

During viral RNA synthesis of a negative-strand RNA virus, the viral RNA-dependent RNA polymerase (vRdRp) must gain access to the sequestered RNA in the nucleocapsid to use it as the template, but at the same time may not disrupt the nucleocapsid assembly. Our structural and mutagenesis studies showed that a flexible structural motif acts as a potential access gate to the sequestered RNA and plays an essential role in viral RNA synthesis. Interactions of this structural motif within the vRdRp may be required for unveiling the sequestered RNA. This mechanism of action allows the sequestered RNA to be released locally without disrupting the overall structure of the nucleocapsid. Since this flexible structural motif is present in the N proteins of many NSVs, release of the sequestered RNA genome by local conformational changes in the N protein may be a general mechanism in NSV viral RNA synthesis.
KEYWORDS: L protein, nucleocapsid, polymerase, replication, transcription
ABSTRACT
During viral RNA synthesis by the viral RNA-dependent RNA polymerase (vRdRp) of vesicular stomatitis virus, the sequestered RNA genome must be released from the nucleocapsid in order to serve as the template. Unveiling the sequestered RNA by interactions of vRdRp proteins, the large subunit (L) and the phosphoprotein (P), with the nucleocapsid protein (N) must not disrupt the nucleocapsid assembly. We noticed that a flexible structural motif composed of an α-helix and a loop in the N protein may act as the access gate to the sequestered RNA. This suggests that local conformational changes in this structural motif may be induced by interactions with the polymerase to unveil the sequestered RNA, without disrupting the nucleocapsid assembly. Mutations of several residues in this structural motif—Glu169, Phe171, and Leu174—to Ala resulted in loss of viral RNA synthesis in a minigenome assay. After implementing these mutations in the viral genome, mutant viruses were recovered by reverse genetics and serial passages. Sequencing the genomes of the mutant viruses revealed that compensatory mutations in L, P, and N were required to restore the viral viability. Corresponding mutations were introduced in L, P, and N, and their complementarity to the N mutations was confirmed by the minigenome assay. Introduction of the corresponding mutations is also sufficient to rescue the mutant viruses. These results suggested that the interplay of the N structural motif with the L protein may play a role in accessing the nucleotide template without disrupting the overall structure of the nucleocapsid.
IMPORTANCE During viral RNA synthesis of a negative-strand RNA virus, the viral RNA-dependent RNA polymerase (vRdRp) must gain access to the sequestered RNA in the nucleocapsid to use it as the template, but at the same time may not disrupt the nucleocapsid assembly. Our structural and mutagenesis studies showed that a flexible structural motif acts as a potential access gate to the sequestered RNA and plays an essential role in viral RNA synthesis. Interactions of this structural motif within the vRdRp may be required for unveiling the sequestered RNA. This mechanism of action allows the sequestered RNA to be released locally without disrupting the overall structure of the nucleocapsid. Since this flexible structural motif is present in the N proteins of many NSVs, release of the sequestered RNA genome by local conformational changes in the N protein may be a general mechanism in NSV viral RNA synthesis.
INTRODUCTION
Negative-strand RNA viruses (NSVs) include a number of important human pathogens, such as Ebola, avian influenza, and respiratory syncytial viruses. Understanding the mechanism of their replication assists in the development of new therapies. One unique feature that separates NSVs from other viral pathogens is that their genomic RNAs are sequestered in the nucleocapsid during viral RNA synthesis. NSVs encode their own viral RNA-dependent RNA polymerase (vRdRp). During viral RNA synthesis by vRdRp, the assembled nucleocapsid serves as the template, not the released RNA genome (1, 2). The mechanism by which vRdRp gains access to the sequestered genomic RNA to copy complementary RNAs is an unsolved question.
Vesicular stomatitis virus (VSV) is a prototypic NSV. VSV belongs to the Rhabdovirus family and its genome encodes five viral proteins, nucleocapsid (N), phosphoprotein (P), matrix (M), glycoprotein (G), and the large protein (L). vRdRp of VSV is composed of the L and P proteins, whereas the nucleocapsid, assembled by polymerization of the N proteins, serves as the template. The crystal structure of a nucleocapsid-like particle (NLP) shows that the assembly of VSV nucleocapsid requires extensive interactions of a long N-terminal arm and a large loop in the C-terminal domain of the N protein between four neighboring subunits (3, 4). The RNA is sequestered between the N-terminal and C-terminal domains that are formed mostly with α-helices. Some of the bases in the sequestered RNA face the interior of the N protein such that they could not be copied without first being exposed. One possible way is to induce an open N conformation as observed in the structure of some RNA-free N subunits (5, 6). However, that will also require untangling the interactions between the N subunits, which does not seem feasible. Another possible way is to induce a local conformational change at a proposed access gate in the N protein, which will not disrupt the overall structure of the nucleocapsid. This proposed access gate is one of helices in the N-terminal domain, helix 5, which covers the sequestered RNA. If vRdRp can induce a conformational change of helix 5, the sequestered RNA would be exposed to serve as the nucleotide template. The structure of the L protein has been solved (7), as well as the nucleocapsid binding domain of the P protein bound to the nucleocapsid (8). The orientation of the P domain bound to the nucleocapsid suggests that nucleocapsid bound vRdRp faces the access gate of the sequestered RNA. In this position, vRdRp may open the helix-5 access gate to release the sequestered RNA.
In this report, we tested the requirement of L-N interplay for viral RNA synthesis. Mutagenesis studies confirmed that the structural motif consisting of the helix 5 and the following loop may play a role in supporting viral RNA synthesis. Since the helix 5-loop motif is at one of the most flexible regions in the N protein, vRdRp could readily induce a conformation change in this structural motif to unveil the sequestered RNA when bound to the nucleocapsid. The local structural change induced by vRdRp will not disrupt the overall structure of the nucleocapsid.
