

Abstract
Rodent models of traumatic brain injury (TBI) reproduce secondary injury sequela and cognitive impairments observed in patients afflicted by a TBI. Impaired neurotransmission has been reported in the weeks following experimental TBI, and may be a contributor to behavioral dysfunction. The soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, the machinery facilitating vesicular docking and fusion, is a highly-conserved mechanism important for neurotransmission. Following TBI, there is a reduction in both the formation of the SNARE complex and the abundance of multiple SNARE proteins, including the chaperone protein cysteine string protein α (CSPα). Treatment with lithium in naïve rats reportedly increases the expression of CSPα. In the context of TBI, brain-injured rats treated with lithium exhibit improved outcome in published reports, but the mechanisms underlying the improvement are poorly understood. The current study evaluated the effect of lithium administration on the abundance of SNARE proteins and SNARE complex formation, hemispheric tissue loss, and neurobehavioral performance following controlled cortical impact (CCI). Sprague Dawley rats were subjected to CCI or sham injury, and treated daily with lithium chloride or vehicle for up to 14 days. Administration of lithium after TBI modestly improved spatial memory at 14 days post-injury. Semi-quantitative immunoblot analysis of hippocampal lysates revealed that treatment with lithium attenuated reductions in key SNARE proteins and SNARE complex formation at multiple time points post-injury. These findings highlight that treatment with lithium increased the abundance of synaptic proteins that facilitate neurotransmission and may contribute to improved cognitive function after TBI.
Keywords: Traumatic brain injury, Synapse, SNARE, Hippocampus, Cognition, Vesicle
1. Introduction
Numerous clinical and experimental studies demonstrate that TBI results in impaired function, which can persist years after the injury (Dixon et al., 1999; Levin, 1998; Lundin et al., 2006; Pierce et al., 1998; Truelle et al., 2010). In recapitulating pathology observed in TBI patients, the primary injury, and cascade of secondary injury mechanisms can produce cellular dysfunction and loss, impair neurotransmission, and ultimately, lead to the manifestation of motor and cognitive dysfunction in experimental models of TBI (Bramlett et al., 2016; Dietrich et al., 1994; Dixon et al., 1996, 1999; Gao et al., 2011; Hall et al., 2008; Shear et al., 2010, 2016; Smith et al., 1995; Titus et al., 2013). Impairments in neurotransmitter release after experimental TBI have been reported in multiple brain regions, including the cortex, hippocampus and striatum (Dixon et al., 1995a,b, 1996, 1997; Hinzman et al., 2012; Shin et al., 2011; Wagner et al., 2009) and can contribute to the onset of neurobehavioral dysfunction.
Impairments in neurotransmitter release may be attributed to alterations in intrasynaptic machinery necessary for vesicular translocation and docking at the synaptic membrane (Sollner et al., 1993; Sudhof and Rizo, 2011). Formation of the highly-conserved soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex is important for synaptic vesicle fusion with the plasma membrane, and the subsequent neurotransmitter release into the synaptic cleft. Multiple studies utilizing genetically modified animals highlight the relationship between reductions in SNARE proteins and impaired SNARE complex formation, altered intrasynaptic vesicular properties, and reduced neurotransmission (Burre et al., 2010; Chandra et al., 2005; Greten-Harrison et al., 2010; Janezic et al., 2013; Schoch et al., 2001; Vardar et al., 2016; Washbourne et al., 2002). Cysteine string protein alpha (CSPα) and alpha synuclein (α-synuclein) have been shown to be important proteins critical for the formation of the SNARE complex (Burre et al., 2010; Chandra et al., 2005; Sharma et al., 2011). We previously reported TBI reduced the hippocampal abundance of multiple SNARE protein monomers and SNARE complexes in a time-dependent and protein-dependent manner (Carlson et al., 2016). Additionally, ultrastructural analysis of hippocampal synapses at 1 week post-injury, indicted changes in SNARE proteins, including CSPα, occurred concurrently with reductions in synaptic vesicle density and altered intrasynaptic vesicular distribution (Carlson et al., 2016). CSPα was reduced within one day after TBI and preceded reductions in SNARE complex formation. A therapeutic strategy aimed to promote SNARE complex formation could contribute to functional recovery following TBI.
Lithium has been widely used as a treatment for psychiatric illnesses, but the mechanism underlying the therapeutic effect is not fully understood. Previous reports demonstrate treatment with lithium can increase the expression of CSPα in vitro and in vivo (Cordeiro et al., 2000, 2003). Considering TBI reduces CSPα abundance, treatment with lithium may promote expression of CSPα and, potentially, stabilize or restore SNARE protein abundance and SNARE complex formation in the injured brain. Lithium attenuates motor and cognitive impairments after TBI, but the mechanisms promoting this recovery are poorly understood (Dash et al., 2011; Yu et al., 2012, 2013; Zhu et al., 2010). This study is the first to evaluate the effect of lithium treatment on SNARE protein and SNARE complex abundance.
In this study, we examined the effect of daily lithium treatment on motor and cognitive function, overt cell loss, and SNARE protein abundance during the two weeks following TBI. This time course was selected based upon our previous work showing that TBI impairs SNARE complex formation during this post-injury window (Carlson et al., 2016). Treatment with lithium produced modest improvements in spatial memory, but did not improve acute motor performance or reduce overt cell loss. The current findings highlight that treatment with lithium increases the abundance of multiple SNARE proteins, including CSPα and α-synuclein, and promotes SNARE complex formation at multiple time points after injury. These findings provide the first evidence that post-injury treatment with lithium increases the abundance of SNARE proteins and promotes SNARE complex formation in the hippocampus following TBI.
2. Materials and methods
2.1. Animals
All experimental procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee in accordance with the guidelines established by the National Institutes of Health in the Guide for the Care and Use of Laboratory Animals. Animals were housed up to 2 rats per cage in the University of Pittsburgh vivarium with a 12:12 light/dark photoperiod (light on at 7:00 am) and provided food and water ad libitum. Adult male Sprague Dawley rats (Harlan, Indianapolis, IN) aged 10–14 weeks weighing 275–375 g were utilized in this study.
2.2. Controlled cortical impact
Rats were anesthetized using 4% isoflurane with a 2:1 N2O/O2 mixture in a ventilated anesthesia chamber. Following endotracheal intubation, the rats were mechanically ventilated with a 2% isoflurane mixture. Animals were placed in a stereotaxic frame in the supine position and body temperature was monitored by rectal thermistor probe and maintained at 37 °C with a heating pad. Following a midline incision, the soft tissues were reflected. A 7 mm craniotomy was completed between bregma and lambda and centered 5 mm lateral of the sagittal suture to expose the dura mater over the right parietal cortex. Animals were randomly assigned to receive either sham or controlled cortical impact (CCI) injury. Sham animals were subjected to all surgical procedures except the induction of the CCI injury. The CCI injury was completed using a small bore (1.975 cm) double-acting stroked-constrained pneumatic cylinder with a 5.0 cm stroke. A beveled impactor tip (6 mm in diameter) was set to produce a tissue deformation of 2.7 mm at a velocity of 4 m/s with a dwell time of 150 msec. Following sham or CCI injury, the scalp was sutured, anesthesia stopped, and the righting time of each animal was monitored and recorded. Once ambulatory, the animals were returned to their home cage.
