

Despite successful suppression of HIV replication with antiretroviral therapy, current treatments are unable to eradicate the latent virus reservoir, and treatment interruption almost invariably results in the reactivation of HIV even after decades of virus suppression. Homeostatic proliferation of latently infected cells is one mechanism that could maintain the latent reservoir. To understand the impact of homeostatic mechanisms on virus reactivation and reservoir size, we experimentally depleted CD4+ T cells in ART-treated SIV-infected rhesus macaques and monitored their homeostatic rebound. We find that depletion-induced proliferation of CD4+ T cells is insufficient to reactivate the viral reservoir in vivo. Furthermore, the proportion of SIV DNA+ CD4+ T cells remains unchanged during reconstitution, suggesting that the reservoir is resistant to this mechanism of expansion at least in this experimental system. Understanding how T cell homeostasis impacts latent reservoir longevity could lead to the development of new treatment paradigms aimed at curing HIV infection.
KEYWORDS: SIV, SIV reservoir, homeostatic proliferation
ABSTRACT
A major barrier to human immunodeficiency virus (HIV) eradication is the long-term persistence of latently infected CD4+ T cells harboring integrated replication-competent virus. It has been proposed that the homeostatic proliferation of these cells drives long-term reservoir persistence in the absence of virus reactivation, thus avoiding cell death due to either virus-mediated cytopathicity or immune effector mechanisms. Here, we conducted an experimental depletion of CD4+ T cells in eight antiretroviral therapy (ART)-treated, simian immunodeficiency virus (SIV)-infected rhesus macaques (RMs) to determine whether the homeostatically driven CD4+ T-cell proliferation that follows CD4+ T-cell depletion results in reactivation of latent virus and/or expansion of the virus reservoir. After administration of the CD4R1 antibody, we observed a CD4+ T cell depletion of 65 to 89% in peripheral blood and 20 to 50% in lymph nodes, followed by a significant increase in CD4+ T cell proliferation during CD4+ T cell reconstitution. However, this CD4+ T cell proliferation was not associated with detectable increases in viremia, indicating that the homeostatic activation of CD4+ T cells is not sufficient to induce virus reactivation from latently infected cells. Interestingly, the homeostatic reconstitution of the CD4+ T cell pool was not associated with significant changes in the number of circulating cells harboring SIV DNA compared to results for the first postdepletion time point. This study indicates that, in ART-treated SIV-infected RMs, the homeostasis-driven CD4+ T-cell proliferation that follows experimental CD4+ T-cell depletion occurs in the absence of detectable reactivation of latent virus and does not increase the size of the virus reservoir as measured in circulating cells.
IMPORTANCE Despite successful suppression of HIV replication with antiretroviral therapy, current treatments are unable to eradicate the latent virus reservoir, and treatment interruption almost invariably results in the reactivation of HIV even after decades of virus suppression. Homeostatic proliferation of latently infected cells is one mechanism that could maintain the latent reservoir. To understand the impact of homeostatic mechanisms on virus reactivation and reservoir size, we experimentally depleted CD4+ T cells in ART-treated SIV-infected rhesus macaques and monitored their homeostatic rebound. We find that depletion-induced proliferation of CD4+ T cells is insufficient to reactivate the viral reservoir in vivo. Furthermore, the proportion of SIV DNA+ CD4+ T cells remains unchanged during reconstitution, suggesting that the reservoir is resistant to this mechanism of expansion at least in this experimental system. Understanding how T cell homeostasis impacts latent reservoir longevity could lead to the development of new treatment paradigms aimed at curing HIV infection.
INTRODUCTION
Infection with human immunodeficiency virus (HIV) continues to be a massive global health problem, with an estimated one million deaths worldwide in 2017 affecting mainly developing countries in sub-Saharan Africa (1). Despite the major declines in morbidity and mortality associated with HIV infection that followed the introduction of highly effective antiretroviral therapy (ART), there is still not a cure for this infection. The inability to eradicate HIV infection with current therapeutic regimens is thought to be related to the presence of a pool of latently infected cells harboring integrated replication-competent virus (often referred to as the virus reservoir) which persist indefinitely in ART-treated HIV-infected individuals (2–4). This persistent reservoir of latently infected cells is composed primarily of resting memory CD4+ T cells and is responsible for the relatively rapid rebound of viremia to pretreatment levels which follows interruption of ART (5–8). Kinetic studies have suggested that even in the absence of any residual virus replication, the natural decay of the reservoir of latently infected cells will last 60 years or more in ART-treated HIV-infected individuals (2, 3, 9). As such, lifelong administration of ART remains necessary, thus lowering the quality of life of HIV-infected individuals and creating a major burden on both individual and public health care programs (10–13). For these reasons, eradicating or at least functionally curing HIV infection would have a remarkable impact on global health.
Resting memory CD4+ T cells and, in particular, central memory CD4+ T cells (Tcm) are the main cell type harboring the persistent HIV/simian immunodeficiency virus (SIV) reservoir under ART (3, 14). This latent reservoir is established as early as day 3 postinfection and persists for the lifetime of the subject (15, 16). Potential mechanisms responsible for the reservoir persistence include residual virus replication under ART with low-level infection of new cells (17, 18), long life span of latently infected cells (19), and homeostatic proliferation of latently infected cells that occurs in the absence of virus reactivation (20, 21). Homeostatic proliferation is a particularly compelling mechanism for latent reservoir maintenance, as stimulation of resting CD4+ T cells from HIV-infected patients with IL-7 results in only minimal HIV reactivation compared with that of T cell receptor (TCR) stimulation (22). In addition, in vitro experiments have shown latently infected cells expand and are maintained in response to stimulation with homeostatic cytokines such as interleukin-7 (IL-7), IL-15, and IL-2 (14, 23). In this scenario, the latently infected, proliferating CD4+ T cells avoid cell death induced by either the virus-mediated cytopathic effect or immune effector mechanisms. To what extent homeostatic cell proliferation (i) induces virus reactivation from latently infected cells in vivo and (ii) impacts the size of the latent CD4+ T cell reservoir remains a largely unanswered question in the field of HIV cure. In this study, we used the established experimental system of in vivo CD4+ T cell depletion in ART-treated SIV-infected rhesus macaques (RMs) (24–27) to directly investigate the role of homeostatic proliferation on the stability and size of the latent virus reservoir.
