (Figure 3a). In the U profile of C. intestinalis, PCoA of methylation distances separated introduced Canadian populations and putative native European populations. The three recently introduced Canadian populations, HF (Halifax), MR (Murray River), and YM (Yarmouth), were grouped together, while the two geographically close native populations from Europe (SL and SC) were completely distinguished from each other (Figure 3b). In the M profile, individuals were less clumped but clustered in a similar pattern as that of the U profile (Figure 3).
Figure 3.
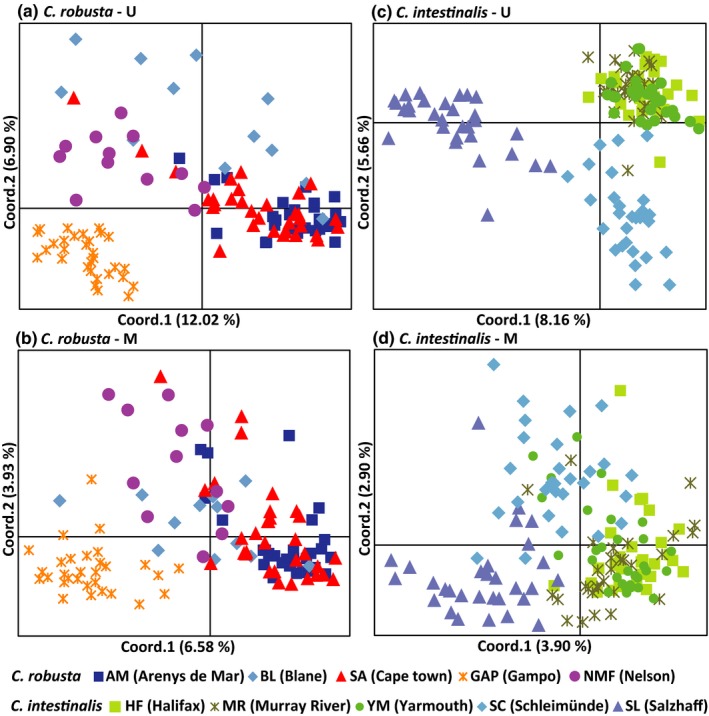
Principal coordinates analysis (PCoA) of U and M profiles for Ciona robusta and C. intestinalis. The first two coordinates are displayed with the indication of the percentage of variance explained in brackets. Populations collected from the same regions are represented by different shades of the same colors
Mantel tests showed significant correlation between genetic differentiation (Table 6) with methylation differentiation (Table 5A) in both U (R = 0.700, p = 0.017) and M (R = 0.663, p = 0.008) profiles, suggesting that genetic variation contributes to methylation variation observed in this study. In the pRDA mode, four environmental factors and 15 microsatellites were screened using the forward selection method for further analyses (Tables 7 and 8). The partitioning of total methylation variation showed that genetic variation and environmental factors could explain 11.8% (p = 0.001) and 10.5% (p = 0.001), respectively, of which 8.4% could be explained by both factors (Figure 4). Furthermore, 2.1% (p = 0.001) and 3.4% (p = 0.001) of the observed methylation variation can be solely explained by environmental factors and genetic variation, respectively (Figure 4).
Table 6.
Estimates of population genetic differentiation (pairwise ΦPT) in Ciona robusta based on 152 microsatellites
| AM | BL | SA | GAP | |
|---|---|---|---|---|
| AM | ||||
| BL | 0.027* | |||
| SA | 0.030** | 0.047* | ||
| GAP | 0.223** | 0.193** | 0.222** | |
| NMF | 0.225** | 0.172** | 0.216** | 0.106** |
*p < 0.05; **p < 0.01.
Table 7.
Screening results for environmental factors by the forward selection method
| Variables | R 2 | R 2 Cum | R adj 2 Cum | F | p‐Value |
|---|---|---|---|---|---|
| MinS | 0.060 | 0.060 | 0.052 | 7.256 | 0.001 |
| MinT | 0.027 | 0.087 | 0.071 | 3.304 | 0.001 |
| MaxS | 0.025 | 0.112 | 0.088 | 3.128 | 0.001 |
| AveS | 0.024 | 0.137 | 0.105 | 3.095 | 0.001 |
Table 8.
Screening results for microsatellites by the forward selection method
| Variables | R 2 | R 2 Cum | R adj 2 Cum | F | p‐Value |
|---|---|---|---|---|---|
| Cin27 | 0.064 | 0.064 | 0.056 | 7.767 | 0.001 |
| Cin189 | 0.025 | 0.089 | 0.073 | 3.026 | 0.001 |
| Cin104 | 0.018 | 0.107 | 0.083 | 2.300 | 0.001 |
| Cin182 | 0.014 | 0.121 | 0.089 | 1.745 | 0.001 |
| Cin211 | 0.011 | 0.133 | 0.093 | 1.426 | 0.002 |
| Cin106 | 0.011 | 0.144 | 0.096 | 1.406 | 0.004 |
| Cin141 | 0.011 | 0.155 | 0.099 | 1.363 | 0.001 |
| Cin126 | 0.011 | 0.165 | 0.102 | 1.350 | 0.005 |
| Cin72 | 0.010 | 0.176 | 0.105 | 1.321 | 0.013 |
| Cin17 | 0.010 | 0.186 | 0.107 | 1.268 | 0.015 |
| Cin162 | 0.010 | 0.196 | 0.110 | 1.280 | 0.017 |
| Cin60 | 0.010 | 0.206 | 0.112 | 1.282 | 0.019 |
| Cin76 | 0.010 | 0.215 | 0.115 | 1.281 | 0.012 |
| Cin179 | 0.009 | 0.225 | 0.116 | 1.211 | 0.039 |
| Cin160 | 0.009 | 0.234 | 0.118 | 1.203 | 0.041 |
Figure 4.
