

Abstract
Enantioselectivity increases with increasing carbonyl electrophilicity in 2-propanol-mediated reductive couplings of aldehydes with branched aryl-substituted allylic acetates to form products of carbonyl anti-(α-aryl)allylation. This unusual phenomenon is caused by aldehyde coordination to diastereomeric kinetic vs thermodynamic carbonyl binding sites that deliver enantiomeric products. Exploiting this effect, anti-diastereo- and enantioselective (α-aryl)allylations of fluoral hydrate and difluoroacetaldehyde ethyl hemiacetal were developed.
Graphical Abstract
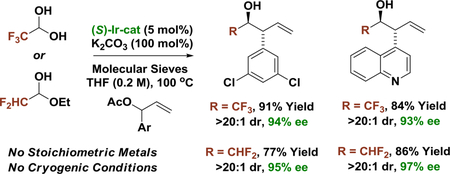
In the course of exploring alcohol-mediated carbonyl reductive couplings, our laboratory has developed a broad suite of catalytic enantioselective allylations and propargylations.1 Whereas uniformly high levels of diastereo- and enantioselectivity are observed across diverse reactions of this type,1,2 for branched aryl-substituted allylic acetate pronucleophiles, enantioselectivities were highly variable: while high levels of asymmetric induction were observed in 2-propanol-mediated reductive couplings of paraformaldehyde,3 enantioselectivities in additions to higher aldehydes were found to increase with increasing carbonyl electrophilicity - spanning a range of 1 to 94% ee. An investigation into this intriguing effect was undertaken and has led to two major outcomes: (a) a novel mechanism for enantioinduction was uncovered wherein aldehyde coordination to diastereomeric kinetic vs thermodynamic binding sites of the iridium catalyst results in formation of enantiomeric products, and (b) the development of catalytic enantioselective anti-(α-aryl)allylations of aqueous fluoral and difluoroacetaldehyde ethyl hemiacetal.
Only isolated examples of enantioselective carbonyl (α-aryl)allylation appear in the literature, which involve stoichiometric chirality transfer from auxiliaries.4 Notwithstanding a single report of asymmetric Sakurai-type allylations of glyoxamides,5e no systematic studies of catalytic enantioselective carbonyl (α-aryl)allylation exist and, for the isolated examples that have been described, modest levels of asymmetric induction are typically observed.5 Catalytic enantioselective (α-aryl)allylations of fluoral and difluoroacetaldehyde are unknown and, despite the importance of fluorinated substructures in pharmaceutical and agrochemical ingredients,6 nearly all related enantioselective metal-catalyzed fluoral additions require anhydrous conditions or conditions involving in situ generation of gaseous fluoral.6i Finally, although difluoromethyl groups are unique in their ability to serve as lipophilic hydrogen-bond donors,7 there is a surprising paucity of catalytic methods for formation of enantiomerically enriched CHF2-bearing stereocenters.8 Here, we show that commercially available 75 wt% solutions of fluoral hydrate in water or 90 wt% solutions of difluoroacetaldehyde ethyl hemiacetal in ethanol can be exploited in highly anti-diastereo- and enantioselective carbonyl (α-aryl)allylations. To illustrate the preparative utility of this method, it was applied to syntheses of di- and trifluoromethylated derivatives of the FDA approved alkaloid d-hyoscyamine (dextro-atropine).
