

Despite viral suppression by antiretrovirals, HIV proteins continue to be detected in infected cells and neurologic complications remain common in infected people. Although HIV is unable to infect neurons, viral proteins, including gp120 and Tat, can enter neurons and can cause neuronal degeneration and neurocognitive impairment. Neuronal health is dependent on the functional integrity of mitochondria, and damaged mitochondria are subjected to mitochondrial control mechanisms. Multiple lines of evidence suggest that specific elimination of damaged mitochondria through mitophagy and mitochondrial dynamics play an important role in CNS diseases. Here, we show that in human primary neurons, gp120 and Tat favor the balance of mitochondrial dynamics toward enhanced fragmentation through the activation of mitochondrial translocation of DRP1 to the damaged mitochondria. However, mitophagy fails to go to completion, leading to neuronal damage. These findings support a role for altered mitophagy in HIV-associated neurological disorders and provide novel targets for potential intervention.
KEYWORDS: HIV gp120, HIV Tat, human primary neurons, mitochondrial fragmentation, mitochondrial damage, mitophagy, autophagy
ABSTRACT
HIV enters the central nervous system (CNS) during the early stages of infection and can cause neurological dysfunction, including neurodegeneration and neurocognitive impairment. The specific autophagy responsible for removal of damaged mitochondria (mitophagy) and mitochondrial dynamics constitute neuronal mitochondrial quality control mechanisms and are impaired in neurodegenerative disorders and numerous other diseases. The release of HIV proteins gp120 and Tat from infected cells is thought to play an important role in HIV-associated neurocognitive disorders (HAND), but the mechanism(s) leading to impairment are poorly understood. Here, we report that exposure of human primary neurons (HPNs) to HIV gp120 and Tat accelerates the balance of mitochondrial dynamics toward fission (fragmented mitochondria) and induces perinuclear aggregation of mitochondria and mitochondrial translocation of dynamin-related protein 1 (DRP1), leading to neuronal mitochondrial fragmentation. HIV gp120 and Tat increased the expression of microtubule-associated protein 1 light chain 3 beta (LC3B) protein and induced selective recruitment of Parkin/SQSTM1 to the damaged mitochondria. Using either a dual fluorescence reporter system expressing monomeric red fluorescent protein and enhanced green fluorescent protein targeted to mitochondria (mito-mRFP-EGFP) or a tandem light chain 3 (LC3) vector (mCherry-EGFP-LC3), both HIV proteins were found to inhibit mitophagic flux in human primary neurons. HIV gp120 and Tat induced mitochondrial damage and altered mitochondrial dynamics by decreasing mitochondrial membrane potential (ΔΨm). These findings indicate that HIV gp120 and Tat initiate the activation and recruitment of mitophagy markers to damaged mitochondria in neurons but impair the delivery of mitochondria to the lysosomal compartment. Altered mitochondrial dynamics associated with HIV infection and incomplete neuronal mitophagy may play a significant role in the development of HAND and accelerated aging associated with HIV infection.
IMPORTANCE Despite viral suppression by antiretrovirals, HIV proteins continue to be detected in infected cells and neurologic complications remain common in infected people. Although HIV is unable to infect neurons, viral proteins, including gp120 and Tat, can enter neurons and can cause neuronal degeneration and neurocognitive impairment. Neuronal health is dependent on the functional integrity of mitochondria, and damaged mitochondria are subjected to mitochondrial control mechanisms. Multiple lines of evidence suggest that specific elimination of damaged mitochondria through mitophagy and mitochondrial dynamics play an important role in CNS diseases. Here, we show that in human primary neurons, gp120 and Tat favor the balance of mitochondrial dynamics toward enhanced fragmentation through the activation of mitochondrial translocation of DRP1 to the damaged mitochondria. However, mitophagy fails to go to completion, leading to neuronal damage. These findings support a role for altered mitophagy in HIV-associated neurological disorders and provide novel targets for potential intervention.
INTRODUCTION
Early in the epidemic, human immunodeficiency virus type 1 (HIV-1) was found to infect the central nervous system (CNS) and to be associated with many neurological diseases, including dementia (1). With the availability of combination antiretroviral therapy (ART), HIV-associated dementia (HAD) has become infrequent, but minor cognitive impairment continues to be common even in those fully suppressed on ART (2, 3). Although much research has identified the frequency and extent of neurological dysfunction and markers associated with HIV-associated neurocognitive disorders (HAND), the molecular and cellular pathways driving HAND remain unclear.
Recently, we and others have identified that macroautophagy (autophagy) is dysregulated in human brains affected by HAND and in animal models of HAND (4–7). In response to nutrient deprivation, cells upregulate autophagy as a survival process that recycles cytoplasmic constituents like subcellular organelles and proteins to supply essential macromolecules to the cell. However, cells also utilize nutrient-independent basal autophagy as a quality control mechanism that eliminates misfolded and/or toxic protein aggregates and damaged organelles (8). Mitophagy is a selective form of autophagy that removes dysfunctional mitochondria to maintain efficient cellular metabolism and to reduce cellular stress triggered by increased oxidative stress (8–10). Mitochondria are indispensable for neuronal survival and function and for generating energy through oxidative phosphorylation. Mitochondria also function in fatty acid metabolism, cell death, and calcium regulation and control (11, 12). Multiple mitochondrial quality control mechanisms are activated based on the intensity of mitochondrial damage and are essential for maintaining mitochondrial function and integrity (11).
Mitochondria form a dynamic network that constantly undergoes rearrangement and turnover. By regulating the interconnection and size of the mitochondrial network, the cell can control energy production and many other mitochondrial processes (5, 13). While specific GTPases are responsible for fusion and fission of the mitochondria, the size of the mitochondrial network is regulated by de novo mitochondrial biogenesis and mitophagy, through which autophagosomes deliver mitochondria to lysosomes for hydrolytic degradation. Mitochondria exposed to biological stress undergo perinuclear aggregation and recruitment of dynamin-related GTPase (Drp1) prior to initiation of mitochondrial fission and mitophagy (11, 14–16). The subsequent elimination of damaged mitochondria by asymmetric mitochondrial fragmentation and mitophagy promotes cellular health and survival (8, 15).