Nonhuman primate models of HIV infection, particularly SIVmac infection of RMs, have been used to validate new treatments and vaccines in a preclinical setting as well as to test hypotheses regarding HIV pathogenesis and persistence (28, 29). Our laboratory previously has examined the effects of antibody-mediated CD4+ T cell depletion in multiple experiments in order (i) to understand the homeostatic reconstitution of CD4+ cells in uninfected rhesus macaques (RMs) and sooty mangabeys (SMs) (24); (ii) to understand the impact of CD4+ T cell depletion in nonpathogenic SIV-infected SMs (25); and (iii) to determine how CD4+ T cell depletion in SIV-infected RMs impacts the level of virus replication, the pattern of infected cells, and the overall pathogenesis of the infection (26, 27, 30). Overall, the latter experiments indicated that experimental CD4+ T cell depletion in SIV-infected RMs results in increased virus replication, expanded cellular tropism (which involves tissue macrophages and microglial cells), and faster disease progression. However, experimental CD4+ T cell depletion was never conducted in ART-treated SIV-infected RMs.
In the current study, we used antibody-mediated depletion of CD4+ T cells in ART-suppressed SIV-infected RMs to investigate whether and to what extent the homeostatic proliferation of CD4+ T cells that follows CD4+ T cell depletion is (i) sufficient to induce detectable virus reactivation from latently infected cells and (ii) capable of maintaining the size of the latent virus reservoir under ART. Consistent with previous experiments, CD4 depletion resulted in significant loss of CD4+ T cells in peripheral blood and lymph nodes (LN), which was followed by a substantial increase in CD4+ T cell proliferation (measured as expression of the marker Ki-67). However, this CD4+ T cell proliferation was not associated with detectable increases in plasma viremia, indicating that the homeostatic activation of CD4+ T cells is not sufficient to induce virus reactivation from latently infected cells. Interestingly, the homeostatic reconstitution of the CD4+ T cell pool did not result in increases in the number of circulating cells harboring SIV DNA compared to the first postdepletion time point. As such, this study indicates that the homeostasis-driven CD4+ T cell proliferation that follows experimental CD4+ T cell depletion occurs in the absence of detectable reactivation of latent virus and does not increase the size of the virus reservoir as measured in circulating cells.
RESULTS
Study design.
In order to understand the impact of CD4+ T cell homeostasis on the stability and the size of the virus reservoir, we administered 50 mg/kg of body weight of the CD4-depleting antibody CD4R1 to eight ART-treated SIV-infected Indian-origin RMs (Fig. 1A). Of note, four of the animals started ART at week 8 postinfection, and four others started ART at 8 to 9 months postinfection. All RMs were treated with a three-drug ART regimen (tenofovir, 20 mg/kg/day; emtricitabine, 40 mg/kg/day; and dolutegravir, 2.5 mg/kg/day), administered daily by subcutaneous injection. As expected, upon ART initiation, all RMs exhibited a decrease in viral load (Spearman coefficient, −0.87; P = 0.03), and most animals controlled viremia within 2 to 4 months, with the animals placed on ART earlier controlling viremia more rapidly (Fig. 1B). Importantly, viremia was undetectable in all RMs within 10 months of ART initiation (Fig. 1B). Similar to ART-treated HIV-infected humans, all RMs showed a partial reconstitution of both percentage and absolute number of CD4+ T cells during ART (Fig. 1C and D).
FIG 1.

Experimental design and ART treatment. (A) Study design. All animals were infected with SIVmac239, 4 animals were treated with antiretroviral therapy (ART) at 2 months postinfection (early chronic treated animals), and another 4 animals were treated with ART at 10 months postinfection (late chronic treated animals). After ∼10 months of ART, when virus was fully suppressed, the CD4R1 anti-CD4-depleting antibody was administered in all animals (black arrow). Both early chronic and late chronic treated animals were monitored for 4 and 7 months, respectively. Small blood draws were collected every 2 weeks from all animals to monitor CD4 counts. Large blood draws (red arrows), rectal biopsy specimens (green arrows), and fine needle aspirates or lymph node biopsy specimens (blue arrows) were collected pre-CD4 depletion and post-CD4 depletion, at 4 or 5 months after CD4 depletion. (B) Plasma viral loads for individual animals using the standard assay (limit, 60 copies/ml plasma). (C) CD4 counts (cells/μl whole blood). (D) Percentage of CD4+ T cells. Open symbols represent early chronic treated animals, and closed symbols represent late chronic treated animals. Each symbol represents a different animal, and the line represents the median.
CD4+ T cell depletion and reconstitution.
Two months after viral loads had been suppressed below the limit of detection, CD4R1 antibody (clone OKT4 with RM IgG1 heavy chain) was administered as a single 50-mg/kg dose by a slow bolus injection that was not followed by any adverse reactions. As shown in Fig. 2, and consistent with previous studies, CD4R1 treatment resulted in a significant depletion of circulating CD4+ T cells, both in absolute number (Fig. 2A) and as a fraction of CD3+ T cells (Fig. 2B), with a nadir of CD4+ T cells ranging between 2.3% and 22.4% of the baseline predepletion levels (Fig. 2C). Flow cytometric analysis of naive and memory CD4+ T cell subsets showed that CD4+ T cell depletion was somewhat more pronounced for naive T cells, which led to a modest increase in the relative proportion of memory T cell subsets (Fig. 2D to G). The level of CD4+ T cell depletion was also assessed in peripheral lymph nodes by fine-needle aspirate and in the rectum by rectal biopsy specimens (RB). We found that the frequency of CD4+ T cells in LN declined significantly by an average of 49% compared to predepletion levels (P = 0.0313 by Wilcoxon rank sum) (Fig. 2H and J), while RB showed a trend toward decline of total CD4+ T cells (Fig. 2I and K, respectively). As expected based on previous studies (24, 26, 27), the depletion of peripheral blood CD4+ T cells was followed by a slow increase in their absolute numbers (Fig. 2A) and proportion within the CD3+ T cells (Fig. 2B) over the 4 months of postdepletion follow up.
FIG 2.

Dynamics of CD4+ T cell depletion and reconstitution. (A to C) CD4+ T cells expressed as counts (cells/μl whole blood) (A), frequency in PBMC (B), and a percentage of baseline CD4 counts (C). (D to G) CD4+ T cell memory subsets were measured during CD4 depletion and reconstitution, including naive (D), central memory (E), transitional memory (F), and effector memory T cells (G). (H to K) Tissue measures of CD4+ T cells. The frequency of CD4+ T cells in lymph nodes (H) and rectal biopsy specimens (I). The percentage of CD4+ T cells as a percentage of baseline in lymph nodes (J) and rectal biopsy specimens (K). Open symbols represent early chronic treated animals, and closed symbols represent late chronic treated animals. Each symbol represents a different animal, and the line represents the median. Statistical differences were determined using Wilcoxon rank sum test (*, P < 0.05).
