

Abstract
Background:
To determine if variations exist in the KSHV host receptor EPHA2’s coding region that affect KSHV infectivity and/or KS prevalence among South African HIV-infected patients.
Methods:
A retrospective candidate gene association study was performed on 150 patients which were randomly selected from a total of 756 HIV-infected patients and grouped according to their KS status and KSHV serodiagnosis; namely group 1: KS+/KSHV+; group 2: KS−/KSHV+; group 3: KS−/KSHV−. Peripheral blood DNA was used to extract DNA and PCR amplify and sequence the entire EPHA2 coding region, which was compared to the NCBI reference through multiple alignment.
Results:
100% (95% CI 92.9-100%) of the KS positive patients, and 31.6% (95% CI 28.3-35.1%) of the KS negative patients were found to be KSHV seropositive. Aggregate variation across the entire EPHA2 coding region identified an association with KS (OR=6.6 (95% CI 2.8, 15.9), p=2.2×10−5). This was primarily driven by variation in the functionally important protein tyrosine kinase domain (Pkinase-Tyr; OR=4.9 (95% CI 1.9, 12.4), p=0.001) and the sterile-α-motif (SAM; OR=13.8 (95% CI 1.7, 111.6), p=0.014). Mutation analysis revealed two novel, non-synonymous heterozygous variants (c.2254T>C: OR undefined, adj. p=0.02; and c.2990G>T: OR undefined, adj. p=0.04) in Pkinase-Tyr and SAM, respectively, to be statistically associated with KS; and a novel heterozygous transition (c.2727C>T: OR=6.4 (95% CI 1.4, 28.4), adj. p = 0.03) in Pkinase-Tyr to be statistically associated with KSHV.
Conclusions:
Variations in the KSHV entry receptor gene EPHA2 affected susceptibility to KSHV infection and KS development in a South African HIV-infected patient cohort.
Keywords: HIV, Kaposi’s sarcoma, Kaposi’s sarcoma-Associated Herpesvirus, Receptor EPHA2, epidemiology, South Africa
1. Introduction
Kaposi’s sarcoma (KS1) is an Acquired Immunodeficiency Syndrome (AIDS)-defining disease, the prevalence of which has massively escalated since the advent of the Human Immunodeficiency Virus (HIV) epidemic. Currently, KS is the most common AIDS-related malignancy worldwide and of particular significance in sub-Saharan Africa where KS is one of the most common human malignancies with an estimated 37,214 new cases in 2012 [1–4]. The etiological agent of KS is the oncogenic Kaposi’s sarcoma-Associated Herpes Virus (KSHV) (also named human herpesvirus-8 (HHV-8)), which is mainly transmitted via saliva [5]. KSHV seroprevalence is particularly high in Southern Africa with some studies indicating >50% [3,6–8]. Although KSHV is necessary for KS development, it is not sufficient for oncogenesis. Precipitating factors, such as HIV-related immune suppression (i.e. low CD4 count), promote the oncogenic transformation of spindle cells which is driven by the expression of viral oncoproteins encoded by KSHV following infection of endothelial cells [9]. Beside the enormous impact of HIV infection, several other risk factors have been proposed to play a role in the progression to KS in KSHV-infected individuals, such as environmental influences (alumino-silicate rich volcanic soils), parasites, diet, herbs, and drugs (antimalarials) [10,11]. However, none of these risk factors have been proven to date to directly influence KS progression. While HIV co-infection remains the most important trigger of KS development, and although both KSHV and HIV prevalence is exceptionally high in sub-Saharan Africa, not all co-infected patients develop KS, pointing to a potential underlying genetic predisposition [3,6,12–16].
While genetic association studies so far have focused on immune-modulatory genes [17–21], the Eph Receptor A2 protein (EPHA2) tyrosine kinase receptor is a promising candidate for investigation of KSHV-induced KS as it potentially acts on two levels, namely susceptibility to KSHV infection, and susceptibility to KS development.
EPHA2 has recently been identified as a host receptor utilised by KSHV for entry into and trafficking within endothelial cells [22,23]. Additionally, EPHA2 has been implicated in oncogenesis: EPHA2 is upregulated on the mRNA and protein level in a wide variety of cancer cell lines and tissues, and EPHA2 signalling has been implicated in cell transformation, tumour maintenance and progression, angiogenesis and metastasis [22,24–28]. Although vital in the uptake mechanism of KSHV and significantly involved in oncogenesis, little is known about the pathological consequences of sequence variants in the EPHA2 gene on KSHV infection and/or KS development. To date, polymorphisms within EPHA2 have only been identified in association with cataract pathogenesis [29–34]. Therefore, this study aimed to investigate potential sequence variants in the protein coding region of the EPHA2 gene in South African HIV-infected patients. We hypothesised that if such variants exist and translate into amino acid changes, particularly in known functional domains, they may predispose affected individuals to KSHV infection and/or KS oncogenesis. Here we report that aggregate variation (having ≥1 rare variant with Minor Allele Frequency (MAF) < 5%) from the reference sequence (NM_004431) across the entire EPHA2 coding region, particularly driven by variation within the functionally important and conserved protein tyrosine kinase (Pkinase-Tyr) and sterile-α-motif (SAM) domains, is associated with susceptibility to KS. Moreover, we found that three novel, non-synonymous variants in the Pkinase-Tyr and SAM domains are associated with KSHV infection (c.2727C>T) or KS development (c.2254T>C and c.2990G>T).