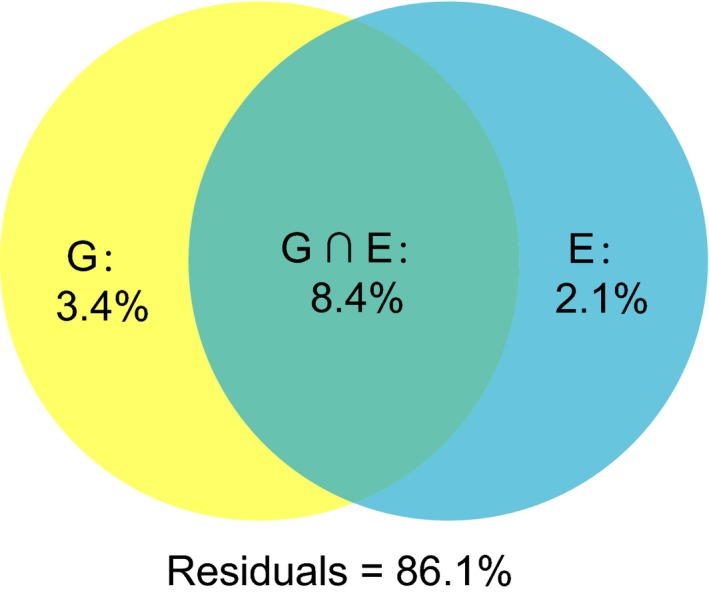
Results of variation partitioning analysis performed to assess the relative contribution of genetic variation and environmental effects to the total observed DNA methylation variation
3.3. Environment‐related subepiloci
SAM analyses detected that 24 (4.0%) and 20 (2.4%) subepiloci were correlated with at least one of the six environmental factors for C. robusta and C. intestinalis, respectively (Table 9). BAYESCAN identified six and 10 outliers for C. robusta and C. intestinalis, respectively (Figure 5; Table 9). Three environment‐related subepiloci in C. robusta and two in C. intestinalis were also identified as outliers in BAYESCAN (Figure 6; Table 9). The frequencies of environment‐related subepiloci varied along with the environmental factors and showed significant difference among populations (Figure 6). For C. robusta, seven subepiloci were temperature‐related, of which five were correlated with the minimum temperature. We detected 20 salinity‐related subepiloci in C. robusta, and 85% were related to the mean minimum salinity (Table 9). Furthermore, seven subepiloci were related to more than one salinity parameter, and three subepiloci were associated with both temperature‐ and salinity‐related parameters. For C. intestinalis, nine subepiloci were temperature‐related and 16 were correlated with salinity. Interestingly, 15 of 16 salinity‐related epiloci in C. intestinalis were correlated with the mean maximum salinity, mean minimum salinity, and average salinity at the same time, and five of nine temperature‐related subepiloci were also associated with salinity (Table 9).
Table 9.
Epiloci detected by SAM and/or BAYESCAN
| Epiloci | ID | MaxT‐related | MinT‐related | AveT‐related | MaxS‐related | MinS‐related | AveS‐related | BAYESCAN identified |
|---|---|---|---|---|---|---|---|---|
| Ciona robusta | ||||||||
| P1.157 | u1 | 1 | 1 | 1 | y | |||
| P1.227 | u2 | 1 | ||||||
| P1.230 | u3 | 1 | 1 | |||||
| P1.286 | u4 | 1 | 1 | |||||
| P1.313 | u5 | 1 | ||||||
| P1.341 | u6 | 1 | ||||||
| P1.389 | u7 | 1 | ||||||
| P2.163 | u8 | 1 | ||||||
| P2.179 | u9 | 1 | ||||||
| P2.197 | u10 | 1 | ||||||
| P2.229 | u11 | 1 | ||||||
| P2.240 | u12 | 1 | ||||||
| P2.241 | u13 | 1 | ||||||
| P2.308 | u14 | y | ||||||
| P2.322 | u15 | 1 | 1 | |||||
| P5.158 | u16 | 1 | ||||||
| P5.296 | u17 | 1 | 1 | |||||
| P5.334 | u18 | y | ||||||
| P5.479 | u19 | 1 | 1 | 1 | y | |||
| P6.245 | u20 | y | ||||||
| P6.260 | u21 | 1 | ||||||
| P6.306 | u22 | 1 | 1 | 1 | y | |||
| P6.313 | u23 | 1 | 1 | 1 | ||||
| P1.227 | m1 | 1 | ||||||
| P1.389 | m2 | 1 | ||||||
| P6.309 | m3 | 1 | ||||||
| P6.367 | m4 | 1 | ||||||
| Ciona intestinalis | ||||||||
| P1.200 | u1 | 1 | 1 | 1 | 1 | y | ||
| P1.445 | u2 | y | ||||||
| P2.177 | u3 | 1 | 1 | 1 | ||||
| P2.229 | u4 | 1 | 1 | 1 | ||||
| P2.265 | u5 | 1 | ||||||
| P4.190 | u6 | y | ||||||
| P4.220 | u7 | 1 | ||||||
| P4.312 | u8 | y | ||||||
| P4.362 | u9 | y | ||||||
| P4.382 | u10 | 1 | 1 | 1 | 1 | |||
| P4.396 | u11 | 1 | 1 | 1 | ||||
| P5.189 | u12 | 1 | 1 | 1 | ||||
| P5.261 | u13 | 1 | 1 | 1 | ||||
| P5.300 | u14 | 1 | 1 | 1 | 1 | |||
| P5.465 | u15 | y | ||||||
| P6.171 | u16 | 1 | 1 | |||||
| P6.209 | u17 | y | ||||||
| P6.214 | u18 | 1 | 1 | |||||
| P6.250 | u19 | 1 | 1 | y | ||||
| P6.273 | u20 | 1 | 1 | 1 | ||||
| P6.298 | u21 | y | ||||||
| P6.388 | u22 | 1 | 1 | 1 | 1 | |||
| P6.398 | u23 | 1 | 1 | 1 | ||||
| P6.406 | u24 | y | ||||||
| P1.198 | m1 | 1 | 1 | 1 | ||||
| P2.357 | m2 | 1 | 1 | 1 | ||||
| P2.410 | m3 | 1 | 1 | 1 | ||||
| P5.312 | m4 | 1 | 1 | 1 | ||||
Each locus is listed with ID, the association with temperature‐ and salinity‐related parameters, identified by BAYESCAN or not.
Figure 5.
