

SERINC5 is a member of a family of multipass transmembrane proteins that inhibit the infectivity of retroviruses, including HIV-1. These proteins are incorporated into virions and inhibit infection of target cells unless counteracted by viral antagonists such as HIV-1 Nef. The only other biological function with which these proteins have been associated is the formation of serine-containing membrane lipids. Here we show that SERINC5 is a glycosylated protein and that N-glycosylation is important for its steady-state expression. In the absence of N-glycosylation, SERINC5 is prone to proteasomal degradation. Nonetheless, N-glycosylation per se is required neither for the ability of SERINC5 to inhibit HIV-1 infectivity nor for its sensitivity to antagonism by Nef.
KEYWORDS: glycosylation, Nef, SERINC5, human immunodeficiency virus
ABSTRACT
SERINC5 is an inhibitor of retroviral infectivity that is counteracted by viral proteins, including HIV-1 Nef. Inhibition of infectivity by SERINC5 is associated with its incorporation into virions. Nef counteracts this inhibition, presumably by removing SERINC5 from sites of virion assembly at the plasma membrane. While evaluating the virion incorporation of SERINC5, we observed that a relatively high molecular weight form was preferentially present in virions. We used various glycosidases to establish that virion-associated SERINC5 is modified by N-linked, complex glycans, whereas the majority of SERINC5 in cells is of relatively low molecular weight and is modified by high-mannose glycans. Sequence alignment of SERINC family proteins led us to identify a conserved N-glycosylation site, N294, in SERINC5. We mutated this site to evaluate its effect on glycosylation, the restrictive activity of SERINC5, and the sensitivity of SERINC5 to antagonism by Nef. Our results demonstrate that N294 is the major site of N-glycosylation in SERINC5. Although N-glycosylation was required neither for restrictive activity nor for sensitivity to Nef per se, we observed a decrease in the steady-state expression of glycosylation-deficient SERINC5 (the N294A mutant) compared to the wild-type protein. Expression of this mutant was partly restored by treatment of cells with MG132 (a proteasome inhibitor) but not with bafilomycin A1 (a lysosomal inhibitor). We conclude that although not required for restrictive activity or Nef sensitivity, N-linked glycosylation is important for maintaining the steady-state expression of SERINC5 and that nonglycosylated SERINC5 is likely subjected to a quality control mechanism that induces its proteasomal degradation.
IMPORTANCE SERINC5 is a member of a family of multipass transmembrane proteins that inhibit the infectivity of retroviruses, including HIV-1. These proteins are incorporated into virions and inhibit infection of target cells unless counteracted by viral antagonists such as HIV-1 Nef. The only other biological function with which these proteins have been associated is the formation of serine-containing membrane lipids. Here we show that SERINC5 is a glycosylated protein and that N-glycosylation is important for its steady-state expression. In the absence of N-glycosylation, SERINC5 is prone to proteasomal degradation. Nonetheless, N-glycosylation per se is required neither for the ability of SERINC5 to inhibit HIV-1 infectivity nor for its sensitivity to antagonism by Nef.
INTRODUCTION
The accessory proteins of primate lentiviruses have key roles in the counteraction of host proteins and processes that are deleterious to viral replication (1, 2). Nef is particularly important for pathogenesis in vivo (3–5). By coopting the vesicular trafficking machinery of the cell, Nef downregulates various cell surface molecules, including major histocompatibility complex class I (MHC-I) and CD4, thereby protecting infected cells from clearance by cytotoxic T and NK cells (6–9). Nef-deficient viruses are also intrinsically less infectious than their Nef-encoding counterparts (10–14). This phenomenon remained unexplained until the discovery that SERINC proteins inhibit infectivity and are counteracted and downregulated by Nef (15–17).
SERINCs belong to a conserved gene family in eukaryotes. These proteins have 10 or 11 predicted transmembrane domains, and they derive their name from their first noted function, SERine INCorporator, as they were reported to play a role in the incorporation of serine into membrane lipids (18). Unlike other known human immunodeficiency virus (HIV) restriction factors, SERINCs are not interferon inducible and hence can be categorized as a constitutive or intrinsic cellular defense. SERINC5 (S5) and S3 are incorporated into virions and inhibit the efficiency of target cell infection (16, 17). Nef from HIV-1 and simian immunodeficiency virus (SIV), Glycogag from murine leukemia virus (MLV), and S2 from EIAV can each counteract the restrictive activity of the SERINC proteins; this counteraction is associated with a decrease in the cell surface expression of the SERINCs and a decrease in their incorporation into the virions (15–17, 19).
The five known members of the human SERINC family share 17% amino acid identity and have similar predicted topologies. However, they differ in the extent to which they restrict HIV-1 infectivity and are susceptible to Nef. SERINC5 seems to be the most potent restrictor and accounts for the majority of the Nef effect (at least in T cell lines); in contrast, SERINC3 has modest restrictive activity, and SERINC2 is devoid of restrictive activity (16, 17). The study of chimeras between the nonrestrictive SERINC2 and the restrictive SERINC5 led to a partial mapping of SERINC5's restrictive activity (20). While the ability of SERINC5 to restrict HIV-1 infectivity is conserved among vertebrates, sensitivity to Nef is not. The study of chimeras between the Nef-sensitive human SERINC5 and Nef-resistant frog SERINC5 (from Xenopus tropicali) led to the identification of a specific intracellular loop as a Nef-responsive determinant in SERINC5 (21).
The mechanism by which the SERINC proteins inhibit infectivity remains uncertain, but the current model is that virion-associated SERINC affects virion-target cell fusion. Since HIV-1 envelope glycoproteins (Envs) differ in their sensitivity to SERINC5 and SERINC3, SERINCs seem likely to act directly on Env, perhaps by destabilizing the trimer (22). SERINC5 renders HIV-1 particularly sensitive to neutralization by antibodies that recognize the membrane proximal external region (MPER) of gp41, such as 4E10 and 10E8 (23); these results support the notion that the SERINCs can affect the conformation of Env. Recent work has further suggested that SERINC5 affects the ability of virions to form fusion pores with target cell membranes (22). Since the virion derives its envelope lipids from the producer cell and SERINCs participate in the synthesis of serine-containing lipids, these proteins could plausibly affect viral infectivity by modifying the composition of the virion membrane; however, analysis of cellular and viral lipids in the absence or presence of SERINC5 revealed no differences in lipid composition (24).
While studying the incorporation of human SERINC5 into HIV-1 virions, we observed a difference in the apparent molecular weights of cellular SERINC5 (cS5) and virion-incorporated SERINC5 (vS5). cS5 ran in denaturing polyacrylamide gels (SDS-PAGE) as a doublet, with the major species running at a relatively low molecular weight (40 kDa) and the minor species running at a relatively high molecular weight (55 kDa). In contrast, vS5 consisted almost entirely of the high-molecular-weight 55-kDa form. We hypothesized that the high-molecular-weight form was glycosylated and that glycosylated SERINC5 was preferentially incorporated into virions. Using various glycosidases, we confirm that S5 is an N-glycosylated protein and that this form is selectively incorporated into virions. Sequence comparison of human SERINC proteins led to the identification of a conserved N-linked glycosylation site: N294 in SERINC5. Mutational analysis of this residue demonstrated that N-glycosylation is important for the steady-state expression of SERINC5; in the absence of glycosylation, SERINC5 is prone to proteasomal degradation. However, when wild-type SERINC5 and the N-glycosylation mutant (N294A) are compared at equal expression levels, glycosylation is strictly required neither for restrictive activity nor for counteraction by HIV-1 Nef.
RESULTS
A high-molecular-weight form of SERINC5 is preferentially incorporated into virions.