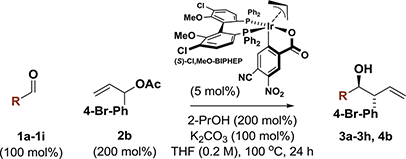
In an initial set of experiments, para-substituted benzaldehydes 1a-1f were subjected to 2-propanol-mediated reductive couplings with 1-(4-bromophenyl)allyl acetate 2b using the cyclometallated π-allyliridium C,O-benzoate complex modified by (S)-Cl,MeO-BIPHEP as catalyst (Table 1). An unusual correlation between the electronic properties of the aldehyde and enantioselectivity emerged. Whereas electron-rich aldehydes such as p-(dimethylamino)benzaldehyde 1a and anisal-dehyde 1b provided products 3a and 3b in nearly racemic form, the degree of asymmetric induction gradually improved with increasing aldehyde electrophilicity. Ultimately, using aqueous fluoral hydrate 1i as the carbonyl electrophile in the presence of 4Å molecular sieves, the product of carbonyl (α-aryl)allylation 4b was formed in 88% yield with complete anti-diastereoselectivity in 94% enantiomeric excess (Table 1, entry 9). In the absence of molecular sieves, only trace quantities of reductive coupling product 4b were observed. As our iridium catalyst is tolerant of aqueous organic conditions,1 the primary role of molecular sieves would appear to involve dehydration of fluoral hydrate.
Table 1.
Correlation between Aldehyde Electrophilicity and Enantioselectivity in the anti-(α-Aryl)allylation of Aromatic Aldehydes and Fluoral Hydrate.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | σ | 1a-1i | 3a-3h | Yield% | dr | ee% |
| 1 | 4-Me2N-Ph | -0.83 | 1a | 3a | 16 | 4:1 | 1 |
| 2 | 4-MeO-Ph | -0.27 | 1b | 3b | 54 | 9:1 | 5 |
| 3 | 4-Me-Ph | -0.17 | 1c | 3c | 69 | 4:1 | 12 |
| 4 | Ph | 0.00 | 1d | 3d | 74 | 4:1 | 15 |
| 5 | 4-F-Ph | 0.06 | 1e | 3e | 88 | 3:1 | 16 |
| 6 | 4-Br-Ph | 0.23 | 1f | 3f | 88 | 4:1 | 23 |
| 7 | 3-(6-Br-Pyr) | - | 19 | 3g | 82 | 5:1 | 37 |
| 8 | 2-(6-Br-Pyr) | - | 1h | 3h | 75 | 4:1 | 65 |
| 9b | CF3 | - | 1i | 4b | 88 | >20:1 | 94 |
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
4Å molecular sieves 300 wt%). Fluoral hydrate in water (75 wt%).
Conditions identified for 2-propanol-mediated reductive anti-(α-aryl)allylation of fluoral hydrate 1i were applied to branched aryl-substituted allylic acetates 2a-2l (Table 2). The products of carbonyl (α-aryl)allylation 4a-4l were formed in good to excellent yields as single diastereomers with uniformly high levels of enantioselectivity. The syn-diastereomer was not observed upon 1H NMR analysis of crude reaction mixtures. Beyond products 4b-4e derived from parasubstituted α-aryl allyl acetates, adducts 4f, 4g and 4h, which incorporate ortho-substituted, 3,5-disubstituted and 3,4-disubstituted aryl moieties, respectively, could be formed in a highly stereoselective fashion. Finally, allyl donors bearing heteroaromatic rings 2i-2l provided the corresponding products of carbonyl (α-aryl)allylation 4i-4l in good to excellent yields and uniformly high stereoselectivities. The absolute stereochemical assignment of adducts 4a-4k is made in analogy to that established for compound 4l, which was determined by single crystal X-ray diffraction analysis.
Table 2.
Enantioselective Iridium-Catalyzed anti-(α-Aryl)allylation of Fluoral Hydrate 1i to Form CF3-Bearing β-Aryl Alcohols 4a-4l.a
 |
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
Encouraged by the preceding results, optimal conditions for carbonyl anti-(α-aryl)allylation were applied to difluoroacetaldehyde (Table 3). The feasibility of this process was uncertain, as difluoroacetaldehyde is only available as the ethyl hemiacetal in 90 wt% solutions in ethanol, and ethanol could itself engage in transfer hydrogenative carbonyl addition. In the event, using the same set of branched aryl-substituted allylic acetates 2a-2l, the CHF2-bearing β-aryl alcohols 5a-5l were formed in good to excellent yields with complete anti-diastereoselectivity and uniformly high levels of enantioselectivity. The products were accompanied by small amounts of the ethanol coupling side products (3–14% yield), which were easily removed upon chromatographic isolation of the principal reaction products. The absolute stereochemical assignment of adducts 5a-5k is made in analogy to that established for compound 5l, which was determined by single crystal X-ray diffraction analysis.