Mitochondrial dynamics and mitophagy play a crucial role in neurodegenerative diseases and aging. In neurons, the translocation of Parkin to damaged mitochondria principally occurs within the somatodendritic compartment, a compartment rich in mature lysosomes, which allows efficient mitophagy to occur (17, 18). The mechanisms of neurodegeneration are still not well understood, but recent studies show that HIV proteins impair clearance pathways like autophagy. HIV proteins gp120 and Tat are thought to mediate neuronal toxicity and increase oxidative stress pathways. HIV gp120 has been shown to induce autophagy in cardiomyocytes via the N-methyl-d-aspartate (NMDA) receptor (19), whereas the HIV Tat protein can induce depolarization of neurons, NMDA receptor dysregulation, and disruption of calcium homeostasis (20, 21). Tat also has been found to decrease neuronal mitochondrial size in TAT transgenic mice, to impair mitochondrial membrane potential (ΔΨm) in rat cortical neurons, and to alter neuronal autophagy by modulating autophagosome-lysosome fusion (5).
In the research presented here, we hypothesized that neuronal mitophagy plays an important role in HAND and that HIV proteins gp120 and Tat would dysregulate mitophagy. Our data reveal that in human primary neurons (HPNs), gp120 and Tat favor the balance of mitochondrial dynamics toward enhanced fragmentation through the activation of mitochondrial translocation of DRP1 to the damaged mitochondria. However, mitophagy fails to go to completion, leading to neuronal damage.
RESULTS
Internalization of HIV gp120 and Tat in human neurons induces mitochondrial fission and increases mitophagosome formation.
Our first set of experiments were designed to examine the localization of gp120 and Tat following internalization into HPNs. HPNs were treated with 100 ng/ml of gp120, Tat, or both proteins for 24 h. Heat-inactivated HIV gp120, which does not internalize, and 0.5 μM dextran sulfate, which blocks the binding and uptake of Tat, were used to demonstrate the specificity of HIV gp120 and Tat protein internalization (22–24). Protein internalization in microtubule-associated protein 2 (MAP2)-positive neurons was observed by immunostaining with antibodies against the neuronal somatodendritic marker MAP2 and antibodies against gp120 and Tat and examining stained cells by confocal microscopy. Whereas no gp120 or Tat staining was observed in neuronal cells treated with vehicle (Fig. 1A to D), heat-inactivated gp120 (Fig. 1E and F), or Tat plus dextran sulfate (Fig. 1G and H), internalized gp120 and Tat proteins were found to localize at the perinuclear region in the area of mitochondria (Fig. 1I to L).
FIG 1.

Perinuclear localization of internalized HIV gp120 and Tat in MAP2-positive human primary neurons. Confocal laser scanning microscopy analysis of human primary neurons treated with vehicle (A to D), 100 ng/ml heat-inactivated HIV gp120 (E, F), 100 ng/ml HIV Tat and 0.5 μM dextran sulfate (G, H), 100 ng/ml HIV gp120 recombinant protein (I, J), or 100 ng/ml HIV Tat recombinant protein (K, L) for 24 h. Neurons were fixed and stained for MAP2 somatodendritic neuronal marker antibody (green) and gp120 or Tat antibodies (magenta). Internalization of HIV gp120 and Tat was detected with specific antibodies against gp120 and Tat. DAPI (4′,6-diamidino-2-phenylindole) was used to stain nuclei (blue). White arrows indicate localization of HIV gp120 and Tat in perinuclear areas where mitochondria are present. Scale bar, 10 μm.
Having observed localization of HIV proteins around mitochondria, our next series of experiments were designed to identify a possible effect of gp120 and Tat on mitophagy. HPNs were treated with gp120 and Tat for 6 h and 24 h and stained with anti-TOM20 and anti-light chain 3B-II (LC3B-II) antibodies. Whereas vehicle-treated cells maintained a typical healthy tubular network of mitochondria, perinuclear mitochondrial clustering and distinct mitochondrial fission (mitochondrial fragmentation) were observed in neurons following internalization of gp120, Tat, or a combination of both proteins (Fig. 2A to H). Additionally, in HIV protein-treated cells, autophagosomes with LC3B-II staining (mitophagosomes) colocalized with mitochondria.
FIG 2.

HIV gp120 and Tat induce mitochondrial fission and mitophagosome formation. Confocal laser microscopy analysis showing a healthy tubular network of mitochondria in vehicle-treated cells (A, E) and mitochondrial fragmentation in HIV gp120- and Tat-treated neurons (B to D, F to H) 6 h posttreatment with 100 ng/ml HIV gp120 (B), Tat (C), or both (D) and 24 h posttreatment with 100 ng/ml HIV gp120 (F), Tat (G), or both (H). Neurons were immunostained with TOM20 mitochondrial marker (green) and LC3B-II autophagosome marker (red). In the enlarged images of boxed areas, solid white arrows indicate typical tubular healthy mitochondria and dashed white arrows indicate fragmented mitochondria. Colocalization of mitochondria with LC3 autophagosomes (mitophagosomes) appears yellow. Scale bars, 10 μm.
To further assess the effects of gp120 and Tat on mitophagy, we examined the lipidation of LC3B-I to LC3B-II and degradation of SQSTM1/P62 (SQSTM1). Comparing the 6-h and 24-h time points, both HIV proteins induced progressive, time-dependent increases in LC3B-II and SQSTM1 (Fig. 3A to D). At 6 h, gp120 and Tat treatment resulted in mean 3-fold and 2.7-fold increases in LC3B-II lipidation and mean 1.7-fold and 1.6-fold increases in SQSTM1, respectively (P < 0.03 for all comparisons to controls). Combination treatment with both viral proteins did not result in an additive effect (Fig. 3A and B). Carbonyl cyanide 3-chlorophenylhydrazone (CCCP), a known inducer of mitophagy, was used as a positive control. At 24 h posttreatment, gp120 and Tat increased LC3B-II lipidation by 4.3-fold and 4.5-fold (mean values) and SQSTM1 by 1.8-fold and 2.3-fold, respectively. The combination of both HIV proteins induced a mean 5.5-fold increase in LC3B-II lipidation and a mean 2.7-fold increase in SQSTM1 (P < 0.03 for all comparisons to controls) (Fig. 3C and D). The increase in LC3B-II lipidation following gp120 and Tat treatment is indicative of autophagosome formation and mitophagy initiation in neuronal cells. However, the concomitant accumulation of SQSTM1 in damaged mitochondria suggests that there is a potential block in mitophagy, resulting in delayed mitochondrial degradation.
FIG 3.

