

Abstract
Late-onset Alzheimer's disease (LOAD) or sporadic AD is the most common form of AD. The precise pathogenetic changes that trigger the development of AD remain largely unknown. Large-scale genome-wide association studies (GWASs) have identified single-nucleotide polymorphisms in multiple genes which are associated with AD; most notably, these are ABCA7, bridging integrator 1(B1N1), triggering receptor expressed on myeloid cells 2 (TREM2), CD33, clusterin (CLU), complement receptor 1 (CRI), ephrin type-A receptor 1 (EPHA1), membrane-spanning 4-domains, subfamily A (MS4A) and phosphatidylinositol binding clathrin assembly protein (PICALM) genes. The proteins coded by the candidate genes participate in a variety of cellular processes such as oxidative balance, protein metabolism, cholesterol metabolism and synaptic function. This review summarizes the major gene loci affecting LOAD identified by large GWASs. Tentative mechanisms have also been elaborated in various studies by which the proteins coded by these genes may exert a role in AD pathogenesis have also been elaborated. The review suggests that these may together affect LOAD pathogenesis in a complementary fashion.
Keywords: Alzheimer's disease, genome-wide association study, Heart and Aging Research in Genomic Epidemiology, LOAD, single nucleotide polymorphism, Translational Genomics Research Institute
Introduction
The first case of Alzheimer's disease (AD) was described more than 100 years ago, but the precise pathogenetic changes leading to the development of AD are still a matter of considerable controversy. Based on the age of onset and heredity, AD is classified into early-onset AD (EOAD), late-onset AD (LOAD) and familial AD. LOAD or sporadic AD is the most common form of AD, accounting for about 90 per cent of cases and usually occurring after the age of 65 yr1. Neurofibrillary tangles of phosphorylated tau protein and senile plaques composed of amyloid β (Aβ)-protein are the two characteristic pathological hallmarks of AD; however, there exists controversy in how well these correlate with AD phenotype as some AD brains on post-mortem examination reveal minimal plaques and tangles2.
The protein apolipoprotein E (ApoE) is the only well-established genetic risk factor for LOAD. The APOE gene consists of four exons and three introns, with a total of 3597 base pairs, and is mapped to chromosome 19. ApoE is polymorphic with three major isoforms, ApoE2, ApoE3 and ApoE4. High frequency of the APOE4 allele is found in patients with AD than in the general population3. ApoE4 is known to inhibit neurite outgrowth, disrupt neuronal cytoskeleton4, stimulate tau phosphorylation and cause neurodegeneration5. However, neither is the APOE4 variant present in all AD cases nor is it absolutely essential for AD pathogenesis6. Multiple rare mutations in the amyloid precursor protein gene (APP), PSEN1 gene and PSEN2 gene cause early-onset AD7. However, a large case-control study (3940 cases and 13,373 controls) reported that common variants in these genes were not likely to make strong contributions to susceptibility for LOAD8.
Recent efforts have been focussed on conducting genome-wide association studies (GWASs) to identify newer risk genes for LOAD. Multi-stage meta-analytic reports by different groups documented the association of single-nucleotide polymorphisms (SNPs) in 10 genes with AD; these being ABCA7, bridging integrator 1 gene (BIN1), triggering receptor expressed on myeloid cells gene (TREM), CD33, clusterin gene (CLU), complement receptor 1 gene (CR1), ephrin type-A receptor 1 gene (EPHA1), CD2AP, membrane-spanning 4-domains, subfamily A (MS4A) gene cluster and phosphatidylinositol binding clathrin assembly protein gene (PICALM)9,10,11,12.
In 2009, Lambert et al13 published an open letter of two-stage GWAS performed on AD subjects and controls. The three-city study identified two new susceptibility loci: CLU and CR1. They also detected evidence for the association of PICALM with AD13. A collaborative consortium from Europe and the USA [European AD Initiative 1 (EADI 1)] also performed a GWAS over 16,000 individuals with AD and controls. They identified two novel loci CLU and PICALM, significantly associated with AD. They also observed one more associated locus BIN114. In 2010, Seshadri et al15 performed a three-stage analysis of GWAS data to identify additional loci associated with LOAD. In their gene discovery phase, they concluded that BIN1 showed association with AD in GWAS. They also confirmed the association of two reported loci; CLU and PICALM with LOAD15. Hollingworth et al10 undertook a combined analysis of four independent genome-wide studies- GERAD1, TGEN1, ADNI and EADI1 - to identify new susceptibility loci of AD. Their data provided significant evidence for the association of ABCA7, MS4A gene cluster with AD at stage one. In stage two, they observed association of more suggestive loci; CD33 and EPHA1 with AD10. To identify newer susceptibility loci for AD, the AD Genetic Consortium (ADGC) group conducted a three-staged association study on AD patients and provided compelling evidence for the association of MS4A4A, EPHA1 and CD33 with AD. They also replicated previous associations of CR1, CLU and PICALM with LOAD11. Advances in sequencing techniques of entire genomes identified rare variants in those patients, in whom linkage analysis cannot be done. TREM2 is one of the variants that increase the risk of AD12.