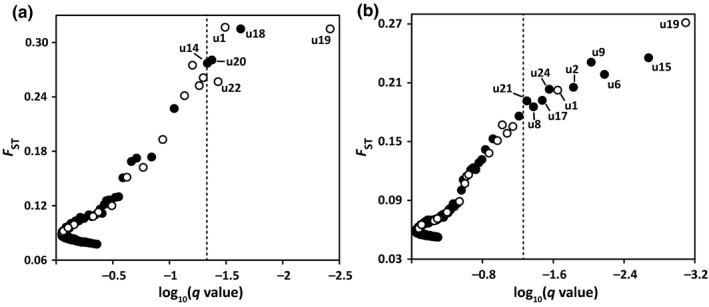
Epiloci under selection in Ciona robusta (a) and C. intestinalis (b) conducted by BAYESCAN. The dashed line donates the q‐value of 0.05. The open dots indicate that epiloci were correlated with environmental factors in the spatial analysis method (SAM)
Figure 6.
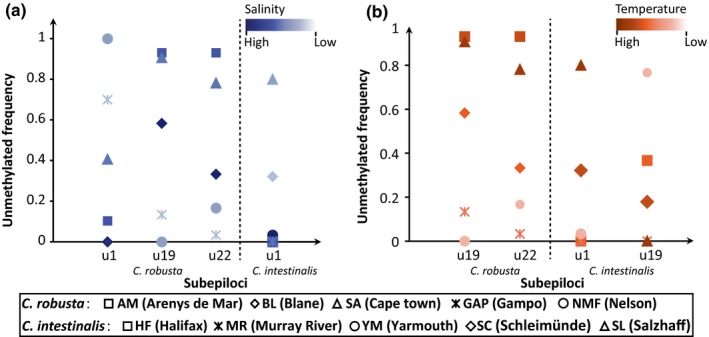
The unmethylation variation of u‐subepiloci identified by both the spatial analysis method (SAM) and BAYESCAN in different populations
4. DISCUSSION
In this study, we explored the population methylation patterns of C. robusta and C. intestinalis at the genome level. Our results revealed highly consistent patterns in both invasive species. We detected a high degree of intrapopulation methylated polymorphism for both invasive species. In addition, both C. robusta and C. intestinalis showed significant population methylation differentiation and both genetic variation and varied local environments could partly explain the DNA methylation variation of C. robusta. We identified subepiloci presumably under selection and some of them were significantly correlated with environmental factors such as temperature and/or salinity. All results in our study suggest the putative contribution of DNA methylation variation to environmental response during biological invasions.
4.1. Methylation diversity within populations
For C. robusta and C. intestinalis, a great proportion of subepiloci was polymorphic, especially for m‐subepiloci (79.03%–92.40% in C. robusta, 81.39%–94.39% in C. intestinalis). The ratios of polymorphic loci were higher than those in other species, such as the tubeworm F. enigmaticus where only 50% of the methylation‐susceptible loci were polymorphic (Ardura et al., 2017). A study on a rare floodplain herb Viola elatior showed that the polymorphism among populations ranged from 22.7% to 61.8% for u‐subepiloci and varied from 30.6% to 61.8% for methylated subepiloci (Schulz, Eckstein, & Durka, 2014). Spontaneous epi‐mutations, which generally occur as the consequence of incorrect replication of the methylation pattern, may be one important reason for the high level of methylation polymorphism, as epi‐mutation rate can be several orders of magnitude higher than that of genetic mutations (Becker et al., 2011; van der Graaf et al., 2015; Hagmann et al., 2015; Schmitz et al., 2011). In addition, previous studies showed that Ciona ascidians are among the most genetically diverse animal investigated to date: The allelic polymorphism across the entire genome was 1.2% in C. robusta and the genomic average synonymous diversity was estimated to 0.05 per site in C. intestinalis (Dehal et al., 2002; Tsagkogeorga, Cahais, & Galtier, 2012). The high genetic variation can cause the absence of restriction enzyme sites, resulting in the high degree of methylated polymorphism.
The high level of intrapopulation methylation diversity can also be seen from the high values of Shannon index. Recent studies showed that environmental stresses could increase random methylation changes, leading to the high methylation divergence among individuals in the same populations (Huang et al., 2017; Morán et al., 2013; Richards et al., 2012). In a salt‐enriched experiment of brown trout (Salmo trutta L.), genomewide methylation changes were triggered after feeding on salt‐enriched diets for 2 days, and the largest methylation differentiation was observed at the fourth day after treatment (Morán et al., 2013). Interestingly, our recent study showed that Ciona ascidians could respond to rapid changing environments in the methylation profile, such as after 1 hr of high temperature exposure or after 3 hr of low salinity challenge, and environmental changes increased intrapopulation methylation variation (Huang et al., 2017). Many studies clearly showed that environment‐induced DNA methylation variation was heritable, thus such a type of DNA methylation variation can be relatively steadily maintained in populations (see the review by Verhoeven et al., 2016). Among Ciona populations collected from a broad geographical scale, multiple methylation features among populations, including the large number of population‐private epiloci, the high variation of polymorphic epiloci, and high values of Shannon index among populations, suggest that local environments may leave detectable methylation signatures at the whole genome level, particularly under the circumstance that Ciona species geographically spread less than one century ago (Zhan et al., 2015).
4.2. Remarkable methylation differentiation among populations
In our study, significant population methylation differentiation was found among populations for both C. robusta and C. intestinalis. The remarkable methylation differentiation can be influenced by various factors, including genetic background, spontaneous epi‐mutations, environmental induction and natural selection, and drift (Richards, 2006; Richards et al., 2017). For C. robusta, our results based on pRDA indicated that part of DNA methylation variation was explained by genetic variation. In addition to separate mechanisms shaping and maintaining genetic and DNA methylation variation, genetic and stable DNA methylation variation can be shaped simultaneously by the same local environmental pressures or neutral processes, leading to the correlation between genetic and methylation patterns (Preite et al., 2015). In the results of pRDA, more than 85% of the observed DNA methylation variation was not explained by available environmental factors and genetic variation, suggesting that the generation and/or maintenance of DNA methylation variation are complex processes and more variables including environmental and molecular ones may participate in such complex processes. More local environmental factors may influence DNA methylation variation at the population level, leading to a high degree of DNA methylation differentiation among populations collected from different environments. In addition, several recent studies suggest substantial genetic impacts on DNA methylation variation (Dubin et al., 2015; Roadmap Epigenetics Consortium et al., 2015). However, the contribution of genetic variation to DNA methylation variation was not as high as expected in this study. Two possible reasons may explain the observed patterns here: (a) genetic information based on 152 microsatellite markers was not enough to reflect the genomewide genetic variation among populations of C. robusta, and (b) two methods for both genetic and methylation analyses did not capture the same set of variation in the genome. These two possible reasons may result in poor correlation between detected genetic and DNA methylation variation in multiple analyses.