HIV gp120 and Tat increase LC3II lipidation and P62 expression 6 h posttreatment with 100 ng/ml HIV gp120, Tat, or both (A) and 24 h posttreatment with 100 ng/ml HIV gp120, Tat or both (C). CCCP was used as the positive control. Neuronal cell lysates were extracted with mitochondrial lysis buffer, clarified by centrifugation, and analyzed by Western blotting using antibodies against LC3B and SQSTM1. Beta-actin (ACTB) was used as an internal loading control. (B and D) The relative expression of LC3B-II and SQSTM1 (P62) was normalized to that of beta-actin. Each data point was normalized to the corresponding result for vehicle-treated cells and analyzed by Image J software. Student's t test was performed to test for statistical significance. Data are presented as mean values ± standard deviations (SD) (n = 3 independent donors). *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant.
HIV gp120 and Tat induce translocation of DRP1 to mitochondria and mitochondrial fragmentation.
The elimination of damaged mitochondria is coordinated by asymmetric fragmentation. Fragmentation is powered by dynamin-related protein 1 (DRP1), which hydrolyzes GTP to remove damaged mitochondria (25), an important process for mitochondrial biogenesis, quality control, transport, and apoptosis (26, 27). Thus, it was important to determine if gp120 and Tat proteins trigger DRP1-mediated fission in HPNs. In these experiments, we examined whether the two HIV proteins altered DRP1 expression in neuronal cells. Following exposure to HIV gp120 and Tat, DRP1 expression was increased under all conditions, with mean increases for gp120, Tat, and gp120-Tat of 3.9-, 4.1- and 6.3-fold, respectively (P < 0.01 for all comparisons to controls) (Fig. 4A and B). We next examined by confocal microscopy whether HIV gp120 or Tat induced the translocation of DRP1 to mitochondria. Comparison of HIV protein-treated neurons to controls showed increased mitochondrial translocation of DRP1 to TOM20-stained mitochondria (Fig. 4C to F, merged yellow puncta). Together, these results indicate that internalized HIV proteins promote mitochondrial fragmentation through DRP1 translocation.
FIG 4.

HIV gp120 and Tat trigger mitochondrial translocation of dynamin-related protein 1 (DRP1), leading to mitochondrial fission and altered mitochondrial dynamics. (A) Western blot analysis of neuronal lysates showing increased DRP1 expression in neurons treated with HIV gp120 and Tat for 24 h. (B) The relative intensities of DRP1 proteins were normalized to the results for beta-actin (ACTB). Each data point was normalized to the results for treatment with vehicle and analyzed using Image J software. Student's t test was performed to test the statistical significance. Data are presented as mean values ± SD (n = 3 independent donors). **, P < 0.01; ***, P < 0.001. (C to F) Confocal laser microscopy analysis shows increased expression and translocation of DRP1 to TOM20-stained fragmented mitochondria. Neurons were immunostained with antibodies specific to DRP1 (red) and TOM20 (green). In the enlarged images of boxed areas, white arrows indicate colocalization (yellow) of DRP1 with TOM20-stained mitochondria. Scale bars, 10 μm.
HIV gp120 and Tat induce Parkin translocation to damaged mitochondria.
When mitochondria become damaged and lose membrane potential, Parkin translocates to depolarized mitochondria. The PINK1 kinase activity promotes the translocation of Parkin to damaged mitochondria, resulting in Parkin-mediated mitophagy (8, 15, 27). Therefore, we examined mitochondrial Parkin translocation and the immune complex formation of PINK1 and Parkin using confocal microscopy. Translocation of Parkin from the cytosol to mitochondria and colocalization with PINK1 in the perinuclear area were observed in neurons exposed to gp120 and Tat but not in vehicle-treated cells (Fig. 5A to D). Additionally, we observed increased translocation of Parkin to damaged, fragmented mitochondria in gp120- and Tat-treated neurons but not in vehicle-treated cells (Fig. 6A to D). These results suggest that in the presence of gp120 and Tat, Parkin is recruited to PINK1-expressing damaged mitochondria.
FIG 5.

HIV gp120 and Tat increase PINK1-Parkin immune complex formation and translocation to the damaged mitochondria. (A to D) Confocal laser microscopy analysis shows increased expression, translocation to perinuclear areas, and immune association of PINK1-Parkin complexes (indicated by the dashed white arrows in enlarged images of boxed areas) with the damaged mitochondria in HIV gp120- and Tat-treated neurons. In contrast, vehicle-treated neurons display low, diffuse, cytoplasmic expression of PINK1 and Parkin proteins, as indicated by the solid white arrows. Scale bars, 10 μm.
FIG 6.

Induction of mitophagy in human primary neurons treated with HIV gp120 and Tat is associated with increased expression and translocation of Parkin to damaged mitochondria. (A) Healthy tubular network of mitochondria (green) and diffuse cytoplasmic expression of Parkin (red). (B to D) Confocal laser microscopy showing translocation of Parkin (red) to the damaged fragmented mitochondria stained with TOM20 (green). In the enlarged images of boxed areas, solid white arrows indicate mitochondrial tubular network and dashed white arrows indicate colocalization of Parkin (red) with TOM20-stained (green) fragmented mitochondria (yellow). Scale bars, 10 μm.
Following the translocation of Parkin from the cytosol to mitochondria, there is polyubiquitination of mitochondrial substrates and recruitment of SQSTM1, which mediates the aggregation of dysfunctional mitochondria similarly to its aggregation of polyubiquitinated proteins (27). Following treatment of HPNs with gp120 and Tat, SQSTM1 was recruited to mitochondria after Parkin translocation (Fig. 7A to D). In total, these data demonstrate that, following exposure of neurons to gp120 and Tat, PINK1 is recruited to damaged mitochondria, leading to translocation of activated Parkin, recruitment of the LC3-binding protein SQSTM1, and mitochondrial aggregation, a preliminary step in mitochondrial fragmentation.
FIG 7.
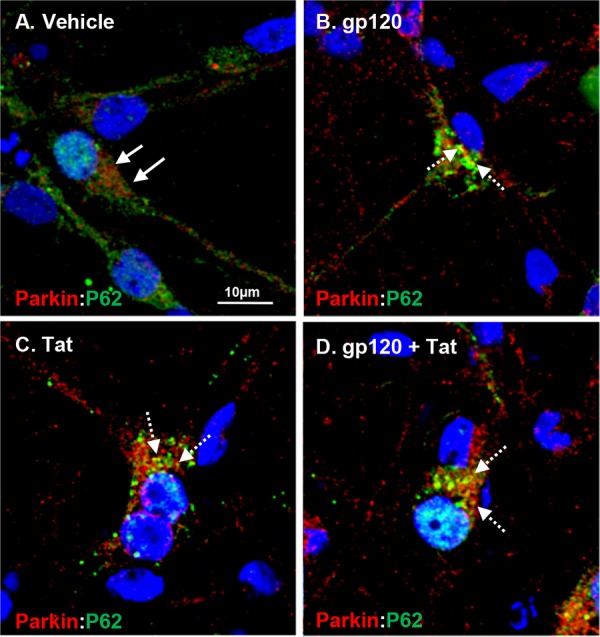
HIV gp120 and Tat treatments induce increased expression of P62/SQSTM1 and increased localization with Parkin-targeted mitochondria. (A) Vehicle-treated neurons display diffuse cytoplasmic localization of Parkin (red) and SQSTM1 (P62) (green), as indicated by the solid white arrows. (B to D) Increased colocalization (yellow) of P62 and Parkin in neurons treated with HIV gp120 and Tat or both, as indicated by the dashed white arrows. Scale bar, 10 μm.