Fig. 1 gives a schematic representation of the multiple research groups who worked to find new susceptibility genes for LOAD and also the different loci which affect LOAD pathogenesis.
Fig. 1.

Schematic representation of multiple organizations who worked to find new genome-wide association study loci and how different loci are connected with each other. The gene loci found as a result of meta-analyses belong to three broad functional categories: immune response, synaptic function and cholesterol metabolism. GWAS, genome wide association studies; GERAD1, genetic and environmental risk for Alzheimer's disease consortium 1; EADI1, European Alzheimer's disease initiative 1; CHARGE, Cohorts for Heart and Aging research in genomic epidemiology; TGRI, Translational Genomics Research Institute; ADGC, Alzheimer's disease genetic consortium; LOAD, late onset Alzheimer's disease.
Alzheimer's disease (AD) pathogenesis as the cumulative effect of multiple genetic risk factors
Large-scale GWASs have identified SNPs in ten genes: ABCA7, BIN1, TREM2, CD33, CLU, CR1, EPHA1, MS4A, CD2AP and PICALM which may participate in the pathogenesis of AD by several functional pathways that are affected9,10,11,12. These genes may be categorized on the bases of their involvement in cellular pathways:
-
(i)
Immune response and inflammation: CR1, MS4A family, EPHA1, CD33, TREM9,10,12.
- (ii)
-
(iii)
Endocytosis and synaptic function: PICALM, BIN1, CD2AP and EPHA19,10.
It is hypothesized that these gene SNPs identified by GWAS influence their respective interconnected cellular processes to cause AD. The exact pathogenesis of AD is still unclear, and it is possible that not all of the above processes are deranged in each case of LOAD. Either of the three may dominate in or solely contribute to LOAD in individual patients. Further, the exact links between the pathways still need to be worked out. However, the common pathways through which these act are widely believed to be the amyloidogenic pathway and the tau hyper-phosphorylation pathway1.
Fig. 2 represents how the various genetic risk factors may be interconnected and contribute to LOAD risk by ultimately inducing amyloid and hyper-phosphorylated tau protein accumulation.
Fig. 2.
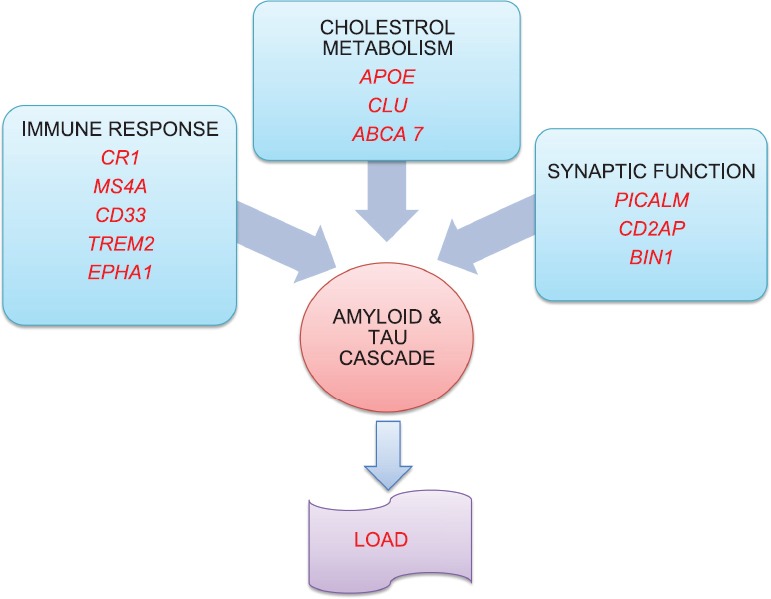
Interconnected responsible pathways to cause amyloid and tau accumulation. Gene involved in AD pathogenesis can be broadly grouped into 3 categories; immune response (CR1, MS4A, TREM2, CD33, EPHA1), cholesterol metabolism (APOE, CLU, ABCA7), synaptic function (PICALM, CD2AP, BIN1). The cumulative effect of all these genes is manifested through the final common pathway of amyloid and tau cascade.
Functional significance of new genetic loci associated with LOAD
Genes associated with lipid metabolism: CLU codes for the secretory hetero-dimeric 75-80 kDa CLU also known as apolipoprotein J16. This gene encodes a 2 kb mRNA which translates into a 449 amino acid primary polypeptide chain17. CLU is a highly conserved chaperone protein that is found in the cell cytosol under some stress conditions18. It is expressed in most mammalian tissues19, and has been reported to be involved in neurodegeneration and hypoxic-ischaemic neuronal death20. Elevated level of CLU has been found in post-mortem AD brains and also in the brains of ApoE4 carriers21. CLU is involved in the regulation of Aβ. This has been demonstrated in guinea pig brain perfusion model where apolipoprotein J interacts with the soluble form of Aβ in a specific and reversible manner and forms complexes in the brain, facilitating the transport of soluble Aβ across the blood-brain barrier22. In transgenic mouse model (clu− and clu+), it has been seen that Aβ deposits in clu− mice are significantly reduced as compared to clu+ which indicates that CLU has a role in Aβ fibril formation and neurotoxicity23. Plasma CLU level was reported to be associated with rapid clinical progression in AD, suggesting its possible use as a biomarker of AD24. GWASs found a significant negative association [odds ratio (OR)=0.86] between an SNP within the CLU, rs11136000 and the risk of having AD14. This association was found in both APOE4 carriers and non-carriers15.