Current models suggest that SERINC5 (S5) incorporates into HIV-1 virions to inhibit infectivity and is excluded from virions by Nef. To study these phenomena, we first determined the amount of plasmid for SERINC5 expression that was nonsaturating. We transfected HEK293 Tet-On TRE-HIV-Δnef cells, which contain a nef-negative HIV-1 genome under the control of a doxycycline-inducible promoter, in a dose-response format with the pBJ5-S5-iHA expression plasmid (encoding SERINC5 with a hemagglutinin [HA] tag in an extracellular loop; 100 ng up to 1 μg), either with or without a Nef expression plasmid (pCINL; 400 ng). After 24 h, the cells were treated with doxycycline to induce viral gene expression. Eight hours later, whole-cell extracts (WCE) and virions were prepared for analysis by immunoblotting. Figure 1A shows the expression levels of SERINC5 in cells (cS5) and corresponding levels of virion-incorporated SERINC5 (vS5), in the absence and presence of Nef. cS5 increased with the increasing amounts of transfected plasmid; however, the signal saturated at the two or three highest plasmid amounts (lanes 4 to 6 and 9 to 11 of the upper panels, which show the cells). Hereafter, 30 and 100 ng of S5 expression plasmids, which were nonsaturating amounts, were used for transfections. In the absence of Nef, SERINC5 was incorporated into the virions in a dose-dependent manner (lanes 8 to 11 of the lower panels, which show the virions). The expression of Nef decreased virion incorporation of SERINC5 (lanes 3 to 6 of the lower panels), as has been shown previously by others (25, 26).
FIG 1.

SERINC5 runs as a doublet in SDS-PAGE of whole cells, but the minor, high-molecular-weight species of that doublet is selectively incorporated into virions and excluded by Nef. (A) HEK293 Tet-On TRE-HIV-ΔNef cells were transfected with increasing amounts (100, 350, 700, and 1,000 ng) of pBJ5-S5-iHA either with or without the addition of 400 ng of a Nef expression construct (pCI-NL). Twenty-four hours later, the production of virions was induced by the addition of 0.5 μg/ml of doxycycline for 8 h, after which the cells and supernates were harvested, processed, and immunoblotted for S5-iHA, Nef, p24 (capsid), and GAPDH. (B) HEK293 cells were transfected with 30 or 100 ng of pBJ5-S5-iHA (or an empty vector control) together with 1.5 μg of either pNL43 or pNL43ΔNef. Twenty-four hours later, the cells and supernates were harvested and immunoblotted as described for panel A. (C) SERINC3/5 double KO Jurkat-TAg cells were transfected with either 30 or 100 ng of pBJ5-S5-iHA (or an empty vector control) together with 1.5 μg of either pNL43 or pNL43ΔNef. Forty-eight hours later, the cells and supernates were processed and immunoblotted as described above.
We observed that cS5 ran as a doublet, with the major band running at 40 kDa and a minor band at 55 kDa. Surprisingly, the virion preparations revealed a different scenario: virion S5 (vS5) migrated as a single species of approximately 55 kDa, which is the size of the minor species of cS5. Apparently, the minor, higher-molecular-weight species of cS5 is selectively incorporated into virions as vS5. Notably, while the 55-kDa species of vS5 was modulated by Nef, Nef had little or no effect on either 40-kDa or 55-kDa cS5.
To generalize these observations to a system used more routinely to produce infectious virions, we transfected HEK293 cells with either pNL43 (a proviral clone encoding Nef) or pNL43ΔNef (an isogenic clone lacking an intact nef), either with or without cotransfection of pBJ5-S5-iHA. Twenty-four hours later, the cells and virions were isolated and immunoblotted as described above. As shown in Fig. 1B, cS5 was again detected as a doublet (lanes 2 and 3 and lanes 5 and 6 of the upper panels, which show the cells), with a major species of about 40 kDa and a minor species of 55 kDa. Again, the major species in virions, vS5, migrates with an apparent molecular weight of 55 kDa (lanes 2, 3, 5, and 6 in the lower panels, which show the virions). This 55-kDa species was relatively excluded from virions when Nef was expressed (compare NL4.3 to NL4.3ΔNef).
To ensure that these differences extended to T cells, we transfected SERINC3/5 double knockout (KO) Jurkat TAg cells with pBJ5-S5-iHA, again together with either pNL43 or pNL43ΔNef, and performed immunoblotting for cS5 in whole-cell extracts and vS5 in virion pellets obtained after partial purification through a sucrose cushion. As seen in Fig. 1C, the lower-molecular-weight species was again the most abundant in WCEs (lanes 2, 3, 5, and 6 in the top panels, which show the cells), while the higher-molecular-weight species was selectively incorporated into virions and was relatively excluded from virions by Nef (lanes 2, 3, 5, and 6 in the lower panels, which show the virions). In summary, three different experimental systems, including one that is T cell line-based, all indicate that a high-molecular-weight SERINC5 species is selectively incorporated into virions.
SERINC5 is modified by N-linked complex glycans.
We hypothesized that the high-molecular-weight form of SERINC5 is a consequence of a posttranslational modification, specifically glycosylation. To determine whether the observed species of SERINC5 were differentially glycosylated, we treated virion-incorporated SERINC5 (vS5) obtained from the doxycycline-inducible HIV expression system with various glycosidases (Fig. 2A, top panel). To maximize the amount of vS5, Nef was not coexpressed. Virion proteins were treated with endoglycosidase H (Endo-H), PNGase F, O-glycosidase, and neuraminidase (sialidase). Endo-H removes high-mannose glycans, which are added to proteins in the endoplasmic reticulum (ER). Subsequent trimming and addition reactions in the Golgi body yield complex glycans, which are resistant to Endo-H. PNGase F removes most N-linked glycans, whereas O-glycosidase removes O-linked glycans (glycans attached to serines and threonines) if they are not modified by sialic acid. We observed minimal decrease in the apparent molecular weight of vS5 treated with Endo-H (lanes 3 and 4) and essentially no change after treatment with O-glycosidase (lanes 7 and 8). We observed a small decrease in the apparent molecular weight of vS5 after treatment with neuraminidase, which removes terminal sialic acids (lanes 9 and 10). The most dramatic effect was caused by treatment with PNGase F (lanes 5 and 6). Compared to the untreated sample (lane 5), the PNGase F-treated S5 was reduced to <40 kDa in apparent molecular weight (lane 6), probably reflecting the size of the fully deglycosylated protein. These results indicate that 55-kDa vS5 is N-glycosylated and that the majority of the glycans are complex (insensitive to Endo-H). To independently assess these glycosidase reactions, we reprobed the virion blots for HIV-1 Env using an antibody to gp120 (Fig. 2A, bottom panel). gp120 was highly sensitive to PNGase F and partly sensitive to Endo-H, consistent with the presence of high-mannose glycans (27). Neuraminidase also caused a minor decrease in the apparent molecular weight of gp120, while O-glycosidase caused a paradoxical increase in apparent molecular weight, an effect that was reproducible and suggests that gp120 is probably modified by O-linked glycans. With the possible exception of the effect of O-glycosidase, these results are largely consistent with the known glycosylation status of gp120 and support the validity of the data with respect to SERINC5 (27).
FIG 2.