2.3. Administration of Lithium
A dose of 1 mmol/ml/kg lithium chloride was selected based upon published reports (Dash et al., 2011; Yu et al., 2012; Zhu et al., 2010). Treatment with lithium chloride (Sigma, St. Louis, MO) or vehicle (U.S.P. saline) was initiated at 5 min post-injury by intraperitoneal injection, and continued daily until time of euthanasia at 1, 3, 7, or 14 days post-injury. For assessment of neurobehavioral function only, a dose of 1.5 mmol/ml/kg was evaluated in sham animals to ensure that a higher concentration of lithium was not toxic or impair neurobehavioral performance.
2.4. Vestibular motor function
A total of 66 rats (n = 16–17 per group) were used to evaluate vestibulomotor function. Beam balance and beam walking assessments were completed on days 1–5 post-injury with training completed on day 0 prior to injury. Gross vestibulomotor function was evaluated using a beam balance task in which the animal was placed on a suspended, narrow wooden beam (1.5 cm width) 30 in. above a padded surface. The latency on the beam, up to 60 s, was measured. Three trials per day per animal were performed with a 30 s rest period between trials. The modified beam walking task evaluated finer components of vestibulomotor function and coordination. On the training day, prior to injury, the animal was trained to escape a loud white noise and bright light by traversing the narrow wooden beam (2.5 cm × 100 cm) to enter a darkened goal box at the opposite end. Four pegs (4 cm high and 3 mm diameter) were equally spaced along the center of the beam to increase task difficulty. If the rats fell off the beam or did not cross in the allotted 60 s, the noise and light were stopped and the rat was placed in the goal box. Average latency to traverse the beam for three trials per day was measured.
2.5. Spatial learning
The rats subjected to vestibulomotor testing were also evaluated for spatial learning and retention of spatial memory. Spatial learning was evaluated using the Morris water maze task using a video-tracking system (AnyMaze, Stoelting, Inc. Wood Dale, IL) as previously described (Dixon et al., 2016). A circular tank (180 cm diameter and 45 cm high) was filled with 26 ± 1 °C water to a height of 30 cm to conceal a transparent circular platform (10 cm diameter and 29 cm high) located in a fixed position in the tank at a distance of 45 cm from the wall. Visual cues outside of the maze aided the rat in locating the escape platform. Animals underwent testing beginning on day 10 post-injury, without previous training or exposure to the MWM. Testing continued for 4 consecutive days (10–13 days post-injury) with each animal completing 4 trials per day. Rats were randomly placed in the water against the tank wall, and released to freely swim in the tank to find the hidden platform, for a maximum of 120 s. If the animal was unable to locate the platform within the designated time, it was manually directed to the platform. The animal remained on the platform for 30 s and then was placed into an incubator between trials. Following a 4 min intertrial interval, the animal was returned to the maze for the next trial. On day 14 post-injury, the animal was tested using a probe trial paradigm in which the hidden platform was removed. The location of the hidden platform was designated as the target quadrant. The number of entries, latency to first entry, and distance to first entry in the target quadrant were assessed.
2.6. Assessment of cortical contusion volume
A total of 24 randomly selected rats (n = 6 per group) received an overdose of Fatalplus (intraperitoneally, 100 mg/kg sodium pentobarbital) at 14 days post-injury following assessment of spatial memory. Animals were transcardially perfused with saline, followed by a mixture of 10% neutral-buffered formalin (Fisher Scientific, Waltham, MA). The brains were processed, embedded in paraffin, cut into 5 μm thick coronal sections at every 1 mm and stained with hematoxylin and eosin. Brain hemispheric tissue loss was calculated by measuring ipsilateral and contralateral hemispheric areas of each slice, then multiplying the slice areas by the slice interval thickness and summing all slices, as previously described (Dixon et al., 2016). Ipsilateral tissue loss is expressed as percent contralateral and calculated as follows: (contralateral-ipsilateral)/contralateral × 100.
2.7. Tissue preparation for Western blot analysis
A total of 96 rats (n = 6 per group at each time point) received an overdose of Fatal-plus (100 mg/kg sodium pentoparbital) and the brains dissected at 1, 3, 7, or 14 days post-injury. In the current study, we expanded upon previous findings (Carlson et al., 2016) to include a 3 day time point to gain a better understanding of potential acute changes after CCI. Animals subjected to behavioral testing were not utilized for immunoblotting in order to eliminate possible confounds of behavioral testing on protein abundance. After removal of the brain, the ipsilateral hippocampus was rapidly dissected on a chilled ice plate, immediately snap frozen in liquid nitrogen and stored at −80 °C. Hippocampal samples were homogenized in lysis buffer (0.1 M NaCl, 0.01 M Tris-Cl (pH 7.6), 0.001 M EDTA) with protease inhibitor cocktail kit (Pierce, Rockford, IL). The homogenized whole cell samples were centrifuged at 12,000 ×g at 4 °C for 10 min and the supernatants collected. The protein concentration was determined by a BCA protein assay kit (Thermo Scientific, Pittsburgh, PA) using a 96-well microplate reader (Biotek, Winooski, VT).
2.8. Immunoblotting
To assess the abundance of monomeric SNARE proteins in the hippocampus, whole cell lysates were boiled for 10 min prior to SDS-PAGE. SNARE complex abundance was assessed in unboiled protein samples to retain high molecular weight complexes and were defined as SNAP-25 immunoreactive material >50 kDa that was absent from boiled samples as previously described (Carlson et al., 2016; Chandra et al., 2005). Protein samples (20 μg) and molecular weight markers (Bio-Rad, Hercules, CA) were separated using SDS-PAGE. The resolved proteins were electrophoretically transferred to a PVDF membrane (Invitrogen, Carlsbad, CA). The blots were incubated in 5% nonfat dry milk in tris-buffered saline (blocking solution) for 1 h. Primary antibodies were diluted in blocking solution and incubated overnight at 4 °C. The primary antibodies utilized were: rabbit anti-CSPα (Thermo-Fisher, Rockford, IL), mouse anti-SNAP-25 (Biolegend, San Diego, CA), mouse anti-syntaxin-1 (Sigma-Aldrich, St. Louis, MO), rabbit anti-VAMP2 (Millipore, Temecula, CA), mouse anti-α-synuclein (BD Transduction, San Jose, CA), rabbit anti-synaptophysin (Abcam, Cambridge, MA), and mouse anti-β-actin (Sigma-Aldrich). The following day, the membranes were washed with 1× TBS buffer, incubated in blocking solution containing directly conjugated horse radish peroxidase secondary antibodies for 1 h. Proteins were visualized using a chemiluminescence detection system (Supersignal, Pierce). To normalize for protein content, the membranes were stripped and reprobed with mouse anti-β actin monoclonal antibody (Sigma-Aldrich). Sham and CCI-injured samples for both treatment groups were loaded in the same gel for comparison. Blots were imaged with the Chemidoc Imager (BioRad). Optical density of each protein of interest was measured using ImageJ (National Institutes of Health). The optical density of β-actin was measured for each lane and used for normalization for each sample. Values are presented as the ratio of optical densities of samples as a percentage of sham-injured vehicle-treatment for each time point. Data are expressed as the group means ± standard error of the mean (SEM).