Table 3.
Enantioselective Iridium-Catalyzed anti-(α-Aryl)allylation of Difluoroacetaldehyde Ethyl Hemiacetal 1j to Form CHF2-Bearing β-Aryl Alcohols 5a-5l.a
 |
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
The unusual dependence of enantioselectivity on carbonyl electrophilicity merits discussion, as this phenomenon appears quite unique (Figure 1).9 In earlier studies on carbonyl allylation, an inversion in enantioselectivity was observed in response to the steric features of the allyl donor. Specifically, whereas carbonyl allylations10a,b and crotylations10c,d display matching enantiofacial preferences, an inversion in enantiofacial preference is observed in carbonyl tert-prenylations11 when using the same enantiomer of the chiral cyclometallated π-allyliridium C,O-benzoate catalyst. These data suggest that for small allyl donors, carbonyl addition occurs predominantly through the transition structure indicated on the left-hand side of Figure 1. As steric demand of the allyl donor increases, the allyl fragment prefers to reside in a more open environment, causing addition to occur through the diastereomeric transition structure indicated on the right-hand side of Figure 1. In carbonyl anti-(α-aryl)allylations, the steric demand of the (aryl)allyl appears to lie between these extremes. Carbonyl binding to the more accessible coordination site is kinetically preferred (Figure 1, left) and, for sufficiently electrophilic aldehydes, binding triggers carbonyl addition. However, for less electrophilic aldehydes, binding to the kinetically more accessible site (Figure 1, left) less frequently triggers carbonyl addition and the alternate diastereomeric binding mode can be sampled. Consequently, carbonyl addition through the diastereomeric transition structure (Figure 1, left) begins to compete, resulting in an erosion in enantioselectivity. For anti-(α-aryl)allylations, the diastereomeric transition structure (Figure 1, left) may be thermodynamically preferred or of comparable thermodynamic stability compared to the kinetically more accessible binding mode.
Figure 1.

Stereochemical Model for Iridium-Catalyzed Enantioselective Carbonyl anti-(α-Aryl)allylation.
To illustrate the utility of this method, carbonyl anti-(α-aryl)allylation was applied in syntheses of di- and trifluoro-methylated derivatives of the FDA approved alkaloid d-hyoscyamine (dextro-atropine) (Scheme 1).12 Specifically, adduct 4a was converted to the TBS ether and subjected to modified Johnson-Lemieux conditions for alkene oxidative cleavage.13 The resulting α,β-stereogenic carboxylic acid 6a was exposed to tropine under Shiina’s esterification conditions14 to form the TBS-protected ester 7a in good yield, albeit with some epimerization. Finally, treatment of 7a with HF-pyridine provided CF3-hyoscyamine 8a. The CHF2-bearing anti-(α-aryl)allylation product 5a was converted to CHF2-hyoscyamine 8b in an analogous manner.
Scheme 1.

Enantioselective Syntheses of Di- and Trifluoro-methylated Derivatives of d-Hyoscyamine (dextro-Atropine) 8a and 8b.a
aYields of material isolated by silica gel chromatography. See Supporting Information for further experimental details. b25 oC. c(2-Pr)2NEt (100 mol%).
In summary, we report direct methods for 2-propanolmediated anti-diastereo- and enantioselective (α-aryl)allylations of fluoral hydrate and difluoroacetaldehyde ethyl hemiacetal using readily accessible branched allylic acetate pronucleophiles. These processes take advantage of a unique mechanism for enantioinduction involving competition between diastereomeric kinetic vs thermodynamic carbonyl binding modes. As illustrated by the formation of di- and trifluoromethylated derivatives of d-hyoscyamine (dextro-atropine) 8a and 8b, these methods broaden access to diverse CF3- and CHF2- bearing building blocks, which should assist the small molecule medicinal and agrochemical discovery enterprise.