2. Materials and methods
2.1. Study participants
This case-control study included the following three groups of participants (n=50 per group) that were randomly sampled from a larger study of 756 participants based on KS diagnosis and KSHV serology status: group 1: KS+/KSHV+; group 2: KS−/KSHV+; group 3: KS−/KSHV−. Total sample size was determined based on a priori sample size calculation as a function of the desired power (= 0.8), α error probability (= 0.05), estimated MAF (=0.271, based on the genotypic frequency of a previously reported EPHA2 SNP (rs66786160, [30]) in the 1000 Genomes African population), and estimated odds ratio as a proxy for effect size (=2, based on ORs reported in previous KS and KSHV genetic association studies [17–21]) for Fisher’s exact tests [17–21,30]. As this was an exploratory study of a candidate gene, we did not take correction for multiple testing into account at this stage but did apply this correction post hoc and reported p values as both unadjusted and adjusted. Both male and female HIV-infected patients from any Sub-Saharan African population group residing in the Western Cape, South Africa, who were over 18 years old, were enrolled in this study. Patients with KS were recruited from the Radiation Oncology Unit at Groote Schuur Hospital, where they were receiving treatment for KS. Patients without a clinical diagnosis of KS were recruited from the Infectious Diseases Unit at Groote Schuur Hospital and from Khayelitsha Hospital in the Western Cape, South Africa. All patients had a thorough clinical examination by experienced clinicians to document the occurrence of typical KS cutaneous lesions, mucosal lesions and lymphoedema. When indicated, skin biopsy and chest X-ray supported the diagnosis of KS. Demographic information including sex, age and population group was recorded in addition to clinical information from patient records including HIV status, latest CD4 count and ART treatment status.
2.2. Ethics statement
This study was conducted according to the Declaration of Helsinki, with the informed consent of each participant; all protocols were approved by the Human Research Ethics Committee, Health Sciences Faculty, University of Cape Town (Approval HERC/REF: 136/2013, 057/2013 and 729/2014).
2.3. Serology testing
KSHV serostatus was determined by ELISAs coated with recombinant ORF73 and K8.1 antigens using plasma isolated from patient blood samples. According to previously determined assay specifications, the cut off OD value for the K8.1 ELISA was calculated as “mean of negative controls +0.95”, while the cut off OD value for the ORF73 ELISA was calculated as “mean of negative controls + 0.35” [35]. As a proportion of discordant results were expected because seroconversion to different KSHV antigens is documented to vary amongst individuals [36], participants were considered KSHV seropositive if antibodies to either antigen were detected, as this analytical strategy yields 100% sensitivity and 95.8% specificity [35].
2.4. Mutation analysis
EPHA2 is encoded by a 31,773 base pair gene located on chromosome 1p36 (NCBI Accession number NG_021396) consisting of 17 exons, interspersed with large intronic regions, making up a number of conserved domains [37]. Gene-specific primers used for PCR were designed in order to flank the exons of the coding region of EPHA2, based on previous studies [29,33]. Genomic DNA extracted from patient blood samples (50ng per 25μl reaction) was amplified (35 cycles) in a GeneAmp® 2700 thermal cycler (Applied Biosystems) using the EPHA2 gene-specific primers (0.2 μM) and FastStart Taq DNA polymerase (Roche, 1U). PCR amplicons were visualised on 1% agarose gels to exclude non-specific PCR products, before purification and dideoxy sequencing in both directions with the gene-specific primers used for PCR (Stellenbosch Central Analytical Facility, South Africa). Computational processing of the sequence data was performed on the University of Cape Town Information and Communication Technology Services High Performing Computing Cluster using bioinformatics programmes encompassed in the European Molecular Biology Open Software Suite (EMBOSS) [38], followed by utilizing ClustalW2 multiple alignment (EMBL) [39] to compare the sequences to the EPHA2 reference sequence (NM_004431.3). DNA sequence variants that were predicted to be non-synonymous through in silico translation were further assessed for predicted functional consequences using the PolyPhen-2 prediction tool [40].
2.5. Statistical analysis
Statistical testing of demographic data included Fisher’s exact tests for categorical variables: sex, population group and ART status; and two-way ANOVA for continuous variables: age and CD4 count using GraphPad Prism Version 5.00. Confidence intervals for proportion data were calculated using the Wilson method. Statistical analysis for association of variants with KSHV seroconversion and KS prevalence was restricted to variants with MAF > 3%, due to the statistical power our sample size allowed. MAFs were calculated within the test groups of participants based on 150 participants for the analysis of KSHV susceptibility (100 KSHV+ patients of group 1 and 2 versus 50 KSHV− patients of group 3) and 100 participants for the analysis of KS prevalence (50 KS+ (KSHV+) patients of group 1 versus 50 KS− (KSHV+) patients of group 2). Single Nucleotide Variants (SNVs) in Linkage Disequilibrium (LD) (R2 >0.6) were removed prior to statistical analyses. The R2 valued of 0.6 was chosen as the cutoff to remove SNVs in LD as this was considered conservative enough to keep alleles that have some independence and stringent enough to remove those whose effects would be linked, as in previous studies [41]. Fisher’s exact tests were used to assess the associations between SNVs and case-control status, as previously described [42]. The Bonferroni method was used for post-hoc adjustment of p-values to correct for multiple testing and represented as “adjusted P values,” with p values <0.05 considered statistically significant.
In order to determine whether less common variation was associated with KSHV and KS status, particularly since using a rather small patient cohort of n=150, we performed an aggregate analysis as in previous studies [43,44] in which we considered whether each participant carried ≥1 or 0 EPHA2 SNVs with MAF < 5%. Aggregate scores were determined for all SNVs across EPHA2 and for missense, synonymous and untranslated region (UTR) variants. Additionally, aggregate scores were determined by functional domain (Pkinase-Tyr, SAM, 5‘-UTR, 3‘-UTR and fibronectin type 3 domains). Associations between aggregate scores and case-control status were determined using logistic regression.