2.9. Statistical analyses
Data are presented as mean ± standard error of the mean (SEM). Statistical comparisons of beam balance latency and beam walking latency were completed using a repeated measures one-way analyses of variance (ANOVA) followed by Tukey's multiple comparison tests, when appropriate. Statistical comparison of spatial learning was completed using a two-way ANOVA, followed by Tukey's multiple comparison tests, when appropriate. Probe trial metrics were compared using a one-way ANOVA followed by Tukey's multiple comparison tests, when appropriate. Immunoblot data were compared at each time point using a one-way ANOVA followed by Tukey's multiple comparison test, when appropriate. The p values reported in the text are the results of the post-hoc analyses, unless stated. Statistical tests were completed using Graphpad (Graphpad, La Jolla, CA). A p value < 0.05 was considered statistically significant for all tests.
3. Results
3.1. Lithium modestly improves cognitive performance, but does not attenuate motor impairment or hemispheric tissue loss after TBI
We first examined the efficacy of lithium to improve neurobehavioral function after CCI. Vestibulomotor function was evaluated by beam balance and beam walking tasks on days 1–5 post-injury. In both tasks, CCI + Veh and CCI + Li rats exhibited significant impairments in the duration of beam balancing and beam walking latencies during the testing period, compared to Sham + Veh and Sham + Li rats (p < 0.05; Fig. 1A, B). Treatment with lithium did not improve motor function in either task during the testing period compared to treatment with vehicle.
Fig. 1.
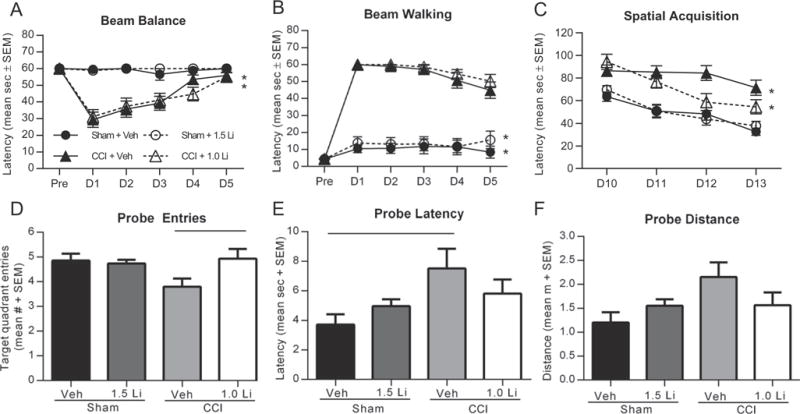
Administration of lithium modestly attenuates cognitive impairments after controlled cortical impact (CCI). (A,B) Assessment of vestibulomotor function with the beam balance and beam walking tasks on days 1–5 post-injury revealed significant impairments after CCI in beam latencies as compared to sham injury (*p < 0.05). Treatment with lithium did not improve motor function in either task. (C) CCI injury resulted in significant increases in spatial acquisition latency times in the Morris water maze task on days 10–13 post-injury compared to sham injury, independent of treatment status (*p < 0.01). Treatment with lithium after CCI resulted in a modest reduction in latency time compared to treatment with vehicle, but it did not reach significance. (D) In the probe trial on day 14 post-injury, assessment of entries into target platform revealed a significant increase in CCI + Li rats compared to CCI + Veh rats (p < 0.05). (E) Assessment of latency to target quadrant entry revealed a significant increase in time for CCI + Veh rats to locate the target quadrant, compared to Sham + Veh rats (p < 0.05). (F) Assessment of distance to first quadrant entry showed a modest increase in swim distance to first target quadrant entry for CCI + Veh rats compared to sham-injured rats, but it did not reach significance. (n = 14–17 per group).
Spatial acquisition was completed on days 10–13 using the Morris water maze. CCI injury resulted in significantly increased latencies to the submerged platform compared to sham injury, independent of treatment status (p < 0.01; Fig. 1C). Retention of spatial memory was assessed using multiple metrics of the probe trial completed on day 14 post-injury. Analysis of the number of target quadrant entries revealed a significant increase in CCI + 1.0 Li rats compared to CCI + Veh rats (p < 0.05; Fig. 1D). Analysis of latency to first entry in target quadrant revealed a significant increase in CCI + Veh rats, compared to Sham + Veh rats (p < 0.05; Fig. 1E). Analysis of distance to first entry in target quadrant showed CCI + Veh rats exhibited a modest increase in distance compared to sham injured rats, but did not reach significance (main ANOVA effect p = 0.056; Fig. 1F).
Previous studies report lithium treatment after experimental TBI reduces cortical cell loss in mice (Yu et al., 2012; Zhu et al., 2010). Hemispheric tissue loss was assessed as a measure of overt cell loss. CCI produced a significant increase in hemispheric tissue loss, in both treatment groups, compared to both sham groups (p < 0.001; Fig. 2A, B). Treatment with lithium after CCI did not attenuate tissue loss.
Fig. 2.
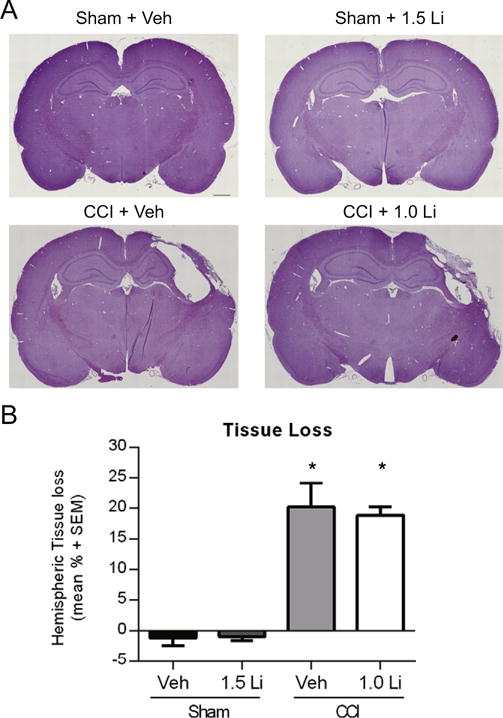
Administration of lithium did not attenuate hemispheric tissue loss. (A) Representative images of hematoxylin and eosin stained coronal brain sections after sham and controlled cortical impact (CCI). (A) CCI produced pronounced tissue loss at the epicenter of injury at 14 days post-injury. Treatment with lithium did not qualitatively reduce the extent of cortical cell loss. Scale bar equals 1 mm. (B) Assessment of hemispheric tissue loss showed that CCI + Veh and CCI + Li rats exhibited a significant increase in tissue loss, compared to Sham + Veh and Sham + Li rats (p < 0.001). Treatment with lithium did not attenuate tissue loss at 14 days post-injury. (n = 6 per group).