HIV gp120 and Tat induce incomplete mitophagy.
During autophagic flux, autophagosomes fuse to the lysosomal compartment to ensure that encapsulated cargo is degraded. To assess autophagic flux in neurons treated with gp120 and Tat, we used an mCherry-EGFP-LC3 vector (15). When the vector is delivered to the lysosomal compartment along with mitochondria, the GFP signal is bleached due to the acidic lysosomal pH, while the mCherry red signal can be observed in both autophagosomes and acidic autolysosomes. Visualization of red fluorescence indicates completion of autophagic flux, as described previously (15). Neurons transfected with the plasmid vector and treated with gp120 and Tat for 24 h were analyzed by confocal microscopy. Neurons treated with gp120, Tat, and gp120-Tat displayed significant increases (P < 0.03) in yellow puncta and few red puncta, indicating accumulation of autophagosomes due to incomplete autophagy (Fig. 8C to E). Vehicle-treated cells showed a basal level of autophagy (Fig. 8A). In contrast, neuronal cells treated with antimycin A, a known inducer of autophagic flux in neurons (28), showed a significant increase (P < 0.001) in red puncta compared to their occurrence in untreated cells, indicating completion of autophagy and increased formation of autolysosomes (Fig. 8B).
FIG 8.

Treatment with gp120 and Tat induces incomplete autophagy. (A to E) Autophagic flux was monitored using a dual-fluorescence mCherry-EGFP-LC3 vector. HPNs transiently expressing mCherry-EGFP-LC3 plasmid were treated with HIV gp120 and Tat for 24 h. A concentration of 10 μM antimycin A was used as a positive control. Cells were then fixed and analyzed using confocal microscopy. Scale bar, 10 μm. (F) Quantitative analysis of the numbers of autophagosome puncta (yellow) and the remaining autolysosome puncta (red) per cell. Student's t test was performed to test for statistical significance. Data are presented as mean values ± SD (n = 4 independent donors; n ≥ 10 cells per condition). *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant.
In order to specifically assess the progression of mitophagy, we used an RFP-EGFP chimeric plasmid, pAT016, encoding a mitochondrion-targeting signal sequence, as described previously (15). Visualization of an RFP signal in the lysosomes indicates completion of mitophagy. As seen in Fig. 9A, neurons displayed a basal level of mitophagy under normal conditions. Neurons treated with antimycin A showed complete mitophagy, as indicated by visualization of the RFP signal in the lysosomes (Fig. 9B). In contrast, neurons treated with gp120, Tat, or the combination of the two proteins displayed a predominant yellow signal, indicating that there was incomplete delivery of mitochondria to the lysosomal compartment and a blockage of autophagic flux (Fig. 9C to E).
FIG 9.

Treatment with gp120 and Tat induces incomplete mitophagy. (A to E) Mitophagic flux was monitored using the p-mito-mRFP-EGFP reporter (pAT016). HPNs transiently expressing mito-mRFP-EGFP plasmid were treated with HIV gp120, Tat, or both for 24 h and then fixed and analyzed using confocal microscopy. Neurons were treated with 10 μM antimycin A for 24 h as a positive control for the induction of complete mitophagy. The diagram graphically demonstrates the change in fluorescence from no mitophagy (merge of green and red giving a yellow signal) to complete mitophagy (red). Scale bar, 10 μm.
To further examine whether mitophagy goes to completion following gp120 and Tat treatment, we examined the lipidation of LC3B-I to LC3B-II in the presence of bafilomycin A1, a lysosome-tropic agent that inhibits autophagosome-lysosome fusion and blocks the degradation of LC3B-II, resulting in the accumulation of LC3B-II (29). In these experiments, neurons were treated with gp120, Tat, or the combination of both proteins in the presence or absence of two different concentrations of bafilomycin A1. Under these conditions, we observed no increase in LC3B-II in the presence of HIV gp120 or Tat or both at the two different concentrations of bafilomycin A1 compared with its levels following bafilomycin A1 treatment alone, indicating that, similarly to bafilomycin A1, gp120 and Tat inhibit autophagic flux (Fig. 10A to D). These data indicate that on exposure of neurons to HIV gp120 or Tat, mitophagy fails to go to completion, resulting in neuron dysfunction.
FIG 10.

Treatment with gp120 and Tat induces blockage of mitophagic flux in neurons. (A to D) Protein expression levels of LC3B-II were analyzed by immunoblotting with LC3B antibody in neuronal cell lysates treated with HIV gp120, Tat, or both for 24 h and with 100 nM (A) or 30 nM bafilomycin A1 (BAFA1) (C) for 8 h prior to collection. Beta-actin (ACTB) was used as an internal loading control. (B, D) The relative expression of LC3B-II was normalized to the expression of ACTB. Each data point was normalized to the results for treatment with vehicle and analyzed by Image J software. Student's t test was performed to test for statistical significance. Data are presented as mean values ± SD (n = 3 independent donors). *, P < 0.05; n.s., not significant.
HIV gp120 and Tat impair mitochondrial function by decreasing mitochondrial membrane potential.
Mitochondria are the main organelles responsible for synthesizing energy for metabolic and cellular processes throughout the body and in the CNS. The mitochondrial membrane potential (ΔΨm) is an important marker to assess the functional state of mitochondria. Neurons utilize Parkin-mediated mitophagy to remove severely damaged mitochondria that have a decreased ΔΨm. Our next experiment examined whether gp120 and Tat induce a loss of mitochondrial membrane potential as a result of the observed mitochondrial damage. Neurons were treated with gp120 and Tat for 24 h and the cell cultures were preloaded with tetramethylrhodamine, ethyl ester (TMRE), for 30 min to label mitochondria prior to collection and analysis by fluorescence-activated cell sorting (FACS). Consistent with our earlier findings, gp120 and Tat significantly decreased ΔΨm in treated neurons (P < 0.05) (Fig. 11A to F). Thus, HIV-induced mitochondrial damage and loss of mitochondrial membrane potential are associated with DRP1-mediated mitochondrial fission.