3. Results
Clinical and demographic information concerning the abovementioned participant groups (total sample size n=150, with n=50 per group) is summarised in Table 1. All patients were HIV-infected as determined serologically. Age did not differ significantly between the three groups: median age was 36, 39 and 40 for groups 1, 2 and 3, respectively. Although the final cohort of patients had a slight overrepresentation of males (55.3%) compared to females (44.7%), the sex ratio in the three groups was similar. Population group distribution was heavily skewed towards black Africans (93.3%) and included only a minority of mixed ancestry (5.3%) and Caucasian (1.3%) individuals which was consistent across the three patient groups. Most recent CD4 counts were recorded at the time of patient recruitment. CD4 counts did not statistically differ between patient groups, although they were lowest in group 1 (KS+/KSHV+) and highest in group 3 patients (KS−/KSHV−). All KS+ patients (group 1) received ART with an average time of 298 ± 483 days before KS diagnosis, whereas a significantly smaller number of KS− patients (groups 2 and 3) were on ART medication (p<0.0001) at the time of recruitment.
Table 1:
Clinical and demographic information of the three patient groups making up the study cohort.
| GROUP 1 | GROUP 2 | GROUP 3 | |
|---|---|---|---|
| KS+ | KS− | KS− | |
| KSHV+ | KSHV+ | KSHV− | |
| Sample size | 50 | 50 | 50 |
| Age, median in years (IQR) | 36 (31-42) | 39 (31-46) | 40 (30-47) |
| Sex, count (%) | |||
| Male | 29 (58) | 26 (52) | 28 (56) |
| Female | 21 (42) | 24 (48) | 22 (44) |
| Population group, number (%) | |||
| Black | 48 (96) | 45 (90) | 47 (94) |
| Mixed ancestry | 2 (10) | 3 (6) | 3 (6) |
| Caucasian | 0 (0) | 2 (4) | 0 (0) |
| On ART at time of blood draw | |||
| Yes | 50 (100) | 34 (68) | 35 (70) |
| No | 0 (0) | 16 (32) | 15 (30) |
| Duration of ART at time of KS diagnosis, mean in days (SD) | 298 (483) | n/a | n/a |
| CD4 count, median in cells/μl (IQR) | 238 (122-341) | 245 (103-470) | 334 (166-511) |
N=150 participants. IQR, inter-quartile range; ART, antiretroviral therapy.
Since none of the demographic parameters presented in Table 1 significantly differed, the three patient groups were considered suitable for further candidate gene association analysis. Fifty plasma samples from patients with clinically diagnosed KS were assessed by K8.1 and ORF73 ELISAs and all (95% CI 92.9-100%) were found to be KSHV seropositive as expected (Table 2A), while 31.6% (95% CI 28.3-35.1%) of the total patient cohort without KS (706 patients) were found to be KSHV seropositive (Table 2B). KSHV status was tested only once per patient with the timing relative to diagnosis of HIV infection being highly variable.
Table 2:
ELISA results indicating KSHV serostatus for samples taken from patients with A. clinically diagnosed KS (50 patients) and B. without KS (706 patients).
| A | K8.1 | B | K8.1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| + | − | Total | + | − | Total | ||||
| ORF73 | + | 40 | 0 | 40 | ORF73 | + | 95 | 42 | 137 |
| − | 10 | 0 | 10 | − | 86 | 483 | 569 | ||
| Total | 50 | 0 | 50 | Total | 181 | 525 | 706 | ||
A patient was classified as KSHV+ if the OD values for K8.1 and ORF73 fell above the cut off for either ELISA assay (indicated by cells with shaded background). KSHV seroprevalence was 100% (95% CI 92.9-100%) in A. and 31.6% (95% CI 28.3-35.1%) in B.
Sequencing of the EPHA2 coding region of the randomly selected final cohort of 150 patients (falling into the three categories based on KS diagnosis and KSHV serology status as outlined above) resulted in a total of 57 unique variants across the 3,964 base pair EPHA2 coding region, 22 of which were predicted to result in amino acid (AA) changes. While the majority (35) of these variants are recorded in the NCBI database of single nucleotide polymorphisms (dbSNP), 22 novel variants were identified in our cohort. The total variation across the coding region of EPHA2 is represented schematically in Figure 1. Exons 3, 5, 11 and 17 contain the highest number of variants uniformly found across all patients compared to the reference sequence. Taking the size of each exon into account, exon 11 can be identified as harbouring the highest average rate of variation per bp (0.0062 variants/bp), followed by exons 17 (0.0033 variants/bp), 12 (0.0030 variants/bp) and 5 (0.0023 variants/bp). Moreover, KS+ patients (group 1) appeared to have an increased number of variations in the Pkinase-Tyr domain (exons 12-15) when compared to KS− patients (groups 2 and 3) (Figure 1).
Figure 1: Schematic representation of the number of identified variants per exon in EPHA2.