Supplementary Material
Acknowledgments.
The Robert A. Welch Foundation (F-0038), the NIH (RO1-GM069445) and the UT Austin Center for Green Chemistry and Catalysis are acknowledged for partial support of this research. The Deutsche Forschungsgemeinschaft (DFG) is acknowledged for postdoctoral fellowship support (JT).
Footnotes
The authors declare no competing financial interest.
Supporting Information Available: Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra. HPLC traces of racemic and enantiomerically enriched products. Crystallographic data for compounds 4l and 5l. This material is available free of charge via the internet at http://pubs.acs.org
REFERENCES
- (1).For a recent review, see: Kim SW; Zhang W; Krische MJ Acc. Chem. Res 2017, 50, 2371.
- (2).For a recent review of umpoled allylations, see: Spielman K; Niel G; de Figueiredo RM; Campagne J-M Chem. Soc. Rev 2018, 47, 1159.
- (3).Garza VJ; Krische MJ J. Am. Chem. Soc 2016, 138, 3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Riediker M; Duthaler RO Angew. Chem., Int. Ed. Engl 1989, 28, 494. [Google Scholar]; (b) Hafner A; Duthaler RO; Marti R; Rihs G; Rothe-Streit P; Schwarzenbach FJ Am. Chem. Soc 1992, 114, 2321. [Google Scholar]; (c) Sebelius S; Szabó KJ Eur. J. Org. Chem 2005, 2539. [Google Scholar]; (d) Vogt M; Ceylan S; Kirschning A Tetrahedron 2010, 66, 6450. [Google Scholar]; (e) Haddad TD; Hirayama LC; Singaram BJ Org. Chem 2010, 75, 642. [DOI] [PubMed] [Google Scholar]; (f) De Sio V; Massa A; Scettri A Org. Biomol. Chem, 2010, 8, 3055. [DOI] [PubMed] [Google Scholar]; (g) Li L-L; Tao Z-L; Han Z-Y; Gong L-Z Org. Lett 2017, 19, 102. [DOI] [PubMed] [Google Scholar]; (h) Tekle-Smith MA; Williamson KS; Hughes IF; Leighton JL Org. Lett 2017, 19, 6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Nakajima M; Saito M; Hashimoto S Chem. Pharm. Bull 2000, 48, 306. [DOI] [PubMed] [Google Scholar]; (b) Zanoni G; Gladiali S; Marchetti A; Piccinini P; Tredici I; Vidari G Angew. Chem. Int. Ed 2004, 43, 846. [DOI] [PubMed] [Google Scholar]; (c) Zhu S-F; Yang Y; Wang L-X; Liu B; Zhou Q-L Org. Lett 2005, 7, 2333. [DOI] [PubMed] [Google Scholar]; (d) Hemelaere R; Carreaux F; Carboni B; Chem. Eur. J 2014, 20, 14518. [DOI] [PubMed] [Google Scholar]; (e) Evans DA; Aye Y; Wu J Org. Lett 2006, 8, 2071. [DOI] [PubMed] [Google Scholar]; (f) Bai B; Zhu H-J; Pan W Tetrahedron 2012, 68, 6829. [Google Scholar]; (g) Miura T; Nishida Y; Morimoto M; Murakami MJ Am. Chem. Soc 2013, 135, 11497. [DOI] [PubMed] [Google Scholar]; (h) Tan Z; Wan X; Zang Z; Qian Q; Deng W; Gong H Chem. Commun. 2014, 50, 3827. [DOI] [PubMed] [Google Scholar]