SERINC5 is N-glycosylated: the higher-molecular-weight species in virions contains complex glycans, while the lower-molecular-weight species that predominates in transfected cells contains immature, high-mannose glycans. (A) HEK293 Tet-On TRE-HIV1-ΔNef cells were transfected with either pBJ5-S5-iHA or an empty vector. Twenty-four hours later, the cells were treated with 0.5 μg/ml of doxycycline for 8 h to induce virus production. The virion pellets obtained by centrifugation through 20% sucrose cushions were suspended in 1× TCEP reducing buffer and then treated with the indicated glycosidases at 37°C for 1 h. The reaction products were first immunoblotted for SERINC5 (HA; top panel), and then the membrane was stripped and reprobed with anti-gp120 (bottom panel). The star indicates a nonspecific band. (B) HEK293 cells were transfected with 100 ng of pBJ5-S5-iHA and 1.5 μg of pNL43ΔNef; 24 h later, the cells and virions were isolated as described above. The virions were treated with PNGase F (or not) for 1 h at 37°C and immunoblotted for SERINC (HA). (C) SERINC3/5 double KO cells were transfected with pNL43ΔNef and 100 ng of pBJ5-S5-iHA. Virions were isolated as described above. The virions were treated with PNGase F as described for panel A and immunoblotted for SERINC5 (HA). (D) The cells from the experiment shown in panel B were suspended in 1× TCEP reducing buffer, and whole-cell extracts were prepared as described in Materials and Methods. An aliquot of the whole-cell extracts was treated with endoglycosidase H (Endo-H) or PNGase F for 1 h at 37°C. The “input” is untreated lysate left at 4°C. The reaction products were blotted for SERINC5 (HA).
We confirmed glycosylation using virions produced from HEK293 cells and SERINC3/5 double KO Jurkat TAg cells. As shown in Fig. 2B and C, vS5 from both these expression systems was sensitive to PNGaseF, confirming that the high-molecular-weight, virion-associated form of SERINC5 is N-glycosylated. We also treated WCEs of HEK293 cells expressing S5 (i.e., cS5) with Endo-H and PNGase F (Fig. 2D). Treatment with Endo-H decreased the apparent molecular weight of the lower-molecular-weight form of cS5 but not the higher form, indicating that the lower form contains high-mannose glycans, while the higher form contains complex glycans (compare lane 1 with lane 3). Treatment with PNGase F yielded essentially one species of under 40 kDa (lane 4). In summary, these data indicate that SERINC5 is modified by N-glycosylation; the data also indicate that the predominant lower-molecular-weight form found in transfected cells contains high-mannose glycans, while the higher-molecular-weight form found in virions contains complex glycans.
SERINC5 is glycosylated at residue N294.
To identify potential N-linked glycosylation sites in SERINC5, we analyzed the SERINC5 amino acid sequence using NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/). Two potential sites were identified: N113 and N294. According to the TOPCONS simulated topological model (http://topcons.cbr.su.se) (28), N113 is within intracellular loop 2 of SERINC5, whereas N294 is present in extracellular loop 7. Residue N294 is also very close to the site (290E-291H) within which insertion of an HA tag in pBJ5-S5-iHA renders SERINC5 detectable at the cell surface by flow cytometry (17), further supporting the notion that this residue is extracellular. To assess the extent of conservation of this glycosylation site in other SERINC family members, we aligned the primary sequences of all five human SERINC family members (Uniprot IDs: S1, Q9NRX5-1; S2, Q96SA4-1; S3, Q13530-1; S4, A6NH21-1; S5, Q86VE9-4). Figure 3A depicts a part of the sequence alignment, highlighting the potential N-linked glycosylation motifs. Each SERINC protein contains a potential site for N-glycosylation in this region (sequence NxS/T; underlined) (Fig. 3A).
FIG 3.

SERINC5 is glycosylated at residue N294. (A) Multiple-sequence alignment of the human SERINC family members using Clustal Omega software. The sequences were obtained from Uniprot (www.uniprot.org). The consensus asparagine (N) residue is within the underlined N-glycosylation motif (NxS/T). (B) Asparagine 294 (N294) of S5-iHA was mutated to alanine. pBJ5-S5-iHA (100 ng) and the pBJ5-(N294A)S5-iHA (100 ng) were used to transfect HEK293 Tet-On TRE-HIV-ΔNef cells that were treated with doxycycline to induce viral gene expression as described for Fig. 1A; alternatively, the same constructs were used to transfect HEK 293 cells together with pNL43ΔNef. The supernates were harvested and processed to obtain virions. The virion preparations were treated with PNGase F or with O-glycosidase as indicated. (C) HEK293 cells were transfected to express wild-type S5-iHA (30 ng of pBJ5-S5-iHA) or (N294A)S5-iHA [100 ng of pBJ5-(N294A)S5-iHA] without any proviral DNA. The cells were either left untreated or treated with DMSO or brefeldin A in DMSO for 4 h, after which they were harvested, and the cell lysates were immunoblotted for SERINC (HA).
We mutated N294 in SERINC5 to evaluate both its role in glycosylation and the functional significance of this modification. We expressed the (N294A)S5-iHA mutant in HEK293 Tet-On TRE-HIV-ΔNef cells and in HEK293 cells together with pNL43ΔNef. After partially purifying the virions, we treated the virion proteins with PNGase F (Fig. 3B). Untreated (N294A)S5-iHA protein migrated in SDS-PAGE at a size similar to that of the PNGase F-treated wild-type protein (compare lane 4 with 3 and lane 10 with 9). In contrast to the wild-type protein, treatment with PNGase F did not affect the migration of the (N294A)S5-iHA in SDS-PAGE (compare lanes 4 and 5 and lanes 10 and 11), indicating a lack of N-linked glycosylation and suggesting that N294 is the primary if not the sole site of N-linked glycosylation in SERINC5. To provide further evidence of glycosylation at this site, we treated wild-type S5-iHA- and (N294A)S5-iHA-expressing cells with brefeldin A, which blocks the transport of proteins from the ER to the Golgi complex. As shown in Fig. 3C, brefeldin A caused loss of the higher-molecular-weight species (compare lane 5 with lane 7) in the case of wild-type SERINC5, supporting the notion that this species contains complex glycans that are added in the Golgi complex. Again, the higher-molecular-weight species was not detected when the N294A mutant was expressed. Notably, we observed an increase in the band density of (N294A)S5-iHA after treatment of the cells with brefeldin A (compare lane 6 with lane 8), suggesting possible stabilization of the mutant protein under these conditions.
Lack of N-glycosylation decreases the steady-state expression of SERINC5 but does not affect intrinsic restrictive activity or sensitivity to Nef.
Since the glycosylated form of SERINC5 is preferentially incorporated into virions, we asked whether glycosylation is required for either the restrictive activity of SERINC5 or for its sensitivity to Nef. We expressed wild-type S5-iHA and (N294A)S5-iHA in HEK293 cells together with either pNL43 or pNL43ΔNef and measured virion infectivity using HeLa-P4.R5 indicator cells in an infectious center assay. The infectivity of each virion preparation was measured as described previously (29), and the number of infectious centers was normalized to the concentrations of p24 capsid antigen measured by enzyme-linked immunosorbent assay (ELISA) (Fig. 4A). HEK293 cells express endogenous SERINCs, and nef-negative virions are 2- to 5-fold less infectious than wild-type virions in our experience, even in the absence of expression of exogenous SERINC (data not shown). To attribute restrictive activity and counteraction by Nef only to the SERINC5 expressed experimentally by transfection, we calculated the infectivity of each sample relative to the “no exogenous SERINC5” control for each viral genotype, i.e., we set the infectivity of both the wild-type and nef-negative virion preparations in the absence of exogenous SERINC5 to 1.0. Wild-type S5-iHA and (N294A)S5-iHA restricted the infectivity of ΔNef virions similarly at both amounts of plasmids tested (30 and 100 ng). On the other hand, (N294A)S5-iHA appeared more sensitive to Nef; i.e., the infectivity of wild-type (Nef-positive) virion preparations was higher when (N294A)S5-iHA was expressed than when wild-type SERINC5 was expressed.
FIG 4.