FIG 11.

HIV-1 gp120 and Tat alter mitochondrial dynamics by decreasing mitochondrial membrane potential (ΔΨm) in human primary neurons. (A to E) Live human primary neurons were stained with TMRE to label active mitochondria and with LIVE/DEAD aqua blue dead cell stain to determine the viability of cells in the presence or absence of gp120, Tat, or both. The mitochondrial uncoupler CCCP (20 μM) was used as the positive control. (B to D) Neurons treated with HIV gp120 and Tat for 24 h show a significant decrease in TMRE staining compared with the results for vehicle-treated neurons (A). (F) The accompanying graph shows the percentages of TMRE-positive cells. Student's t test was performed to test the statistical significance. Data are presented as mean values ± SD (n = 4 independent donors). *, P < 0.05; ***, P < 0.001.
DISCUSSION
HIV Tat and HIV gp120 are known to be neurotoxic (30). However, the mechanism(s) associated with these neurotoxic effects are unclear. Here, we demonstrate that these HIV proteins initiate the activation and recruitment of mitophagy markers to damaged mitochondria in neurons but impair the delivery of mitochondria to the lysosomal compartment, resulting in incomplete mitophagy and neuronal damage.
Since HIV does not infect neurons, the neuronal dysfunction associated with HIV is mediated through the effects of viral proteins and cytokines released by infected cells, including microglia and astrocytes (31–34). The use of antiretroviral drugs with increased CNS penetrance has decreased the incidence of HAD and HAND. However, antiretroviral treatment may induce mitochondrial dysfunction and toxicity and premature aging in infected individuals (35–38). HIV gp120 binds the chemokine receptors CXCR4 and CCR5, resulting in its internalization (39, 40). It then forms a vesicular complex with mannose-binding lectin (MBL) (41), binds microtubules (42), and undergoes anterograde or retrograde axonal trafficking (22, 23, 41, 43). Thus, internalized HIV gp120 impairs mitochondrial transport and dynamics (42), which negatively impacts synaptic energy distribution. Following internalization of Tat by neuronal cells (24), the neurotoxic effects are mediated through interactions with NMDA receptors, synaptic and dendritic pruning (44), induction of apoptosis (45), Ca2+ dysregulation (46, 47), and oxidative stress (47). Tat and gp120 in neurons have been shown to alter mitochondrial morphology and function (48, 49) and to promote abnormal autophagy through disruption of the endolysosomal and autophagy pathways.
Our findings presented here describe an important role for mitophagy in regulating the mitochondrial toxicity and neuronal dysfunction associated with HIV infection. Our data indicate that, following internalization into primary human neurons, HIV gp120, Tat, and the combination of the two proteins all induce LC3B-II lipidation and autophagosome formation. We also show that gp120 and Tat initiate perinuclear accumulation and aggregation of damaged mitochondria, followed by mitochondrial fission induced by the translocation of DRP1 to damaged mitochondria. Combined exposure to gp120 and Tat induces the maximum accumulation of autophagosomes and increased mitochondrial fission. Additionally, mitochondrial fragmentation is initiated, as shown by the recruitment of PINK1 to damaged mitochondria, leading to translocation of Parkin, recruitment of SQSTM1, and mitochondrial aggregation. Thus, although mitophagy is initiated, the accumulation of SQSTM1 is indicative of mitophagy failing to go to completion. Furthermore, we show that gp120 and Tat impair mitochondrial function by decreasing mitochondrial membrane potential, which is associated with DRP1-mediated fission. In total, these findings provide an important mechanism to explain how exposure to HIV gp120 and Tat promote mitochondrial dysfunction through aberrant mitophagy that adversely affects neurons.
Elimination of damaged mitochondria through mitophagy is important for maintaining cell homeostasis (8, 15, 25, 50). Microorganisms, including viruses, have developed molecular strategies to hijack autophagy-associated proteins to promote persistent infection (15, 51). HIV proteins secreted from infected cells of the CNS can be internalized by neurons, lead to modifications in mitochondrial fission/fusion processes, and induce mitochondrial dysfunction (52).
Previously published studies have indicated that PINK1/Parkin-mediated pathways are important for mitochondrial function. Parkin-targeted neuronal mitochondria are localized in perinuclear areas to help with elimination of dysfunctional mitochondria in the lysosomal compartment (17, 18, 27, 53). Here, we show that exposure of human primary neurons to HIV gp120 and Tat induces the translocation of Parkin to mitochondria, a hallmark of mitophagy. Following the translocation of Parkin from the cytosol to fragmented, damaged mitochondria, SQSTM1 is recruited to damaged mitochondria and functions as a mediator of mitochondrial aggregation. This aggregation of the damaged mitochondria to the perinuclear area serves as a mechanism to cluster and prevent the spreading of nonfunctional mitochondria to other cellular compartments, including neuronal synapses, and favors the bioenergetically active mitochondria (54). SQSTM1 serves as a link between LC3 and ubiquitinated substrates and is efficiently degraded by autophagy. The increase in SQSTM1 following treatment of neurons with gp120 and Tat suggests that clearance of the protein is impaired as a result of inhibition of autophagic flux. Our studies using reporter plasmids expressing dual fluorescence mCherry-EGFP-LC3 and monomeric red fluorescent protein-enhanced green fluorescent protein targeted to mitochondria (mito-mRFP-EGFP) further support the finding that internalization of gp120 and Tat leads to incomplete mitophagy and impaired elimination of autophagosomes and mitochondria in the lysosomal compartment. The combination of gp120 and Tat promoted additive inhibition of autophagic flux.
Neurons have a high energetic demand that depends heavily on the mitochondrial function (55, 56). Mitochondrial injury and damage are associated with a loss of mitochondrial membrane potential (57). Here, we demonstrate that neurons incubated with HIV gp120 and Tat exhibit a significant reduction of TMRE-positive cells, indicative of Parkin-targeted mitochondria having a decreased ΔΨm.
In summary, we have shown in human primary neurons that HIV gp120 and Tat alter mitochondrial dynamics, resulting in incomplete mitophagy and failure to clear damaged mitochondria. There is compelling evidence supporting mitochondrial dysfunction and impaired mitophagy as having a central role in normal aging and the development of neurodegenerative disorders. Our study is a step forward in unraveling the molecular basis of the association of mitophagy with HIV and can contribute to the design of novel approaches to counter the accelerated aging and neurocognitive disorders associated with HIV infection of the brain.