RESULTS
The flexible structural motif in the N protein.
When the N and P proteins of VSV were expressed in Escherichia coli, a nucleocapsid-like particle (NLP) could be purified (9). The crystal structure of the NLP revealed that the N protein has an extended N-terminal arm and a large C-terminal loop (3). The nucleocapsid is assembled by extensive interactions between the N subunits to sequester the viral RNA in the core (4). Once the nucleocapsid is assembled, the overall structure is fairly stable except for two regions as shown in Fig. 1. Based on the B factors, the C-terminal loop and the helix 5/subsequent loop (helix 5-loop) are the most flexible regions in the NLP. The structural motif composed of the helix 5-loop forms the access gate to the sequestered RNA. In the NLP, the sequestered RNA is inaccessible if the helix 5-loop gate is in place. On the other hand, the flexible C-terminal loop is shown to interact with the nucleocapsid-binding domain of the P protein (8). This interaction, however, still maintains the sequestering of the viral RNA. During the viral RNA synthesis, vRdRp must gain access to the sequestered RNA in order to use it as the nucleotide template. Since the helix 5-loop motif covers the sequestered RNA and is flexible, the sequestered RNA may be locally released if a conformational change of the helix 5-loop motif could be induced by vRdRp, which could be accomplished without interrupting the nucleocapsid assembly.
FIG 1.
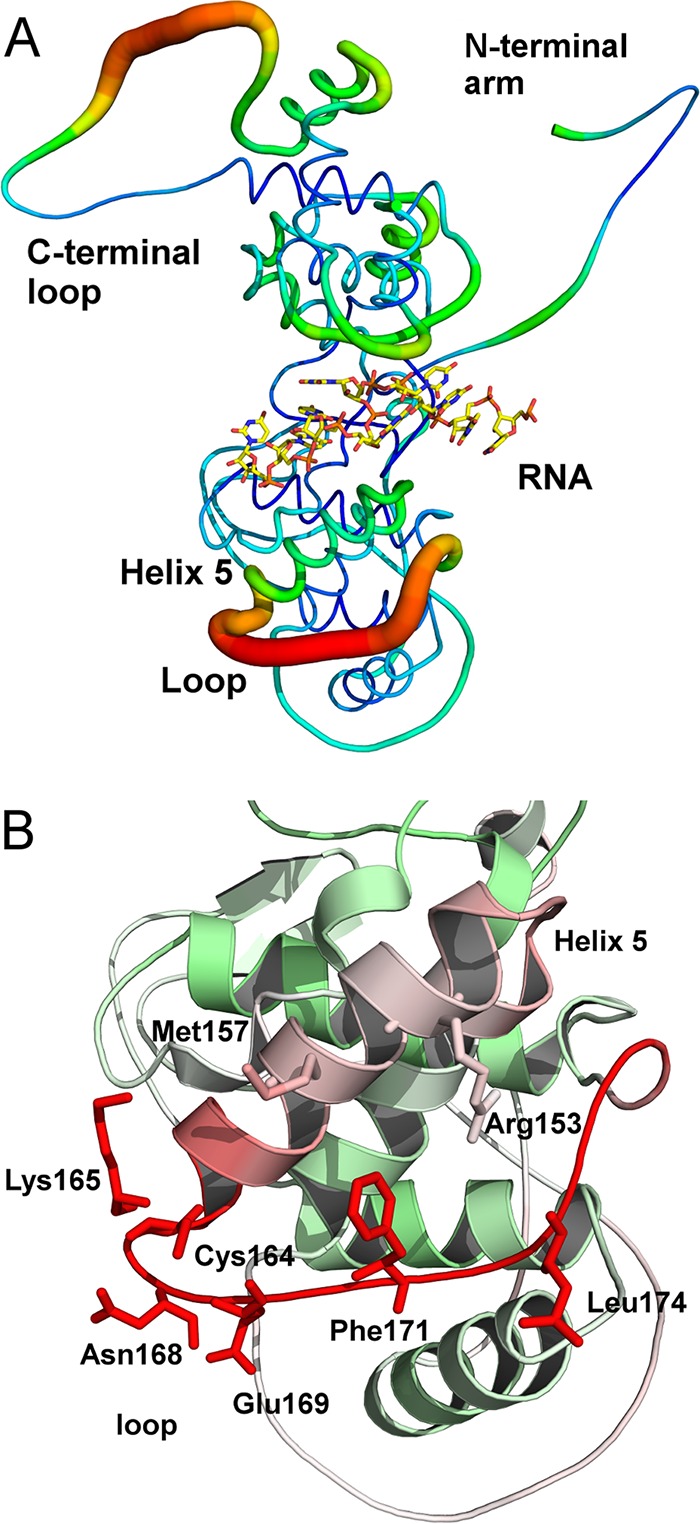
Flexibility in the N protein. (A) A B-factor putty drawing of the N protein structure in the nucleocapsid-like particle. The regions colored red also have a large diameter, which corresponds to higher B factors and are associated with higher structural flexibility. The sequestered RNA was shown as a stick model. (B) Closeup look at the helix 5-loop motif. The red color of the models also corresponds to high B factors. Selected residues are shown as stick models and labeled. The drawings were prepared with PyMOL (49).
Role of the helix 5-loop motif.
As shown in Fig. 1, several residues are exposed on the surface of the helix 5-loop motif. Residues Arg153, Met157, Cys164, Lys165, and Asn168 are located on the helix 5, whereas Glu169, Phe171, and Leu174 are located on the loop. An alanine-scanning mutagenesis was carried out to verify the role of each residue in viral RNA synthesis. After each mutation was introduced in the plasmid expressing the N protein, the activity of viral RNA synthesis was quantitated in a minigenome assay (10) (Fig. 2). Mutations of most residues in helix 5 did not significantly change the activity, but obvious reduction was observed by the Met157Ala mutation. On the other hand, mutation of residues on the loop dramatically reduced the activity of viral RNA synthesis. This observation strongly suggests that the loop in this structural motif may play an important role in supporting viral RNA synthesis by vRdRp.