ABCA7 is a member of the superfamily of ATP-binding cassette (ABC) transporters, which transport various molecules across extra- and intra-cellular membranes. These transporters are divided into eight distinct subfamilies. ABCA7 is a member of the ABC1 subfamily25. This gene codes for a membrane protein which is expressed in the myelolymphatic tissues, brain and trachea26. Analysis of isolated foetal human brain cells has shown that microglia express the highest level of ABCA7 mRNA27. This gene is also involved in AD pathogenesis28. It regulates the phagocytosis of apoptotic cell debris inside the brain. Protein products of these loci bind with APOA1 and contribute to the apolipoprotein-mediated phospholipid efflux mechanism in cells29.
In stage 1 meta-analysis of GERAD1, TGEN1, ADNI and EADI1, evidence was found for the positive association (OR=1.22) of SNP of ABCA7 (rs3764650) with AD. This has further been proven in stages 2 and 3 meta-analysis10. Another SNP variant of ABCA7, i.e. rs3752246, was found to be associated with AD in stage 2 meta-analysis (OR=1.17). However, association of rs3764650 with ABCA7 expression was not observed30.
Genes associated with inflammatory response: CD33 is located on chromosome 19q13.3 in humans and codes for the 67kDa CD33 protein31. CD33 belongs to the sialic acid-binding immunoglobulin-like lectins (Siglecs) family32. It is expressed in microglial cells in the human brain33. The Siglecs family mediates cell-cell interaction through glycan recognition34. They also play an important role in the regulation of functions of innate and adaptive immune cell systems35. CD33 is expressed by haematopoietic and phagocytic cells and participates in adhesion processes of human primary immune cells36. It appears to inhibit the production of pro-inflammatory cytokines [such as interleukin-1β, tumour necrosis factor alpha (TNF-α)] by monocytes37. Being an inhibitory receptor in immune response, it also regulates cell growth and survival and also induces apoptosis38.
CD33 inhibits Aβ clearance in LOAD39. It has been seen that levels of CD33-positive microglial cells are increased in brains of AD patients, and play a direct role in the progression of AD. The CD33 SNP rs3865444, which confers protection against AD, has been seen to be associated with reductions in both CD33 expression and insoluble Aβ42 levels in AD brain33. Various SNPs of CD33 such as rs3826656 and rs3865444 are found to be associated with AD40.
CR1 found on chromosome 1q32 codes for the complement regulatory protein, CR1 or CD35 which is expressed widely on a number of blood cells41 and can also be found dissolved in the blood plasma42. CR1 induces phagocytosis by forming a complex with C3b/C4b. Extracellular domain of CR1 is composed of long homologous repeats (LHRs). Genetic duplications and deletions result in increased number of LHR regions, which result in the formation of four co-dominant alleles of CR1. Frequencies of the four alleles vary only slightly between populations43. The increased number of LHRs means that the larger alleles have additional C3b/C4b-binding sites44.
The classical complement pathway has been long known to play a protective role in AD by acceleration of clearance of the Aβ plaques. Aβ interacts with C1q of the classical complement pathway45. This results in the activation of the membrane attack complex comprising C3b/C4b, which results in activation of glial cells46. CR1 helps in this process by providing multiple C3b/C4b-binding sites47. Lambert et al13 found an SNP variant of CR1, rs6656401 (OR=1.12) with a strong association with LOAD.
EPHA1 also known as eph is located on chromosome 7q34.1. The protein product belongs to the tyrosine kinase receptor family48 and the ephrin receptor subfamily. The ligand for the EphA receptor is ephrin-A, which is anchored to the cell membrane through a glycosylphosphatidylinositol linkage49. Eph receptors and ephrins are expressed in endothelial and epithelial cells50, and guide the migration of cells during embryonic development and also have a role in cytoskeletal organization of neuronal processes51. They play a role in synaptic development and plasticity52. Additional roles in apoptosis and inflammation exist53. AD patients with an allele of EPHA1 (A allele) having enhanced rate of cerebral metabolism for glucose in the right lateral occipitotemporal gyrus and inferior temporal gyrus may not have hippocampal atrophy54. Combined result of the meta-analysis of the GERAD consortia with the ADGC GWAS shows that the rs11767557 SNP of the EPHA1 gene is negatively associated with AD (OR=0.90)10.