The phase of CD4+ T cell reconstitution following antibody-mediated depletion of these cells is typically characterized by a marked but transient increase in the level of CD4+ T cell proliferation (24, 26, 27). To assess this phenomenon in the current experiment, we next measured longitudinally in peripheral blood, lymph node biopsy specimens, and rectal biopsy specimens the frequency of CD4+ T cells expressing the proliferation marker Ki-67 and the activation marker PD-1. As expected based on previous studies, we observed a significant increase in the percentage of Ki67+ CD4+ T cells, with the highest levels found at weeks 4 and 6 post-CD4 depletion (P = 0.003 by Friedman test) (Fig. 3A). The percentage of circulating Ki67+ CD4+ T cells returned to baseline between 2 and 3 months after CD4+ depletion. Of note, only a modest, nonsignificant increase in the percentage of Ki67+ CD4+ T cells was observed in LN and RB after CD4+ T cell depletion (Fig. 3B and C). The latter data, however, need to be interpreted with caution, since these tissues were sampled only at week 1 and 16 postdepletion, thus corresponding to two time points at which only modest changes in the levels of Ki67+ CD4+ T cells were also observed in peripheral blood (Fig. 3A). The observed increase in the percentage of Ki67+ CD4+ T cells was more pronounced in memory CD4+ T cell subsets as opposed to naive CD4+ T cells (TN), with transitional memory (TTM) cells exhibiting the highest mean levels, followed by effector memory (TEM) cells and then central memory T cells (TCM; week 4 means: TTM, 9.89%; TEM, 7.48%; TCM, 6.2%; TN, 1.2%) (Fig. 3D to G). We also found that CD4+ T cell depletion was followed by a gradual increase in PD-1 expression in CD4+ T cells from peripheral blood mononuclear cells (PBMCs) and LN, but no change in PD-1 expression in RB was seen (Fig. 3H to J). We also measured the fraction of CD4+ T cells expressing CD25 and HLA-DR and observed no significant change during CD4 depletion and reconstitution (data not shown).
FIG 3.

Proliferation of CD4+ T cells after CD4 depletion. Expression of Ki67 on CD4+ T cells was measured in PBMC (A), LN (B), and RB (C) during CD4 depletion and reconstitution. (D to G) Ki67 was measured within each CD4+ T cell memory subset: naive (D), central memory (E), transitional memory (F), and effector memory (G). Expression of PD-1 on CD4+ T cells was measured in PBMC (H), LN (I), and RB (J) during CD4 depletion and reconstitution. Open symbols represent early chronic treated animals, and closed symbols represent late chronic treated animals. Each symbol represents a different animal, and the line represents the median.
Depletion-induced CD4+ T cell proliferation does not result in a detectable increase in virus production.
A key question to be addressed by this study is whether and to what extent homeostatic proliferation of CD4+ T cells that are latently infected with SIV results in reactivation of virus transcription and virion production. To this end, we longitudinally investigated the levels of plasma viremia before and after CD4+ T cell depletion and, in particular, during the phase of increased CD4+ T cell proliferation that follows CD4 depletion. We first measured plasma viral loads using a standard SIVmac239-specific quantitative PCR (qPCR) with a limit of detection similar to that of standard HIV qPCR assays (below 60 copies/ml of plasma) and found no detectable levels of viremia in any of the eight CD4-depleted animals (Fig. 4A). To determine whether the CD4+ T cell proliferation that follows CD4 depletion induced low levels of virus production, we next repeated our measurement using an ultrasensitive viral load assay with a limit of detection of 3 copies per ml of plasma (31). While the ultrasensitive viral load assay showed very low but detectable levels of plasma viremia in all animals, no significant difference in virus production was noted after CD4 depletion on the levels of residual viremia (Fig. 4B). Overall, these data indicate that strong activation of the homeostatic proliferation of CD4+ T cells as induced by CD4 depletion is not sufficient per se to induce any significant reactivation of latent virus.
FIG 4.

CD4 depletion does not induce increases in SIVmac239 viral loads. Viral load was measured using both the standard sensitivity (60 copies/ml of plasma) (A) and the ultrasensitive (3 copies/ml of plasma) viral load assay (B). Open symbols represent early chronic treated animals, and closed symbols represent late chronic treated animals. Each symbol represents a different animal, the solid line represents the median, and the horizontal dotted line represents the limit of detection (60 or 3 copies/ml of plasma).
Changes in the size of the latent virus reservoir during CD4+ T cell depletion and reconstitution.
To examine the effects of antibody-mediated CD4+ T cell depletion and the consequent CD4+ T cell homeostatic proliferation on the size of the latent SIV reservoir, we measured the levels of total cell-associated SIV DNA both predepletion and at multiple time points during CD4+ T cell reconstitution (weeks 1, 2, 6, and 16 to 20). Similar to previous reports in HIV-infected humans (3, 9, 14, 21, 32) and in SIV-infected RMs (33–36), prior to CD4 depletion we observed medians (ranges) of 838.13 (268.97 to 2,204.23) and 3,258.38 (824.98 to 10,947.63) SIV copies per million cells in PBMCs and CD4+ T cells, respectively (Fig. 5A and B). As expected, following CD4 depletion, the levels of SIV copies significantly declined by 4.8-fold in PBMCs (P = 0.0016 by Friedman test with multiple comparisons), thus reflecting the lower percentage of CD4+ T cells within the PBMC population (Fig. 5A). Importantly, we also observed that during the phase of CD4+ T cell reconstitution the level of cell-associated SIV DNA per million circulating CD4+ T cells trended toward decline between week 2 and weeks 16 to 20 at an average of 9-fold (Fig. 5B). This result indicates either that SIV DNA-negative cells contribute more to the pool of CD4+ T cells that proliferate following CD4 depletion or that SIV DNA-positive CD4+ T cells are less likely to survive the homeostatic cell cycle than SIV DNA-negative cells.
FIG 5.