3.2. Lithium increases the abundance of CSPα and α-synuclein after TBI
We sought to first examine the effect of lithium on the SNARE chaperone protein CSPα. CCI + Veh rats exhibited a significant reduction in CSPα (34 kDa) abundance at each time point, compared to Sham + Veh rats and Sham + Li rats at 7 and 14 days post-injury (p < 0.05; Fig. 3A). Treatment with lithium delayed the reduction in CSPα after CCI, as CCI + Li rats exhibited significantly lower abundance of CSPα at only 7 and 14 days post-injury, compared to Sham + Veh rats (p < 0.05). Assessment of α-synuclein (18 kDa) revealed a significant reduction in Sham + Li rats at 1 day, compared to Sham + Veh rats (p < 0.05, Fig. 3B). CCI-injured rats, independent of treatment, exhibited significantly reduced α-synuclein abundance at 1, 3, and 7 days post-injury compared to Sham + Veh rats (p < 0.01). At 3 and 7 days post-injury, CCI + Li rats showed significantly increased α-synuclein abundance compared to CCI + Veh rats (p < 0.01).
Fig. 3.
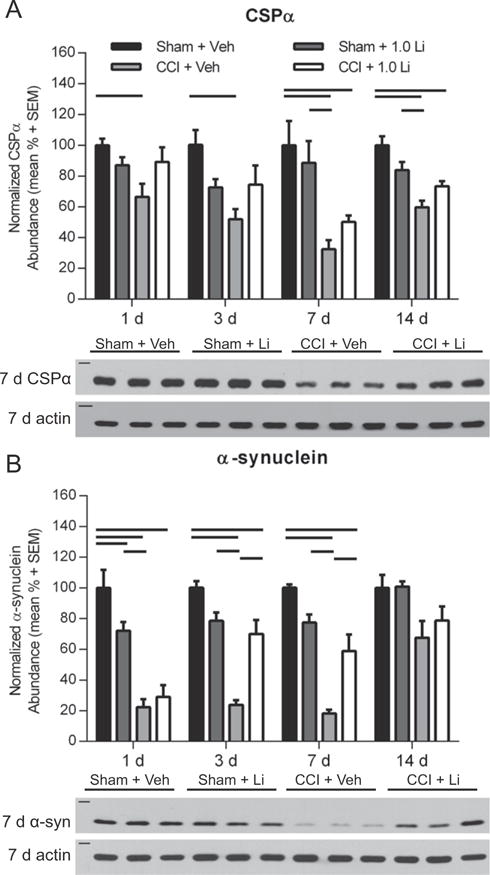
Administration of lithium increased CSPα and α-synuclein abundance in the hippocampus after controlled cortical impact (CCI). (A) Representative Western blot image of CSPα in the hippocampus at 7 days post-injury (n = 3 per group shown). Assessment of CSPα (34 kDa, marker is 37 kDa) revealed that CCI + Veh rats exhibited a significantly lower abundance at 1, 3, 7, and 14 days post-injury, compared to Sham + Veh rats and at 7 and 14 days post-injury compared to Sham + Li rats (p < 0.05). CCI + Li rats exhibited significantly decreased CSPα abundance at 7 and14 days post-injury compared to Sham + Veh rats (p < 0.05). (B) Representative Western blot image of α-synuclein (α-syn; 18 kDa, marker is 20 kDa) at 7 days post-injury (n = 3 per group shown). Sham + Li rats exhibited significantly reduced α-synuclein at 1 day post-injury (p < 0.01). At 1, 3 and 7 days post-injury, α-synuclein abundance was significantly reduced in both CCI + Veh and CCI + Li rats compared to Sham + Veh rats (p < 0.01). At 3 and 7 days post-injury, CCI + Veh rats showed reduced α-synuclein abundance compared to Sham + Li rats (p < 0.01) and CCI + Li rats (p < 0.01). At 14 days post-injury, the abundance of α-synuclein in CCI-injured rats were not significantly different from sham rats, independent of treatment. Proteins of interest were normalized to actin (42 kDa, marker is 50 kDa). (n = 6 sham and n = 6 CCI-injured per treatment group per time point).
3.3. Lithium increases the abundance of monomeric SNAP-25 and syntaxin-1 after TBI
We next examined the effect of lithium treatment on the abundance of SNAP-25 and syntaxin-1 monomers. Comparison of Sham + Veh and Sham + Li rats revealed a significant difference in SNAP-25 (25 kDa) monomer abundance at 3 days post-injury (p < 0.05; Fig. 4A). The abundance of SNAP-25 in CCI + Veh rats was significantly reduced at 1, 3, 7 and 14 days post-injury compared to Sham + Veh rats (p < 0.05) and was significantly reduced at 7 days post-injury compared to Sham + Li rats (p < 0.05). CCI + Li rats exhibited reduced SNAP-25 levels at 3 days post-injury compared to Sham + Veh rats (p < 0.05). At 7 days post-injury, CCI + Li rats showed significantly increased SNAP-25 monomer abundance compared to CCI + Veh rats (p < 0.05). The abundance of SNAP-25 monomers in CCI + Li rats was not significantly different from Sham + Veh and Sham + Li rats at 1, 7 and 14 days post-injury. Assessment of syntaxin-1 (35 kDa) monomers revealed that CCI + Veh rats exhibited significantly reduced levels at 3, 7, and 14 days post-injury as compared to Sham + Veh rats (p < 0.05) and at 3 and 7 days post-injury compared to Sham + Li rats (p < 0.05; Fig. 4B). The abundance of syntaxin-1 in CCI + Li rats was significantly reduced at 3 and 7 days post-injury compared to Sham + Veh rats (p < 0.05). CCI + Li rats exhibited significantly increased syntaxin-1 abundance compared to CCI + Veh rats at 14 days post-injury (p < 0.05).
Fig. 4.