DNA methylation variation, at least part of it, can be independent from genetic variation in C. robusta. The direct evidence supporting this view comes from our pRDA analysis. In addition, the lowest population genetic differentiation was observed between two European populations AM (Arenys de Mar) and BL (Blanes), but the epigenetic pairwise ΦPT value of this population pair was much higher than genetic differentiation estimators in both U and M profiles. Although a slight discrepancy of population genetic patterns can be detected when different markers are used to characterize genetic differentiation, the patterns of variation still appeared to be congruent among different types of markers (e.g., Maguire, Peakall, & Saenger, 2002). These two lines of evidence suggest that genetic component alone cannot explain the patterns observed in this study, and environmental factors played a role in shaping DNA methylation differentiation among populations.
The autonomous proportion of DNA methylation variation can be generated either randomly or environmentally induced, and both types can be environment‐related (van der Graaf et al., 2015; Hagmann et al., 2015). In a clonal fish Chrosomus eosneogaeus, random DNA methylation variation has been recorded in natural populations to cope with unpredictably changing environments (Leung et al., 2016). Studies on Arabidopsis thaliana showed that the stochastic changes in environment‐related methylation could be maintained over several generations and the accumulation of DNA methylation variance finally led to differentiation among populations (van der Graaf et al., 2015; Hagmann et al., 2015; Schmitz et al., 2011). Stress‐related methylation differentiation has been documented in experimental populations (Gao et al., 2010; Huang et al., 2017; Morán et al., 2013; Verhoeven et al., 2010). The stability of stress‐related DNA methylation variation varied from several hours to several generations (Herman, Spencer, Donohue, & Sultan, 2014; Huang et al., 2017). If the same advantageous phenotype arose every generation repeatedly by transient methylated modifications, such changes are expected to contribute to the interpopulation methylation differentiation in different environments (Schulz et al., 2014). The environment‐related DNA methylation variation may have an effect to maximize individual fitness to local ecological conditions. Studies of three acroporid corals showed that DNA methylation variation may influence their tolerance to the thermal stress and ocean acidification (Dimond & Roberts, 2016). In wild baboons Papio cynocephalus, a large number of differentially methylated regions were found near metabolism‐related genes, suggesting the epigenetic regulation for catering to different resource availability (Lea et al., 2016). For invasive species, recent works showed the correlation between methylation differentiation and diverse phenotypes in different invaded habitats, suggesting the potential role of DNA methylation variation in shaping different phenotypes to respond to varied environmental stresses (Gao et al., 2010; Richards et al., 2012). Despite that only a small proportion of DNA methylation variation was explained solely by collected environmental factors, fitness effects of some loci may be sufficient to make Ciona populations adapt to local environments. Our previous study also showed that a limited number of CpG loci and/or genes were involved in environmental adaptation (Huang et al., 2017; Pu & Zhan, 2017).
4.3. Environment‐related epiloci
We detected that subepiloci of C. robusta (4.0%) and C. intestinalis (2.4%) significantly correlated with changes of environmental factors by SAM. Similarly, 14 epiloci (4.5%) were identified to significantly correlate with ecoclimatic variables in the study of Orchids Dactylorhiza (Paun et al., 2010). However, the proportion of environmental‐related subepiloci in our study was much less than that in red grouse Lagopus lagopus scotica associated with gastrointestinal parasite load (13.6%; Wenzel & Piertney, 2014). Likewise, 12% of polymorphic epiloci in Spartina alterniflora were found to be correlated with oil exposure (Robertson, Schrey, Shayter, Moss, & Richards, 2017). The different ratios among studies may result from the difference of stresses and degree of stresses. In concert, findings in related studies indicated that environmental factors modulated the establishment and maintenance of methylation modifications to specific stress‐related genes (Paun et al., 2010; Pu & Zhan, 2017). Although epiloci related with temperature and salinity in C. robusta and C. intestinalis were identified, it was still unknown whether these environment‐related epiloci were identical in certain groups responding to temperature or salinity in this study. This comparison cannot be clarified by using fragments acquired from MSAP, because fragments from different species with the same sizes may represent different sequences (Caballero et al., 2008), even they are the products of the same selective primers.
In this study, subepiloci with significant methylation differentiation among populations were identified by BAYESCAN, which may be involved in local adaptation (Foll & Gaggiotti, 2008). The overlap at subepiloci of SAM with BAYESCAN suggests that these environment‐related subepiloci may contribute to the divergence of methylation pattern among populations. This hypothesis was confirmed by another specific study in populations of C. robusta, where we detected the correlation between methylation patterns of key genes and environmental factors (Pu & Zhan, 2017). Significant correlation was observed between methylation levels and water temperature at several CpG sites in heat shock protein 90 (HSP90), while one CpG site in Na+‐K+‐2Cl− cotransporter (NKCC) showed significant association with salinity (Pu & Zhan, 2017). As the technique of MSAP can only produce anonymous fragments with unknown genetic contexts, we cannot identify whether the outliers detected in these two studies play a consistent role in the process of response to environmental changes. All these results demonstrated that varied local environments may have important influence on the methylation patterns in populations. Therefore, we infer that DNA methylation may enhance invaders’ adaptive capacity to different habitats by extending the flexibility of a genotype to respond differentially under variable environmental conditions.