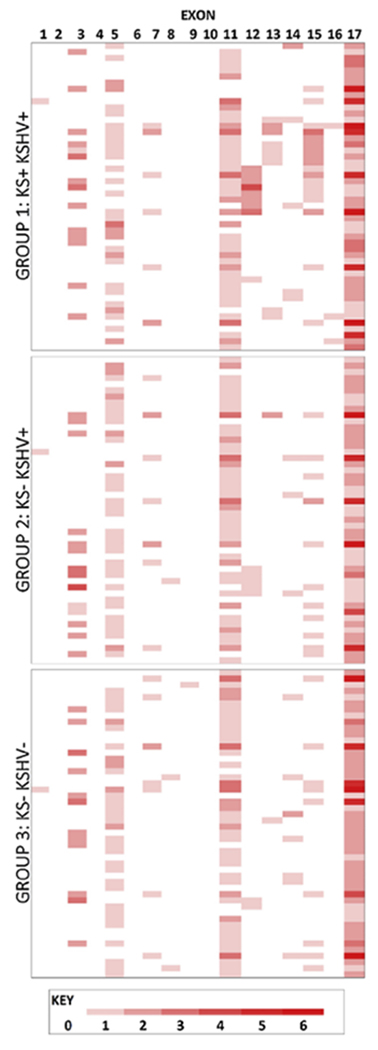
Groups 1, 2 and 3 refer to the patient groups detailed in Methods and described in Table 1, each consisting of 50 patients represented by rows. Columns represent the exons of EPHA2. Cells are coloured depending on the number of variants identified according to the key below the figure.
Aggregate variation across EPHA2 was associated with increased risk of KS. Specifically, 37 (74%) KS cases (group 1) had one or more SNV versus only 15 (30%) of KS−/KSHV+ controls (group 2) (OR=6.6 (95% CI: 2.8, 15.9), p=2.2×10−5, Supplementary Table S1). Missense variants, rather than synonymous variants or variants in the UTR, appeared to be the primary drivers of this association (OR=4.9 (95% CI: 2.0, 11.7), p=0.0004). When aggregate variation was considered within each of the functional domains of EPHA2, it was observed that having one or more SNV in the Pkinase-Tyr domain or SAM domain was associated with increased risk of KS (OR=4.9 (95% CI: 1.9, 12.4), p=0.001 and OR=13.8 (95% CI: 1.7, 111.6), p=0.014, respectively).
After removing SNVs based on LD and with MAF < 3% (Table 3), 8 individual variants were assessed statistically for association with KS development by comparing occurrence in KS+/KSHV+ (group 1) patients versus KS−/KSHV+ (group 2) patients. The variant at mRNA positions 2254 is predicted to result in a novel, non-conservative AA change from Leucine to Proline at AA position 700 and is in LD with another SNV at mRNA position 2257 predicted to result in an AA substitution from Aspartate to Alanine at the next AA position, 701, in the conserved Pkinase-Tyr domain. These heterozygous variants were found to co-occur in 8 (16%) KS+ patients (group 1) (with an additional group 1 patient showing only the variant at mRNA position 2257) and not to occur at all (0%) in KS− patients in group 2 (OR undefined, adj. p=0.04, Table 3). Additionally, several other variants occurring in the Pkinase-Tyr domain (spanning mRNA positions 1990-2766), were found to be overrepresented in the KS+ (group 1) patients (Table 3). Within the SAM domain, a heterozygous G>T variant at mRNA position 2990, predicted to result in a substitution of Asparagine for Lysine at AA position 945, was found to be significantly more frequent among KS+/KSHV+ (group 1) patients (18%) compared to KS−/KSHV+ (group 2) patients (0%) (OR undefined, adj. p=0.02, Table 3). Both the variant at mRNA position 2254 and at position 2990 were found to be ‘probably damaging’ when assessed for functional impact using the PolyPhen-2 prediction tool.
Table 3:
Associations of individual variants within the EPHA2 coding region with KS status.
| mRNA level | AA level | No. Of KS+/KSHV+ patients (Group 1, n=50) |
No. Of KS−/KSHV+ patients (Group 2, n=50) |
P value / Adjusted P value | OR (95% CI) | MAF |
|---|---|---|---|---|---|---|
| 2990 G>K |
945 K>N |
9 (18%) | 0 (0%) | 0.003/ 0.021* | Undefined | 0.045 |
| 2254 T>Y |
700 L>P |
8 (16%) | 0 (0%) | 0.006/ 0.04* | Undefined | 0.04 |
| 2688 G>S rs7652 80326 |
845 A>P |
6 (12%) | 0 (0%) | 0.027/ 0.16 | Undefined | 0.03 |
| 2727 C>Y |
858 R>C |
14 (28%) | 7 (14%) | 0.14/ 0.698 | 2.4 (0.9, 6.6) | 0.105 |
| 2325 G>S rs7470 58254 |
724 A>P |
5 (10%) | 1 (2%) | 0.204/ 0.818 | 5.4 (0.6, 48.4) | 0.03 |
| 2217 A>M rs7633 07879 |
688 M>L |
2 (4%) | 5 (10%) | 0.436/ 1 | 0.4 (0.1, 2) | 0.035 |
| 915 C>S |
254 L>V |
2 (4%) | 5 (10%) | 0.436/ 1 | 0.4 (0.1, 2) | 0.035 |
| 2047 T>Y rs3402 1505 |
631 M>T |
7 (14%) | 5 (10%) | 0.76/ 1 | 1.5 (0.4, 5) | 0.065 |
SNVs in significant LD (R2>0.6) and with MAF < 3% were removed from the analysis before Fisher’s exact statistical tests were performed between KS+/KSHV+ (group 1) and KS−/KSHV+ (group 2) patients. Resulting P values and adjusted P values corrected for multiple comparison are shown. SNVs are presented in order of most significant P values, and statistically significant associations at the p ≤ 0.05 level are indicated with *. Position in mRNA and protein is according to the EPHA2 NCBI references NM_004431.3 and NP_004422.2, respectively, and nucleotides and AA are named according to IUPAC standards. Where available, dbSNP identifiers are given, and italics indicated that the reported SNV at that position is a different nucleotide change to that reported here. Ambiguous bases represent heterozygous variations. Protein change is predicted by in silico translation of the DNA sequence.