The nonglycosylated (N294A)S5-iHA is expressed at steady state relatively less efficiently, but its restrictive activity and sensitivity to Nef are intrinsically similar to those of the wild-type protein. Zero, 30, or 100 ng of pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA was used to transfect HEK293 cells together with 1.5 μg of pNL43 or pNL43ΔNef proviral plasmids. Twenty-four hours after transfection, the cells and virions were isolated for measurement of infectivity, immunoblotting, and flow cytometry. (A) Virion infectivity. After partial purification through a 20% sucrose cushion, virions were used to infect HeLa P4.R5 indicator cells for 48 h. The infectious centers were developed using X-Gal and then counted using image analysis software. The data were normalized to p24 content as determined by ELISA. The relative infectivity was calculated by dividing the infectious centers/p24 for each sample by the corresponding no-SERINC5 control for each viral genotype (wild type or Δnef), which was set for each virus at 1.0. The magenta and blue boxes in the infectivity data indicate the conditions under which the expressions of the wild-type S5-iHA and (N294A)S5-iHA mutant were similar (also indicated by similar-color-coded stars in panel B). Individual datum points shown are the averages from two fully independent experiments; within each experiment, each datum point was derived from duplicate measurements of both infectious centers and p24 concentration. The error bars indicate plus and minus 1 standard deviation. (B) Exclusion of S5 from virions by Nef. Cells and virions from one of the experiments used to generate the data shown in panel A were analyzed by immunoblotting for the indicated proteins. (C) Flow cytometric analysis of SERINC5 surface downregulation. The virion producer cells from one of the experiments used to generate the data in panel A were stained with anti-HA antibody to detect surface SERINC5 expression and with 2G12 antibody (anti-HIV-1 gp120) to detect the Env glycoprotein on the cell surface, identifying virus-expressing cells. Two color plots of Env versus SERINC5 intensities are shown with the percentage of cells in each quadrant indicated. (D) Flow cytometric data from two independent experiments, including the data shown in panel C. The cells expressing Env were gated, and the mean fluorescence intensities (MFIs) of the SERINC5-iHA strains were determined using FlowJo software and then plotted using GraphPad Prism software; error bars are plus/minus the standard deviation.
We considered that the apparently greater Nef sensitivity of (N294A)S5-iHA might be due to its relatively lower expression (as seen in Fig. 4B). When we compared datum points at which similar amounts of SERINC5 were expressed (boxes in Fig. 4A and corresponding color-coded stars in Fig. 4B), the Nef sensitivity of (N294)S5-iHA and that of wild-type S5-iHA appeared similar. Figure 4B shows an immunoblot of the cells and virions in a representative experiment. (N294A)S5-iHA was expressed less efficiently than the wild-type SERINC5 (compare lanes 5 and 6 with lanes 9 and 10), suggesting that lack of glycosylation destabilizes the protein. Despite its relatively low expression and lack of glycosylation, (N294A)S5-iHA was incorporated into virions (lane 9 and 10) and was relatively excluded from virions by Nef (lanes 7 and 8); similar effects were detected in the case of wild-type SERINC5, which as before migrated at relatively high apparent molecular weight (lanes 2, 3, 5, and 6).
We used flow cytometry to measure the expression of (N294A)S5-iHA on the surface of the virion producer cells, which were identified by their expression of Env at the cell surface (Fig. 4C and D). (N294A)S5-iHA was less well expressed at the cell surface than wild-type S5-iHA, consistent with its decreased expression as detected at the whole-cell level by immunoblotting. Both (N294A)S5-iHA and the wild-type S5-iHA were downregulated by Nef, consistent with their similar Nef sensitivity in the infectivity assays.
To confirm that the intrinsic sensitivity of Serinc5 to Nef is not markedly dependent on glycosylation, we used an extended dose-response format [10, 30, 100, and 300 ng of plasmid expressing either wild-type S5-iHA or (N294A)S5-iHA; Fig. 5]. Figure 5A shows the range of S5-iHA expression levels in cell lysates from a representative experiment, and the bar graph in Fig. 5B shows the SERINC-HA band intensities quantified by densitometry and normalized to the loading control GAPDH (glyceraldehyde-3-phosphate dehydrogenase) band from virion producer cells from three independent experiments. For Fig. 5C, instead of graphing infectivity versus the amount of plasmid (as we did in Fig. 4A), we graphed the infectivity versus the loading control-normalized band intensity for SERINC-HA in the immunoblot of the virion producer cells from three independent experiments. Using nonlinear regression analysis, we fit semilog curves to the data sets and compared the slopes using the extra sum-of-squares F test (GraphPad Prism 7.0), which indicated that the S5-iHA and (N294A)S5-iHA slopes were not significantly different (P = 0.8238). These data suggest that the activities of wild-type S5-iHA and (N294A)S5-iHA proteins are similar against virions produced in the presence of Nef.
FIG 5.
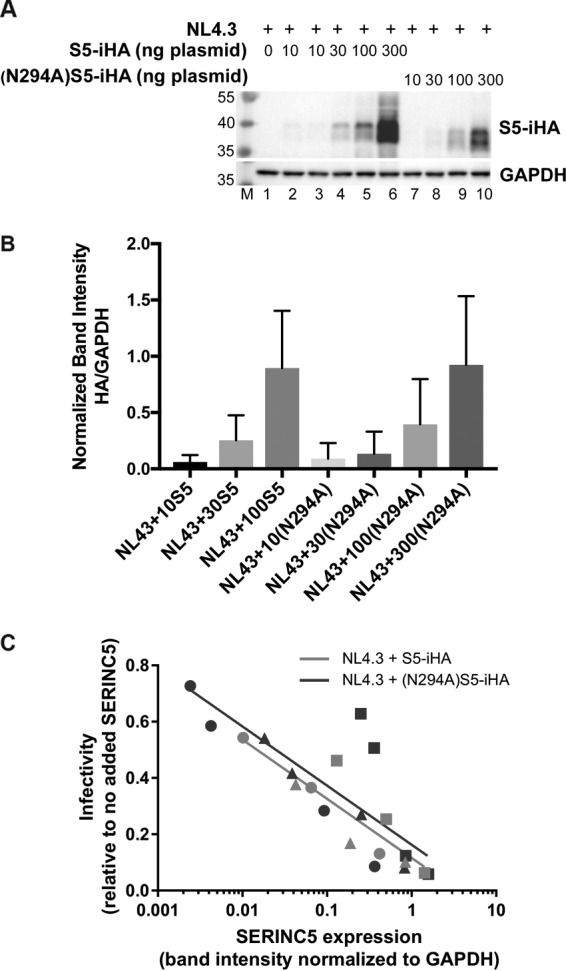
Extended dose-response experiment evaluating the sensitivity to Nef of nonglycosylated (N294A)S5-iHA as a function of protein expression. Zero, 10, 30, 100, or 300 ng of pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA was used to transfect HEK293 cells together with 1.2 μg of pNL43 or pNL43ΔNef proviral plasmids. Twenty-four hours after transfection, the cells and virions were isolated for immunoblotting and measurement of infectivity. (A) Immunoblot showing the expression of wild-type S5-iHA and (N294A)S5-iHA after transfection of cells with the indicated amounts of plasmids. (B) The expression of S5-iHA, (N294A)S5-iHA, and GAPDH was quantified using Image Lab software (Bio-Rad). The band intensities for HA in the immunoblots were normalized to GAPDH and plotted using GraphPad Prism 7.0 software. The 300 ng pBJ5-S5-iHA condition was excluded due to image saturation. The bar graph represents data from three independent experiments; error bars are the standard deviations. (C) Viral infectivity (infectious centers/p24 relative to the no-SERINC5 control) was measured as described in the legend of Fig. 4A but is graphed versus the GAPDH-normalized band intensity for SERINC 5-HA. Data are from three independent experiments (shown using different symbols: circle, square, or triangle); within each experiment, each datum point was derived from duplicate measurements of infectious centers and p24 concentration and a single measurement of SERIN5-iHA band intensity. Data for the 300 ng wild-type S5-iHA were excluded due to image saturation. Best-fit trend lines are shown for NL43 with either wild-type S5-iHA (pink line) or (N294A)S5-iHA (blue line). Trend lines were generated using nonlinear regression analysis with the least-squares method fitting to semilog lines. The slopes of the two lines were compared using the extra sum-of-squares F test (GraphPad Prism 7.0) and were not significantly different (P = 0.82).