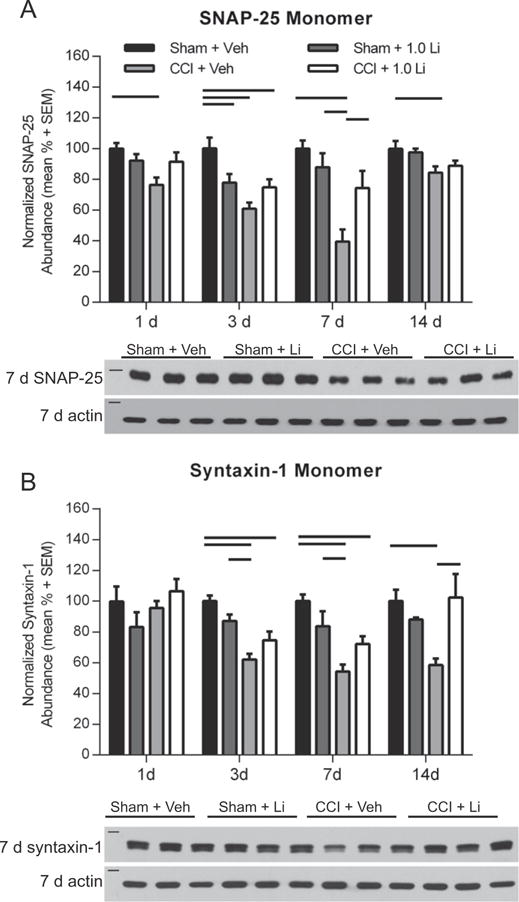
Administration of lithium increased monomeric SNAP-25 and syntaxin-1 abundance in the hippocampus after controlled cortical impact (CCI). (A) Representative Western blot image of SNAP-25 monomers in the hippocampus at 7 days post-injury (n = 3 per group shown). Assessment of SNAP-25 (25 kDa, marker is 25 kDa) revealed that Sham + Li rats exhibited significantly lower abundance compared to Sham + Veh rats at 3 days post-injury (p < 0.05). CCI + Veh rats showed significantly lower abundance of SNAP-25 at 1, 3, 7 and 14 days post-injury, compared to Sham + Veh rats (p < 0.05) and at 7 days post-injury compared to Sham + Li rats (p < 0.05). CCI + Li rats exhibited reduced SNAP-25 abundance compared to Sham + Veh rats at 3 days post-injury (p < 0.05). CCI + LI rats exhibited increased SNAP-25 abundance compared to CCI + Veh rats at 7 days post-injury (p < 0.05). (B) Representative Western blot image of syntaxin-1 monomers (35 kDa, marker is 37 kDa) at 7 days post-injury (n = 3 per group shown). Assessment of syntaxin-1 revealed no difference between groups at 1 day post-injury. CCI + Veh rats exhibited significantly lower abundance at 3, 7 and 14 days post-injury, compared to Sham + Veh rats and at 3 and 7 days post-injury compared to Sham + Li rats (p < 0.05). CCI + LI rats showed reduced syntaxin-1 levels compared to Sham + Veh rats at 3 and 7 days post-injury (p < 0.05). At 14 days post-injury, CCI + LI rats had increased syntaxin-1 abundance compared to CCI + Veh rats (p < 0.05). Proteins of interest were normalized to actin (42 kDa, marker is 50 kDa). (n = 6 sham and n = 6 CCI-injured per treatment group per time point).
3.4. Lithium increases the abundance of VAMP2 and synaptophysin after TBI
Assessment of VAMP2 (18 kDa) revealed a significant difference between Sham + Li rats and Sham + Veh rats at 7 days post-injury (p < 0.05; Fig. 5A). The abundance of VAMP2 in CCI + Veh rats was significantly reduced at 3, 7 and 14 days post-injury compared to Sham + Veh rats (p < 0.05), and at 7 and 14 days compared to Sham + Li rats (p < 0.01). CCI + Li rats exhibited significantly reduced VAMP2 levels at 1 and 3 days post-injury compared to Sham + Veh rats (p < 0.05). Treatment with lithium after CCI significantly increased VAMP2 abundance at 7 and 14 days post-injury, compared to vehicle treatment after CCI (p < 0.05). The abundance of the presynaptic protein synaptophysin (35 kDa) was assessed as an indication of relative synaptic density. At 3, 7 and 14 days post-injury, the abundance of synaptophysin in CCI + Veh rats was significantly reduced compared to Sham + Veh rats (p < 0.05; Fig. 5B), and at 7 and 14 days post-injury compared to Sham + Li rats (p < 0.01). The abundance of synaptophysin in CCI + Li rats was significantly reduced at 7 days post-injury compared to Sham + Veh rats (p < 0.05). Treatment with lithium after CCI significantly increased synaptophysin levels compared to vehicle treatment after CCI at 3, 7 and 14 days post-injury.
Fig. 5.
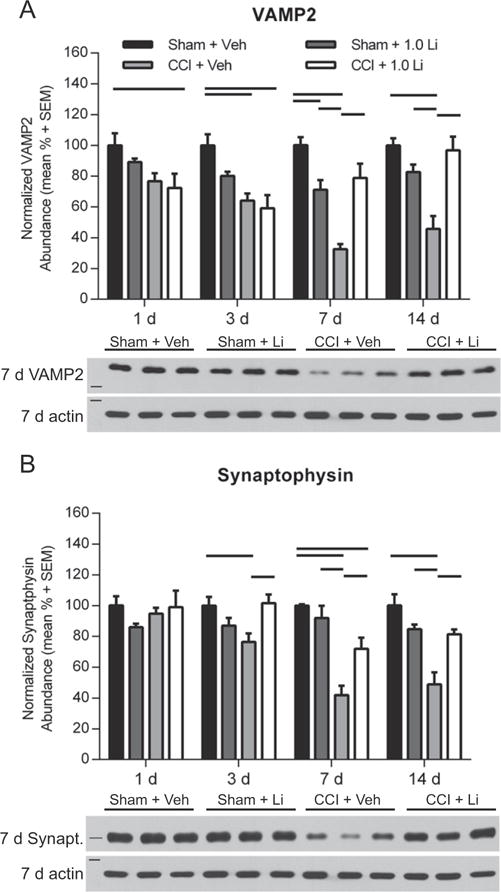
Administration of lithium increased VAMP2 and synaptophysin abundance in the hippocampus after controlled cortical impact (CCI). (A) Representative Western blot image of VAMP2 (18 kDa, marker is 15 kDa) in the hippocampus at 7 days post-injury (n = 3 per group shown). Sham + Li rats exhibited reduced VAMP2 abundance compared to Sham + Veh rats at 7 days post-injury (p < 0.05). CCI + Veh rats exhibited significantly reduced VAMP2 abundance compared to Sham + Veh rats at 3, 7 and 14 days post-injury (p < 0.05) and Sham + Li rats at 7 and 14 days post-injury (p < 0.01). CCI + Li rats showed reduced VAMP2 abundance compared to Sham + Veh rats at 1 and 3 days post-injury (p < 0.05). CCI + Li rats had increased VAMP2 levels compared to CCI + Veh at 7 and 14 days post-injury. (B) Representative Western blot image of synaptophysin (synapt.; 35 kDa, marker is 37 kDa) at 7 days post-injury (n = 3 per group shown). Assessment of synaptophysin revealed no differences between groups at 1 day post-injury. CCI + Veh rats exhibited significantly lower abundance at 3, 7 and 14 days post-injury, compared to Sham + Veh and at 7 and 14 days post-injury compared Sham + Li rats (p < 0.05). CCI + Li rats exhibited reduced synaptophysin levels compared to Sham + Veh rats at 7 days post-injury (p < 0.05). CCI + Li rats had increased synaptophysin levels compared to CCI + Veh rats at 3, 7 and 14 days post-injury (p < 0.05) Proteins of interest were normalized to actin (42 kDa, marker is 50 kDa). (n = 6 sham and n = 6 CCI-injured per treatment group per time point).