Aggregate tests did not find statistically significant associations between EPHA2 SNVs and KSHV status. However, there was a trend indicating that having one or more missense SNV was associated with increased risk of KSHV infection. Specifically, 40 (40%) KSHV cases had one or more missense SNV compared to only 12 (24%) KSHV negative controls (OR=2.1 (95% CI: 0.98, 4.5), p=0.06, Supplementary Table S2). To test individual variants for association with KSHV infection, we compared the occurrence of 4 SNVs in KSHV+ groups to KSHV− patients which we identified after removing SNVs based on LD and with MAF < 3% (Table 4). We discovered a novel heterozygous C>T variant in the conserved Pkinase-Tyr domain at mRNA position 2727, which was found to be statistically significant (OR=6.4 (95% CI: 1.4, 28.4), adj. p=0.03), occurring in 21 (21%) KSHV+ patients (14 in group 1, and 7 in group 2) and in only 2 (4%) KSHV− (group 3) patients (Table 4). This variant is predicted to result in a non-conservative substitution of a Cysteine for Arginine at AA position 858 which is predicted to be “probably damaging” using the PolyPhen-2 prediction tool. The SNV at mRNA position 2727 was also found to be overrepresented in KS+/KSHV+ patients (14 patients (28%) in group 1 compared to KS−/KSHV+ patients (7 patients (14%) in group 2) but this was not statistically significant (Table 3). The presence of this variant in KS−/KSHV+ (group 2) patients, while to a lesser extent than in KS+/KSHV+ (group 1) patients, indicated that the occurrence of this variant in KS+/KSHV+ (group 1) patients may be a consequence of increased susceptibility to KSHV infection and thereby an indirect association with KS development.
Table 4:
Associations of individual variants within the EPHA2 coding region with susceptibility to KSHV infection.
| SNV | AA level | No. of KSHV+ patients (Group 1+2, n=100) | No. of KSHV− patients (Group 3, n=50) | P value / Adjusted P Value | OR (95% CI) | MAF |
|---|---|---|---|---|---|---|
| 2727 C>Y | 858 R>C |
21 (21%) | 2 (4%) | 0.007/ 0.028* | 6.4 (1.4, 28.4) | 0.077 |
| 2990 G>K | 945 K>N |
9 (9%) | 2 (4%) | 0.338/ 1 | 2.4 (0.5, 11.4) | 0.037 |
| 2217 A>M rs763307879 |
688 M>L |
7 (7%) | 2 (4%) | 0.718/ 1 | 1.8 (0.4, 9) | 0.03 |
| 2047 T>Y rs34021505 |
631 M>T |
12 (12%) | 7 (14%) | 0.796/ 1 | 0.8 (0.3, 2.3) | 0.067 |
SNVs in significant LD (R2>0.6) and with MAF < 3% were removed from the analysis before Fisher’s exact statistical tests were performed between KSHV+ (the sum of groups 1 and 2) patients and KSHV− (group 3) patients. Resulting P values and adjusted P values corrected for multiple comparison are shown. SNVs are presented in order of most significant P values, and statistically significant associations at the p ≤ 0.05 level are indicated with *. Position in mRNA and protein is according to the EPHA2 NCBI references NM_004431.3 and NP_004422.2, respectively, and nucleotides and AA are named according to IUPAC standards. Where available, dbSNP identifiers are given, and italics indicated that the reported SNV at that position is a different nucleotide change to that reported here. Ambiguous bases represent heterozygous variations. Protein change is predicted by in silico translation of the DNA sequence.
4. Discussion
Genetic variants in receptors for viral entry and/or oncogenesis have been documented to have functional consequences for pathogenicity. The genetic factors underlying susceptibility to KSHV infection and KS development are not fully understood. We therefore set out to identify sequence variants in EPHA2, being both an entry receptor for KSHV [22,23] as well as being upregulated in various tumours, including KS [22,25–28], in South African HIV-infected patients, and to determine the association of any identified EPHA2 variants with susceptibility to KSHV infection and/or KS development.
As AIDS-related KS is by far the most common form of the KSHV-associated malignancies and particularly affects individuals in Sub-Saharan Africa due to the HIV/AIDS epidemic [3], we restricted the recruitment of patients to HIV-infected individuals from this geographical region, presenting at hospitals in the Western Cape province of South Africa. In support with the reported disproportionately high KSHV seroprevalence in sub-Saharan Africa (>50%) compared to world prevalence rates (<10%), which has not significantly changed since the onset of the HIV/AIDS epidemic [16], we determined a 31.6% (95% CI 28.3-35.1%) KSHV seropositivity in our total KS− patient cohort (Table 2), presenting the first assessment of KSHV seroprevalence in the Western Cape Province of South Africa. This is in agreement with earlier studies conducted in Soweto, Johannesburg and Kwa-Zulu Natal, which have indicated that KSHV seroprevalence is between 30-40% [3].
Despite the high HIV/KSHV seroprevalence in sub-Saharan Africa, not all co-infected patients develop KS. To elucidate a potential underlying genetic predisposition due to variants in the EPHA2 protein, we performed aggregate variation across the entire EPHA2 coding region to assess a retrospective candidate gene association with KSHV infectivity and/or KS prevalence. Since mother-to-child transmission via saliva is thought to be the primary route of KSHV transmission [5], the extent of later sexual transmission that could be confounding for KSHV infection is thought to be minimal [45,46]. All patients recruited to this study were adults between 19 and 72 years of age (Interquartile Range: 31-47, see Table 1); therefore, it can be assumed that their exposure to and infection with KSHV has been concluded at the time of recruitment.