MG-132 but not bafilomycin A1 partially rescues the expression of (N294A)-S5iHA.
Since the (N294A)S5-iHA mutant was expressed relatively poorly, we hypothesized that glycosylation might contribute to the stability of SERINC5. To test this, (N294A)S5-iHA or wild-type S5-iHA was expressed by plasmid transfection in HEK293 cells in the presence or absence of a proteasomal inhibitor (MG-132) or a lysosomal inhibitor (bafilomycin A1). The transfected cells were treated with the inhibitors for 4 to 6 h before analysis of WCEs by Western blotting (Fig. 6). We observed that MG-132, but not bafilomycin A1, partially rescued the expression of (N294A)S5-iHA. Notably, the expression of wild-type SERINC5 was not substantially increased by either MG-132 or bafilomycin A1, suggesting that wild-type protein is rapidly degraded by neither the proteasome nor the lysosome. These results regarding (N294A)S5-iHA suggest that the absence of glycosylation destabilizes SERINC5, potentially due to improper folding that renders the protein susceptible to an ER-based quality control mechanism and leads to proteasomal degradation.
FIG 6.
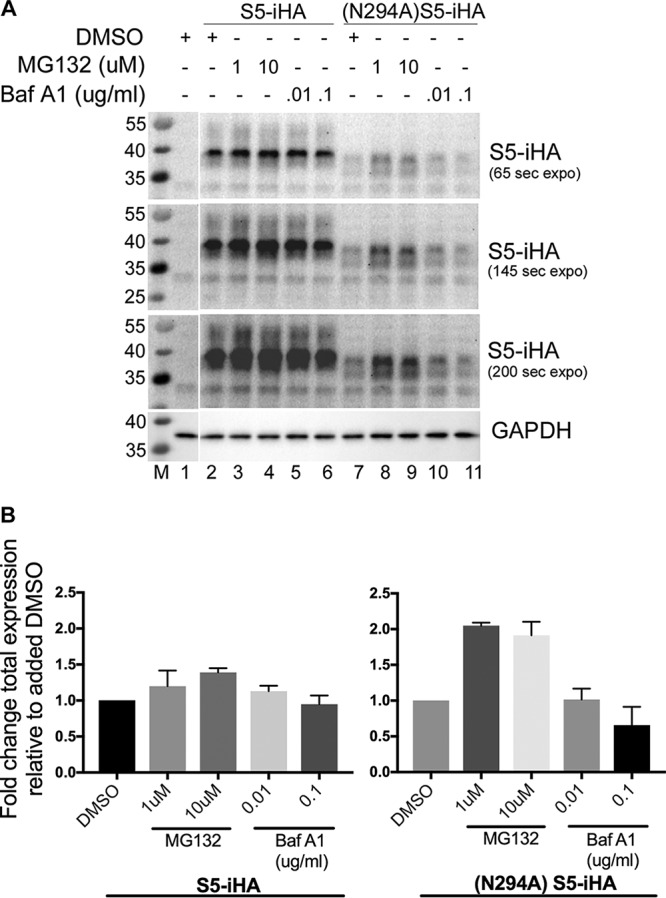
MG-132, rather than bafilomycin A1, partially rescues the steady-state expression of (N294A)S5-iHA. (A) HEK293 cells were transfected with either 50 ng of pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA. Twenty-four hours after transfection, the culture medium was changed to include MG-132 (a proteasome inhibitor) or bafilomycin A1 (a lysosome inhibitor) at the indicated concentrations for 4 to 6 h. The cells were harvested, suspended in 1× TCEP reducing buffer, sonicated, and immunoblotted for SERINC5 (HA) and GAPDH. (B) The experiment shown in panel A was repeated, and the band intensities for S5-iHA and (N294A)S5-iHA from both experiments were quantified using ImageLab software (Bio-Rad). The values for the SERINC proteins were normalized to GAPDH and plotted using GraphPad Prism software; error bars are the standard deviations. The expression of wild-type S5-iHA and (N294A)S5-iHA in the presence of DMSO only were each set to 1.0.
The subcellular localizations of wild-type and N294A SERINC5 are similar.
Mutation of glycosylation sites can lead to protein retention and degradation in the ER (30, 31). To evaluate whether (N294A)S5 is retained in the ER relative to the wild-type protein, we transfected HeLa P4.R5 cells to express wild-type S5-iHA or (N294A)S5-iHA, and the subcellular distributions of the proteins were evaluated using immunofluorescence microscopy (Fig. 7). We used a relatively small amount of plasmid to minimize potential artifacts of overexpression. S5-iHA and (N294A)S5-iHA were similarly distributed: each concentrated around the nucleus and distributed diffusely throughout the cytoplasm, consistent with localization to the nuclear envelope and the ER, and to a lesser extent the plasma membrane. (N294A)S5-iHA was not appreciably retained in the ER relative to the wild-type protein, consistent with its detection at the cell surface and in virions.
FIG 7.

Subcellular localization of wild-type SERINC5 and (N294A)SERINC5. HeLa P4.R5 cells were transfected with 25 ng of either pcDNA, pBJ5-S5-iHA, or pBJ5-(N294A)S5-iHA. The next day, the cells were fixed, permeabilized, and stained with mouse anti-HA IgG followed by rhodamine-X-conjugated donkey anti-mouse IgG. Images were acquired on a wide-field fluorescence microscope (Olympus) as a z-series and processed by a nearest-neighbor deconvolution algorithm using SlideBook version 4.1 software. A midcell single plane was exported, and the final images were assembled and adjusted identically using Adobe Photoshop software.
DISCUSSION
SERINCs are multipass transmembrane proteins that restrict retroviral infectivity. One family member, SERINC5, appears to account for the majority of the effect of the viral Nef protein on the infectivity of HIV-1: by counteracting SERINC5, Nef enhances virion infectivity. SERINC5 and its antiretroviral activity are well conserved among eukaryotes. Remarkably, even SERINC5 from frogs inhibits the infectivity of HIV-1 (21). In addition to HIV-1 Nef, SERINC5 antagonists include glycoGag of murine leukemia virus and S2 of equine infectious anemia virus. Current evidence suggests that SERINC5 acts in virions to inhibit virion-target cell fusion, and the viral antagonists appear to decrease the incorporation of SERINC5 into virions via a mechanism related to clathrin- and AP2-mediated SERINC5 endocytosis. Here we observed that a mature, glycosylated form of SERINC5 is selectively incorporated into HIV-1 virions, but glycosylation is not required for intrinsic restrictive activity or for sensitivity to Nef.
Our data indicate that SERINC5 is modified by the addition of complex glycans at essentially a single site, N294. This mature, glycosylated form is nearly the only form found in virions when wild-type SERINC5 is expressed, presumably because it is the form that reaches the plasma membrane. A smaller, immature form modified by high-mannose glycans is instead the predominant form of SERINC5 detected in cells. Importantly, although detected in cells of the T lymphoid line Jurkat as well as in HEK293 cells, the predominance of the high-mannose form in cellular SERINC5 might be a consequence of an overexpression condition associated with transient transfection. A similar situation was observed in the case of restriction factor BST-2 (tetherin), and conclusions regarding the mechanism of antagonism by Vpu based on detection of the high-mannose form that dominates transfected HEK293 cells (proteasomal degradation) were not the same as those based on detection of the mature, endogenous protein (endolysosomal trafficking and degradation) (26, 32–34). Unfortunately, we are unaware of reagents that enable detection of the endogenous SERINC proteins. Nonetheless, distinguishing the mature from the immature glycosylated forms of SERINC5 is likely to be important in mechanistic studies of Nef and other viral antagonists. For example, a recent study suggests that Nef induces lysosomal degradation of SERINC5 (35, 36). Such degradation might be most apparent when measuring the mature, 55-kDa, post-ER form of the protein, rather than when measuring the 40-kDa, immature, high-mannose form. In this regard, our data do not show evidence of a Nef-mediated decrease in the 55-kDa mature form of SERINC5 at the whole-cell level (Fig. 1A). Instead, at least when Nef was expressed from a proviral plasmid introduced by transfection, we observed a paradoxical increase in the immature, high-mannose form of SERINC5 (Fig. 1B and C and 4A).