3.5. Lithium attenuates SNARE complex reductions after TBI
Lastly, we examined the effect of lithium treatment on SNARE complex abundance by assessing high-molecular weight SNAP-25 immunoreactivity in unboiled lysates. SNAP-25 complex abundance in CCI-injured animals, independent of treatment, showed a modest reduction at 3 days post-injury, but it did not reach significance (Fig. 6A). Analysis of SNAP-25 complexes revealed a significant reduction in CCI + Veh rats at 7 days post-injury compared to Sham + Veh and Sham + Li rats (p < 0.05) and at 14 days post-injury compared to Sham + Veh rats (p < 0.01; Fig. 6A). The abundance of SNAP-25 complexes in CCI + Li rats was not significantly different from sham animals at any time point assessed.
Fig. 6.
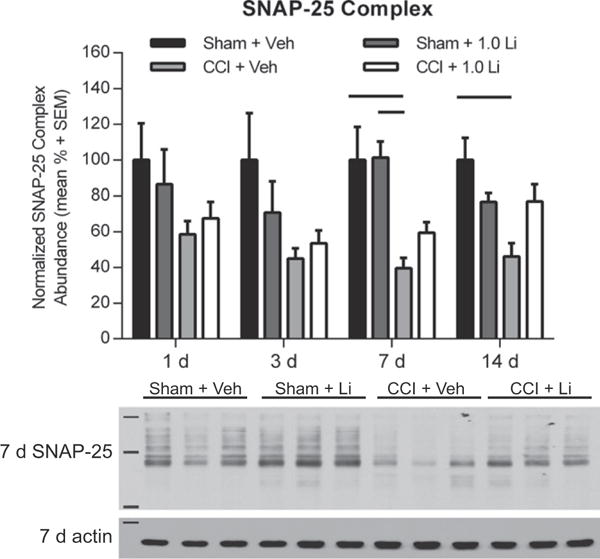
Administration of lithium increased SNAP-25 complex formation in the hippocampus after controlled cortical impact (CCI). (A) Representative Western blot image of SNAP-25 complexes (markers are 37, 75, and 250 kDa) in the hippocampus at 7 days post-injury s (n = 3 per group shown). Assessment of SNAP-25 complexes revealed no differences between groups at 1 and 3 days post-injury. CCI + Veh rats exhibited significantly reduced SNAP-25 complexes compared to Sham + Veh and Sham + Li rats at 7 days (p < 0.05) and at 14 days post-injury, compared to Sham + Veh rats (p < 0.01). Proteins of interest were normalized to actin (42 kDa, marker is 50 kDa). (n = 6 sham and n = 6 CCI-injured per treatment group per time point).
4. Discussion
The current study evaluated the effect of lithium on SNARE protein abundance, overt cell loss, and neurobehavioral function following TBI. We demonstrate for the first time that lithium treatment increased SNARE protein abundance and enhanced SNARE complex formation in the hippocampus at multiple time points after TBI. These findings suggest a novel mechanism by which lithium may promote recovery in the injured brain.
Treatment with lithium improves motor performance in the weeks following TBI, but the magnitude of recovery is injury severity dependent. After CCI, lithium-treated mice exhibited injury severity dependent improvements in beam walking task performance at 7, 14, and 21 days post-injury and reduced efficacy with increased depth of injury (Yu et al. 2012, 2013). However, in a rat model of CCI, daily treatment with lithium (1 mmol/kg/d) did not improve acute motor function (Dash et al., 2011). In the current study, daily treatment with lithium (1 mmol/kg/d) did not improve acute motor function using tasks previously shown to be sensitive to therapeutic interventions after CCI (Dixon et al., 2016).
Lithium administration improves cognitive function after TBI. In a mouse model of CCI, daily injections of 1 mmol/kg/d lithium improved spatial acquisition and memory 14–16 days post-injury (Zhu et al., 2010). In a rat model of CCI, daily treatment of lithium for 5 days improved spatial acquisition performance on days 22–24 and spatial memory on day 25 post-injury. In the current study, daily treatment with lithium (1 mmol/kg/d) did not improve spatial acquisition performance, but modestly improved spatial memory at 14 days following TBI. While these findings were not as robust as previous reports, the data corroborate previous studies showing lithium improves cognitive performance after TBI.
Lithium's mood-stabilizing effects are well characterized in psychiatric disorders, but debate persists about the actions of lithium in the context of neurodegeneration. Lithium can attenuate cell loss through neuroprotective signaling cascades, increase the expression of BDNF, and attenuate oxidative stress (Fukumoto et al., 2001; Jacobsen and Mork, 2004; Kim et al., 2011; Son et al., 2003; Wada et al., 2005). Inhibition of GSK-3beta and increased Wnt signaling is protective against ischemic injury, Alzheimer's disease, tauopathies, Huntington's disease, and Parkinson's disease (De Ferrari et al., 2003; Fiorentini et al., 2010; Noble et al., 2005; Wei et al., 2001; Youdim and Arraf, 2004). Lithium treatment after TBI yields severity and species dependent reductions in contusion volume and hippocampal cell loss (Dash et al., 2011; Yu et al., 2012, 2013; Zhu et al., 2010). We observed no hemispheric tissue loss attenuation, which aligns with previous findings at 28 days post-injury (Dash et al. 2011). In mice subjected to CCI, lithium administration did not reduce lesion volume at 3 weeks post-injury; however, increased lithium concentrations reduced lesion volume (Yu et al., 2012). In this study, lithium did not reduce hemispheric tissue loss which highlights that the beneficial effects of lithium after TBI are not solely attributed to neuroprotection.
The current data show lithium treatment increases the abundance of synaptic proteins important for neurotransmission. Lithium increases the abundance of CSPα mRNA and protein in the naïve rat brain, but not SNAP-25, syntaxin 33 and 35, α-synuclein, or synaptophysin abundance (Cordeiro et al., 2000, 2003; Scarr and Dean, 2012). Increasing CSPα expression after TBI is important for restoring SNARE complex formation, as targeted knockout of CSPα impairs SNARE complex formation (Chandra et al., 2005; Sharma et al., 2011). However, it was unknown if lithium could increase the abundance of CSPα and additional SNARE proteins in the context of TBI.
TBI reduces the abundance of the highly conserved SNARE complex in the hippocampus at 1 and 2 weeks post-injury (Carlson et al., 2016). Additionally, TBI altered the levels of multiple SNARE proteins, including CSPα after TBI (Carlson et al., 2016). The importance of CSPα and α-synuclein in SNARE complex formation is highlighted by CSPα knockout mice that exhibit impaired SNARE complex formation (Burre et al., 2010; Chandra et al., 2005; Sharma et al., 2011), and that supplementation of α-synuclein restored SNARE complex formation (Chandra et al., 2005). We corroborate and expand upon our previous findings by showing vehicle treatment reduces CSPα abundance at 1, 3, 7, and 14 days after CCI. Compared with vehicle treatment, lithium treatment resulted in increased CSPα abundance at 1 and 3 days after CCI. Lithium may delay the loss of CSPα, as it was reduced at 7 and 14 days post-injury. Interestingly, treatment with lithium had a robust effect on α-synuclein abundance at 3 and 7 days post-injury, compared to vehicle treatment after CCI. It is plausible that recovery of CSPα and α-synuclein together after lithium treatment promote SNARE complex formation after TBI. In naïve rats, lithium supplemented chow provided for 4 weeks did not influence α-synuclein abundance in cortical homogenates (Scarr and Dean, 2012); however, this contradiction could be attributed to the method of lithium administration, duration of lithium treatment, and differential actions of lithium in the injured brain compared to the naïve brain. The current findings highlight that lithium treatment increases hippocampal CSPα and α-synuclein after TBI.