FIG 2.

Role of each exposed residue. (A) Illustration of the minigenome construct and a list of alanine mutations for the surface-exposed residues in the helix 5-loop motif. Cys164 was also mutated to an alanine (green) as a control. (B) Luciferase activities in the minigenome assays. The mutations of the N protein in pN and controls are labeled. (C) Protein expression levels of N mutants in the minigenome assays as determined by Western blotting.
To ensure ubiquitously distributed protein concentrations, the protein expression level of N mutants was measured by Western blot (Fig. 2C). Most N mutants showed a similar level of protein expression, except for L174A, which had a 33% expression level compared to the wild-type (wt) N protein. For most of the N mutants, the protein expression level was not the main factor responsible for reduction of viral RNA synthesis. In case of L174A, the reduction in N protein expression may not play a major role in the reduction of viral RNA synthesis.
Nucleocapsid assembly of N mutants.
To examine the effects of N mutations on the assembly of the nucleocapsid, these mutations were introduced in our expression system of a NLP (9). The crystal structure of the NLP revealed the molecular interactions in the nucleocapsid (3), and NLP recapitulates the assembly of nucleocapsid as found in VSV virion (11). The N mutants were coexpressed with the P protein in E. coli and NLPs were purified as described previously (9). The elution position of the N mutant complexes in the size exclusion chromatography confirmed that the N mutants formed the same NLP as the wild-type N protein (Fig. 3A). Electron micrographs of negative-stain images of the NLPs clearly showed the ring-like structure of mutant NLPs, which contains 10 N subunits and is the same as that of the wild-type NLP (Fig. 3B).
FIG 3.

(A) Elution profile of NLPs from an S200 size exclusion column. The light blue line corresponds to wt NLP, the red line corresponds to E169A, the dark green line corresponds to F171A, and the light green line corresponds to L174A. (B) TEM images, class averages, and selected particles of the E169A, F171A, and L174A. Class averages were prepared with EMAN2, and particles were selected to show that the mutants still assemble in the same way as the wt rings (10 subunits in each assembly).
Specific effects of N mutants on viral RNA synthesis.
There are three types of viral RNA synthesized during VSV infection: mRNA during transcription, and cRNA/vRNA during replication. To dissect which specific types of viral RNA synthesis were affected by the N mutations, each species of the viral RNAs were quantitated by RT-qPCR in the minigenome assay (Fig. 4). The minigenome contains the leader and trailer sequences of the full-length genome and the luciferase reporter gene. As shown in Fig. 4, the levels of mRNA and cRNA were reduced the most when N mutants were included in the minigenome assays. Since vRNA could also be produced by the minigenome plasmid, the level of vRNA in this assay would be the mixture of two sources. Based on the changes in the vRNA levels, the level of mRNA was changed to a large degree compared to the wild type, consistent with the reduction in luciferase activities. For L174A mutant that has a reduced level of protein expression, the vRNA level was similar to that when the wt N was included in the minigenome assay, suggesting that effects on viral RNA synthesis was not due to the protein level of the N mutant.
FIG 4.

RT-qPCR quantification of mRNA, cRNA, and vRNA from the minigenome assay. Reduction in viral RNA levels of the E169A, F171A, and L174A mutants compared to the wt N, respectively, was calculated using 2−ΔΔCT values. The log2 of 2−ΔΔCT is plotted on the vertical axis to show the relative reduction of RNA, and the shade for each viral RNA species is shown.
Compensatory mutations in other viral proteins.
If the loop residues are involved in supporting viral RNA synthesis by vRdRp, it may be expected that compensatory mutations in other viral proteins can restore the activity of viral RNA synthesis by vRdRp. To identify the compensatory mutations, each of the mutations in the loop was introduced in the N gene of the genome of the parent VSV and mutant viruses were rescued by reverse genetics (12). At first, the mutant viruses were passaged five times in BHK-21 cells at 31°C at a low MOI of 0.01 to avoid the potential formation of defective interfering (DI) particles. In the last round, mutant viruses were harvested 24 h postinfection, and virus titers were determined by plaque assays. Subsequently, mutant viruses passaged at 31°C were used to infect BHK-21 cells at 37°C with a multiplicity of infection (MOI) of 1.0, followed by three more passages using an MOI of 0.01 and titering by plaque assays. Serial passages were continued until the virus titers reached 107 PFU/ml (similar to wt VSV). The viruses from the last round were plaque purified in HeLa cells, and the genome was sequenced for each mutant virus (Table 1). Compensatory mutations were identified in the L protein of all three mutant viruses, confirming the critical role of the helix-loop motif in the interplay between the N and L protein to support viral RNA synthesis. In addition, a compensatory mutation was also found in the P protein in mutant Niii (Leu174Ala). This is also consistent with the role the helix-loop motif in supporting vRdRp activities because the interactions of P with N contribute to the activity of viral RNA synthesis (13, 14). More interestingly, mutations were also identified in the N protein (Table 1). This suggests that optimal correlations between L and N are required for efficient viral growth. No mutations were found in M and G proteins.
TABLE 1.
Mutations identified in rescued viruses passaged at 37°C
| Virus | Passage | N gene(s) | P gene | L gene |
|---|---|---|---|---|
| Ni (E169A) | 14 | M166I, R179H | I753M | |
| Nii (F171A) | 26 | L110V | R998Q | |
| Niii (L174A) | 18 | L174V | S264Y | R728K |
We noticed that the mutant viruses passaged at 31°C did not grow well when they were first transferred to 37°C. This indicates that the initial compensatory mutations meet the minimum requirements to restore the viral RNA synthesis. Further compensatory mutations are required to fully restore the activity of vRdRp or effective virus assembly at 37°C.