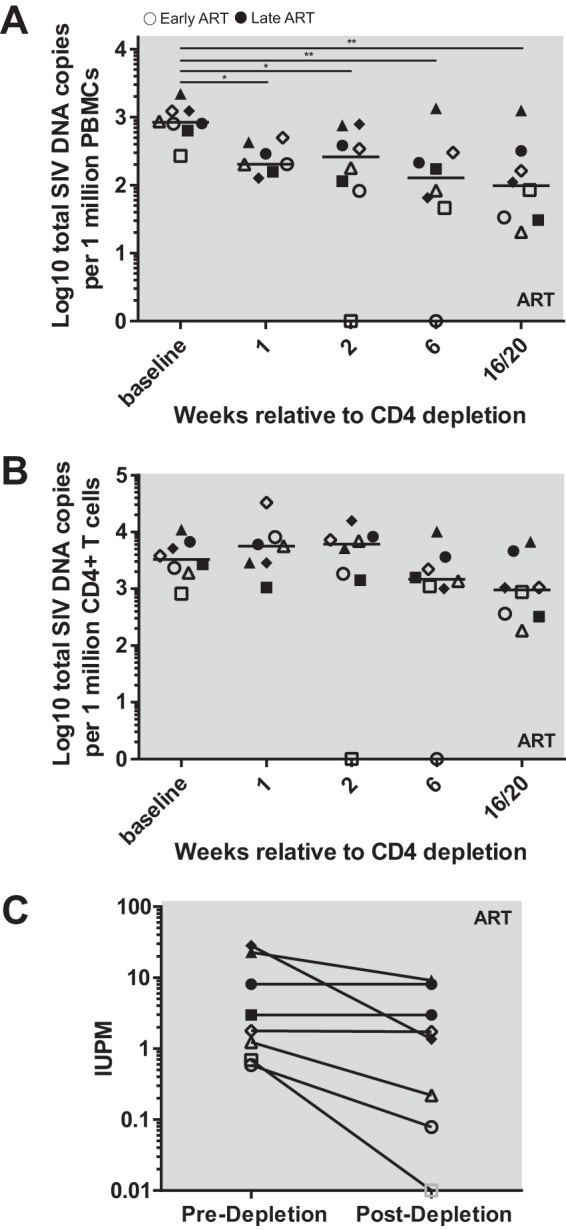
SIV reservoir dynamics after CD4 depletion. Total cell-associated SIV DNA was measured in PBMC (A) and calculated in CD4+ T cells (B). (C) Quantitative SIV outgrowth assay. Open symbols represent early chronic treated animals, and closed symbols represent late chronic treated animals. Each symbol represents a different animal. Gray outlined animals were undetectable, the solid line represents the median, and horizontal dotted lines represent the limit of detection (60 or 3 copies/ml of plasma). Statistical differences were determined using the Friedman test (*, P < 0.05; **, P < 0.01).
To determine whether the observed declines in total cell-associated SIV DNA observed after CD4 depletion resulted in a commensurate decay in the proportion of replication-competent virus, we next performed in a subset of the same samples, as based on cell availability, a quantitative viral outgrowth assay (SIV QVOA.) As shown in Fig. 5C, the infectious units per million (IUPM) were an average of 0.5 log lower at week 16 or 28 after CD4 depletion than the predepletion baseline. Taken together, these data show that antibody-mediated CD4 depletion results in decreases in the total and replication-competent SIV reservoir.
DISCUSSION
The mechanisms by which the reservoir of latently infected CD4+ T cells persists indefinitely under fully suppressive ART and then rapidly reactivates after treatment interruption are not fully understood. Defining the molecular mechanisms responsible for the reservoir persistence will facilitate the development of HIV cure strategies that either permanently block HIV reactivation or rely on latency reversal in order to make the viral reservoir vulnerable to immune effector mechanisms (i.e., shock and kill). In the current study, we focused on the role played by homeostatic proliferation of CD4+ T cells as a mechanism of reservoir persistence and used the established experimental model of antibody-mediated depletion of CD4+ T cells to induce a transient but high level of homeostatic proliferation of CD4+ T cells in a group of eight ART-suppressed SIV-infected RMs. We wish to emphasize, as this study represents a typical example of an experiment of high relevance to the field of HIV latency and cure, that for obvious ethical reasons this study would be very difficult to conduct in HIV-infected humans but can be performed in the highly relevant model of ART-treated SIV-infected RMs.
Specifically, in this study we sought to determine whether and to what extent the homeostatic proliferation of CD4+ T cells that follows CD4+ T cell depletion is (i) sufficient to induce detectable virus reactivation from latently infected cells and (ii) capable of maintaining or even expanding the size of the latent virus reservoir under ART. We found that homeostatic CD4+ T cell proliferation (i) was not associated with detectable increases in plasma viremia, indicating no significant virus reactivation from the reservoir latently infected cells, and (ii) did not result in increases in the fraction of circulating CD4+ T cells harboring total cell-associated SIV DNA during the phase of CD4+ T cell reconstitution after depletion. This study supports the hypothesis that, in ART-treated SIV-infected RMs, the homeostasis-driven CD4+ T cell proliferation that follows experimental CD4+ T cell depletion occurs in the absence of detectable reactivation of latent virus and does not seem to expand the size of the virus reservoir as measured in circulating cells.
The observation that no virus reactivation is measurable during strong CD4+ T cell homeostatic proliferation is fully consistent with the hypothesis that this physiological mechanism of immune function plays a key role in maintaining the reservoir, as these latently infected cells can expand without being targeted by either the virus cytopathic effect or the host immune system. Whether our observations in the setting of homeostatic proliferation induced by CD4+ T cell depletion are recapitulated in clinical or preclinical settings in which homeostatic proliferation is not triggered in such a dramatic fashion, or in which other types of CD4+ T cell proliferations are taking place (i.e., direct T cell activation through T cell receptor stimulation, proinflammatory cytokine exposure, etc.), is not addressed by the current study and therefore remains to be ascertained. Of note, the fact that homeostatic CD4+ T cell proliferation is not sufficient per se to reverse latency in vivo is consistent with a recent in vitro study of latency reversal indicating that the homeostatic cytokine interleukin-7 (IL-7) alone is insufficient to reactivate virus in culture (22). Our observations are also consistent with a recent report indicating that some latently infected, resting CD4+ T cells require several rounds of a very strong stimulation in order to allow the reactivation of virus production (21).