4.4. Technical issues
MSAP is a method based on PCR amplification of selected restriction fragments, which can be affected by both methylation modifications and genetic mutations of restriction sites. Therefore, MSAP data matrices inevitably include part of genetic information. The ambiguous definition of type IV fragments in MSAP makes it difficult to distinguish between genetic and methylation variation. Although the polymorphism level of epiloci and population methylation differentiation may be affected by the mixed genetic variation, the role of local environmental factors in shaping DNA methylation variation can still be inferred from results of pRDA and SAM analyses. In order to reduce the confusion in data interpretation between type II and type III, the “Mixing Scoring 1” approach was used in our study to combine both type II (HPA+/MSP−) and type III (HPA−/MSP+) as methylated epiloci. The use of this strategy resulted in more conservative results but some degree of loss of methylation variation among individuals. As MSAP has its technical disadvantages (Schulz et al., 2013), whole epigenome sequencing‐based methods are needed in future studies to obtain more accurate epigenetic information.
AUTHOR CONTRIBUTIONS
A.Z. conceived the study; P.N. and A.Z. designed the experiment. P.N. conducted the experiments and analyzed the data; S.L., Y.L., W.X., and X.H. did both laboratory and field works; P.N. and A.Z. wrote the manuscript, and all authors contributed to the revisions; all authors reviewed and commented on the manuscript.
DATA ACCESSIBILITY
All raw MSAP data and transformed MSAP data for both species, and raw SSRs data for C. robusta used in this study have been deposited into Dryad (https://doi.org/10.5061/dryad.q8v97).
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (31622011; 31772449) to AZ, and Youth Innovation Promotion Association, Chinese Academy of Science (2018054) to SL.
Ni P, Li S, Lin Y, Xiong W, Huang X, Zhan A. Methylation divergence of invasive Ciona ascidians: Significant population structure and local environmental influence. Ecol Evol. 2018;8:10272–10287. 10.1002/ece3.4504
REFERENCES
- Alonso, C. , Pérez, R. , Bazaga, P. , Medrano, M. , & Herrera, C. M. (2016). MSAP markers and global cytosine methylation in plants: A literature survey and comparative analysis for a wild‐growing species. Molecular Ecology Resources, 16, 80–90. 10.1111/1755-0998.12426 [DOI] [PubMed] [Google Scholar]
- Ardura, A. , Zaiko, A. , Morán, P. , Planes, S. , & Garcia‐Vazquez, E. (2017). Epigenetic signatures of invasive status in populations of marine invertebrates. Scientific Reports, 7, 42193 10.1038/srep42193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, C. , Hagmann, J. , Müller, J. , Koenig, D. , Stegle, O. , Borgwardt, K. , & Weigel, D. (2011). Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature, 480, 245–249. 10.1038/nature10555 [DOI] [PubMed] [Google Scholar]
- Bock, D. G. , Caseys, C. , Cousens, R. D. , Hahn, M. A. , Heredia, S. M. , Hübner, S. , … Rieseberg, L. H. (2015). What we still don't know about invasion genetics. Molecular Ecology, 24, 2277–2297. 10.1111/mec.13032 [DOI] [PubMed] [Google Scholar]
- Bonin, A. , Bellemain, E. , Eidesen, P. B. , Pompanon, F. , Brochmann, C. , & Taberlet, P. (2004). How to track and assess genotyping errors in population genetics studies. Molecular Ecology, 13, 3261–3273. 10.1111/j.1365-294X.2004.02346.x [DOI] [PubMed] [Google Scholar]
- Bossdorf, O. , Richards, C. L. , & Pigliucci, M. (2008). Epigenetics for ecologists. Ecology Letters, 11, 106–115. [DOI] [PubMed] [Google Scholar]
- Bouchemousse, S. , Bishop, J. D. D. , & Viard, F. (2016). Contrasting global genetic patterns in two biologically similar, widespread and invasive Ciona species (Tunicata, Ascidiacea). Scientific Reports, 6, 24875 10.1038/srep24875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti, R. , Gissi, C. , Pennati, R. , Caicci, F. , Gasparini, F. , & Manni, L. (2015). Morphological evidence that the molecularly determined Ciona intestinalis type A and type B are different species: Ciona robusta and Ciona intestinalis . Journal of Zoological Systematics and Evolutionary Research, 53, 186–193. 10.1111/jzs.12101 [DOI] [Google Scholar]
- Caballero, A. , Quesada, H. , & Rolán‐Alvarez, E. (2008). Impact of amplified fragment length polymorphism size homoplasy on the estimation of population genetic diversity and the detection of selective loci. Genetics, 179, 539–554. 10.1534/genetics.107.083246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton, J. T. (1996). Pattern, process, and prediction in marine invasion ecology. Biological Conservation, 78, 97–106. 10.1016/0006-3207(96)00020-1 [DOI] [Google Scholar]
- Carver, C. E. , Mallet, A. L. , & Vercaemer, B. (2006). Biological synopsis of the solitary tunicate Ciona intestinalis. Canadian Manuscript Report of Fisheries and Aquatic Sciences No., 2746, 55.