MATERIALS AND METHODS
Chemical reagents and antibodies.
The chemicals used were carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Millipore-Sigma), bafilomycin A1 (Enzo Life Sciences), lipofectamine 2000 (Thermo Fisher Scientific), antimycin A (Sigma-Aldrich), and those in the TMRE-mitochondrial membrane potential assay kit (Abcam). The primary antibodies used included rabbit monoclonal anti-DRP1 (Cell Signaling), rabbit polyclonal anti-Parkin (Abcam), mouse monoclonal anti-Parkin (Santa Cruz Biotechnology), rabbit monoclonal anti-LC3B (Cell Signaling; Novus Biologicals), mouse monoclonal anti-SQSTM1/P62 (Abcam), mouse monoclonal anti-TOM20 (BD Biosciences), rabbit polyclonal anti-TOM20 (Santa Cruz Biotechnology), mouse monoclonal anti-actin (Sigma), mouse monoclonal anti-PINK1 (Abcam), and chicken monoclonal anti-MAP2 (Novus Biologicals) antibodies. The secondary antibodies used for immunofluorescence experiments were Alexa Fluor 488-, 568-, or 647-conjugated donkey anti-mouse, anti-rabbit, or anti-goat IgG (Thermo Fisher Scientific). HIV-1 IIIB gp120 (number 11784) and HIV-1 IIIB Tat (number 2222) recombinant proteins and mouse monoclonal antibodies against HIV-1 gp120 (number 2343) and HIV-1 Tat (number 1974) were obtained from the NIH AIDS Reagent Program. To address the specificity of gp120 staining, we used heat-inactivated gp120 protein that is unable to be internalized in neurons. Heat inactivation was performed at 65°C for 1 h. To address the specificity of Tat staining, 0.5 μM dextran sulfate (Sigma-Aldrich) was used to block the binding and uptake of Tat protein in neuronal cells. The HIV protein concentrations used in this study are consistent with the concentrations used in other studies, where their physiological roles were examined in tissues, and the localized concentration is expected to be slightly greater than that detected in serum from HIV-infected individuals (32, 58–61). The 100-ng/ml dose of HIV-1 gp120 and Tat proteins was selected from a dose-response experiment.
DNA constructs.
The mito-mRFP-EGFP vector was a kind gift from Seong-Jun Kim and Aleem Siddiqui, UC San Diego. The mCherry-EGFP-LC3B vector was a kind gift from Constanza J. Cortes and Albert R. La Spada, UC San Diego.
Cell culture, immunofluorescence, and imaging.
For isolation of human primary neurons (HPNs), forebrain fetal tissue was obtained from Advanced Bioscience Resources (Alameda, CA) and from the University of Washington School of Medicine according to the University of California San Diego Institutional Review Board guidelines and processed as previously described (41, 62). The brain tissue used for these studies was obtained from fetuses at a gestational age of 90 to 130 days. However, after 2 weeks in culture, neuronal cells are fully differentiated and have properties of neurons obtained from adult brain tissue (41, 62–64). Neurons were seeded at 105 cells per well on poly-d-lysine- and laminin-coated glass coverslips (Fisher Scientific) in 24-well plates and kept in neurobasal medium (Life Technologies Corporation) enriched with B27 (Life Technologies), 2 mM Glutamax (Life Technologies Corporation), and 10 μg/ml gentamicin (Life Technologies Corporation) for 2 weeks, with the medium changed every 3 days (41). HPNs were transfected using lipofectamine 2000 reagent (Life Technologies Corporation). For immunostaining experiments, a 4% paraformaldehyde (PFA) solution with sucrose in phosphate-buffered saline (PBS) was used for cell fixation and 0.25% Triton X-100 solution in PBS was used for cell permeabilization (41). Cells were incubated with the specific primary antibodies, followed by Alexa Fluor-conjugated secondary antibodies. Microscopic images were obtained using an Olympus FluoView FV1000 confocal microscope and minimally processed using Adobe Photoshop.
Western blot analysis.
For immunoblot experiments, neurons were seeded at 5 × 105 per well in poly-d-lysine- and laminin-coated 6-well dishes (Fisher Scientific) and maintained as described above. Extracted cell lysates were clarified by centrifugation, subjected to SDS-PAGE as described previously (15), and then transferred to nitrocellulose (NC) or polyvinylidene difluoride (PVDF) membranes using the Bolt Western blotting system (Thermo Fisher Scientific). Membranes were blocked with 5% bovine serum albumin (BSA; Sigma-Aldrich) in PBS supplemented with 0.1% Tween 20 (Sigma-Aldrich) and incubated with primary antibodies, and then the WesternBreeze immunodetection kit (Thermo Fisher Scientific) was used.
Translocation and mitophagy treatment.
Cells were treated with 20 μM CCCP or 100 ng/ml HIV gp120, Tat, the combination of both, or vehicle. CCCP, a potent mitochondrial oxidative phosphorylation uncoupler, was used as a positive control.
TMRE-mitochondrial membrane potential assay.
A mitochondrial membrane potential assay kit (Abcam) was used according to the supplier's instructions to quantify modifications in mitochondrial membrane potential. The assay uses TMRE (tetramethylrhodamine) dye to label healthy mitochondria in living cells. TMRE reagent labels active mitochondria but fails to accumulate with depolarized, damaged mitochondria that have a decreased mitochondrial membrane potential. We used this assay in combination with the LIVE/DEAD fixable aqua dead cell stain to determine cell viability. HPNs were treated with or without HIV recombinant proteins for 24 h and stained with TMRE for 15 min, followed by aqua DEAD cell stain for 30 min. CCCP was used as a positive control. Live cells were collected and analyzed by flow cytometry.
Statistical analysis.
Paired Student's t tests were used to analyze the statistical significance for all data. Statistical analysis was performed using GraphPad Prism 5 (La Jolla, CA). A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We acknowledge the support of the National Institute of Neurological Disorders and Stroke of the NIH (grants number R01 NS084912 and R01 NS104015 to S.A.S.) and the National Institute of Mental Health (NIH/NIMH) (grant number 5R25MH-081482-09 through an Interdisciplinary Research Fellowship in NeuroAIDS and the National Institute of Drug Abuse [NIH/NIDA] through a TMARC developmental pilot grant, number P50DA026306, both to C.T.-D.). Additional support was from the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (impaactnetwork.org).
Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) Network was provided by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under awards number UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC), and UM1AI106716 (IMPAACT LC), with cofunding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Jennifer Santini, UCSD Neuroscience Core and Light Microscopy Facility, funded by NINDS P30NS04710 core grant, for confocal microscopy technical assistance. We are grateful to Constanza J. Cortes and Albert R. La Spada, UC San Diego, for providing the mCherry-EGFP-LC3 vector construct and Seong-Jun Kim and Aleem Siddiqui, UC San Diego, for the mito-mRFP-EGFP plasmid construct. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV gp120, HIV Tat recombinant proteins and HIV gp120 and Tat antibodies. We thank Grant R. Campbell, Pratima Rawat, Seong-Jun Kim, Mohsin Khan, and Jihyung Lee, UC San Diego, for technical assistance and useful scientific discussions.
The authors declare no competing financial interest.
REFERENCES
- 1.Spudich S, Gonzalez-Scarano F. 2012. HIV-1-related central nervous system disease: current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb Perspect Med 2:a007120. doi: 10.1101/cshperspect.a007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group, HNRC Group. 2011. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gannon P, Khan MZ, Kolson DL. 2011. Current understanding of HIV-associated neurocognitive disorders pathogenesis. Curr Opin Neurol 24:275–283. doi: 10.1097/WCO.0b013e32834695fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alirezaei M, Kiosses WB, Fox HS. 2008. Decreased neuronal autophagy in HIV dementia: a mechanism of indirect neurotoxicity. Autophagy 4:963–966. doi: 10.4161/auto.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fields J, Dumaop W, Eleuteri S, Campos S, Serger E, Trejo M, Kosberg K, Adame A, Spencer B, Rockenstein E, He JJ, Masliah E. 2015. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: implications for HIV-associated neurocognitive disorders. J Neurosci 35:1921–1938. doi: 10.1523/JNEUROSCI.3207-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spector SA, Zhou D. 2008. Autophagy: an overlooked mechanism of HIV-1 pathogenesis and neuroAIDS? Autophagy 4:704–706. doi: 10.4161/auto.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ojha CR, Lapierre J, Rodriguez M, Dever SM, Zadeh MA, DeMarino C, Pleet ML, Kashanchi F, El-Hage N. 2017. Interplay between autophagy, exosomes and HIV-1 associated neurological disorders: new insights for diagnosis and therapeutic applications. Viruses 9:E176. doi: 10.3390/v9070176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Youle RJ, Narendra DP. 2011. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolman NJ, Chambers KM, Mandavilli B, Batchelor RH, Janes MS. 2013. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 9:1653–1662. doi: 10.4161/auto.24001. [DOI] [PubMed] [Google Scholar]
- 10.Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grenier K, McLelland GL, Fon EA. 2013. Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front Neurol 4:100. doi: 10.3389/fneur.2013.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nunnari J, Suomalainen A. 2012. Mitochondria: in sickness and in health. Cell 148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westermann B. 2010. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 14.Kim S, Kim HY, Lee S, Kim SW, Sohn S, Kim K, Cho H. 2007. Hepatitis B virus X protein induces perinuclear mitochondrial clustering in microtubule- and dynein-dependent manners. J Virol 81:1714–1726. doi: 10.1128/JVI.01863-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. 2013. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog 9:e1003722. doi: 10.1371/journal.ppat.1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan M, Syed GH, Kim SJ, Siddiqui A. 2015. Mitochondrial dynamics and viral infections: a close nexus. Biochim Biophys Acta 1853:2822–2833. doi: 10.1016/j.bbamcr.2014.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai Q, Zakaria HM, Simone A, Sheng ZH. 2012. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 22:545–552. doi: 10.1016/j.cub.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Simone FI, Darbinian N, Amini S, Muniswamy M, White MK, Elrod JW, Datta PK, Langford D, Khalili K. 2016. HIV-1 Tat and cocaine impair survival of cultured primary neuronal cells via a mitochondrial pathway. J Neuroimmune Pharmacol 11:358–368. doi: 10.1007/s11481-016-9669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng L, Zhang Z, Xu K, Qi G. 2013. HIV-1 gp120 induces autophagy in cardiomyocytes via the NMDA receptor. Int J Cardiol 167:2517–2523. doi: 10.1016/j.ijcard.2012.06.067. [DOI] [PubMed] [Google Scholar]
- 20.Li W, Li G, Steiner J, Nath A. 2009. Role of Tat protein in HIV neuropathogenesis. Neurotox Res 16:205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]
- 21.Kovalevich J, Langford D. 2012. Neuronal toxicity in HIV CNS disease. Future Virol 7:687–698. doi: 10.2217/fvl.12.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed F, MacArthur L, De Bernardi MA, Mocchetti I. 2009. Retrograde and anterograde transport of HIV protein gp120 in the nervous system. Brain Behav Immun 23:355–364. doi: 10.1016/j.bbi.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wenzel ED, Bachis A, Avdoshina V, Taraballi F, Tasciotti E, Mocchetti I. 2017. Endocytic trafficking of HIV gp120 is mediated by dynamin and plays a role in gp120 neurotoxicity. J Neuroimmune Pharmacol 12:492–503. doi: 10.1007/s11481-017-9739-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ. 2000. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 6:1380–1387. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- 25.Chan DC. 2012. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 26.Strack S, Cribbs JT. 2012. Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J Biol Chem 287:10990–11001. doi: 10.1074/jbc.M112.342105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebrahimi-Fakhari D, Saffari A, Wahlster L, DiNardo A, Turner D, Lewis TL Jr, Conrad C, Rothberg JM, Lipton JO, Kolker S, Hoffmann GF, Han MJ, Polleux F, Sahin M. 2016. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep 17:2162. doi: 10.1016/j.celrep.2016.10.051. [DOI] [PubMed] [Google Scholar]