Recently, our laboratory explored the ability of CD8+ T cells to suppress residual virus production using antibody-mediated depletion of CD8+ T cells in ART-treated SIV-infected rhesus macaques (33). In this experiment, experimental CD8+ lymphocyte depletion resulted in robust increases in residual viremia in 13 out of 13 RMs, with a recovery of CD8+ T cells being closely associated with declines in viremia below the limits of detection in standard assays. In that experiment, however, we could not exclude the hypothesis that the homeostatic proliferation of CD4+ T cells occurring in response to removal of a large proportion of CD8+ T cells could be responsible for the increased level of viremia. The results presented in the current study provide unique insight into our previous work, demonstrating definitively that homeostatic proliferation of CD4+ T cells in response to lymphocyte depletion is insufficient to cause the increases in residual virus replication of up to ∼3 logs over the standard level observed after CD8 depletion. As such, the current set of results provides strong evidence in favor of the hypothesis that, in the setting of ART treatment, CD8+ T cells provide a suppressive effect on virus production from latently infected CD4+ T cells.
The observation that the level of total cell-associated SIV DNA per million circulating CD4+ T cells trends toward decline during the phase of CD4+ T cell reconstitution after antibody-mediated depletion is surprising, as the depletion-induced CD4+ T cell proliferation should also result in the expansion of integrated viral genomes. The lack of an increase in SIV DNA-positive CD4+ T cells during homeostatic reconstitution suggests either that (i) uninfected (i.e., SIV DNA-negative) CD4+ T cells contribute more to the pool of proliferating CD4+ T cells following CD4 depletion or that (ii) infected (i.e., SIV DNA-positive) CD4+ T cells are less likely to survive the homeostatic proliferation. The latter possibility is somewhat less likely, as putative mechanisms of cell death, such as direct virus-mediated cytopathic effect and host immune effector functions, would be triggered by the activation of virus expression in latently infected cells, which is not consistent with the observed lack of viremia blips after CD4+ T cell depletion (Fig. 4). However, we cannot rule out the possibility that the homeostatic signal that leads to cell proliferation does in fact induce activation of virus transcription in latently SIV-infected CD4+ T cells, but the expression of virus gene products results in either aborted cell cycle (i.e., vpr induction of cell cycle arrest) or induction of cell death (via either direct cytopathicity or immune effector mechanisms) prior to sufficient virus production to be detected as plasma viremia. An intriguing additional possibility is that the homeostatic proliferation in response to CD4+ T cell depletion is less effective in cells that express inhibitory markers, such as PD-1 and CTLA-4, that are known to be upregulated on latently SIV-infected cells (37, 38). Finally, it is possible that under long-term ART there is an in vivo selection for latently SIV-infected cells that are intrinsically less sensitive to activation signals, therefore increasing their in vivo longevity as resting cells. Whatever the mechanism responsible for the lack of increase in SIV DNA-positive cells during the homeostatic proliferation of CD4+ T cells which follows CD4 depletion, it does not seem that this phenomenon is more or less pronounced in cells infected with replication-competent virus, as inducible virus declines at about the same average magnitude (∼1 log).
The current pilot study is, to the best of our knowledge, the first ever of its kind, i.e., exploring the effects of CD4+ T cell depletion in ART-treated SIV-infected macaques. We must emphasize that this study has some limitations, which, in turn, suggest important avenues for further exploration. First, the study includes a relatively small number of animals (n = 8) in which ART was initiated either early (day 56 postinfection) or late (8 to 9 months postinfection) after SIVmac239 inoculation. As such, the presented results will have to be confirmed in larger groups of animals and possibly when ART is initiated very early after infection (i.e., day 3 to 10 postinfection), i.e., in a setting in which the virus reservoir under ART is smaller and potentially more vulnerable to the effect of specific interventions aimed at reducing its size. Second, the study does not include an analytical treatment interruption, and therefore we could not determine the impact of CD4 depletion and reconstitution on the time to and magnitude of rebound viremia, which are key functional measures of the efficacy of any intervention on the size of the viral reservoir. Third, at the time of this study we did not have the technology to perform multiparametric sorting of relatively small CD4+ T cell subpopulations in either peripheral blood or lymphoid tissues for subsequent high-resolution analysis of the level of SIV DNA and RNA in these cells. As such, the current study does not address how CD4 depletion and reconstitution impacted the relative proportion of SIV DNA-positive cells within memory CD4+ T cell subsets or CD4+ T cells expressing markers such as PD-1 and CTLA-4. Finally, while the study clearly shows that CD4 depletion during ART treatment induces homeostatic activation of CD4+ T cells with absent or limited virus reactivation, we do not know what specific cell signaling pathways allow the proliferation of CD4+ T cells without reactivation of virus expression. We envision that future studies of CD4+ depletion in ART-treated SIV-infected RMs will provide important additional information on how homeostatic proliferation of CD4+ T cells impacts the size of the virus reservoir.
MATERIALS AND METHODS
Animals.
Eight Indian-origin RMs were enrolled in this study. Four were intrarectally (i.r.) infected with a 10,000 50% tissue culture infectious dose (TCID50) of SIVmac239 (194-11R, 207-11R, 268-11R, and 315-11R), while four were infected intravenously (i.v.) with 3,000 TCID50 of SIVmac239 (REf16, RFv15, RVz15, and RZe15). Three animals were female and five animals were male, varying in age from 3 to 5 years old at the time of infection. All animals were housed at the Yerkes National Primate Research Center of Emory University. These studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (39) and were approved by the Emory University (AWA no. A3180-01) Institutional Animal Care and Use Committee (IACUC). All animals were anesthetized before the performance of any procedure, and proper steps were taken to ensure the welfare and minimize the suffering of all animals in the proposed studies.
Antiretroviral therapy.
All RMs were put on ART, 4 animals initiated ART at 8 weeks after SIV infection (REf16, RFv15, RVz15, and RZe15), and 4 animals initiated ART after 21 weeks of infection (194-11R, 207-11R, 268-11R, and 315-11R). These groups of animals are referred to here as early and late chronic treated, respectively. The ART regimen was 20 mg/kg/day of tenofovir (PMPA; Gilead, Foster City, CA), 40 mg/kg/day of emtricitabine (FTC; Gilead), and 2.5 mg/kg/day of dolutegravir (DTG; ViiV, Middlesex, UK) administered daily as a subcutaneous injection. The ART combination was made under sterile laboratory conditions in 2-liter stocks and frozen at −20°C until use. Complete suppression (no viral blips) of plasma viremia to below the limit of detection (60 copies/ml of plasma) was achieved within 4 months in the early chronic treated animals and within 8 months in the late chronic treated animals. All animals were kept on ART for 10 months before CD4 depletion for consistency, and ART was administered daily until the end of the study.
Depletion of CD4+ T cells.