Verification of the compensatory mutations.
Compensatory mutations identified in the rescued mutant viruses were introduced in the plasmids expressing N, P, and L proteins, respectively, and the minigenome assays were repeated. As shown in Fig. 5, the compensatory mutation in the L protein alone is not sufficient to restore the activity of viral RNA synthesis with the Ala mutations of Glu169 and Leu174 introduced in the N protein. In the case of Phe171, the mutation to Ala did not result in a large reduction of viral RNA synthesis, and the single compensatory mutation of Arg998Gln in the L protein is sufficient to restore the activity. Additional mutation of Leu110Val in the N protein did not further enhance the activity of viral RNA synthesis, implying this mutation may be related only to the assembly of the virus. On the other hand, all compensatory mutations are required for Glu169 and Leu174 mutations in order to fully restore the activity. The minigenome assays confirmed that the Leu174Val mutation is required for full restoration of the polymerase activity.
FIG 5.

Verification of compensatory mutations. Luciferase activities were measured in minigenome assays. The horizontal axis is labeled as follows: controls, P169a (pN-E169A, pP, and pL), P169b (pN-E169A, pP, and pL-I753M), P169c (pN-M166I/E169A/R179H, pP, an pL-I753M), P171a (pN-F171A, pP, and pL), P171b (pN-F171A, pP, an pL-R998Q), P171c (pN-L110V/F171A, pP, and pL-R998A), P174a (pN-L174A, pP, and pL), P174b (pN-L174A, pP, and pL-R728K), P174c (pN-L174V, pP-S264Y, and pL-R728K), and P174d (pN-L174A, pP-S264Y, and pL-R728K). Mutations introduced in the plasmids that express the corresponding proteins or the wt plasmids are indicated in parentheses.
Confirmation of compensatory mutations required for virus replication.
To verify that the identified compensatory mutations are sufficient to construct a viable mutant virus, the mutations from the three mutant viruses were introduced in the genome of the parent VSV, and virus rescue was carried out subsequently. Mutant viruses corresponding to all N mutations were successfully rescued and grew to similar titers as the parent VSV and wt VSV (Fig. 6). This result confirms that mutations identified in these mutant viruses can produce viable viruses when they are introduced in the parent virus.
FIG 6.

Growth curves of mutant viruses. Compensatory mutations identified in Ni, Nii, and Niii were introduced in the genome of the parent VSV (mcP VSV), and the mutant viruses were rescued (mcP VSV-Ni, mcP VSV-Nii, and mcP VSV-Niii; Table 1). Confluent BHK-21 cells were infected by each marked virus with an MOI of 0.01. Samples were collected from each infection at the given time points, and the virus titers were determined by plaque assays.
DISCUSSION
The mechanism of viral RNA synthesis by NSV vRdRp is unique because the template is the nucleocapsid, not the free RNA genome. Since the genomic RNA is sequestered in the nucleocapsid, vRdRp must induce structural changes in the N protein in order to gain access to the sequestered RNA. Another important requirement is that the integrity of the nucleocapsid must be retained after the viral RNA synthesis is terminated. The size of vRdRp (7) suggests that the sequestered RNA may be locally released by vRdRp, without disrupting the overall structure of the nucleocapsid.
The atomic structure of the nucleocapsid and the N protein has been determined for a number of NSVs, and in the form of NLP, empty NLP, or the N subunit (3–6, 15–34). As shown by the vast amount of structural data, the common scheme of nucleocapsid assembly of NSVs involves extensive cross-subunit interactions by termini or loops extended from the core of the N protein (35). These extensive interactions make the overall structure of the nucleocapsid very stable. This raises the question of how vRdRp may gain access to the sequestered RNA. Based on an open conformation found in the RNA-free N subunit (5, 6), one possible mechanism is to induce the open conformation of the N protein by vRdRp. However, this mechanism would require a large conformational change (up to a 30° rotation) of the domains in the N protein and disruption of the cross-subunit interactions by the extended polypeptides. This not only requires broad interactions between vRdRp and the nucleocapsid but also may result in disassembly of the nucleocapsid. The open conformation of the N protein is stabilized by the chaperone P protein to serve as the precursor for nucleocapsid assembly. The assembly of the nucleocapsid may not be a reversible process after it has been completed and the P protein been released from the assembled nucleocapsid.
In this study, we focused on a structural motif of the helix 5-loop in the N protein of VSV that constitutes the access gate to the sequestered RNA. This structural motif is one of the two flexible regions in the nucleocapsid, suggesting its conformational changes are more easily induced by interactions with vRdRp. Structural changes of the helix 5-loop are expected to be local and would not disrupt the nucleocapsid assembly. Previous studies showed that residues on helix 5 directly interact with the sequestered genomic RNA and are essential for RNA encapsidation and viral RNA synthesis (3, 36). The structure of the nucleocapsid binding domain of the P protein bound in the NLP indicated that vRdRp would face the helix 5-loop motif when it binds the nucleocapsid (8), implying a direct contact between them. Our mutagenesis studies proved that residues Met157, Glu169, Phe171, and Leu174 in the helix 5-loop motif are essential for the activity of viral RNA synthesis. The N mutations do not interrupt the assembly of the nucleocapsid and appear to mainly affect the synthesis of mRNA and cRNA. Compensatory mutations in vRdRp (L and P proteins), as well as those in the N protein, can restore the activity of viral RNA synthesis. These mutations are also sufficient to generate a viable mutant virus when introduced in the wt viral genome. Since the initial mutations we introduced in the N protein were alanines, which eliminates the potential interactions of the wt N residues with other proteins, residues resulted from compensatory mutations may not directly reestablish interactions with the mutated Ala but rather interactions with the helix 5-loop motif. This is also consistent with the observation that additional mutations in the N protein are required to fully restore the activity of vRdRp. Nevertheless, our study confirmed that potential interactions of the helix 5-loop motif with vRdRp may be required for viral RNA synthesis in VSV. A similar flexible structural motif is also found in the N protein of paramyxoviruses (Nipah virus, parainfluenza virus 5, and mumps virus), suggesting a common mechanism for accessing sequestered RNA in NSV nucleocapsid (5, 26, 37).