MS4A encodes several proteins including CD20. This gene family is further divided into at least 12 subgroups from MS4A1 to MS4A1255. CD20 expressed by B-lymphocytes56 forms a hetero-tetrameric complex on the cell membrane that regulates Ca2+ influx downstream57. This regulation of calcium signalling may have an important role in neurodegeneration and AD pathogenesis58. Several members of this cluster (such as MS4A1, MS4A2 and MS4A4B) have an important role in immunity59. MS4A4B appears to have a role in Th1 development, CD8+ memory T-cell function and modulation of regulatory T-cell signalling60. MS4A2 mediates interactions with IgE-bound antigens that lead to cellular responses such as the degranulation of mast cells61.
Meta-analysis data of GWAS by ADGC suggested two SNPs of the MS4A gene cluster: rs610932 and rs670139 to be associated with LOAD10. Another independent GWAS study on the Spanish population revealed the association of rs1562990 SNP of MS4A with AD62.
TREM2 codes for a membrane glycoprotein, consisting of an extracellular immunoglobulin-like domain and a cytoplasmic tail that is involved in receptor signalling complex along with the DAP12 and TYRO binding proteins63. This protein functions in the immune response and may be involved in chronic inflammation64. In brain cells, TREM2 is primarily expressed on microglia65,66. Microglia stimulate the proliferation of CD4+ T-cells, as well as the secretion of TNF and CCL267. Microglia have phagocytic role on amyloid plaques68. In a study, reduced phagocytic activity was found in microglial cells to phagocytose β amyloid fragment of AD brain in TREM2 knockdown mice in comparison with mice expressing TREM269. A rare missense mutation (rs75932628) in the TREM2 results in an R47H substitution which has been found to confer a significant risk of AD. This may be because of the inability of the brain to clear Aβ toxicity65.
Genes associated with endocytosis: PICALM codes for PICALM which can influence the risk of AD through modulation of APP processing via AP2-dependent clathrin-mediated endocytotic pathways, resulting in changes in Aβ level70. PICALM initiates clathrin polymerization at sites of coated pit formation71. It was seen in cell culture experiments that clathrin-mediated endocytosis (CME) retrieved full length APP from the cell surface, thus promoting the intracellular accumulation of amyloid72. In the endosome, full length APP is cleaved in to Aβ by β-secretase (BACE) and this is released into the brain interstitial fluid. Increased number of endosomes formed by CME drives more APP into the cell73, resulting in an increase of Aβ production74. Synaptic vesicles limit the dispersion of neurotransmitter at the pre-synaptic plasma membrane. It was seen in live cell image of hippocampal neurons that synaptic vesicle containing VAMP2 on surface helped in diffusing neurotransmitters along the axonal membrane75. PICALM may also be involved in directing the trafficking of VAMP276. The SNP of PICALM which has been found to be most significantly protective against LOAD is rs3851179 (OR=0.86)14.
BIN1 codes for Myc box-dependent-interacting protein 1. It is a nucleo-cytoplasmic tumour suppressor adaptor protein77. Isoforms of this protein expressed in the central nervous system are involved in synaptic vesicle endocytosis78. The BIN1 is identified as the most important genetic susceptibility locus in LOAD after APOE79. Higher BIN1 expression has been reported to be linked with later age at onset and shorter disease duration80. Although the mechanisms are still not fully understood, data suggest that BIN1 affects AD risk primarily by modulating tau pathology. BIN1 also affects other cellular functions including endocytosis/trafficking, inflammation, calcium homoeostasis and apoptosis79. Seshadri et al15 combined the data from CHARGE, TGEN, EADI1 and GERAD1 groups and analyzed by a three-stage sequential meta-analysis. They reported the association (OR=1.13) of the BIN1 SNP rs744373 with LOAD15. Another independent study- The Washington Heights-Inwood Columbia Aging Project and the Estudio Familiar de Influencia Genetica de Alzheimer study also showed positive associations of the BIN1 SNP rs7561528 with LOAD in the ε4 carrier state81.
CD2AP codes for CD2-associated protein which is a scaffolding molecule that regulates the actin cytoskeleton82. It plays a role in receptor-mediated endocytosis. CD2AP contributes to APP metabolism and subsequent Aβ generation83. It regulates the encounter of APP and BACE1 in axonal and dendritic endosomes84. GERAD1, EADI1, deCODE and AD-IG GWAS datasets observed independent evidence for the association of CD2AP gene loci with AD (OR=1.11 for rs9349407 SNP)10.
Racial variation of Alzheimer's disease susceptibility genes
Survival after the diagnosis of AD varies amongst different races, ranging from 3 to 9 years. African American and Latino AD patients have better survival than Caucasian patients and genetic background plays an important role in the progression of AD85. Most GWASs and replication studies of AD have been done in populations of European descent, and non-European genetic studies of new AD-susceptibility loci are limited. Studies that evaluated the association of CLU and CR1 with AD in Asian populations are limited86. Many AD-associated SNPs of CLU, PICALM and BIN1 were not necessarily identical in Caribbean Hispanic individuals compared with a European American data set81. Meta-analytic data showed that CLU, PICALM and CR1 were associated with LOAD in Caucasians subjects, but a study found that investigated SNPs of CR1, CLU and PICALM were not associated with AD in a Polish population87. A study found that in the Korean population, the PICALM is the only AD susceptibility loci in addition to APOE88. ADGC assembled multiple data sets for meta-analysis representing African American older subjects. The data showed another SNP (rs115550680) of ABCA7 (OR=1.79) was associated with AD in comparison to European ancestry89.