Reductions in monomeric SNARE proteins impair SNARE complex formation (Burre et al., 2010; Chandra et al., 2005; Sharma et al., 2011; Vardar et al., 2016) and alter synaptic function in cultured neurons and hippocampal slice cultures (Burre et al., 2010; Washbourne et al., 2002). Ablation of SNAP-25 directly impairs depolarization-stimulated exocytosis in cultured neurons (Washbourne et al., 2002), highlighting the link between SNARE monomeric proteins and neurotransmission. Following vehicle treatment, monomeric SNAP25 was reduced at all time points, and syntaxin-1 and VAMP2 were reduced at 3, 7 and 14 days post-injury. Treatment with lithium attenuated the loss of these proteins at 1 and 2 weeks post-injury, time points in which lithium increased SNARE complex abundance.
We replicated previous findings (Carlson et al., 2016) that CCI significantly impairs SNARE complex formation at 1 and 2 weeks post-injury. While it did not reach significance, CCI injury produced a modest reduction in SNARE complex formation at 3 days post-injury. Treatment with lithium attenuated TBI-induced reductions in SNARE complex abundance at 1 and 2 week post-injury. Considering SNARE complex formation is an important process in neurotransmitter release, increased SNARE complex formation may improve neurotransmission in the injured brain.
An argument could be made that SNARE protein reductions are reflective of synaptic loss. Ultrastructural quantification showed maximal hippocampal synaptic loss at 2 days and approached 50% recovery by 2 weeks post-injury (Scheff et al., 2005), highlighting the dynamic process of synaptic loss and plasticity after TBI. While we cannot disregard the influence of synaptic loss, the varying time courses of individual SNARE protein reductions suggest these findings are not solely reflective of post-traumatic synaptic loss. If this was the case, maximal SNARE protein loss would be expected at 3 days post-injury. No protein assessed exhibited maximal reductions at 3 days post-injury. These observations suggest that changes in SNARE protein abundances could also be attributed to additional mechanisms, such as degradation, impaired axonal transport or reduced expression after TBI. Hippocampal synapses exhibit altered synaptic vesicle distribution at 1 week post-injury (Carlson et al., 2016). It is also plausible that increased SNARE protein levels after lithium treatment could be reflective of increased vesicular packing density in the injured synapse. Conditions of stress have been shown to alter intrasynaptic vesicular packing density (Magarinos et al., 1997), but the effect of lithium treatment after TBI on packing density is unknown. Defining the precise mechanism by which lithium increases SNARE protein and SNARE complex abundance is not possible in the current study. It is plausible lithium-mediated increases in CSPα or α-synuclein can stabilize SNARE protein levels and improve SNARE complex formation or may promote the expression of multiple SNARE proteins in the injured brain. Future studies are warranted to understand the mechanisms by which TBI reduces SNARE protein abundance and lithium promotes SNARE complex formation to optimize therapeutic strategies to promote synaptic function in the injured brain.
In summary, administration of lithium increases the abundance of SNARE proteins and promotes SNARE complex formation after TBI. These findings highlight that treatment with lithium increases SNARE complex formation after TBI, which may improve synaptic function in the injured brain. This study provides novel evidence that lithium improves formation of the synaptic SNARE complex which may improve synaptic and cognitive function after TBI.
Abbreviations
- α-synuclein α-syn
alpha synuclein
- CCI
controlled cortical impact
- CSPα
cysteine string protein α
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- TBI
traumatic brain injury
Footnotes
Funding: This work was supported by the National Institutes of Health [NIH-NS40125 (CED), NIH-NS060672 (CED), 1-F32NS090748 (SWC)] the VA Healthcare System [VAI01RX001127 (CED)] and The Walter L. Copeland Fund of The Pittsburgh Foundation [UN2014-73618 (SWC)].
References
- Bramlett HM, Dietrich WD, Dixon CE, Shear DA, Schmid KE, Mondello S, Wang KK, Hayes RL, Povlishock JT, Tortella FC, Kochanek PM. Erythropoietin treatment in traumatic brain injury: operation brain trauma therapy. J Neurotrauma. 2016;33:538–552. doi: 10.1089/neu.2015.4116. [DOI] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SW, Yan H, Ma M, Li Y, Henchir J, Dixon CE. Traumatic brain injury impairs soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex formation and alters synaptic vesicle distribution in the hippocampus. J Neurotrauma. 2016;33:113–121. doi: 10.1089/neu.2014.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Cordeiro ML, Umbach JA, Gundersen CB. Lithium ions enhance cysteine string protein gene expression in vivo and in vitro. J Neurochem. 2000;74:2365–2372. doi: 10.1046/j.1471-4159.2000.0742365.x. [DOI] [PubMed] [Google Scholar]
- Cordeiro ML, Gundersen CB, Umbach JA. Dietary lithium induces regional increases of mRNA encoding cysteine string protein in rat brain. J Neurosci Res. 2003;73:865–869. doi: 10.1002/jnr.10707. [DOI] [PubMed] [Google Scholar]
- Dash PK, Johnson D, Clark J, Orsi SA, Zhang M, Zhao J, Grill RJ, Moore AN, Pati S. Involvement of the glycogen synthase kinase-3 signaling pathway in TBI pathology and neurocognitive outcome. PLoS One. 2011;6:e24648. doi: 10.1371/journal.pone.0024648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J Neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Bao J, Johnson KM, Yang K, Whitson J, Clifton GL, Hayes RL. Basal and scopolamine-evoked release of hippocampal acetylcholine following traumatic brain injury in rats. Neurosci Lett. 1995a;198:111–114. doi: 10.1016/0304-3940(95)11979-7. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Liu SJ, Jenkins LW, Bhattachargee M, Whitson JS, Yang K, Hayes RL. Time course of increased vulnerability of cholinergic neurotransmission following traumatic brain injury in the rat. Behav Brain Res. 1995b;70:125–131. doi: 10.1016/0166-4328(95)80002-6. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Bao J, Long DA, Hayes RL. Reduced evoked release of acetylcholine in the rodent hippocampus following traumatic brain injury. Pharmacol Biochem Behav. 1996;53:679–686. doi: 10.1016/0091-3057(95)02069-1. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Ma X, Marion DW. Reduced evoked release of acetylcholine in the rodent neocortex following traumatic brain injury. Brain Res. 1997;749:127–130. doi: 10.1016/s0006-8993(96)01310-8. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, DeKosky ST. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16:109–122. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Bramlett HM, Dietrich WD, Shear DA, Yan HQ, Deng-Bryant Y, Mondello S, Wang KK, Hayes RL, Empey PE, Povlishock JT, Tortella FC, Kochanek PM. Cyclosporine treatment in traumatic brain injury: operation brain trauma therapy. J Neurotrauma. 2016;33:553–566. doi: 10.1089/neu.2015.4122. [DOI] [PubMed] [Google Scholar]