We observed that the glycosylation-deficient SERINC5 mutant, (N294A)S5-iHA, was expressed relatively poorly at steady state relative to the wild-type protein, a condition that could be partly reversed by inhibiting the proteasome. These observations are consistent with the roles of glycosylation in protein folding and quality control at the ER and in protein trafficking (30). For example, mutational loss of N-linked glycosylation in the case of the ATP-binding cassette transporter, ABCA3, is associated with a decrease in protein expression that is reversed by inhibition of the proteasome; these effects are reminiscent of our data regarding SERINC5 (31). Notably, the glycosylation mutant of ABCA3 was partially retained in the ER. Increased ER retention of (N294A)S5-iHA relative to the wild-type protein is not obvious in our immunofluorescence data, and the mutant reaches the plasma membrane as measured by flow cytometry and as assessed by its ability to incorporate into virions. Nonetheless, a modest degree of ER retention could occur.
Finally, we observed that in comparison to the wild-type protein, the (N294A)S5-iHA mutant was similarly active at restricting infectivity, and it was similarly sensitive to antagonism by HIV-1 Nef. Our assessments of sensitivity to Nef included measurements of virion infectivity, Nef-mediated downregulation of SERINC5 from the cell surface, and Nef-mediated exclusion of SERINC5 from virions. Our conclusion that the virologic effect of SERINC5 is independent of glycosylation requires careful assessment of protein expression. At face value, i.e., without consideration of the differences in expression, our initial plasmid dose-response data could be interpreted as showing that (N294A)S5-iHA restricts infectivity as well as the wild-type protein, but it is relatively more sensitive to Nef. Conceivably, in experiments in which exogenous SERINCs are expressed, the dynamic range of restriction might be different from the dynamic range of Nef responsiveness. These considerations warrant careful assessment of protein expression and dose-response effects in quantitative virologic assays designed to measure the properties of SERINC proteins.
MATERIALS AND METHODS
Plasmids, cells, and reagents.
Proviral plasmids pNL43 and pNL43ΔNef were described previously (37, 38). The pBJ5-SERINC5-iHA (pBJ5-S5-iHA) plasmid encodes an internal HA tag inserted between residues 290 and 291 and was the kind gift of Heinrich Gottlinger (17). The N294A mutation was introduced into pBJ5-S5-iHA using the QuikChange site-directed mutagenesis kit (Agilent Technologies), yielding pBJ5-(N294A)S5-iHA, and was verified by Sanger sequencing (Genewiz). The Nef expression plasmid, pCINL, has been described previously (39).
HEK293 Tet-On TRE-HIV △Nef cells, HEK293 cells containing a doxycycline-inducible HIV-1 genome lacking nef and constitutively expressing CD4, have been described (40); these cells were maintained in Tet-Free Dulbecco's modified Eagle medium (DMEM plus 10% Tet-free fetal bovine serum [FBS] and penicillin-streptomycin [P/S] with 1 μg/ml puromycin, 200 μg/ml G418, and 200 μg/ml Zeocin). HEK293 cells and HeLa P4.R5 cells were maintained in complete DMEM (DMEM with 10% FBS and P/S). SERINC3/5 double KO JTAg cells containing clustered regularly interspaced short palindromic repeat(s) (CRISPR)-disrupted open reading frames (ORFs) for SERINC3 and 5 were the kind gift of Heinrich Gottlinger (17) and were maintained in complete RPMI (RPMI with 10% FBS and P/S).
Mouse anti-HA (clone 16B12) and brefeldin A were obtained from BioLegend; sheep anti-gp120 was obtained from the NIH-AIDS Reagent Program (25, 41); mouse anti-p24 was obtained from Millipore; sheep anti-Nef was a gift from Celsa Spina; mouse anti-GAPDH was obtained from GeneTex; mouse anti-tubulin was obtained from Sigma; and DNA-In Jurkat transfection reagent was from MTI-GlobalStem. The glycosidases were obtained from NEB. Tris(2-carboxyethyl)phosphine (TCEP), dimethyl sulfoxide (DMSO), and bafilomycin A1 were obtained from Sigma. MG-132 was obtained from Cell Signaling, and Lipofectamine 2000, PageRuler prestained protein ladder, and PageRuler Plus prestained protein ladder were obtained from Thermo Fisher.
Transfections.
HEK293 cells were seeded at a density of 150,000 cells/ml/well in 12-well plates. They were transfected with the indicated amounts of pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA expression plasmid along with 1.5 μg of proviral plasmids pNL43 or pNL43ΔNef using Lipofectamine 2000 according to the manufacturer's protocol. Twenty-four hours later, the cells and supernatants were harvested. HEK293 Tet-On TRE-HIV-ΔNef cells were seeded at a density of 150,000 cells/ml/well in 12-well plates and were transfected with pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA plasmids either with or without the Nef expression plasmid pCINL. Twenty-four hours after transfection, the cells were treated with 0.5 μg/ml of doxycycline for 8 h, after which the cells and supernates were collected. SERINC3/5 double KO JTAg cells were seeded at a density of 400,000 cells/well in 6-well plates and were transfected with pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA plasmids along with 1.5 μg of proviral plasmids pNL43 or pNL43ΔNef using DNA-In Jurkat transfection reagent according to the manufacturer's protocol. Cells and supernates were harvested after 48 h.
Preparation of samples for immunoblotting.
The cells from each well of 12-well plates were harvested, washed once with cold phosphate-buffered saline (PBS), suspended directly in 200 to 250 μl of 1× TCEP reducing buffer (50 mM Tris-HCl, 2% SDS, 10% glycerol, 50 mM TCEP, bromophenol blue), and either stored at −80°C or processed directly. To avoid aggregation of SERINC5, the cell lysates were not boiled; instead, the DNA was sheared by sonication (5 cycles of 20-s pulse followed by 90-s rest at 4°C using a Bioruptor [Diagenode]). The cell lysates were vortexed for 10 s prior to loading on the gel. For virion purification, the culture supernates were clarified of cells and debris by centrifugation at 400 × g. To avoid contamination from cell debris, only the upper 85% of the supernatant was carefully removed and layered over a 20% sucrose cushion and centrifuged at 23,500 × g for 1 h at 4°C. The supernates were aspirated, and the virion-containing pellet was suspended in 25 μl of 1× TCEP reducing buffer and loaded onto the gel after vortexing for 10 s (without any sonication). The cell lysates and viral pellets were resolved on 10%, 1-mm-thick, denaturing SDS-PAGE gels, transferred onto polyvinylidene difluoride (PVDF) membranes, immunoblotted with the indicated antibodies, and visualized using Western Clarity detection reagent (Bio-Rad). Chemiluminescence was detected using ChemiDoc Imager (Bio-Rad) and quantified using Bio-Rad Image Lab v5.1 software. Primary and secondary antibodies were prepared in antibody dilution buffer, consisting of 2% milk in PBST (PBS with 0.02% Tween 20), as follows: 1:1,000 for mouse anti-HA; 1:500 for mouse anti-p24; 1:3,000 for sheep anti-Nef; 1:3,000 for sheep anti-HIV-1 gp120; 1:5,000 for mouse anti-GAPDH; 1:1,000 for mouse antitubulin; 1:5,000 for horseradish peroxidase (HRP)-conjugated anti-mouse and anti-sheep secondary antibodies.