Potential therapeutic implications of GWAS loci
Novel loci may exert their effects in a number of pathways such as oxidative balance, protein metabolism, cholesterol metabolism and synaptic function90. Genes with moderate to large effects on LOAD risk are valuable targets for therapeutic development. Neuroinflammation is both a cause and a consequence of AD and treatment with anti-inflammatory agents is likely to be successful if initiated before the onset of neurological symptoms91. Similarly, on the lipid metabolism front, the CLU protein may be targeted to reduce AD risk92. Genes associated with endocytosis and synaptic functions are BIN1, PICALM and CD2AP. Modulating these at the gene-expression level using siRNA or antisense techniques is a valid approach.
New developments
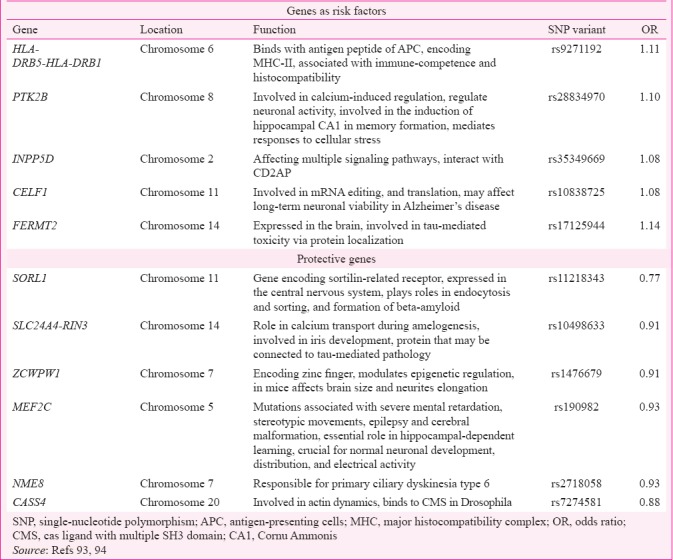
While the present review focuses on the most established gene loci involved in AD pathogenesis as suggested by GWAS, several newer loci have made a foray into the AD scene. Under the supervision and support of International Genomics of AD project, two-stage meta-analysis identified 11 loci which are HLA-DRB5-DRB1 gene, SORL1, PTK2B, SLC24A4, ZCWPW1, INPP5D, MEF2C, CELF1, NME8, CASS4, FERMT2 genes, with suggestive evidence of association with AD93. The Table represents newer loci involved in AD pathogenesis as suggested by GWAS with tentative pathogenic mechanisms94.
Table.
New susceptibility gene loci of Alzheimer's disease
Conclusion
GWASs have revealed the association of new gene loci with AD. The first few identified SNPs from GWAS suggest the involvement of different associated pathways with pathogenesis of AD although the exact mechanisms remain unknown. Modification and advancing the research in these pathways may lead to therapeutic intervention for AD. Many of these GWAS loci may serve as biomarkers of AD. The search for additional genetic risk factors requires more large-scale meta-analysis of GWAS and enhanced statistical power as well as replicating these findings at the molecular level. Exciting times await us in AD genetic research and newer paradigms might open in the near future.
Footnotes
Financial support & sponsorship: None.
Conflicts of Interest: None.