- 29.Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mocchetti I, Bachis A, Avdoshina V. 2012. Neurotoxicity of human immunodeficiency virus-1: viral proteins and axonal transport. Neurotox Res 21:79–89. doi: 10.1007/s12640-011-9279-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma M, Nath A. 1997. Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol 71:2495–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT. 2000. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A 97:11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. 1995. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 34.Nath A. 2002. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis 186(Suppl 2):S193–S198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- 35.Hulgan T, Gerschenson M. 2012. HIV and mitochondria: more than just drug toxicity. J Infect Dis 205:1769–1771. doi: 10.1093/infdis/jis105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heaton RK, Clifford DB, Franklin DR Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group 2010. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology 75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robertson K, Liner J, Meeker RB. 2012. Antiretroviral neurotoxicity. J Neurovirol 18:388–399. doi: 10.1007/s13365-012-0120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Payne BAI, Wilson IJ, Hateley CA, Horvath R, Santibanez-Koref M, Samuels DC, Price DA, Chinnery PF. 2011. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat Genet 43:806–810. doi: 10.1038/ng.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA. 2007. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ 14:296–305. doi: 10.1038/sj.cdd.4402006. [DOI] [PubMed] [Google Scholar]
- 40.Misse D, Cerutti M, Noraz N, Jourdan P, Favero J, Devauchelle G, Yssel H, Taylor N, Veas F. 1999. A CD4-independent interaction of human immunodeficiency virus-1 gp120 with CXCR4 induces their cointernalization, cell signaling, and T-cell chemotaxis. Blood 93:2454–2462. [PubMed] [Google Scholar]
- 41.Teodorof C, Divakar S, Soontornniyomkij B, Achim CL, Kaul M, Singh KK. 2014. Intracellular mannose binding lectin mediates subcellular trafficking of HIV-1 gp120 in neurons. Neurobiol Dis 69:54–64. doi: 10.1016/j.nbd.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avdoshina V, Fields JA, Castellano P, Dedoni S, Palchik G, Trejo M, Adame A, Rockenstein E, Eugenin E, Masliah E, Mocchetti I. 2016. The HIV protein gp120 alters mitochondrial dynamics in neurons. Neurotox Res 29:583–593. doi: 10.1007/s12640-016-9608-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bachis A, Aden SA, Nosheny RL, Andrews PM, Mocchetti I. 2006. Axonal transport of human immunodeficiency virus type 1 envelope protein glycoprotein 120 is found in association with neuronal apoptosis. J Neurosci 26:6771–6780. doi: 10.1523/JNEUROSCI.1054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HJ, Martemyanov KA, Thayer SA. 2008. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci 28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aksenova MV, Aksenov MY, Adams SM, Mactutus CF, Booze RM. 2009. Neuronal survival and resistance to HIV-1 Tat toxicity in the primary culture of rat fetal neurons. Exp Neurol 215:253–263. doi: 10.1016/j.expneurol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Self RL, Mulholland PJ, Nath A, Harris BR, Prendergast MA. 2004. The human immunodeficiency virus type-1 transcription factor Tat produces elevations in intracellular Ca2+ that require function of an N-methyl-D-aspartate receptor polyamine-sensitive site. Brain Res 995:39–45. doi: 10.1016/j.brainres.2003.09.052. [DOI] [PubMed] [Google Scholar]
- 47.Aksenov MY, Hasselrot U, Wu G, Nath A, Anderson C, Mactutus CF, Booze RM. 2003. Temporal relationships between HIV-1 Tat-induced neuronal degeneration, OX-42 immunoreactivity, reactive astrocytosis, and protein oxidation in the rat striatum. Brain Res 987:1–9. doi: 10.1016/S0006-8993(03)03194-9. [DOI] [PubMed] [Google Scholar]
- 48.Rozzi SJ, Avdoshina V, Fields JA, Trejo M, Ton HT, Ahern GP, Mocchetti I. 2017. Human immunodeficiency virus promotes mitochondrial toxicity. Neurotox Res 32:723–733. doi: 10.1007/s12640-017-9776-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rozzi SJ, Avdoshina V, Fields JA, Mocchetti I. 2018. Human immunodeficiency virus Tat impairs mitochondrial fission in neurons. Cell Death Discov 4:8. doi: 10.1038/s41420-017-0013-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim SJ, Syed GH, Siddiqui A. 2013. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog 9:e1003285. doi: 10.1371/journal.ppat.1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dreux M, Chisari FV. 2010. Viruses and the autophagy machinery. Cell Cycle 9:1295–1307. doi: 10.4161/cc.9.7.11109. [DOI] [PubMed] [Google Scholar]
- 52.Fields JA, Serger E, Campos S, Divakaruni AS, Kim C, Smith K, Trejo M, Adame A, Spencer B, Rockenstein E, Murphy AN, Ellis RJ, Letendre S, Grant I, Masliah E. 2016. HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders. Neurobiol Dis 86:154–169. doi: 10.1016/j.nbd.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Narendra D, Tanaka A, Suen DF, Youle RJ. 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. 2010. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Attwell D, Laughlin SB. 2001. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 56.Sanchez AB, Varano GP, de Rozieres CM, Maung R, Catalan IC, Dowling CC, Sejbuk NE, Hoefer MM, Kaul M. 2016. Antiretrovirals, methamphetamine, and HIV-1 envelope protein gp120 compromise neuronal energy homeostasis in association with various degrees of synaptic and neuritic damage. Antimicrob Agents Chemother 60:168–179. doi: 10.1128/AAC.01632-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haelterman NA, Yoon WH, Sandoval H, Jaiswal M, Shulman JM, Bellen HJ. 2014. A mitocentric view of Parkinson's disease. Annu Rev Neurosci 37:137–159. doi: 10.1146/annurev-neuro-071013-014317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilbert M, Kirihara J, Mills J. 1991. Enzyme-linked immunoassay for human immunodeficiency virus type 1 envelope glycoprotein 120. J Clin Microbiol 29:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oh SK, Cruikshank WW, Raina J, Blanchard GC, Adler WH, Walker J, Kornfeld H. 1992. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J Acquir Immune Defic Syndr 5:251–256. doi: 10.1097/00126334-199203000-00005. [DOI] [PubMed] [Google Scholar]
- 60.Klasse PJ, Moore JP. 2004. Is there enough gp120 in the body fluids of HIV-1-infected individuals to have biologically significant effects? Virology 323:1–8. doi: 10.1016/j.virol.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 61.Cummins NW, Rizza SA, Badley AD. 2010. How much gp120 is there? J Infect Dis 201:1273–1274 (Letter.) doi: 10.1086/651434 (Reply, 201: 1274–1275, doi:.) [DOI] [PubMed] [Google Scholar]
- 62.Avramut M, Zeevi A, Achim CL. 2001. The immunosuppressant drug FK506 is a potent trophic agent for human fetal neurons. Brain Res Dev Brain Res 132:151–157. doi: 10.1016/S0165-3806(01)00307-8. [DOI] [PubMed] [Google Scholar]
- 63.Ray B, Chopra N, Long JM, Lahiri DK. 2014. Human primary mixed brain cultures: preparation, differentiation, characterization and application to neuroscience research. Mol Brain 7:63. doi: 10.1186/s13041-014-0063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hammond RR, Iskander S, Achim CL, Hearn S, Nassif J, Wiley CA. 2002. A reliable primary human CNS culture protocol for morphological studies of dendritic and synaptic elements. J Neurosci Methods 118:189–198. doi: 10.1016/S0165-0270(02)00126-7. [DOI] [PubMed] [Google Scholar]