- Fiorentini A, Rosi MC, Grossi C, Luccarini I, Casamenti F. Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. PLoS One. 2010;5:e14382. doi: 10.1371/journal.pone.0014382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology. 2001;158:100–106. doi: 10.1007/s002130100871. [DOI] [PubMed] [Google Scholar]
- Gao X, Deng P, Xu ZC, Chen J. Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS One. 2011;6:e24566. doi: 10.1371/journal.pone.0024566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS. Alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma. 2008;25:235–247. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Quintero JE, Gerhardt GA, Lifshitz J. Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury. J Neurotrauma. 2012;29:1197–1208. doi: 10.1089/neu.2011.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JP, Mork A. The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain-derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5-HT and 5-HIAA levels. Brain Res. 2004;1024:183–192. doi: 10.1016/j.brainres.2004.07.065. [DOI] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B, Deltheil T, Kosillo P, Cioroch M, Wagner K, Ansorge O, Bannerman DM, Bolam JP, Magill PJ, Cragg SJ, Wade-Martins R. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A. 2013;110:E4016–E4025. doi: 10.1073/pnas.1309143110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Rane A, Lussier S, Andersen JK. Lithium protects against oxidative stress-mediated cell death in alpha-synuclein-overexpressing in vitro and in vivo models of Parkinson's disease. J Neurosci Res. 2011;89:1666–1675. doi: 10.1002/jnr.22700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HS. Cognitive function outcomes after traumatic brain injury. Curr Opin Neurol. 1998;11:643–646. doi: 10.1097/00019052-199812000-00006. [DOI] [PubMed] [Google Scholar]
- Lundin A, de Boussard C, Edman G, Borg J. Symptoms and disability until 3 months after mild TBI. Brain Inj. 2006;20:799–806. doi: 10.1080/02699050600744327. [DOI] [PubMed] [Google Scholar]
- Magarinos AM, Verdugo JM, McEwen BS. Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci U S A. 1997;94:14002–14008. doi: 10.1073/pnas.94.25.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- Scarr E, Dean B. Altered neuronal markers following treatment with mood stabilizer and antipsychotic drugs indicate an increased likelihood of neurotransmitter release. Clin Psychopharmacol Neurosci. 2012;10:25–33. doi: 10.9758/cpn.2012.10.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Sharma M, Burre J, Sudhof TC. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol. 2011;13:30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- Shear DA, Lu XC, Bombard MC, Pedersen R, Chen Z, Davis A, Tortella FC. Longitudinal characterization of motor and cognitive deficits in a model of penetrating ballistic-like brain injury. J Neurotrauma. 2010;27:1911–1923. doi: 10.1089/neu.2010.1399. [DOI] [PubMed] [Google Scholar]
- Shear DA, Dixon CE, Bramlett HM, Mondello S, Dietrich WD, Deng-Bryant Y, Schmid KE, Wang KK, Hayes RL, Povlishock JT, Kochanek PM, Tortella FC. Nicotinamide treatment in traumatic brain injury: operation brain trauma therapy. J Neurotrauma. 2016;33:523–537. doi: 10.1089/neu.2015.4115. [DOI] [PubMed] [Google Scholar]
- Shin SS, Bray ER, Zhang CQ, Dixon CE. Traumatic brain injury reduces striatal tyrosine hydroxylase activity and potassium-evoked dopamine release in rats. Brain Res. 2011;1369:208–215. doi: 10.1016/j.brainres.2010.10.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12:169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Son H, Yu IT, Hwang SJ, Kim JS, Lee SH, Lee YS, Kaang BK. Lithium enhances long-term potentiation independently of hippocampal neurogenesis in the rat dentate gyrus. J Neurochem. 2003;85:872–881. doi: 10.1046/j.1471-4159.2003.01725.x. [DOI] [PubMed] [Google Scholar]
- Sudhof TC, Rizo J. Synaptic vesicle exocytosis. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus DJ, Furones C, Kang Y, Atkins CM. Age-dependent alterations in cAMP signaling contribute to synaptic plasticity deficits following traumatic brain injury. Neuroscience. 2013;231:182–194. doi: 10.1016/j.neuroscience.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truelle JL, Koskinen S, Hawthorne G, Sarajuuri J, Formisano R, Von Wild K, Neugebauer E, Wilson L, Gibbons H, Powell J, Bullinger M, Hofer S, Maas A, Zitnay G, Von Steinbuechel N, Qolibri Task F. Quality of life after traumatic brain injury: the clinical use of the QOLIBRI, a novel disease-specific instrument. Brain Inj. 2010;24:1272–1291. doi: 10.3109/02699052.2010.506865. [DOI] [PubMed] [Google Scholar]
- Vardar G, Chang S, Arancillo M, Wu YJ, Trimbuch T, Rosenmund C. Distinct functions of syntaxin-1 in neuronal maintenance, synaptic vesicle docking, and fusion in mouse neurons. J Neurosci. 2016;36:7911–7924. doi: 10.1523/JNEUROSCI.1314-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A, Yokoo H, Yanagita T, Kobayashi H. Lithium: potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. J Pharmacol Sci. 2005;99:307–321. doi: 10.1254/jphs.crj05009x. [DOI] [PubMed] [Google Scholar]
- Wagner AK, Drewencki LL, Chen X, Santos FR, Khan AS, Harun R, Torres GE, Michael AC, Dixon CE. Chronic methylphenidate treatment enhances striatal dopamine neurotransmission after experimental traumatic brain injury. J Neurochem. 2009;108:986–997. doi: 10.1111/j.1471-4159.2008.05840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- Wei H, Qin ZH, Senatorov VV, Wei W, Wang Y, Qian Y, Chuang DM. Lithium suppresses excitotoxicity-induced striatal lesions in a rat model of Huntington's disease. Neuroscience. 2001;106:603–612. doi: 10.1016/s0306-4522(01)00311-6. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Arraf Z. Prevention of MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic neurotoxicity in mice by chronic lithium: involvements of Bcl-2 and Bax. Neuropharmacology. 2004;46:1130–1140. doi: 10.1016/j.neuropharm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Yu F, Wang Z, Tchantchou F, Chiu CT, Zhang Y, Chuang DM. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J Neurotrauma. 2012;29:362–374. doi: 10.1089/neu.2011.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Wang Z, Tanaka M, Chiu CT, Leeds P, Zhang Y, Chuang DM. Posttrauma cotreatment with lithium and valproate: reduction of lesion volume, attenuation of blood-brain barrier disruption, and improvement in motor coordination in mice with traumatic brain injury. J Neurosurg. 2013;119:766–773. doi: 10.3171/2013.6.JNS13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu ZF, Wang QG, Han BJ, William CP. Neuroprotective effect and cognitive outcome of chronic lithium on traumatic brain injury in mice. Brain Res Bull. 2010;83:272–277. doi: 10.1016/j.brainresbull.2010.07.008. [DOI] [PubMed] [Google Scholar]