References
- 1.Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213–27. doi: 10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–9. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 3.Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer's disease: A systematic review and meta-analysis. Neuroepidemiology. 2012;38:1–7. doi: 10.1159/000334607. [DOI] [PubMed] [Google Scholar]
- 4.Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE, et al. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro . Science. 1994;264:850–2. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 5.Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell. 2009;5:634–45. doi: 10.1016/j.stem.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grupe A, Abraham R, Li Y, Rowland C, Hollingworth P, Morgan A, et al. Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum MolGenet. 2007;16:865–73. doi: 10.1093/hmg/ddm031. [DOI] [PubMed] [Google Scholar]
- 7.Cruchaga C, Haller G, Chakraverty S, Mayo K, Vallania FL, Mitra RD, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer's disease families. PLoS One. 2012;7:e31039. doi: 10.1371/journal.pone.0031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerrish A, Russo G, Richards A, Moskvina V, Ivanov D, Harold D, et al. The role of variation at aβPP, PSEN1, PSEN2, and MAPT in late onset Alzheimer's disease. J Alzheimers Dis. 2012;28:377–87. doi: 10.3233/JAD-2011-110824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jun G, Naj AC, Beecham GW, Wang LS, Buros J, Gallins PJ, et al. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol. 2010;67:1473–84. doi: 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429–35. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–27. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 14.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–40. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu JT, Tan L. The role of clusterin in Alzheimer's disease: Pathways, pathogenesis, and therapy. Mol Neurobiol. 2012;45:314–26. doi: 10.1007/s12035-012-8237-1. [DOI] [PubMed] [Google Scholar]
- 17.Park S, Mathis KW, Lee IK. The physiological roles of apolipoprotein J/clusterin in metabolic and cardiovascular diseases. Rev Endocr Metab Disord. 2014;15:45–53. doi: 10.1007/s11154-013-9275-3. [DOI] [PubMed] [Google Scholar]
- 18.Trougakos IP. The molecular chaperone apolipoprotein J/clusterin as a sensor of oxidative stress: Implications in therapeutic approaches - A mini-review. Gerontology. 2013;59:514–23. doi: 10.1159/000351207. [DOI] [PubMed] [Google Scholar]
- 19.Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, et al. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci U S A. 1999;96:6745–50. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SE, Jomary C. Clusterin. Int J Biochem Cell Biol. 2002;34:427–31. doi: 10.1016/s1357-2725(01)00155-8. [DOI] [PubMed] [Google Scholar]
- 21.Lidström AM, Bogdanovic N, Hesse C, Volkman I, Davidsson P, Blennow K, et al. Clusterin (apolipoprotein J) protein levels are increased in hippocampus and in frontal cortex in Alzheimer's disease. Exp Neurol. 1998;154:511–21. doi: 10.1006/exnr.1998.6892. [DOI] [PubMed] [Google Scholar]
- 22.Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, et al. Glycoprotein 330/megalin: Probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci U S A. 1996;93:4229–34. doi: 10.1073/pnas.93.9.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeMattos RB, O’dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, et al. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2002;99:10843–8. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry. 2010;67:739–48. doi: 10.1001/archgenpsychiatry.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein I, Sarkadi B, Váradi A. An inventory of the human ABC proteins. Biochim Biophys Acta. 1999;1461:237–62. doi: 10.1016/s0005-2736(99)00161-3. [DOI] [PubMed] [Google Scholar]
- 26.Chan SL, Kim WS, Kwok JB, Hill AF, Cappai R, Rye KA, et al. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro . J Neurochem. 2008;106:793–804. doi: 10.1111/j.1471-4159.2008.05433.x. [DOI] [PubMed] [Google Scholar]
- 27.Kim WS, Guillemin GJ, Glaros EN, Lim CK, Garner B. Quantitation of ATP-binding cassette subfamily - A transporter gene expression in primary human brain cells. Neuroreport. 2006;17:891–6. doi: 10.1097/01.wnr.0000221833.41340.cd. [DOI] [PubMed] [Google Scholar]
- 28.Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikeda Y, Abe-Dohmae S, Munehira Y, Aoki R, Kawamoto S, Furuya A, et al. Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochem Biophys Res Commun. 2003;311:313–8. doi: 10.1016/j.bbrc.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barber RC. The genetics of Alzheimer's disease. Scientifica (Cairo) 2012;2012:246210. doi: 10.6064/2012/246210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varki A, Crocker PR. I-type lectins. J Biol Chem. 1995;270:14243–6. doi: 10.1074/jbc.270.24.14243. [DOI] [PubMed] [Google Scholar]