MATERIALS AND METHODS
Cell culture and viruses.
BSR-T7 (a gift from Biao He, University of Georgia) and BHK-21 cells were maintained in Dulbecco modified Eagle medium (DMEM) containing 5% fetal bovine serum (FBS) with 100 units of penicillin, 20 U of streptomycin (10). BSR-T7 cells were additionally supplemented with Geneticin G418 (1 mg/ml) on every second passage. HeLa cells were maintained in DMEM containing 10% FBS 100 U of penicillin, and 20 U of streptomycin (38). Recombinant vaccinia virus vTF7-3 were prepared in CV-1 cells cultured in Eagle minimum essential medium supplemented with 10% FBS.
Plasmid mutagenesis.
The original plasmids pLuc, pN, pP, and pL were kindly provided by Asit K. Pattnaik at the University of Nebraska—Lincoln. pLuc was constructed in pGEM-3 vector by inserting a leader sequence, the luciferase gene and a trailer sequence (39). pN, pP, and pL were used to express N, P, and L proteins of VSV. All site-directed mutations in N, P, and L genes were constructed using Q5 site-directed mutagenesis kit (New England BioLabs) according to the manufacturer's instructions. The same method was also used to introduce site-directed mutations in the full-length VSV genome.
The wt full-length VSV genome plasmid is a gift from Gail Wertz at the University of Alabama at Birmingham. The full-length VSV (Indiana serotype) genome was modified by inserting the coding sequence of a mCherry protein fused at the N terminus of the P gene (mcP VSV), using the overlap PCR method (Table 2).
TABLE 2.
Primers used for introducing the mCherry gene in the full-length genome of the VSV
| Primer | Nucleotide sequence (5′–3′)a |
|---|---|
| N(+)-F | CAGCCTGATGACATTGAGTATACATCTCTTACTACAGCAGG |
| N-mC(–)-R | TCCTCGCCCTTGCTCACCATGGTGGCGGCGATATCTGTTAGTTTTTTT |
| P(+)-F | ATGGACGAGCTGTACAAGggaggaaacagcggaggaATGGATAATCTCACAAAAGTTCG |
| P-M(–)-R | GCCTATTAAGTCATTATGCCAATTTAAATCTGAGCTTGACGGGC |
| mC(+)-F | TATGAAAAAAACTAACAGATATCGCCGCCACCATGGTGAGCAAGGGCGAGG |
| mC-P(–)-R | ACTTTTGTGAGATTATCCATtcctccgctgtttcctccCTTGTACAGCTCGTCCAT |
The underlined portions indicate the recognition sites of restriction enzymes, BstZ17 I and SwaI, respectively. The Kozak sequence is italicized. Lowercase lettering indicates flexible linker (GGNSGG) (40).
VSV minigenome assay.
BSR-T7 cells seeded to approximately 95% confluence in 12-well plates were infected with vTF7-3 at an MOI of 1 to 5 for 1 h at 37°C. Cells were washed twice with Opti-MEM serum-free medium (Invitrogen) and transfected with 2 μg of pLuc, 1.5 μg of pN, 1 μg of pP, and 0.5 μg of pL by using Lipofectamine 2000 (Invitrogen) (39). The cells were incubated for 6 h at 37°C, and then fresh DMEM with 5% FBS was exchanged. At 24 h posttransfection, cell lysates were assayed for the luciferase activity.
Western blotting.
Western blots were prepared as described previously (40). Lysates were prepared from BSR-T7 cells transfected with N-WT, N-E169A, N-F171A, and N-L174A. Samples were run on 8.5% acrylamide SDS-PAGE gels, and Western blots were probed with specific antibodies for anti-VSV N and developed using Pierce ECL Western blotting substrate (Thermo Fisher Scientific).
N protein expressions and purifications.
All of cDNA mutations were cloned into the expression vector previously shown (7) using a HiFi assembly kit with forward primer (5′-CTTAAGAAGGAGATATACCATGGCTTCTGTTACAGTCAAG-3′) and reverse primer (5′-GATGGCCCATGGTATATCTCCTTCATTGTCAAATTCTGAC-3′) according to the manufacturer's protocol. Positive clones were identified by sequencing and transformed into E. coli strain BL21(DE3). The N rings were expressed and purified as previously described (9). After purification, size exclusion was carried out with a HiLoad 16/100 Superdex 200 Prep-Grade column equilibrated with 50 mM Tris (pH 7.5) and 300 mM NaCl. The second peak, corresponding to the pure NLPs, was analyzed by SDS-PAGE for purity, and then utilized for further transmission electron microscopy (TEM) experiments.
Transmission electron microscopy.
Images were taken at 30K magnification on a JEOL JEM-1400 (120 kV) TEM apparatus using a 2k×2k, 14-μm Ultrascan1000 (Gatan). Samples grids (25 μg/ml concentration of protein) were prepared by adding 2 μl onto a carbon-coated, 400-mesh copper grid. After 1 min, the sample was blotted with Whatman 4 filter paper. A 2-μl buffer wash/blot step was added before staining with 4% uranyl acetate for 30 s and blotting away the excess. All particle picking and 2D class averages were performed in EMAN2 (41), and selected class averages and particles are shown in Fig. 3.