- (6).For selected reviews on methods for the preparation of organofluorine compounds, see: Ma J-A; Cahard D Chem. Rev 2004, 104, 6119.Brunet VA; O’Hagan D Angew. Chem. Int. Ed 2008, 47, 1179.Bobbio C; Gouverneur V Org. Biomol. Chem 2006, 4, 2065.Audouard C; Ma J-A; Cahard D Adv. Org. Synth 2006, 2, 431.Ma J-A; Cahard D Chem. Rev 2008, 108, PR1–PR43.Cao L-L; Gao BL; Ma S-T; Liu Z-P Curr. Org. Chem 2010, 14, 888.Kang YK; Kim DY Curr. Org. Chem 2010, 14, 917.Cahard D; Xu X; Couve-Bonnaire S; Pannecoucke X Chem. Soc. Rev 2010, 39, 558.Nie J; Guo H-C; Cahard D; Ma J-A Chem. Rev 2011, 111, 455.Furuya T; Kamlet AS; Ritter T Nature 2011, 473, 470.Besset T; Schneider C; Cahard D Angew. Chem. Int. Ed 2012, 51, 5048.Studer A Angew. Chem. Int. Ed 2012, 51, 8950.Chen P; Liu G Synthesis 2013, 45, 2919.Liang T; Neumann CN; Ritter T Angew. Chem. Int. Ed 2013, 52, 8214.Barata-Vallejo S; Lantano B; Postigo A Chem. Eur. J 2014, 20, 16806.Campbell MG; Ritter T Chem. Rev 2015, 115, 612.Xu X-H; Matsuzaki K; Shibata N Chem. Rev 2015, 115, 731.Yang X; Wu T; Phipps RJ; Toste FD Chem. Rev 2015, 115, 826.Alonso C; Martinez de Marigorta E; Rubiales G; Palacios F Chem. Rev 2015, 115, 1847.
- (7).Erickson JA; McLoughlin JI J. Org. Chem. 1995, 60, 1626. [Google Scholar]
- (8).(a) Bandini M; Sinisi R; Umani-Ronchi A Chem. Commun. 2008, 4360. [DOI] [PubMed] [Google Scholar]; (b) Liu Y-L; Shi T-D; Zhou F; Zhao X-L; Wang X; Zhou J Org. Lett. 2011, 13, 3826. [DOI] [PubMed] [Google Scholar]; (c) Grassi D; Li H; Alexakis A Chem. Commun. 2012, 48, 11404. [DOI] [PubMed] [Google Scholar]; (d) Aikawa K; Yoshida S; Kondo D; Asai Y; Mikami K Org. Lett. 2015, 17, 5108. [DOI] [PubMed] [Google Scholar]; (e) Banik SM; William Medley J; Jacobsen EN Science 2016, 353, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bos M; Huang W-S; Poisson T; Pannecoucke X; Charette AB; Jubault P Angew. Chem. Int. Ed. 2017, 56, 13319. [DOI] [PubMed] [Google Scholar]; (g) van der Mei FW; Qin C; Morrison RJ; Hoveyda AH J. Am. Chem. Soc. 2017, 139, 9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Unlike the present transformations, in chiral phosphoramide-catalyzed cross-aldol reactions, enantioselectivity increases as aldehydes become either more electron rich or more electron poor, indicative of a change in the stereodetermining event: Denmark SE; Bui T Proc. Nat. Acad. Sci. U.S.A 2004, 101, 5439.
- (10).(a) Kim IS; Ngai M-Y; Krische MJ J. Am. Chem. Soc. 2008, 130, 6340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim IS; Ngai M-Y; Krische MJ J. Am. Chem. Soc. 2008, 130, 14891. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim IS; Han SB; Krische MJ J. Am. Chem. Soc. 2009, 131, 2514. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gao X; Townsend IA; Krische MJ J. Org. Chem. 2011, 76, 2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Han SB; Kim IS; Han H; Krische MJ J. Am. Chem. Soc. 2009, 131, 6916. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Han SB; Kim IS; Han H; Krische MJ J. Am. Chem. Soc. 2010, 132, 12517. [Google Scholar]
- (12).Rita P; Animesh DK Int. Res. J. Pharm. 2011, 2, 11. [Google Scholar]
- (13).Chen YK; Lurain AE; Walsh PJ J. Am. Chem. Soc / 2002, 124, 12225. [DOI] [PubMed] [Google Scholar]
- (14).Shiina I; Ibuka R; Kubota M Chem. Lett 2002, 286. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.