- 33.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–43. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 2014;14:653–66. doi: 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–66. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 36.Jandus C, Simon HU, von Gunten S. Targeting siglecs - a novel pharmacological strategy for immuno- and glycotherapy. Biochem Pharmacol. 2011;82:323–32. doi: 10.1016/j.bcp.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez Y, Herrera MT, Soldevila G, Garcia-Garcia L, Fabián G, Pérez-Armendariz EM, et al. High glucose concentrations induce TNF-α production through the down-regulation of CD33 in primary human monocytes. BMC Immunol. 2012;13:19. doi: 10.1186/1471-2172-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vitale C, Romagnani C, Falco M, Ponte M, Vitale M, Moretta A, et al. Engagement of p75/AIRM1 or CD33 inhibits the proliferation of normal or leukemic myeloid cells. Proc Natl Acad Sci U S A. 1999;96:15091–6. doi: 10.1073/pnas.96.26.15091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gandy S, Heppner FL. Microglia as dynamic and essential components of the amyloid hypothesis. Neuron. 2013;78:575–7. doi: 10.1016/j.neuron.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008;83:623–32. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu D, Niu ZX. The structure, genetic polymorphisms, expression and biological functions of complement receptor type 1 (CR1/CD35) Immunopharmacol Immunotoxicol. 2009;31:524–35. doi: 10.3109/08923970902845768. [DOI] [PubMed] [Google Scholar]
- 42.Krych-Goldberg M, Moulds JM, Atkinson JP. Human complement receptor type 1 (CR1) binds to a major malarial adhesin. Trends Mol Med. 2002;8:531–7. doi: 10.1016/s1471-4914(02)02419-x. [DOI] [PubMed] [Google Scholar]
- 43.Wong WW, Wilson JG, Fearon DT. Genetic regulation of a structural polymorphism of human C3b receptor. J Clin Invest. 1983;72:685–93. doi: 10.1172/JCI111018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert JC, Bettens K, Le Bastard N, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17:223–33. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nayak A, Ferluga J, Tsolaki AG, Kishore U. The non-classical functions of the classical complement pathway recognition subcomponent C1q. Immunol Lett. 2010;131:139–50. doi: 10.1016/j.imlet.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 46.Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer's disease beta-peptide. J Exp Med. 1998;188:431–8. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Furtado PB, Huang CY, Ihyembe D, Hammond RA, Marsh HC, Perkins SJ, et al. The partly folded back solution structure arrangement of the 30 SCR domains in human complement receptor type 1 (CR1) permits access to its C3b and C4b ligands. J Mol Biol. 2008;375:102–18. doi: 10.1016/j.jmb.2007.09.085. [DOI] [PubMed] [Google Scholar]
- 48.Kuang SQ, Bai H, Fang ZH, Lopez G, Yang H, Tong W, et al. Aberrant DNA methylation and epigenetic inactivation of eph receptor tyrosine kinases and ephrin ligands in acute lymphoblastic leukemia. Blood. 2010;115:2412–9. doi: 10.1182/blood-2009-05-222208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 50.Yamazaki T, Masuda J, Omori T, Usui R, Akiyama H, Maru Y, et al. EphA1 interacts with integrin-linked kinase and regulates cell morphology and motility. J Cell Sci. 2009;122:243–55. doi: 10.1242/jcs.036467. [DOI] [PubMed] [Google Scholar]
- 51.Wilkinson DG. Multiple roles of EPH receptors and ephrins in neural development. Nat Rev Neurosci. 2001;2:155–64. doi: 10.1038/35058515. [DOI] [PubMed] [Google Scholar]
- 52.Pitulescu ME, Adams RH. Eph/ephrin molecules - A hub for signaling and endocytosis. Genes Dev. 2010;24:2480–92. doi: 10.1101/gad.1973910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med. 2015;3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang HF, Tan L, Hao XK, Jiang T, Tan MS, Liu Y, et al. Effect of EPHA1 genetic variation on cerebrospinal fluid and neuroimaging biomarkers in healthy, mild cognitive impairment and Alzheimer's disease cohorts. J Alzheimers Dis. 2015;44:115–23. doi: 10.3233/JAD-141488. [DOI] [PubMed] [Google Scholar]
- 55.Liang Y, Buckley TR, Tu L, Langdon SD, Tedder TF. Structural organization of the human MS4A gene cluster on chromosome 11q12. Immunogenetics. 2001;53:357–68. doi: 10.1007/s002510100339. [DOI] [PubMed] [Google Scholar]
- 56.Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120:214–22. doi: 10.1172/JCI40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fengling M, Qingxiang G, Lijia Z, Wei Z. Influx of extracellular calcium participates in rituximab-enhanced ionizing radiation-induced apoptosis in Raji cells. Toxicol Lett. 2012;209:221–6. doi: 10.1016/j.toxlet.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 58.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–50. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 59.Ma J, Yu JT, Tan L. MS4A cluster in Alzheimer's disease. Mol Neurobiol. 2015;51:1240–8. doi: 10.1007/s12035-014-8800-z. [DOI] [PubMed] [Google Scholar]
- 60.Xu H, Yan Y, Williams MS, Carey GB, Yang J, Li H, et al. MS4a4B, a CD20 homologue in T cells, inhibits T cell propagation by modulation of cell cycle. PLoS One. 2010;5:e13780. doi: 10.1371/journal.pone.0013780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howie D, Nolan KF, Daley S, Butterfield E, Adams E, Garcia-Rueda H, et al. MS4A4B is a GITR-associated membrane adapter, expressed by regulatory T cells, which modulates T cell activation. J Immunol. 2009;183:4197–204. doi: 10.4049/jimmunol.0901070. [DOI] [PubMed] [Google Scholar]
- 62.Antúnez C, Boada M, González-Pérez A, Gayán J, Ramírez-Lorca R, Marín J, et al. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer's disease. Genome Med. 2011;3:33. doi: 10.1186/gm249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lue LF, Schmitz CT, Serrano G, Sue LI, Beach TG, Walker DG, et al. TREM2 protein expression changes correlate with Alzheimer's disease neurodegenerative pathologies in post-mortem temporal cortices. Brain Pathol. 2015;25:469–80. doi: 10.1111/bpa.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bouchon A, Dietrich J, Colonna M. Cutting edge: Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–5. doi: 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- 65.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–16. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prinz M, Kettenmann H, Ransom BR. Neuroglia. New York: Oxford University Press; 2013. Microglia lineage and development; pp. 172–84. [Google Scholar]