We identified missense variants and variants within the functionally important Pkinase-Tyr and SAM domains, specifically 2254 T>C (located in the Pkinase-Tyr domain) and 2990 G>T (located in the SAM domain) which were associated with KS, Table 3. Each of these variants was designated a ‘probably damaging’ annotation when assessed for functional impact using the PolyPhen-2 prediction tool. Interestingly, additional individual Pkinase-Tyr domain variants spanning exons 12-15 (particularly 2688 G>C (rs765280326), 2727 C>T, 2325 G>C (rs747058254) and 2047 T>C (rs34021505)), although not significantly associated with KS, were found to be overrepresented among KS patients (group 1), Table 3 and Figure 1. We can speculate that, although rare, their functional impact may be important, represented by the significant association of aggregate Pkinase-Tyr variation with KS. It is plausible that they may enhance EPHA2 Pkinase-Tyr signalling which is essential for the function of the EPHA2 receptor and which has been linked to a metastatic, aggressive phenotype in a number of cancers [24–27]. The variant 2990 G>T (located in the cytoplasmic SAM protein interaction domain), found to be overrepresented among patients with KS (Table 3) may further contribute to oncogenesis by altering the function of the SAM, a protein-protein interaction region suggested to bind adaptor proteins thereby mediating the downstream signalling events triggered by EPHA2 activation [47,48].
Also located in the Pkinase-Tyr domain, the variant at mRNA position 2727, a C>T encoding an Arginine to Cysteine mutation, was identified as being associated with increased susceptibility to KSHV infection (Table 4), occurring predominately in KSHV+ patients (group 1 and 2) compared to KSHV− (group 3) patients. It can be speculated that this variant may not only enhance susceptibility to KSHV infection but also subsequently contribute to enhanced KS development (Table 3). Due to its cytoplasmic location in the Pkinase-Tyr domain it is unlikely that this variant enhances KSHV binding, it may rather enhance downstream EPHA2 signalling that is essential to internalisation of bound KSHV. Interestingly, no significant sequence variation between the analysed patient groups was found in EPHA2’s ligand binding domain, supporting the hypothesis of the importance of the Pkinase-Tyr domain for KSHV-driven KS development.
A number of variants previously reported on the dbSNP were identified in this study (35 out of the total 57), with a further 22 novel variants identified in our South African cohort. Large genotyping studies, such as the 1000 Genomes project and ExAc, have contributed the majority of the genetic variations stored in the dbSNP [49,50]. While these studies include African populations from Nigeria, Kenya, Gambia and Sierra Leone and people with African ancestry residing in America and the Caribbean, Southern African populations are not well represented [49]. It is thought that Southern African populations specifically have exceptionally high levels of genetic diversity due partly to the selective pressure of long term exposure to infectious diseases and due to the lack of founder effects present in populations that have migrated [51,52]. Therefore, it is expected that we would see variants in our South African population that have not yet been recorded in the dbSNP.
However, there were several limitations to this study. The overall number of recruited patients was rather small, restricting the power of the analysis to only be able to detect associations of SNVs with MAF > 3% with KS development and/or KSHV infection. Selection biases may have overestimated the association of EPHA2 variants as only HIV positive patients presenting at clinics were recruited. With the purpose to describe a single variable distribution in our (small) patient cohort as a first step to elucidate a pattern of association between EPHA2 variants and KS and/or KSHV prevalence, we performed a univariate analysis which was not adjusted to other risk factors of KSHV infection and/or KS development. This study therefore lays the basis for further investigation into the impact and functional relevance of EPHA2 variants on KSHV infection and subsequent KS development. While the herein reported association of EPHA2 variants with KSHV infection and KS development requires validation, this may have clinical implications in terms of identifying KSHV infected patients who are susceptible to KS development and highlighting EPHA2 as a potential therapeutic target.
5. Conclusion
This study for the first time describes the identification of sequence variants in EPHA2 in a South African HIV-infected patient cohort in relation to KSHV infection and KS development. Some variants, particularly affecting the Pkinase-Tyr and SAM domains, were predicted to potentially cause substantial alterations to the expression and/or function of EPHA2.
Supplementary Material
Highlights.
KSHV serumconversion among South African HIV-positive patients is >30%
Sequence variations in the KSHV host receptor EPHA2 are associated with KS development and KSHV infectivity
EPHA2 sequence variations are primarily located in the protein tyrosine kinase domain and the sterile-α-motif
Validation of the association of sequence variations in EPHA2 with KS development and KSHV infectivity may have clinical implications in identifying patients who are susceptible to KS development and highlighting EPHA2 as a potential therapeutic target
Acknowledgements
We particularly thank all patient participants in this study.
Funding
This work was supported by grants from the Cancer Association of South Africa (CANSA) to G.S., the South African Medical Research Council (SA-MRC) to A.A.K. and C.S., the National Research Foundation (NRF) of South Africa to G.S. and M.J.B. and the University of Cape Town, South Africa to G.S.