CD4+ cells were depleted using CD4R1 (clone OKT4 with RM IgG1 heavy chain) CD4+ depletion antibody (NIH NHP Reagent Resource Program) administered in a slow intravenous bolus at 50 mg/kg (purity was confirmed by high-performance liquid chromatography). CD4+ T cell depletion was determined by quantification of CD4+ T cell frequency in blood, lymph node, and rectal biopsy specimens using flow cytometry.
Sample collection and tissue processing.
Longitudinal collection and processing of blood, fine needle aspirates (FNA), or lymph node and rectal biopsy specimens were performed as previously described (33, 34, 40, 41). Briefly, 2 ml of blood samples was used for complete blood count (CBC), the remaining blood was centrifuged to collect plasma, and then peripheral blood mononuclear cells (PBMCs) were collected over density gradient centrifugation using Ficoll (Gibco, Massachusetts). Twenty rectal punch biopsy specimens were collected using an anoscope placed a short distance into the rectum. RB were washed with media (RPMI plus 10% fecal calf serum plus penicillin-streptomycin-glutamine; Gibco) 3 times and then incubated for 2 h with collagenase (1 mg/ml; Sigma, Darmstadt, Germany) and DNase (30 μl/150 ml; Roche, Basel, Switzerland) at 37°C. RB were then mechanically processed with a cannula attached to a 30-ml syringe and filtered through a 70-μm filter to remove any tissue debris. Cells were washed once in media and counted (Countess II; Life Technologies, CA). For FNA and LN biopsy specimen collection, the skin around the LN was surgically prepared (hair clipped and surgically scrubbed). For FNAs superficial LN was palpated, and the FNA was collected by passing a 22-gauge needle attached to a 3-ml syringe through the skin and into the LN 4 times. No suction was applied. Samples were directly expelled into media and the needle flushed 3 times. FNAs were then washed and counted. For the LN biopsy procedure, an incision was made over the LN, and LN was excised over clamps by blunt dissection. Fat and connective tissue was removed and the LN mechanically dissociated through a 70-μm strainer and washed with media, and cells were counted. All samples were processed, fixed (1% paraformaldehyde), and analyzed within 24 h of collection. Leftover cells were frozen with freezing medium (fetal calf serum plus 10% dimethyl sulfoxide; Sigma) or stored as a cell pellet for later DNA and RNA analysis.
Immunophenotype by flow cytometry.
Multiparametric flow cytometry was performed on PBMCs, LN mononuclear cells, and cells isolated from RBs with fluorescently labeled monoclonal antibodies cross-reactive in RMs. CCR5 and CCR7 staining was performed at 37°C for 30 min, all other surface stains were performed at room temperature for 30 min, and Ki67 was measured using the fixation/permeabilization solution kit (BD Biosciences, California). All specimens were acquired on an LSR II (BD Biosciences), and the acquired data were analyzed with FlowJo software (Tree Star, Oregon, USA).
Plasma viral load and cell-associated SIVgag DNA.
Standard plasma viral loads (limit of detection, 60 copies/ml plasma) (42) or ultrasensitive plasma viral loads (31) were performed as described previously. Quantification of cell-associated SIV DNA was performed as previously described (33).
SIVmac239 quantitative viral outgrowth assay.
Latently infected cells were quantified by using a limiting dilution culture assay in which peripheral CD4+ T cells were enriched from PBMC using magnetic beads and column purification (Miltenyi Biotec, Bergisch Gladbach, Germany). The CD4+ T cells were stimulated for 20 h with anti-CD3 (clone SP34-2; BD Biosciences) and anti-CD28 (clone CD28.2) antibodies in medium and 100 U/ml IL-2 (Sigma). After being washed, the CD4+ T cells were cocultured with CEMx174 cells in duplicate 5-fold serial dilutions ranging from as many as 1 × 106 cells per well to as few as 320 cells per well. The cells were cultured in media and 100 U/ml IL-2 (Sigma). The ratio of CD4+ T cells to CEMx174 was 1:4 for the 2 highest dilutions. A constant number of 1,000,000 CEMx174 cells was added to all other wells. The cultures were split every 7 days, and fresh medium was added. After 14 days, growth of virus was detected by quantitative reverse transcription-PCR (qRT-PCR). SIV RNA was isolated from 400 μl of culture supernatant using the Zymo viral RNA isolation kit (Zymo Research). The cell culture supernatant underwent centrifugation for 90 min at 21,000 × g before extraction by following the manufacturer's instructions. DNase treatment was performed using an RQ1 RNase-free DNase kit (Promega). A one-step qRT-PCR targeting SIVgag was performed using an Applied Biosystems 7500 real-time PCR system (Applied Biosystems) and the Veriquest probe one-step qRT-PCR master mix (Affymetrix) with the following primers and probe: SIVmacgagFwd, 5′-GCAGAGGAGGAAATTACCCAGTAC-3′; SIVmacgagRev, 5′-CAATTTTACCCAGGCATTTAATGTT-3′; SIVmacgag-probe, 5′-6FAM-TGTCCACCTGCCATTAAGCCCGA-3IBFQ-3′. The frequencies of infected cells were determined by the maximum-likelihood method (43) and were expressed as infectious units per million (IUPM) CD4+ T cells.
Statistical analysis.
All statistical analyses were performed using GraphPad Prism 7.0, correcting for multiple comparisons where appropriate. Significance was attributed as P values of <0.05 (*), <0.01 (**), and <0.005 (***).
ACKNOWLEDGMENTS
We acknowledge the Kean laboratory for providing us with SIVmac239 and the TCID50 data to use in our infections. We also acknowledge the laboratory of Jeffrey Lifson for performing the ultrasensitive viral loads. We thank Barbara Cervasi, Kiran Gill, and Shan Liang for technical support, Stephanie Ehnert and the Yerkes Research Resources technicians for study organization and scheduling, and the YNPRC veterinary staff, especially Sherrie Jean, for caring for the animals.
This study was funded by NIH grants R37 AI066998 to G.S., P51 OD011132 to the YNPRC, and P30 AI050409 to the Emory CFAR.