Infectivity assays.
HEK293 cells were transfected with 1.5 μg of pNL43 or pNL43ΔNef proviral plasmids either with or without 30 or 100 ng of pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA expression plasmids. For the extended dose-response experiments using 10, 30, 100, or 300 ng of S5 expression plasmids, 1.2 μg of pNL43 was used. The next day, the transfected HEK293 cells were harvested for immunoblot analysis as described above and for flow cytometry as described below. The culture supernates were clarified by centrifugation at 400 × g, and the clarified supernates were layered over 20% sucrose cushions and centrifuged at 23,500 × g for 1 h at 4°C in duplicate. One set of the virion pellets was suspended in 1× TCEP reducing buffer and used for immunoblotting, while the other set was suspended in complete DMEM and serial dilutions were used to infect CD4-positive HeLa P4.R5 indicator cells in duplicate, which were seeded in 48-well plates at a cell density of 20,000 cells/well. An aliquot of the DMEM-suspended virions was used for p24 ELISA (ABL). After 48 h, infectivity was measured by enumerating infectious centers in the cultures of the HeLa P4.R5 indicator cells (29): the cells were fixed with 1% formaldehyde and 0.2% glutaraldehyde for 5 min at room temperature and then stained with 4 mM potassium ferrocyanide, 4 mM potassium ferricyanide, 2 mM MgCl2, and 0.4 mg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) for 6 h or overnight. After washing and air drying the wells, the blue cells representing the infectious centers were counted using image analysis software (29). The number of infectious centers was normalized to the content of p24 antigen. Since HEK cells support a nef phenotype even in the absence of exogenous SERINC expression (10, 14), we further normalized the data (expressed initially as infectious centers per nanogram of p24) to the corresponding “nontransfected SERINC5” control for each viral genotype (wild type or Δnef). In the extended dose-response experiments, the normalized infectivity values were graphed versus the intensity of the bands for the SERINC proteins measured in the virion producer cells.
Flow cytometry.
Transfected HEK293 cells were washed with cold PBS and stained with 10 μg/ml of 2G12 (anti-HIV1 gp120) together with a 1:200 dilution of mouse anti-HA (to detect SERINC5-iHA; BioLegend) for 40 min at 4°C, before washing with cold PBS and staining with 1:500 dilution of phycoerythrin (PE)-conjugated anti-mouse IgG (BioLegend) and 1:500 AF647-conjugated anti-human IgG (Jackson ImmunoResearch) for 30 min at 4°C in the dark. After washing twice with PBS, the cells were fixed with 2% paraformaldehyde in PBS, and the data were collected on a BD Accuri flow cytometer and analyzed using FlowJo (Treestar).
Deglycosylation of SERINC5.
To produce virions, HEK293 or SERINC3/5 double KO JTAg cells were transfected with pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA expression plasmids together with 1.5 μg of pNL43ΔNef proviral plasmid. Alternatively, HEK293 Tet-On TRE-HIV-ΔNef cells were transfected with pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA expression plasmids, and viral gene expression was induced with doxycycline. Virions were partially purified as described above. Virion pellets were suspended in 1× TCEP reducing buffer and were treated with various glycosidases (NEB). The deglycosylation reactions were carried out according to the manufacturer's instructions with minor modification: after the addition of 10× glycoprotein denaturing buffer, the samples were not heated (to avoid aggregation of SERINC5) but were instead left at room temperature for 10 min before addition of the remaining reaction components. For a typical PNGase F reaction, 1 μl of 10× denaturing buffer was added to 6 μl of TCEP-suspended virions. The mixture was incubated at room temperature for 10 min, followed by the addition of 1 μl of 10× Glyco 2 buffer, 1 μl of 10% NP-40, and after mixing, 1 μl of PNGase F. The enzyme was replaced with water for the “no enzyme” control. The reaction mixtures were incubated at 37°C for 1 h. The reaction products were resolved on 10% SDS-PAGE gels and immunoblotted to detect SERINC5. After detection of SERINC5 (Fig. 2A), the PVDF membrane was stripped according to the manufacturer's instructions using Western Re-Probe (G-Biosciences), blocked again with 5% milk in PBS, and probed with sheep anti-HIV-1 gp120 antibody.
To evaluate the glycosylation of cell-associated SERINC5, whole-cell extracts of HEK293 cells transfected with pBJ5 S5-iHA were prepared as described above. After sonicating and vortexing the extracts, an aliquot was used for the deglycosylation reactions in the same manner as described above for virions.
Immunofluorescence microscopy.
One day before transfection, 105 HeLa P4.R5 cells were seeded per well onto glass coverslips in a 24-well plate. Cells were transfected with 25 ng of pBJ5-SERINC5-iHA or 25 ng of pBJ5-(N294A)SERINC5-iHA or pcDNA vector alone. Twenty-four hours later, the cells were fixed with 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.2% NP-40 for 7 min at 4°C. After blocking with 3% bovine serum albumin (BSA) plus 5% normal donkey serum in PBS for 30 min at 4°C, the cells were stained with a 1:400 dilution of mouse anti-HA (BioLegend) for 45 min at 4°C. After 4 washes with PBS, the cells were incubated with a 1:500 dilution of rhodamine-X donkey anti-mouse secondary antibody for 30 min in the dark at 4°C. After 5 washes with PBS, coverslips were mounted on glass slides using Prolong Gold (Pierce) and were cured overnight at room temperature. Z-series of images were acquired using a wide-field fluorescence microscope (Olympus) and were processed using a nearest-neighbor deconvolution algorithm (SlideBook version 4.1 software; Intelligent Imaging Innovations, Denver, CO), and the figure was assembled using Adobe Photoshop software.
(N294A)SERINC5 expression rescue experiments.
HEK293 cells were transfected with 50 ng of either pBJ5-S5-iHA or pBJ5-(N294A)S5-iHA without any proviral plasmid DNA as described above. Twenty-four hours after transfection, the culture medium was replaced with medium containing either DMSO, MG-132, or bafilomycin A1 (the last two in DMSO) at the indicated concentrations, and the cells were incubated for 4 to 6 h. At the end of the incubation, the cells were harvested, washed once with PBS, suspended in 1× TCEP reducing buffer, sonicated as described above, and used for immunoblotting.
ACKNOWLEDGMENTS
This work was funded by NIH grants to J.G. (R01 AI129706) and to M.K.L. (K08 AI112394).
We thank Marissa Suarez for performing p24 ELISAs. We thank Heinrich Gottlinger for the pBJ5-S5-iHA plasmid and the Jurkat Tag SERINC 3/5 double KO cells. We thank the members of the Guatelli lab for their suggestions and discussions.
Author contributions: S.S., M.K.L., and J.G. designed the experiments; S.S. performed the experiments; S.S., M.K.L., and J.G. analyzed the data and wrote the manuscript.