D.W. was supported by US federal funds from the National Cancer Institute at the National Institutes of Health (NIH), under Contract No. HHSN261200800001E. G.M. is supported by the Wellcome Trust (098316), the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation (NRF) of South Africa (Grant No 64787), and NRF incentive funding (UID: 85858). This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations
KS (Kaposi’s sarcoma), KSHV (Kaposi’s sarcoma-Associated Herpes Virus), HIV (Human Immunodeficiency Virus), AIDS (Acquired Immunodeficiency Syndrome), Pkinase-Tyr (protein tyrosine kinase domain), SAM (sterile-α-motif), EPHA2 (Eph Receptor A2 protein), MAF (Minor Allele Frequency), SNV (Single Nucleotide Variants), LD (Linkage Disequilibrium), AA (amino acid), dbSNP (database of single nucleotide polymorphisms), UTR (untranslated region)
Conflicts of interest
The authors state no conflict of interest.
References
- 1.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer 2006; 118:3030–44. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F GLOBOCAN 2012 vl.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, Fr. Int. Agency Res. Cancer; 2013. Available from http//globocan.iarc.fr, accessed 25/02/2015. [Google Scholar]
- 3.Mesri EA, Cesarman E, Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer 2010; 10:707–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sitas F, Newton R. Kaposi’s Sarcoma in South Africa. JNCI Monogr 2000; 2000:1–4. [DOI] [PubMed] [Google Scholar]
- 5.Plancoulaine S, Abel L, Beveren M Van, Trégouët D, Joubert M, Tortevoye P, et al. Human herpesvirus 8 transmission from mother to child and between siblings in an endemic population. Lancet 2000; 356:1062–1065. [DOI] [PubMed] [Google Scholar]
- 6.Dedicoat M, Newton R, Alkharsah KR, Sheldon J, Szabados I, Ndlovu B, et al. Mother-to-child transmission of human herpesvirus-8 in South Africa. J Infect Dis 2004; 190:1068–1075. [DOI] [PubMed] [Google Scholar]
- 7.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles M, et al. Identification of Herpesvirus-Like DNA Sequences in AIDS-Associated Kaposi’s Sarcoma. Science (80-) 1994; 266:1865–1869. [DOI] [PubMed] [Google Scholar]
- 8.Wilkinson D, Sheldon J, Gilks CF, Schulz TF. Prevalence of infection with human herpesvirus 8/Kaposi’s sarcoma herpesvirus in rural South Africa. South African Med J 1999; 89:3–6. [PubMed] [Google Scholar]
- 9.Gramolelli S, Schulz TF. The role of Kaposi sarcoma-associated herpesvirus in the pathogenesis of Kaposi sarcoma. J Pathol 2015; 235:368–380. [DOI] [PubMed] [Google Scholar]
- 10.Ruocco E, Ruocco V, Tornesello ML, Gambardella A, Wolf R, Buonaguro FM. Kaposi’s sarcoma: Etiology and pathogenesis, inducing factors, causal associations, and treatments: Facts and controversies. Clin Dermatol 2013; 31:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitby D, Marshall VA, Bagni RK, Miley WJ, G T, Hines-boykin R, et al. Reactivation of Kaposi’s sarcoma-associated herpesvirus by natural products from Kaposi’s sarcoma endemic regions. Int J Cancer 2007; 120:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plancoulaine S, Gessain A, Beveren M Van, Tortevoye P, Abel L Evidence for a Recessive Major Gene Predisposing to Human Herpesvirus 8 (HHV-8) Infection in a Population in Which HHV-8 Is Endemic. J Infect Dis 2003; 8:1944–1950. [DOI] [PubMed] [Google Scholar]
- 13.Cavallin LE, Goldschmidt-Clermont P, Mesri E a. Molecular and Cellular Mechanisms of KSHV Oncogenesis of Kaposi’s Sarcoma Associated with HIV/AIDS. PLoS Pathog 2014; 10:5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guttman-Yassky E, Cohen A, Kra-Oz Z, Friedman-Birnbaum R, Sprecher E, Zaltzman N, et al. Familial clustering of classic Kaposi sarcoma. J Infect Dis 2004; 189:2023–2026. [DOI] [PubMed] [Google Scholar]
- 15.Hengge UR, Ruzicka T, Tyring SK, Stuschke M, Roggendorf M, Schwartz RA, et al. Review Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 1: epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect Dis 2002; 2:281–292. [DOI] [PubMed] [Google Scholar]
- 16.Dedicoat M, Newton R. Review of the distribution of Kaposi’s sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi’s sarcoma. Br J Cancer 2003; 88:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown EE, Fallin D, Ruczinski I, Hutchinson A, Staats B, Vitale F, et al. Associations of classic Kaposi sarcoma with common variants in genes that modulate host immunity. Cancer Epidemiol Biomarkers Prev 2006; 15:926–934. [DOI] [PubMed] [Google Scholar]
- 18.Foster CB, Lehrnbecher T, Samuels S, Stein S, Mol F, Metcalf J a, et al. An IL6 promoter polymorphism is associated with a lifetime risk of development of Kaposi sarcoma in men infected with human immunodeficiency virus. Blood 2000; 96:2562–2567. [PubMed] [Google Scholar]
- 19.Goedert JJ, Martin MP, Vitale F, Lauria C, Whitby D, Qi Y, et al. Risk of classic kaposi sarcoma with combinations of killer immunoglobulin-like receptor and human leukocyte antigen loci: A population-based case-control study. J Infect Dis 2016; 213:432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aissani B, Boehme a K, Wiener HW, Shrestha S, Jacobson LP, Kaslow R a. SNP screening of central MHC-identified HLA-DMB as a candidate susceptibility gene for HIV-related Kaposi’s sarcoma. Genes Immun 2014; 15:424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehrnbecher TL, Foster CB, Zhu S, Venzon D, Steinberg SM, Wyvill K, et al. Variant genotypes of FcγRIIIA influence the development of Kaposi’s sarcoma in HIV-infected men. Blood 2000; 95:2386–2390. [PubMed] [Google Scholar]