- Chwedorzewska, K. J. , & Bednarek, P. T. (2012). Genetic and epigenetic variation in a cosmopolitan grass Poa annua from Antarctic and Polish populations. Polish Polar Research, 33(1), 63–80. 10.2478/v10183-012-0004-5 [DOI] [Google Scholar]
- Colautti, R. I. , & Lau, J. A. (2015). Contemporary evolution during invasion: Evidence for differentiation, natural selection, and local adaptation. Molecular Ecology, 24, 1999–2017. 10.1111/mec.13162 [DOI] [PubMed] [Google Scholar]
- Darling, J. A. , Bagley, M. J. , Roman, J. , Tepolt, C. K. , & Geller, J. B. (2008). Genetic patterns across multiple introductions of the globally invasive crab genus Carcinus . Molecular Ecology, 17, 4992–5007. 10.1111/j.1365-294X.2008.03978.x [DOI] [PubMed] [Google Scholar]
- Dehal, P. , Satou, Y. , Campbell, R. K. , Chapman, J. , Degnan, B. , De Tomaso, A. , … Rokhsar, D. S. (2002). The draft genome of Ciona intestinalis: Insights into chordate and vertebrate origins. Science, 298, 2157–2167. 10.1126/science.1080049 [DOI] [PubMed] [Google Scholar]
- Dimond, J. L. , & Roberts, S. B. (2016). Germline DNA methylation in reef corals: Patterns and potential roles in response to environmental change. Molecular Ecology, 25, 1895–1904. 10.1111/mec.13414 [DOI] [PubMed] [Google Scholar]
- Doney, S. C. , Ruckelshaus, M. , Duffy, J. E. , Barry, J. P. , Chan, F. , English, C. A. , … Talley, L. D. (2012). Climate change impacts on marine ecosystems. Annual Review of Marine Science, 4, 11–37. 10.1146/annurev-marine-041911-111611 [DOI] [PubMed] [Google Scholar]
- Dowen, R. H. , Pelizzola, M. , Schmitz, R. J. , Lister, R. , Dowen, J. M. , Nery, J. R. , … Ecker, J. R. (2012). Widespread dynamic DNA methylation in response to biotic stress. Proceedings of the National Academy of Sciences of the United States of America, 109, E2183–E2191. 10.1073/pnas.1209329109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray, S. , Legendre, P. , & Blanchet, G. (2013). packfor: Forward Selection with permutation (Canoco p.46). R package version 0.0‐8/r109. Retrieved from https://R-Forge.R-project.org/projects/sedar/
- Dubin, M. J. , Zhang, P. , Meng, D. , Remigereau, M.‐S. , Osborne, E. J. , Paolo Casale, F. , … Nordborg, M. (2015). DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife, 4, e05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, E. J. , Gluckman, P. D. , & Dearden, P. K. (2014). Epigenetics, plasticity, and evolution: How do we link epigenetic change to phenotype? Journal of Experimental Zoology Part B: Molecular and Developmental Evolution, 322, 208–220. 10.1002/jez.b.22571 [DOI] [PubMed] [Google Scholar]
- Dybern, B. I. (1967). Settlement of sessile animals on eternite slabs in two polls near Bergen. Sarsia, 29, 137–150. 10.1080/00364827.1967.10411074 [DOI] [Google Scholar]
- Excoffier, L. , Smouse, P. E. , & Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics, 131, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll, M. , & Gaggiotti, O. (2008). A genome‐scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics, 180, 977–993. 10.1534/genetics.108.092221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulneček, J. , & Kovařík, A. (2014). How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genetics, 15, 2 10.1186/1471-2156-15-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, L. , Geng, Y. , Li, B. , Chen, J. , & Yang, J. (2010). Genome‐wide DNA methylation alterations of Alternanthera philoxeroides in natural and manipulated habitats: Implications for epigenetic regulation of rapid responses to environmental fluctuation and phenotypic variation. Plant, Cell and Environment, 33, 1820–1827. 10.1111/j.1365-3040.2010.02186.x [DOI] [PubMed] [Google Scholar]
- Gugger, P. F. , Fitz‐Gibbon, S. , Pellegrini, M. , & Sork, V. L. (2016). Species‐wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Molecular Ecology, 25, 1665–1680. 10.1111/mec.13563 [DOI] [PubMed] [Google Scholar]
- Hagmann, J. , Becker, C. , Müller, J. , Stegle, O. , Meyer, R. C. , Wang, G. , … Weigel, D. (2015). Century‐scale methylome stability in a recently diverged Arabidopsis thaliana lineage. PLoS Genetics, 11, e1004920 10.1371/journal.pgen.1004920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes, N. A. , Fidler, A. E. , Tremblay, L. A. , Pochon, X. , Dunphy, B. J. , & Smith, K. F. (2018). Understanding the role of DNA methylation in successful biological invasions: A review. Biological Invasions, 20, 2285–2300. 10.1007/s10530-018-1703-6 [DOI] [Google Scholar]
- Herman, J. J. , Spencer, H. G. , Donohue, K. , & Sultan, S. E. (2014). How stable “should” epigenetic modifications be? Insights from adaptive plasticity and bet hedging. Evolution, 68, 632–643. 10.1111/evo.12324 [DOI] [PubMed] [Google Scholar]
- Huang, X. , Li, S. , Ni, P. , Gao, Y. , Jiang, B. , Zhou, Z. , & Zhan, A. (2017). Rapid response to changing environments during biological invasions: DNA methylation perspectives. Molecular Ecology, 26, 6621–6633. 10.1111/mec.14382 [DOI] [PubMed] [Google Scholar]
- Jablonka, E. (2012). Epigenetic variations in heredity and evolution. Clinical Pharmacology and Therapeutics, 92, 683–688. 10.1038/clpt.2012.158 [DOI] [PubMed] [Google Scholar]
- Jones, P. A. (2012). Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nature Reviews Genetics, 13, F484–F492. 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- Joost, S. , Kalbermatten, M. , & Bonin, A. (2008). Spatial analysis method (SAM): A software tool combining molecular and environmental data to identify candidate loci for selection. Molecular Ecology Resources, 8, 957–960. 10.1111/j.1755-0998.2008.02162.x [DOI] [PubMed] [Google Scholar]
- Lande, R. (2015). Evolution of phenotypic plasticity in colonizing species. Molecular Ecology, 24, 2038–2045. 10.1111/mec.13037 [DOI] [PubMed] [Google Scholar]
- Law, J. A. , & Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics, 11, 204–220. 10.1038/nrg2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea, A. J. , Altmann, J. , Alberts, S. C. , & Tung, J. (2016). Resource base influences genome‐wide DNA methylation levels in wild baboons (Papio cynocephalus). Molecular Ecology, 25, 1681–1696. 10.1111/mec.13436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C. E. (1999). Rapid and repeated invasions of fresh water by the copepod Eurytemora affinis . Evolution, 53, 1423–1434. 10.1111/j.1558-5646.1999.tb05407.x [DOI] [PubMed] [Google Scholar]
- Lee, C. E. (2002). Evolutionary genetics of invasive species. Trends in Ecology & Evolution, 17, 386–391. 10.1016/S0169-5347(02)02554-5 [DOI] [Google Scholar]
- Leung, C. , Breton, S. , & Angers, B. (2016). Facing environmental predictability with different sources of epigenetic variation. Ecology and Evolution, 6, 5234–5245. 10.1002/ece3.2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebl, A. L. , Schrey, A. W. , Richards, C. L. , & Martin, L. B. (2013). Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integrative and Comparative Biology, 53, 351–358. 10.1093/icb/ict007 [DOI] [PubMed] [Google Scholar]
- Lin, Y. , Chen, Y. , Yi, C. , Fong, J. J. , Kim, W. , Rius, M. , & Zhan, A. (2017). Genetic signatures of natural selection in a model invasive ascidian. Scientific Reports, 7, 44080 10.1038/srep44080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. , Gao, Z. , & Zhan, A. (2015). Introduction and use of non‐native species for aquaculture in China: Status, risks and management solutions. Reviews in Aquaculture, 7, 28–58. 10.1111/raq.12052 [DOI] [Google Scholar]
- Maguire, T. L. , Peakall, R. , & Saenger, P. (2002). Comparative analysis of genetic diversity in the mangrove species Avicennia marina (Forsk.) Vierh. (Avicenniaceae) detected by AFLPs and SSRs. Theoretical and Applied Genetics, 104, 388–398. 10.1007/s001220100724 [DOI] [PubMed] [Google Scholar]
- Meirmans, P. G. , & Van Tienderen, P. H. (2004). GENOTYPE and GENODIVE: Two programs for the analysis of genetic diversity of asexual organisms. Molecular Ecology Notes, 4, 792–794. 10.1111/j.1471-8286.2004.00770.x [DOI] [Google Scholar]
- Morán, P. , Marco‐Rius, F. , Megías, M. , Covelo‐Soto, L. , & Pérez‐Figueroa, A. (2013). Environmental induced methylation changes associated with seawater adaptation in brown trout. Aquaculture, 392–395, 77–83. 10.1016/j.aquaculture.2013.02.006 [DOI] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Minchin, P. R. , … Wagner, H. (2017). vegan: Community Ecology Package. R package version 2.4‐2. Retrieved from https://CRAN.R-project.org/package=vegan.