REFERENCES
- 1.UNAIDS. 2018. Latest statistics on the status of the AIDS epidemic. UNAIDS, Geneva, Switzerland: http://www.unaids.org/en/resources/fact-sheet. [Google Scholar]
- 2.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 3.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 4.Strain MC, Gunthard HF, Havlir DV, Ignacio CC, Smith DM, Leigh-Brown AJ, Macaranas TR, Lam RY, Daly OA, Fischer M, Opravil M, Levine H, Bacheler L, Spina CA, Richman DD, Wong JK. 2003. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence. Proc Natl Acad Sci U S A 100:4819–4824. doi: 10.1073/pnas.0736332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 6.Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med 1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 7.Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, Kovacs JA, Polis MA, Walker RE, Falloon J, Masur H, Gee D, Baseler M, Dimitrov DS, Fauci AS, Lane HC. 1999. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A 96:15109–15114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joos B, Fischer M, Kuster H, Pillai SK, Wong JK, Boni J, Hirschel B, Weber R, Trkola A, Gunthard HF, Swiss HIV Cohort Study. 2008. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci U S A 105:16725–16730. doi: 10.1073/pnas.0804192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaafoura S, de Goer de Herve MG, Hernandez-Vargas EA, Hendel-Chavez H, Abdoh M, Mateo MC, Krzysiek R, Merad M, Seng R, Tardieu M, Delfraissy JF, Goujard C, Taoufik Y. 2014. Progressive contraction of the latent HIV reservoir around a core of less-differentiated CD4(+) memory T cells. Nat Commun 5:5407. doi: 10.1038/ncomms6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhaskaran K, Hamouda O, Sannes M, Boufassa F, Johnson AM, Lambert PC, Porter K, CASCADE Collaboration 2008. Changes in the risk of death after HIV seroconversion compared with mortality in the general population. JAMA 300:51–59. doi: 10.1001/jama.300.1.51. [DOI] [PubMed] [Google Scholar]
- 11.Cooper DA. 2008. Life and death in the cART era. Lancet 372:266–267. doi: 10.1016/S0140-6736(08)61086-7. [DOI] [PubMed] [Google Scholar]
- 12.Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, Ledergerber B, Lundgren J, Neuhaus J, Nixon D, Paton NI, Neaton JD, INSIGHT SMART Study Group. 2008. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 5:e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lohse N, Hansen AB, Pedersen G, Kronborg G, Gerstoft J, Sorensen HT, Vaeth M, Obel N. 2007. Survival of persons with and without HIV infection in Denmark, 1995-2005. Ann Intern Med 146:87–95. doi: 10.7326/0003-4819-146-2-200701160-00003. [DOI] [PubMed] [Google Scholar]
- 14.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sekaly RP. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ananworanich J, Schuetz A, Vandergeeten C, Sereti I, de Souza M, Rerknimitr R, Dewar R, Marovich M, van Griensven F, Sekaly R, Pinyakorn S, Phanuphak N, Trichavaroj R, Rutvisuttinunt W, Chomchey N, Paris R, Peel S, Valcour V, Maldarelli F, Chomont N, Michael N, Phanuphak P, Kim JH, RV254/SEARCH 010 Study Group . 2012. Impact of multi-targeted antiretroviral treatment on gut T cell depletion and HIV reservoir seeding during acute HIV infection. PLoS One 7:e33948. doi: 10.1371/journal.pone.0033948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitney JB, Hill AL, Sanisetty S, Penaloza-MacMaster P, Liu J, Shetty M, Parenteau L, Cabral C, Shields J, Blackmore S, Smith JY, Brinkman AL, Peter LE, Mathew SI, Smith KM, Borducchi EN, Rosenbloom DI, Lewis MG, Hattersley J, Li B, Hesselgesser J, Geleziunas R, Robb ML, Kim JH, Michael NL, Barouch DH. 2014. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 512:74–77. doi: 10.1038/nature13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chun TW, Nickle DC, Justement JS, Large D, Semerjian A, Curlin ME, O'Shea MA, Hallahan CW, Daucher M, Ward DJ, Moir S, Mullins JI, Kovacs C, Fauci AS. 2005. HIV-infected individuals receiving effective antiviral therapy for extended periods of time continually replenish their viral reservoir. J Clin Investig 115:3250–3255. doi: 10.1172/JCI26197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin AR, Siliciano RF. 2016. Progress toward HIV eradication: case reports, current efforts, and the challenges associated with cure. Annu Rev Med 67:215–228. doi: 10.1146/annurev-med-011514-023043. [DOI] [PubMed] [Google Scholar]
- 19.Palmer S, Maldarelli F, Wiegand A, Bernstein B, Hanna GJ, Brun SC, Kempf DJ, Mellors JW, Coffin JM, King MS. 2008. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 105:3879–3884. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray AJ, Kwon KJ, Farber DL, Siliciano RF. 2016. The latent reservoir for HIV-1: how immunologic memory and clonal expansion contribute to HIV-1 persistence. J Immunol 197:407–417. doi: 10.4049/jimmunol.1600343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S, Rosenbloom DI, Keele BF, Ho YC, Siliciano JD, Siliciano RF. 2017. Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: potential role in latent reservoir dynamics. J Exp Med 214:959–972. doi: 10.1084/jem.20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosque A, Famiglietti M, Weyrich AS, Goulston C, Planelles V. 2011. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog 7:e1002288. doi: 10.1371/journal.ppat.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iglesias-Ussel MD, Romerio F. 2011. HIV reservoirs: the new frontier. AIDS Rev 13:13–29. [PubMed] [Google Scholar]