- 22.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma–associated herpesvirus. Nat Med 2012; 18:961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakraborty S, Veettil MV, Bottero V, Chandran B. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc Natl Acad Sci U S A 2012; 109:E1163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol cancer Res 2008; 6:1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 Overexpression Causes Tumorigenesis of Mammary Epithelial Cells 1. Cancer Res 2001; 61:2301–2306. [PubMed] [Google Scholar]
- 26.Thaker PH, Deavers M, Celestino J, Thornton A, Fletcher MS, Landen CN, et al. EphA2 expression is associated with aggressive features in ovarian carcinoma. Clin Cancer Res 2004; 10:5145–5150. [DOI] [PubMed] [Google Scholar]
- 27.Walker-Daniels J, Coffman K, Azimi M, Rhim JS, Bostwick DG, Snyder P, et al. Overexpression of the EphA2 tyrosine kinase in prostate cancer. Prostate 1999; 41:275–280. [DOI] [PubMed] [Google Scholar]
- 28.Hess AR, Seftor EA, Gardner LMG, Carles-Kinch K, Schneider GB, Seftor REB, et al. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: Role of epithelial cell kinase (Eck/EphA2). Cancer Res 2001; 61:3250–3255. [PubMed] [Google Scholar]
- 29.Shiels A, Bennett TM, Knopf HLS, Maraini G, Li A, Jiao X, et al. The EPHA2 gene is associated with cataracts linked to chromosome 1p. Mol Vis 2008; 14:2042–2055. [PMC free article] [PubMed] [Google Scholar]
- 30.Jun G, Guo H, Klein BEK, Klein R, Jie JW, Mitchell P, et al. EPHA2 is associated with age-related cortical cataract in mice and humans. PLoS Genet 2009; 5:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, Zafar AU, et al. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis 2010; 16:511–517. [PMC free article] [PubMed] [Google Scholar]
- 32.Sundaresan P, Ravindran RD, Vashist P, Shanker A, Nitsch D, Talwar B, et al. EPHA2 polymorphisms and age-related cataract in India. PLoS One 2012; 7:e33001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T, Hua R, Xiao W, Burdon KP, Bhattacharya SS, Craig JE, et al. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum Mutat 2009; 30:603–610. [DOI] [PubMed] [Google Scholar]
- 34.Tan W, Hou S, Jiang Z, Hu Z, Yang P, Ye J. Association of EPHA2 polymorphisms and age-related cortical cataract in a Han Chinese population. Mol Vis 2011; 17:1553–8. [PMC free article] [PubMed] [Google Scholar]
- 35.Mbisa GL, Miley W, Gamache CJ, Gillette WK, Esposito D, Hopkins R, et al. Detection of antibodies to Kaposi’s sarcoma-associated herpesvirus: A new approach using K8.1 ELISA and a newly developed recombinant LANA ELISA. J Immunol Methods 2010; 356:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biggar RJ, Engels EA, Whitby D, Kedes DH, Goedert JJ. Antibody reactivity to latent and lytic antigens to human herpesvirus-8 in longitudinally followed homosexual men. J Infect Dis 2003; 187:12–18. [DOI] [PubMed] [Google Scholar]
- 37.Lindberg RA, Hunter T. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol Cell Biol 1990; 10:6316–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet 2000; 16:276–277. [DOI] [PubMed] [Google Scholar]
- 39.Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007; 23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 40.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De R, Holmes M V., Moore JH, Ritchie MD, Verma SS, Asselbergs FW, et al. Dissecting the obesity disease landscape: Identifying gene-gene interactions that are highly associated with body mass index. Int Conf Syst Biol ISB 2014; :124–131. [Google Scholar]
- 42.Clarke GM, Anderson C a, Pettersson FH, Cardon LR, Morris AP, Zondervan KT. Basic statistical analysis in genetic case-control studies. Nat Protoc 2011; 6:121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackelprang RD, Bamshad MJ, Chong JX, Hou X, Buckingham KJ, Shively K, et al. Whole genome sequencing of extreme phenotypes identifies variants in CD101 and UBE2V1 associated with increased risk of sexually acquired HIV-1o Title. PLoS Pathog 2017; 13:e1006703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris A, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol 2010; 34:188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malope BI, MacPhail P, Mbisa G, MacPhail C, Stein L, Ratshikhopha EM, et al. No evidence of sexual transmission of Kaposi’s sarcoma herpes virus in a heterosexual South African population. AIDS 2008; 22:519–526. [DOI] [PubMed] [Google Scholar]
- 46.Butler LM, Were WA, Balinandi S, Downing R, Dollard S, Neilands TB, et al. Human herpesvirus 8 infection in children and adults in a population-based study in rural Uganda. J Infect Dis 2011; 203:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schultz J, Ponting CP, Hofmann K, Bork P. SAM as a protein interaction domain involved in developmental regulation. Protein Sci 1997; 6:249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smalla M, Schmieder P, Kelly M, Ter Laak A, Krause G, Ball L, et al. Solution structure of the receptor tyrosine kinase EphB2 SAM domain and identification of two distinct homotypic interaction sites. Protein Sci 1999; 8:1954–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Consortium T 1000 GP. A global reference for human genetic variation. Nature 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lek M, Karczewski KJ, Minikel E V., Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell MMC, Tishkoff S a. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet 2008; 9:403–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gomez F, Hirbo J, Tishkoff SA. Genetic variation and adaptation in Africa : Implications for human evolution and disease. Cold Spring Harb Perspect Biol 2014; 6:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.