- Paul‐Pont, I. , De Montaudouin, X. , Gonzalez, P. , Soudant, P. , & Baudrimont, M. (2010). How life history contributes to stress response in the Manila clam Ruditapes philippinarum . Environmental Science and Pollution Research, 17, 987–998. 10.1007/s11356-009-0283-5 [DOI] [PubMed] [Google Scholar]
- Paun, O. , Bateman, R. M. , Fay, M. F. , Hedrén, M. , Civeyrel, L. , & Chase, M. W. (2010). Stable epigenetic effects impact adaptation in allopolyploid orchids (Dactylorhiza: Orchidaceae). Molecular Biology and Evolution, 27, 2465–2473. 10.1093/molbev/msq150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall, R. , & Smouse, P. E. (2006). GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6, 288–295. 10.1111/j.1471-8286.2005.01155.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, A. , Gugger, P. F. , Pellegrini, M. , & Sork, V. L. (2015). Genome‐wide signature of local adaptation linked to variable CpG methylation in oak populations. Molecular Ecology, 24, 3823–3830. 10.1111/mec.13230 [DOI] [PubMed] [Google Scholar]
- Preite, V. , Snoek, L. B. , Oplaat, C. , Biere, A. , Van Der Putten, W. H. , & Verhoeven, K. J. F. (2015). The epigenetic footprint of poleward range‐expanding plants in apomictic dandelions. Molecular Ecology, 24, 4406–4418. 10.1111/mec.13329 [DOI] [PubMed] [Google Scholar]
- Prentis, P. J. , Wilson, J. R. U. , Dormontt, E. E. , Richardson, D. M. , & Lowe, A. J. (2008). Adaptive evolution in invasive species. Trends in Plant Science, 13, 288–294. 10.1016/j.tplants.2008.03.004 [DOI] [PubMed] [Google Scholar]
- Pu, C. , & Zhan, A. (2017). Epigenetic divergence of key genes associated with water temperature and salinity in a highly invasive model ascidian. Biological Invasions, 19, 2015–2028. 10.1007/s10530-017-1409-1 [DOI] [Google Scholar]
- R Core Team (2016). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; Retrieved from https://www.R-project.org/. [Google Scholar]
- Rapp, R. A. , & Wendel, J. F. (2005). Epigenetics and plant evolution. New Phytologist, 168, 81–91. 10.1111/j.1469-8137.2005.01491.x [DOI] [PubMed] [Google Scholar]
- Richards, E. J. (2006). Inherited epigenetic variation—revisiting soft inheritance. Nature Reviews Genetics, 7, 395–401. 10.1038/nrg1834 [DOI] [PubMed] [Google Scholar]
- Richards, E. J. (2008). Population epigenetics. Current Opinion in Genetics & Development, 18, 221–226. 10.1016/j.gde.2008.01.014 [DOI] [PubMed] [Google Scholar]
- Richards, C. L. , Alonso, C. , Becker, C. , Bossdorf, O. , Bucher, E. , Colome‐Tatche, M. , … Verhoeven, K. J. F. (2017). Ecological plant epigenetics: Evidence from model and non‐model species, and the way forward. Ecology Letters, 10.1101/ele.12858 [DOI] [PubMed] [Google Scholar]
- Richards, C. L. , Schrey, A. W. , & Pigliucci, M. (2012). Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecology Letters, 15, 1016–1025. 10.1111/j.1461-0248.2012.01824.x [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium , Kundaje, A. , Meuleman, W. , Ernst, J. , Bilenky, M. , Yen, A. , … Kellis, M. (2015). Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–329. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, M. , Schrey, A. , Shayter, A. , Moss, C. J. , & Richards, C. (2017). Genetic and epigenetic variation in Spartina alterniflora following the deepwater horizon oil spill. Evolutionary Applications, 10, 792–801. 10.1111/eva.12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz, R. J. , Schultz, M. D. , Lewsey, M. G. , O'Malley, R. C. , Urich, M. A. , Libiger, O. , … Ecker, J. R. (2011). Transgenerational epigenetic instability is a source of novel methylation variants. Science, 334, 369–373. 10.1126/science.1212959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz, B. , Eckstein, R. L. , & Durka, W. (2013). Scoring and analysis of methylation‐sensitive amplification polymorphisms for epigenetic population studies. Molecular Ecology Resources, 13, 642–653. 10.1111/1755-0998.12100 [DOI] [PubMed] [Google Scholar]
- Schulz, B. , Eckstein, R. L. , & Durka, W. (2014). Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Molecular Ecology, 23, 3523–3537. 10.1111/mec.12835 [DOI] [PubMed] [Google Scholar]
- Seo, K. , & Lee, Y. (2009). A First Assessment of Invasive Marine Species on Chinese and Korean Coasts In Rilov G., & Crooks J. A. (Eds.), Biological invasions in marine ecosystems, Vol. 204 (pp. 577–585). Berlin, Heidelberg: Springer; 10.1007/978-3-540-79236-9 [DOI] [Google Scholar]
- Sherman, C. D. H. , Lotterhos, K. E. , Richardson, M. F. , Tepolt, C. K. , Rollins, L. A. , Palumbi, S. R. , & Miller, A. D. (2016). What are we missing about marine invasions? Filling in the gaps with evolutionary genomics. Marine Biology, 2016(163), 198 10.1007/s00227-016-2961-4 [DOI] [Google Scholar]