Virus rescue.
mcP VSV was used as the parent VSV to construct the mutant virus genome, according to previously described procedures (12, 42–44). Briefly, confluent 60-mm dishes of BSR-T7 cells were infected with vTF7-3 at an MOI of 10. After 45 min, the cells were cotransfected with a plasmid encoding the full-length VSV genome containing various mutations in the corresponding viral genes (4 μg) and pN (6 μg), pP (4 μg) and pL (2 μg) proteins in 20 μl of Lipofectamine 2000. After 48 to 72 h, the culture medium was collected, filtered through a 0.22-μm-pore size syringe filter twice to remove vTF7-3 vaccinia virus. Viral titers were determined by a plaque assay as described previously (45).
Plaque purification of rescued viruses.
Briefly, monolayers of HeLa cells were grown in 12-well tissue culture plates. Cells were infected with inoculums of the mutant virus 10-fold serially diluted in DMEM without serum. After 1 h of incubation at 37°C in 5% CO2, the infection medium was aspirated and washed three times with phosphate-buffered saline (PBS) and then overlaid with 1 ml of 0.8% (wt/vol) agarose in DMEM. After 24 h of incubation, four plaques were picked from at least two different wells. The agarose plugs were individually transferred to 60-mm plates of BHK-21 cells. Virus growth was monitored by observing mCherry using fluorescence microscopy, and virus titers were determined by plaque assays.
RNA extraction and reverse transcription-PCR.
Viral RNAs were extracted from infected cell cultures by using the RNeasy minikit (Qiagen) according to the manufacturer's instructions, and the total RNA was used in reverse transcription to generate cDNA by using random nonamers (Sigma) as described previously (13, 46). Briefly, the 20-μl reaction mixtures contained 10 μl of RNA, 4 μl of 5× first-strand buffer, 1 μl of random nonamers primers (Sigma), 1 μl of a 10 mM deoxynucleoside triphosphate mix, 2 μl of 0.1 M dithiothreitol, 1 μl of RNaseOUT (Thermo Fisher Scientific) inhibitor, and 1 μl of SuperScript II reverse transcriptase (Invitrogen). The samples were incubated at 42°C for 1 h, and the reaction was inactivated by heating at 70°C for 15 min. The PCRs were performed by using Phusion High-Fidelity DNA polymerase (Thermo Fisher).
Real-time qPCR.
Total RNA was extracted, as described above, from BSR-T7 cells from either mock treated, treated, or transfected with the different luciferase minigenomes. The reverse transcriptions were performed using the same protocol as above, but using specific primers for mRNA (5′-CCAGATCGTTCGAGTCGTTTTTTTTTTTTTTTTTTTACACGGCGATCTTGCC-3′), cRNA (5′-GCTAGCTTGAGCTAGGCATCCGCCGATATCTGTTAG-3′), vRNA (5′-GGCCGTCATGGTGGCGAATACGAAGACAAACAAACC-3′), and β-actin (5′-AGCACTGTGTTGGCGTACAG-3′). Real-time PCR was performed using mRNA forward primer (5′-CCAGATCGTTCGAGTCGT-3′) and reverse primer (5′-CGCGGTGGTGTTGTGTTC-3′), cRNA forward primer (5′-GCTAGCTTCAGCTAGGCATC-3′) and reverse primer (5′-AAACAGAAAACCGACACCTG-3′), vRNA forward primer (5′-AGCAGGTTTGTTGTACGC-3′) and reverse primer (5′-GGCCGTCATGGTGGCGAAT-3′), and β-actin forward primer (5′-AGAGCTACGAGCTGCCTGAC-3′) and reverse primer (5′-AGCACTGTGTTGGCGTAGAG-3′) on an QuantStudio 3 real-time PCR system utilizing a PowerUp SYBR green master mix according to the manufacturer's protocol (Thermo Fisher Scientific). All data were analyzed using the 2−ΔΔCT method as previously described, and the mock, template-free, no-RT-PCR, and template-only controls did not yield a CT value (data not included) after 40 cycles, indicating specific amplification of the selected genes (47).
Sequence analysis of viral genomes.
The primers shown in Table 3 were used to amplify the full-length VSV genomes. Each fragment was cloned into pCR2.1 vector containing the LacZ gene (Invitrogen). Automated sequence analysis was carried out by DNAStar 7.0 software.
TABLE 3.
Primers used for amplification of the full-length genome of the VSV in sequencing
| Primer | Nucleotide sequence (5′–3′) |
|---|---|
| AmpVSV1-F | ACGAAGACAAACAAACC |
| AmpVSV1-R | TCTGAAGTGCTCTGGTAC |
| AmpVSV2-F | TTCTAATCTAAAGGCCAC |
| AmpVSV2-R | CTTCAAGTTGCATATCGG |
| AmpVSV3-F | TAGGGAGGATGCAAACGG |
| AmpVSV3-R | TTGTCGGATAGGAGGTTG |
| AmpVSV4-F | CGGATTAATGAGAGCAAG |
| AmpVSV4-R | ACGAAGACCACAAAACCAG |
Determination of growth kinetics.
Multistep growth curves were generated using a protocol similar to that previously described (48). Briefly, BHK-21 cells seeded to approximately 95% confluence in 12-well plates were infected with each of the mutant viruses at an MOI of 0.01. After 1 h of adsorption at 37°C in 5% CO2 with periodic rocking, the inoculum was removed. The cells were washed three times with PBS to remove unbound virus, and then 1 ml of DMEM was added. At the indicated time points (6, 12, 18, 24, 30, and 36 h), the supernatant of each well was collected and stored at −80°C for determination of the virus titer by a plaque assay in a later time.