- 67.Melchior B, Garcia AE, Hsiung BK, Lo KM, Doose JM, Thrash JC, et al. Dual induction of TREM2 and tolerance-related transcript, tmem176b, in amyloid transgenic mice: Implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro. 2010;2:e00037. doi: 10.1042/AN20100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.D’Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging. 2004;25:675–83. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 69.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–57. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu W, Tan L, Yu JT. The role of PICALM in Alzheimer's disease. Mol Neurobiol. 2015;52:399–413. doi: 10.1007/s12035-014-8878-3. [DOI] [PubMed] [Google Scholar]
- 71.Tebar F, Bohlander SK, Sorkin A. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: Localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell. 1999;10:2687–702. doi: 10.1091/mbc.10.8.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo . Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993;268:608–12. [PubMed] [Google Scholar]
- 74.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–9. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, et al. Simultaneous binding of ptdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science. 2001;291:1051–5. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- 76.Harel A, Wu F, Mattson MP, Morris CM, Yao PJ. Evidence for CALM in directing VAMP2 trafficking. Traffic. 2008;9:417–29. doi: 10.1111/j.1600-0854.2007.00694.x. [DOI] [PubMed] [Google Scholar]
- 77.Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat Genet. 1996;14:69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- 78.Wu T, Shi Z, Baumgart T. Mutations in BIN1 associated with centronuclear myopathy disrupt membrane remodeling by affecting protein density and oligomerization. PLoS One. 2014;9:e93060. doi: 10.1371/journal.pone.0093060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan MS, Yu JT, Tan L. Bridging integrator 1 (BIN1): Form, function, and Alzheimer's disease. Trends Mol Med. 2013;19:594–603. doi: 10.1016/j.molmed.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 80.Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM, et al. Expression of novel Alzheimer's disease risk genes in control and Alzheimer's disease brains. PLoS One. 2012;7:e50976. doi: 10.1371/journal.pone.0050976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee JH, Cheng R, Barral S, Reitz C, Medrano M, Lantigua R, et al. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in caribbeanhispanic individuals. Arch Neurol. 2011;68:320–8. doi: 10.1001/archneurol.2010.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tang VW, Brieher WM. FSGS3/CD2AP is a barbed-end capping protein that stabilizes actin and strengthens adherens junctions. J Cell Biol. 2013;203:815–33. doi: 10.1083/jcb.201304143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dunstan ML, Gerrish A, Morgan T, Owens H, Badarinarayan N, Thomas RS, et al. The role of CD2AP in APP processing. Alzheimers Dement. 2016;12:P458–9. [Google Scholar]
- 84.Sannerud R, Annaert W. Bin1 and CD2AP polarize aβ generation in neurons. EMBO Rep. 2017;18:5–7. doi: 10.15252/embr.201643634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mehta KM, Yaffe K, Pérez-Stable EJ, Stewart A, Barnes D, Kurland BF, et al. Race/ethnic differences in AD survival in US Alzheimer's disease centers. Neurology. 2008;70:1163–70. doi: 10.1212/01.wnl.0000285287.99923.3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin YL, Chen SY, Lai LC, Chen JH, Yang SY, Huang YL, et al. Genetic polymorphisms of clusterin gene are associated with a decreased risk of Alzheimer's disease. Eur J Epidemiol. 2012;27:73–5. doi: 10.1007/s10654-012-9650-5. [DOI] [PubMed] [Google Scholar]
- 87.Klimkowicz-Mrowiec A, Sado M, Dziubek A, Dziedzic T, Pera J, Szczudlik A, et al. Lack of association of CR1, PICALM and CLU gene polymorphisms with Alzheimer disease in a Polish population. Neurol Neurochir Pol. 2013;47:157–60. doi: 10.5114/ninp.2013.33825. [DOI] [PubMed] [Google Scholar]
- 88.Chung SJ, Lee JH, Kim SY, You S, Kim MJ, Lee JY, et al. Association of GWAS top hits with late-onset Alzheimer disease in Korean population. Alzheimer Dis Assoc Disord. 2013;27:250–7. doi: 10.1097/WAD.0b013e31826d7281. [DOI] [PubMed] [Google Scholar]
- 89.Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ϵ4, and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309:1483–92. doi: 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bertram L, Tanzi RE. Neurodegeneration: The molecular pathology of dementia and movement disorders. In: Dickson D, Weller R, editors. Genetics of Alzheimer's disease. 2nd ed. West Sussex: Wiley-Blackwell; 2011. pp. 51–91. [Google Scholar]
- 91.Pimplikar SW. Neuroinflammation in Alzheimer's disease: From pathogenesis to a therapeutic target. J Clin Immunol. 2014;34(Suppl 1):S64–9. doi: 10.1007/s10875-014-0032-5. [DOI] [PubMed] [Google Scholar]
- 92.Wilson MR, Zoubeidi A. Clusterin as a therapeutic target. Expert Opin Ther Targets. 2017;21:201–13. doi: 10.1080/14728222.2017.1267142. [DOI] [PubMed] [Google Scholar]
- 93.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Medway C, Morgan K. The genetics of Alzheimer's disease; putting flesh on the bones. Neuropathol Appl Neurobiol. 2014;40:97–105. doi: 10.1111/nan.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]