REFERENCES
- 1.Kirchhoff F. 2010. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8:55–67. doi: 10.1016/j.chom.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Simon V, Bloch N, Landau NR. 2015. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol 16:546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, Lawson VA, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan JS, Cunningham A, Dwyer D, Dowton D, Mills J. 1995. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270:988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 4.Kestler HW III, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC. 1991. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65:651–662. doi: 10.1016/0092-8674(91)90097-I. [DOI] [PubMed] [Google Scholar]
- 5.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. 1995. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med 332:228–232. doi: 10.1056/NEJM199501263320405. [DOI] [PubMed] [Google Scholar]
- 6.Alsahafi N, Richard J, Prévost J, Coutu M, Brassard N, Parsons MS, Kaufmann DE, Brockman M, Finzi A. 2017. Impaired downregulation of NKG2DlLigands by Nef proteins from elite controllers sensitizes HIV-1-Infected cells to antibody-dependent cellular cytotoxicity. J Virol 91:e00109-. doi: 10.1128/JVI.00109-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 8.Garcia JV, Miller AD. 1991. Serine phosphorylation-independent downregulation of cell-surface CD4 by Nef. Nature 350:508–511. doi: 10.1038/350508a0. [DOI] [PubMed] [Google Scholar]
- 9.Veillette M, Coutu M, Richard J, Batraville LA, Dagher O, Bernard N, Tremblay C, Kaufmann DE, Roger M, Finzi A. 2014. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J Virol 89:545–551. doi: 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aiken C, Trono D. 1995. Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J Virol 69:5048–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chowers MY, Spina CA, Kwoh TJ, Fitch NJ, Richman DD, Guatelli JC. 1994. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J Virol 68:2906–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chowers MY, Pandori MW, Spina CA, Richman DD, Guatelli JC. 1995. The growth advantage conferred by HIV-1 nef is determined at the level of viral DNA formation and is independent of CD4 downregulation. Virology 212:451–457. doi: 10.1006/viro.1995.1502. [DOI] [PubMed] [Google Scholar]
- 13.Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB. 1994. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Exp Med 179:101–113. doi: 10.1084/jem.179.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller MD, Warmerdam MT, Page KA, Feinberg MB, Greene WC. 1995. Expression of the human immunodeficiency virus type 1 (HIV-1) nef gene during HIV-1 production increases progeny particle infectivity independently of gp160 or viral entry. J Virol 69:579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa AS, Frezza C, Clish CB, Neil SJ, Lehner PJ. 2015. Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe 18:409–423. doi: 10.1016/j.chom.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, Santoni FA, Pizzato M. 2015. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526:212–217. doi: 10.1038/nature15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Usami Y, Wu Y, Göttlinger HG. 2015. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526:218–223. doi: 10.1038/nature15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inuzuka M, Hayakawa M, Ingi T. 2005. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J Biol Chem 280:35776–35783. doi: 10.1074/jbc.M505712200. [DOI] [PubMed] [Google Scholar]
- 19.Chande A, Cuccurullo EC, Rosa A, Ziglio S, Carpenter S, Pizzato M. 2016. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc Natl Acad Sci U S A 113:13197–13202. doi: 10.1073/pnas.1612044113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulte B, Selyutina A, Opp S, Herschhorn A, Sodroski JG, Pizzato M, Diaz-Griffero F. 2018. Localization to detergent-resistant membranes and HIV-1 core entry inhibition correlate with HIV-1 restriction by SERINC5. Virology 515:52–65. doi: 10.1016/j.virol.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai W, Usami Y, Wu Y, Göttlinger H. 2018. A long cytoplasmic loop governs the sensitivity of the anti-viral host protein SERINC5 to HIV-1 Nef. Cell Rep 22:869–875. doi: 10.1016/j.celrep.2017.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sood C, Marin M, Chande A, Pizzato M, Melikyan GB. 2017. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J Biol Chem 292:6014–6026. doi: 10.1074/jbc.M117.777714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beitari S, Ding S, Pan Q, Finzi A, Liang C. 2017. Effect of HIV-1 Env on SERINC5 antagonism. J Virol 91:e02214-16. doi: 10.1128/JVI.02214-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trautz B, Wiedemann H, Lüchtenborg C, Pierini V, Kranich J, Glass B, Kräusslich HG, Brocker T, Pizzato M, Ruggieri A, Brügger B, Fackler OT. 2017. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J Biol Chem 292:13702–13713. doi: 10.1074/jbc.M117.797332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatch WC, Tanaka KE, Calcelli T, Rashbaum WK, Kress Y, Lyman WD. 1992. Persistent productive HIV-1 infection of a CD4- human fetal thymocyte line. J Immunol 148:3055–3061. [PubMed] [Google Scholar]
- 26.Mitchell RS, Katsura C, Skasko MA, Fitzpatrick K, Lau D, Ruiz A, Stephens EB, Margottin-Goguet F, Benarous R, Guatelli JC. 2009. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via β-TrCP and endo-lysosomal trafficking. PLoS Pathog 5:e1000450. doi: 10.1371/journal.ppat.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu X, Borchers C, Bienstock RJ, Tomer KB. 2000. Mass spectrometric characterization of the glycosylation pattern of HIV-gp120 expressed in CHO cells. Biochemistry 39:11194–11204. doi: 10.1021/bi000432m. [DOI] [PubMed] [Google Scholar]
- 28.Tsirigos KD, Peters C, Shu N, Käll L, Elofsson A. 2015. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 43(W1):W401–W407. doi: 10.1093/nar/gkv485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Day JR, Martínez LE, Sásik R, Hitchin DL, Dueck ME, Richman DD, Guatelli JC. 2006. A computer-based, image-analysis method to quantify HIV-1 infection in a single-cycle infectious center assay. J Virol Methods 137:125–133. doi: 10.1016/j.jviromet.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 30.Helenius A, Aebi M. 2001. Intracellular functions of N-linked glycans. Science 291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 31.Beers MF, Zhao M, Tomer Y, Russo SJ, Zhang P, Gonzales LW, Guttentag SH, Mulugeta S. 2013. Disruption of N-linked glycosylation promotes proteasomal degradation of the human ATP-binding cassette transporter ABCA3. Am J Physiol Lung Cell Mol Physiol 305:L970–L980. doi: 10.1152/ajplung.00184.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrew AJ, Miyagi E, Strebel K. 2011. Differential effects of human immunodeficiency virus type 1 Vpu on the stability of BST-2/tetherin. J Virol 85:2611–2619. doi: 10.1128/JVI.02080-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Früh K, Moses AV. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a β-TrCP-dependent mechanism. J Virol 83:7931–7947. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goffinet C, Allespach I, Homann S, Tervo HM, Habermann A, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, Krijnse-Locker J, Banting G, Kräusslich HG, Fackler OT, Keppler OT. 2009. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5:285–297. doi: 10.1016/j.chom.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Shi J, Xiong R, Zhou T, Su P, Zhang X, Qiu X, Li H, Li S, Yu C, Wang B, Ding C, Smithgall TE, Zheng YH. 2018. HIV-1 Nef antagonizes SERINC5 restriction by downregulation of SERINC5 via the endosome/lysosome system. J Virol 92:00196–00118. doi: 10.1128/JVI.00196-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Zhou T, Yang J, Lin Y, Shi J, Zhang X, Frabutt DA, Zeng X, Li S, Venta PJ, Zheng YH. 2017. Identification of SERINC5-001 as the predominant spliced isoform for HIV-1 restriction. J Virol 91:e00137-17. doi: 10.1128/JVI.00137-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adachi A, Gendelman H, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spina C, Kwoh TJ, Chowers MY, Guatelli JC, Richman DD. 1994. The importance of nef in the induction of human immunodeficiency virus type 1 replication from primary quiescent CD4 lymphocytes. J Exp Med 179:115–123. doi: 10.1084/jem.179.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Craig HM, Pandori MW, Guatelli JC. 1998. Interaction of HIV-1 Nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc Natl Acad Sci U S A 95:11229–11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tokarev A, Stoneham C, Lewinski MK, Mukim A, Deshmukh S, Vollbrecht T, Spina CA, Guatelli J. 2015. Pharmacologic inhibition of Nedd8 activation enzyme exposes CD4-Induced epitopes within Env on cells expressing HIV-1. J Virol 90:2486–2502. doi: 10.1128/JVI.02736-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Page KA, Stearns SM, Littman DR. 1992. Analysis of mutations in the V3 domain of gp160 that affect fusion and infectivity. J Virol 66:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]