ACKNOWLEDGMENT
This study was supported in part by NIH grant R01 AI106307.
REFERENCES
- 1.Emerson SU, Wagner RR. 1972. Dissociation and reconstitution of the transcriptase and template activities of vesicular stomatitis B and T virions. J Virol 10:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emerson SU, Yu Y. 1975. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J Virol 15:1348–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green TJ, Zhang X, Wertz GW, Luo M. 2006. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313:357–360. doi: 10.1126/science.1126953. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, Green TJ, Tsao J, Qiu S, Luo M. 2008. Role of intermolecular interactions of vesicular stomatitis virus nucleoprotein in RNA encapsidation. J Virol 82:674–682. doi: 10.1128/JVI.00935-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yabukarski F, Lawrence P, Tarbouriech N, Bourhis JM, Delaforge E, Jensen MR, Ruigrok RW, Blackledge M, Volchkov V, Jamin M. 2014. Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat Struct Mol Biol 21:754–759. doi: 10.1038/nsmb.2868. [DOI] [PubMed] [Google Scholar]
- 6.Renner M, Bertinelli M, Leyrat C, Paesen GC, Saraiva de Oliveira LF, Huiskonen JT, Grimes JM. 2016. Nucleocapsid assembly in pneumoviruses is regulated by conformational switching of the N protein. Elife 5:e12627. doi: 10.7554/eLife.12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang B, Li Z, Jenni S, Rahmeh AA, Morin BM, Grant T, Grigorieff N, Harrison SC, Whelan SP. 2015. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 162:314–327. doi: 10.1016/j.cell.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green TJ, Luo M. 2009. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc Natl Acad Sci U S A 106:11713–11718. doi: 10.1073/pnas.0903228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green TJ, Macpherson S, Qiu S, Lebowitz J, Wertz GW, Luo M. 2000. Study of the assembly of vesicular stomatitis virus N protein: role of the P protein. J Virol 74:9515–9524. doi: 10.1128/JVI.74.20.9515-9524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das SC, Nayak D, Zhou Y, Pattnaik AK. 2006. Visualization of intracellular transport of vesicular stomatitis virus nucleocapsids in living cells. J Virol 80:6368–6377. doi: 10.1128/JVI.00211-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ge P, Tsao J, Schein S, Green TJ, Luo M, Zhou ZH. 2010. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science 327:689–693. doi: 10.1126/science.1181766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci U S A 92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harouaka D, Wertz GW. 2012. Second-site mutations selected in transcriptional regulatory sequences compensate for engineered mutations in the vesicular stomatitis virus nucleocapsid protein. J Virol 86:11266–11275. doi: 10.1128/JVI.01238-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rahmeh AA, Morin B, Schenk AD, Liang B, Heinrich BS, Brusic V, Walz T, Whelan SP. 2012. Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc Natl Acad Sci U S A 109:14628–14633. doi: 10.1073/pnas.1209147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albertini AA, Wernimont AK, Muziol T, Ravelli RB, Clapier CR, Schoehn G, Weissenhorn W, Ruigrok RW. 2006. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313:360–363. doi: 10.1126/science.1125280. [DOI] [PubMed] [Google Scholar]
- 16.Ye Q, Krug RM, Tao YJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 17.Raymond DD, Piper ME, Gerrard SR, Smith JL. 2010. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc Natl Acad Sci U S A 107:11769–11774. doi: 10.1073/pnas.1001760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA Jr, Jensen MR, Blackledge M, Ruigrok RW, Jamin M. 2011. Structure of the vesicular stomatitis virus N0-P complex. PLoS Pathog 7:e1002248. doi: 10.1371/journal.ppat.1002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Y, Wang W, Ji W, Deng M, Sun Y, Zhou H, Yang C, Deng F, Wang H, Hu Z, Lou Z, Rao Z. 2012. Crimean-Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. Proc Natl Acad Sci U S A 109:5046–5051. doi: 10.1073/pnas.1200808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carter SD, Surtees R, Walter CT, Ariza A, Bergeron E, Nichol ST, Hiscox JA, Edwards TA, Barr JN. 2012. Structure, function, and evolution of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. J Virol 86:10914–10923. doi: 10.1128/JVI.01555-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raymond DD, Piper ME, Gerrard SR, Skiniotis G, Smith JL. 2012. Phleboviruses encapsidate their genomes by sequestering RNA bases. Proc Natl Acad Sci U S A 109:19208–19213. doi: 10.1073/pnas.1213553109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niu F, Shaw N, Wang YE, Jiao L, Ding W, Li X, Zhu P, Upur H, Ouyang S, Cheng G, Liu ZJ. 2013. Structure of the Leanyer orthobunyavirus nucleoprotein-RNA complex reveals unique architecture for RNA encapsidation. Proc Natl Acad Sci U S A 110:9054–9059. doi: 10.1073/pnas.1300035110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong H, Li P, Bottcher B, Elliott RM, Dong C. 2013. Crystal structure of Schmallenberg orthobunyavirus nucleoprotein-RNA complex reveals a novel RNA sequestration mechanism. RNA 19:1129–1136. doi: 10.1261/rna.039057.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ariza A, Tanner SJ, Walter CT, Dent KC, Shepherd DA, Wu W, Matthews SV, Hiscox JA, Green TJ, Luo M, Elliott RM, Fooks AR, Ashcroft AE, Stonehouse NJ, Ranson NA, Barr JN, Edwards TA. 2013. Nucleocapsid protein structures from orthobunyaviruses reveal insight into ribonucleoprotein architecture and RNA polymerization. Nucleic Acids Res 41:5912–5926. doi: 10.1093/nar/gkt268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiao L, Ouyang S, Liang M, Niu F, Shaw N, Wu W, Ding W, Jin C, Peng Y, Zhu Y, Zhang F, Wang T, Li C, Zuo X, Luan CH, Li D, Liu ZJ. 2013. Structure of severe fever with thrombocytopenia syndrome virus nucleocapsid protein in complex with suramin reveals therapeutic potential. J Virol 87:6829–6839. doi: 10.1128/JVI.00672-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alayyoubi M, Leser GP, Kors CA, Lamb RA. 2015. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein-RNA complex. Proc Natl Acad Sci U S A 112:E1792–E1799. doi: 10.1073/pnas.1503941112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo Y, Wang W, Sun Y, Ma C, Wang X, Wang X, Liu P, Shen S, Li B, Lin J, Deng F, Wang H, Lou Z. 2015. Crystal structure of the core region of hantavirus nucleocapsid protein reveals the mechanism for ribonucleoprotein complex formation. J Virol 90:1048–1061. doi: 10.1128/JVI.02523-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Surtees R, Ariza A, Punch EK, Trinh CH, Dowall SD, Hewson R, Hiscox JA, Barr JN, Edwards TA. 2015. The crystal structure of the Hazara virus nucleocapsid protein. BMC Struct Biol 15:24. doi: 10.1186/s12900-015-0051-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eleouet JF, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. doi: 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- 30.Kirchdoerfer RN, Abelson DM, Li S, Wood MR, Saphire EO. 2015. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep 12:140–149. doi: 10.1016/j.celrep.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung DW, Borek D, Luthra P, Binning JM, Anantpadma M, Liu G, Harvey IB, Su Z, Endlich-Frazier A, Pan J, Shabman RS, Chiu W, Davey RA, Otwinowski Z, Basler CF, Amarasinghe GK. 2015. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein-RNA interactions. Cell Rep 11:376–389. doi: 10.1016/j.celrep.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reguera J, Malet H, Weber F, Cusack S. 2013. Structural basis for encapsidation of genomic RNA by La Crosse Orthobunyavirus nucleoprotein. Proc Natl Acad Sci U S A 110:7246–7251. doi: 10.1073/pnas.1302298110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutsche I, Desfosses A, Effantin G, Ling WL, Haupt M, Ruigrok RW, Sachse C, Schoehn G. 2015. Structural virology: near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 348:704–707. doi: 10.1126/science.aaa5137. [DOI] [PubMed] [Google Scholar]
- 34.Guryanov SG, Liljeroos L, Kasaragod P, Kajander T, Butcher SJ. 2016. Crystal structure of the measles virus nucleoprotein core in complex with an N-terminal region of phosphoprotein. J Virol 90:2849–2857. doi: 10.1128/JVI.02865-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Green TJ, Cox R, Tsao J, Rowse M, Qiu S, Luo M. 2014. Common mechanism for RNA encapsidation by negative-strand RNA viruses. J Virol 88:3766–3775. doi: 10.1128/JVI.03483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rainsford EW, Harouaka D, Wertz GW. 2010. Importance of hydrogen bond contacts between the N protein and RNA genome of vesicular stomatitis virus in encapsidation and RNA synthesis. J Virol 84:1741–1751. doi: 10.1128/JVI.01803-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Severin C, Terrell JR, Zengel JR, Cox R, Plemper RK, He B, Luo M. 2016. Releasing the genomic RNA sequestered in the mumps virus nucleocapsid. J Virol pii:JVI.01422-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z, Gabbard JD, Mooney A, Chen Z, Tompkins SM, He B. 2013. Efficacy of parainfluenza virus 5 mutants expressing hemagglutinin from H5N1 influenza A virus in mice. J Virol 87:9604–9609. doi: 10.1128/JVI.01289-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nayak D, Panda D, Das SC, Luo M, Pattnaik AK. 2009. Single-amino-acid alterations in a highly conserved central region of vesicular stomatitis virus N protein differentially affect the viral nucleocapsid template functions. J Virol 83:5525–5534. doi: 10.1128/JVI.02289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryder AB, Buonocore L, Vogel L, Nachbagauer R, Krammer F, Rose JK. 2015. A viable recombinant rhabdovirus lacking its glycoprotein gene and expressing influenza virus hemagglutinin and neuraminidase is a potent influenza vaccine. J Virol 89:2820–2830. doi: 10.1128/JVI.03246-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 42.Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci U S A 92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitt MA. 2010. Generation of VSV pseudotypes using recombinant DeltaG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J Virol Methods 169:365–374. doi: 10.1016/j.jviromet.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Das SC, Pattnaik AK. 2005. Role of the hypervariable hinge region of phosphoprotein P of vesicular stomatitis virus in viral RNA synthesis and assembly of infectious virus particles. J Virol 79:8101–8112. doi: 10.1128/JVI.79.13.8101-8112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flanagan EB, Ball LA, Wertz GW. 2000. Moving the glycoprotein gene of vesicular stomatitis virus to promoter-proximal positions accelerates and enhances the protective immune response. J Virol 74:7895–7902. doi: 10.1128/JVI.74.17.7895-7902.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rasmussen TB, Uttenthal A, de Stricker K, Belak S, Storgaard T. 2003. Development of a novel quantitative real-time RT-PCR assay for the simultaneous detection of all serotypes of foot-and-mouth disease virus. Arch Virol 148:2005–2021. doi: 10.1007/s00705-003-0145-2. [DOI] [PubMed] [Google Scholar]
- 47.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 48.Ammayappan A, Nace R, Peng KW, Russell SJ. 2013. Neuroattenuation of vesicular stomatitis virus through picornaviral internal ribosome entry sites. J Virol 87:3217–3228. doi: 10.1128/JVI.02984-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.PyMOL. The PyMOL molecular graphics system, v1.3. Schrödinger, LLC, Portland, OR. [Google Scholar]