- 24.Engram JC, Cervasi B, Borghans JA, Klatt NR, Gordon SN, Chahroudi A, Else JG, Mittler RS, Sodora DL, de Boer RJ, Brenchley JM, Silvestri G, Paiardini M. 2010. Lineage-specific T-cell reconstitution following in vivo CD4+ and CD8+ lymphocyte depletion in nonhuman primates. Blood 116:748–758. doi: 10.1182/blood-2010-01-263814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klatt NR, Villinger F, Bostik P, Gordon SN, Pereira L, Engram JC, Mayne A, Dunham RM, Lawson B, Ratcliffe SJ, Sodora DL, Else J, Reimann K, Staprans SI, Haase AT, Estes JD, Silvestri G, Ansari AA. 2008. Availability of activated CD4+ T cells dictates the level of viremia in naturally SIV-infected sooty mangabeys. J Clin Investig 118:2039–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Micci L, Alvarez X, Iriele RI, Ortiz AM, Ryan ES, McGary CS, Deleage C, McAtee BB, He T, Apetrei C, Easley K, Pahwa S, Collman RG, Derdeyn CA, Davenport MP, Estes JD, Silvestri G, Lackner AA, Paiardini M. 2014. CD4 depletion in SIV-infected macaques results in macrophage and microglia infection with rapid turnover of infected cells. PLoS Pathog 10:e1004467. doi: 10.1371/journal.ppat.1004467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ortiz AM, Klatt NR, Li B, Yi Y, Tabb B, Hao XP, Sternberg L, Lawson B, Carnathan PM, Cramer EM, Engram JC, Little DM, Ryzhova E, Gonzalez-Scarano F, Paiardini M, Ansari AA, Ratcliffe S, Else JG, Brenchley JM, Collman RG, Estes JD, Derdeyn CA, Silvestri G. 2011. Depletion of CD4(+) T cells abrogates post-peak decline of viremia in SIV-infected rhesus macaques. J Clin Investig 121:4433–4445. doi: 10.1172/JCI46023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans DT, Silvestri G. 2013. Nonhuman primate models in AIDS research. Curr Opin HIV AIDS 8:255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar N, Chahroudi A, Silvestri G. 2016. Animal models to achieve an HIV cure. Curr Opin HIV AIDS 11:432–441. doi: 10.1097/COH.0000000000000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng M, Paiardini M, Engram JC, Beilman GJ, Chipman JG, Schacker TW, Silvestri G, Haase AT. 2012. Critical role of CD4 T cells in maintaining lymphoid tissue structure for immune cell homeostasis and reconstitution. Blood 120:1856–1867. doi: 10.1182/blood-2012-03-418624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Prete GQ, Shoemaker R, Oswald K, Lara A, Trubey CM, Fast R, Schneider DK, Kiser R, Coalter V, Wiles A, Wiles R, Freemire B, Keele BF, Estes JD, Quinones OA, Smedley J, Macallister R, Sanchez RI, Wai JS, Tan CM, Alvord WG, Hazuda DJ, Piatak M Jr, Lifson JD. 2014. Effect of suberoylanilide hydroxamic acid (SAHA) administration on the residual virus pool in a model of combination antiretroviral therapy-mediated suppression in SIVmac239-infected Indian rhesus macaques. Antimicrob Agents Chemother 58:6790–6806. doi: 10.1128/AAC.03746-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perelson AS, Essunger P, Ho DD. 1997. Dynamics of HIV-1 and CD4+ lymphocytes in vivo. AIDS 11(Suppl A):S17–S24. [PubMed] [Google Scholar]
- 33.Cartwright EK, Spicer L, Smith SA, Lee D, Fast R, Paganini S, Lawson BO, Nega M, Easley K, Schmitz JE, Bosinger SE, Paiardini M, Chahroudi A, Vanderford TH, Estes JD, Lifson JD, Derdeyn CA, Silvestri G. 2016. CD8(+) lymphocytes are required for maintaining viral suppression in SIV-infected macaques treated with short-term antiretroviral therapy. Immunity 45:656–668. doi: 10.1016/j.immuni.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mavigner M, Watkins B, Lawson B, Lee ST, Chahroudi A, Kean L, Silvestri G. 2014. Persistence of virus reservoirs in ART-treated SHIV-infected rhesus macaques after autologous hematopoietic stem cell transplant. PLoS Pathog 10:e1004406. doi: 10.1371/journal.ppat.1004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cartwright EK, McGary CS, Cervasi B, Micci L, Lawson B, Elliott ST, Collman RG, Bosinger SE, Paiardini M, Vanderford TH, Chahroudi A, Silvestri G. 2014. Divergent CD4+ T memory stem cell dynamics in pathogenic and nonpathogenic simian immunodeficiency virus infections. J Immunol 192:4666–4673. doi: 10.4049/jimmunol.1303193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Micci L, Ryan ES, Fromentin R, Bosinger SE, Harper JL, He T, Paganini S, Easley KA, Chahroudi A, Benne C, Gumber S, McGary CS, Rogers KA, Deleage C, Lucero C, Byrareddy SN, Apetrei C, Estes JD, Lifson JD, Piatak M Jr, Chomont N, Villinger F, Silvestri G, Brenchley JM, Paiardini M. 2015. Interleukin-21 combined with ART reduces inflammation and viral reservoir in SIV-infected macaques. J Clin Investig 125:4497–4513. doi: 10.1172/JCI81400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGary CS, Deleage C, Harper J, Micci L, Ribeiro SP, Paganini S, Kuri-Cervantes L, Benne C, Ryan ES, Balderas R, Jean S, Easley K, Marconi V, Silvestri G, Estes JD, Sekaly RP, Paiardini M. 2017. CTLA-4(+)PD-1(-) memory CD4(+) T cells critically contribute to viral persistence in antiretroviral therapy-suppressed, SIV-infected rhesus macaques. Immunity 47:776–788. doi: 10.1016/j.immuni.2017.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fromentin R, Bakeman W, Lawani MB, Khoury G, Hartogensis W, DaFonseca S, Killian M, Epling L, Hoh R, Sinclair E, Hecht FM, Bacchetti P, Deeks SG, Lewin SR, Sekaly RP, Chomont N. 2016. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. PLoS Pathog 12:e1005761. doi: 10.1371/journal.ppat.1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 40.Carnathan D, Lawson B, Yu J, Patel K, Billingsley JM, Tharp GK, Delmas OM, Dawoud R, Wilkinson P, Nicolette C, Cameron MJ, Sekaly RP, Bosinger SE, Silvestri G, Vanderford TH. 2018. Reduced chronic lymphocyte activation following interferon alpha blockade during the acute phase of simian immunodeficiency virus infection in rhesus macaques. J Virol 92:e01760-. doi: 10.1128/JVI.01760-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palesch D, Bosinger SE, Mavigner M, Billingsley JM, Mattingly C, Carnathan DG, Paiardini M, Chahroudi A, Vanderford TH, Silvestri G. 2018. Short-term pegylated interferon alpha2a treatment does not significantly reduce the viral reservoir of simian immunodeficiency virus-infected, antiretroviral therapy-treated rhesus macaques. J Virol 92:e00279-18. doi: 10.1128/JVI.00279-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hofmann-Lehmann R, Swenerton RK, Liska V, Leutenegger CM, Lutz H, McClure HM, Ruprecht RM. 2000. Sensitive and robust one-tube real-time reverse transcriptase-polymerase chain reaction to quantify SIV RNA load: comparison of one- versus two-enzyme systems. AIDS Res Hum Retrovir 16:1247–1257. doi: 10.1089/08892220050117014. [DOI] [PubMed] [Google Scholar]
- 43.Rosenbloom DIS, Elliott O, Hill AL, Henrich TJ, Siliciano JM, Siliciano RF. 2015. Designing and interpreting limiting dilution assays: general principles and applications to the latent reservoir for HIV-1. Open Forum Infect Dis 2:ofv123. doi: 10.1093/ofid/ofv123. [DOI] [PMC free article] [PubMed] [Google Scholar]