- Shine, R. (2012). Invasive species as drivers of evolutionary change: Cane toads in tropical Australia. Evolutionary Applications, 5, 107–116. 10.1111/j.1752-4571.2011.00201.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorte, C. J. B. , Jones, S. J. , & Miller, L. P. (2011). Geographic variation in temperature tolerance as an indicator of potential population responses to climate change. Journal of Experimental Marine Biology and Ecology, 400, 209–217. 10.1016/j.jembe.2011.02.009 [DOI] [Google Scholar]
- Stachowicz, J. J. , Terwin, J. R. , Whitlatch, R. B. , & Osman, R. W. (2002). Linking climate change and biological invasions: Ocean warming facilitates nonindigenous species invasions. Proceedings of the National Academy of Sciences of the United States of America, 99, 15497–15500. 10.1073/pnas.242437499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M. M. , & Bird, A. (2008). DNA methylation landscapes: Provocative insights from epigenomics. Nature Reviews Genetics, 9, 465–476. 10.1038/nrg2341 [DOI] [PubMed] [Google Scholar]
- Therriault, T. W. , & Herborg, L. (2008). Predicting the potential distribution of the vase tunicate Ciona intestinalis in Canadian waters: Informing a risk assessment. ICES Journal of Marine Science, 65, 788–794. 10.1093/icesjms/fsn054 [DOI] [Google Scholar]
- Tsagkogeorga, G. , Cahais, V. , & Galtier, N. (2012). The population genomics of a fast evolver: High levels of diversity, functional constraint, and molecular adaptation in the tunicate Ciona intestinalis . Genome Biology and Evolution, 4, 852–861. 10.1093/gbe/evs054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Graaf, A. , Wardenaar, R. , Neumann, D. A. , Taudt, A. , Shaw, R. G. , Jansen, R. C. , … Johannes, F. (2015). Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proceedings of the National Academy of Sciences of the United States of America, 112, 6676–6681. 10.1073/pnas.1424254112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven, K. J. F. , Jansen, J. J. , van Dijk, P. J. , & Biere, A. (2010). Stress‐induced DNA methylation changes and their heritability in asexual dandelions. New Phytologist, 185, 1108–1118. 10.1111/j.1469-8137.2009.03121.x [DOI] [PubMed] [Google Scholar]
- Verhoeven, K. J. F. , VonHoldt, B. M. , & Sork, V. L. (2016). Epigenetics in ecology and evolution: What we know and what we need to know. Molecular Ecology, 25, 1631–1638. 10.1111/mec.13617 [DOI] [PubMed] [Google Scholar]
- Wang, S. , Lv, J. , Zhang, L. , Dou, J. , Sun, Y. , Li, X. , … Bao, Z. (2015). MethylRAD : A simple and scalable method for genome‐wide DNA methylation profiling using methylation‐ dependent restriction enzymes. Open Biology, 5, 150130 10.1098/rsob.150130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, J. M. , Dijkstra, L. H. , & Wallis, G. P. (2000). Biogeography of a southern hemisphere freshwater fish: How important is marine dispersal? Molecular Ecology, 9, 1815–1821. 10.1046/j.1365-294x.2000.01082.x [DOI] [PubMed] [Google Scholar]
- Wenzel, M. A. , & Piertney, S. B. (2014). Fine‐scale population epigenetic structure in relation to gastrointestinal parasite load in red grouse (Lagopus lagopus scotica). Molecular Ecology, 23, 4256–4273. 10.1111/mec.12833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich, A. , Lenz, D. , Jeschek, M. , Chung, T. H. , Rübensam, K. , Göritz, F. , … Fickel, J. (2016). Paternal intergenerational epigenetic response to heat exposure in male Wild guinea pigs. Molecular Ecology, 25, 1729–1740. 10.1111/mec.13494 [DOI] [PubMed] [Google Scholar]
- Zhan, A. , Briski, E. , Bock, D. G. , Ghabooli, S. , & Macisaac, H. J. (2015). Ascidians as models for studying invasion success. Marine Biology, 2015(162). [Google Scholar]
- Zhan, A. , Darling, J. A. , Bock, D. G. , Lacoursiére‐Roussel, A. , MacIsaac, H. J. , & Cristescu, M. E. (2012). Complex genetic patterns in closely related colonizing invasive species. Ecology and Evolution, 2, 1331–1346. 10.1002/ece3.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, A. , Macisaac, H. J. , & Cristescu, M. E. (2010). Invasion genetics of the Ciona intestinalis species complex: From regional endemism to global homogeneity. Molecular Ecology, 19, 4678–4694. 10.1111/j.1365-294X.2010.04837.x [DOI] [PubMed] [Google Scholar]
- Zhan, A. , Ni, P. , Xiong, W. , Chen, Y. , Lin, Y. , Huang, X. , … Gao, Y. (2017). Biological invasions in aquatic ecosystems in China In Fanghao W., Mingxing J., … Aibin Z. (Eds.), Biological invasions and its management in China (pp. 67–96). Dordrecht, The Netherlands: Springer; 10.1007/978-94-024-0948-2 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw MSAP data and transformed MSAP data for both species, and raw SSRs data for C. robusta used in this study have been deposited into Dryad (https://doi.org/10.5061/dryad.q8v97).