

Abstract
Background
MicroRNAs (miRNAs) are endogenous, small (21–25 nucleotide), non-coding RNAs that play important roles in numerous biological processes. Koi carp exhibit diverse color patterns, making it an ideal subject for studying the genetics of pigmentation. However, the influence of miRNAs on skin color regulation and variation in Koi carp is poorly understood.
Results
Herein, we performed small RNA (sRNA) analysis of the three main skin colors in Koi carp by Illumina sequencing. The results revealed 330, 397, and 335 conserved miRNAs (belonging to 81 families) and 340, 353, and 351 candidate miRNAs in black, red, and white libraries, respectively. A total of 164 differentially expressed miRNAs (DEMs) and 14 overlapping DEMs were identified, including miR-196a, miR-125b, miR-202, miR-205-5p, miR-200b, and etc. Target prediction and functional analysis of color-related miRNAs such as miR-200b, miR-206, and miR-196a highlighted putative target genes, including Mitf, Mc1r, Foxd3, and Sox10 that are potentially related to pigmentation. Determination of reference miRNAs for relative quantification showed that let-7a was the most abundant single reference gene, and let-7a and miR-26b was the most abundant combination.
Conclusions
The findings provide novel insight into the molecular mechanisms determining skin color differentiation in Koi carp, and serve as a valuable reference for future studies on tissue-specific miRNA abundance in Koi carp.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-5189-5) contains supplementary material, which is available to authorized users.
Keywords: miRNAs, Skin color, Characterization, Koi carp, Illumina sequencing
Background
Koi carp, a colorful variant of common carp (Cyprinus carpio L.), has been artificially selected for centuries, and is a popular pet and hobby species that exhibits a wide variety of colors and phenotypes [1]. Unlike the slow rate of phenotypic changes occurring in wild populations, the rapid establishment of diverse colors and coloration patterns under artificial selection makes Koi carp an ideal subject for studying the genetics of pigmentation [2]. Determination of skin color is a complicated process in fish, linked to various cellular, genetic, nutritional, physiological, and environmental factors [3]. Exactly how skin color patterns form is a long standing question among biologists. Extensive studies have identified a series of genes involved in the determination of skin pigmentation, including Melanocortin receptor 1 (Mc1r), Microphthalmia-associated transcription factor (Mitf), Tyrosinase (Tyr), Tyrosine related protein-1 (Tyrp1), and Melanocyte-stimulating hormone (Msh) [4–6]. Additional genes related to the determination of skin color in fish remain to be elucidated.
MicroRNAs (miRNAs) are single-stranded, non-coding, highly-conserved 19–24 nucleotides (nt) molecules that regulate gene expression at the post-transcriptional level by directly targeting RNA-inducing silencing complex (RISC) to cognate messenger RNA targets [7, 8]. Crosstalk between miRNAs and mRNAs is important for transcriptional and signal transduction events involved in multiple biological processes such as apoptosis, cell proliferation, cancer, embryo development, and skin pigmentation [9–12]. Therefore, skin-expressed miRNAs might play a vital role in skin differentiation, color formation and skin diseases. Previous studies have investigated the expression and functions of some miRNAs in animal skin. For example, the skin-specific miR-203 was found to define a molecular boundary between proliferative basal progenitors and terminally differentiating suprabasal cells, ensuring proper identify of neighboring layers [13]. In Drosophila, dorsal abdomen pigmentation is decreased when miR-8 lost [14], and coat color in mice is affected when miR-137 is over-expressed [15]. In white alpaca (WA) and brown alpaca (BA) skin, deep sequencing identified 35 and 13 conserved differentially expressed miRNAs (DEMs), indicating potential functions in coat skin color regulation [16]. Meanwhile, Yan et al. identified 13 DEMs between red and white skin in common carp, miR-429 is a potential regulator because it’s silencing resulted in an obvious change in skin pigmentation [12]. Thus, miRNAs could be involved in the regulation of skin color. However, the molecular and cellular mechanisms regulating skin color variation in fish, especially a variety of colors appeared on a single fish, such as Koi carp (Fig. 1), remain poorly understood.
Fig. 1.

Photograph showing the three different skin color types in Koi carp. R, red; B, black; W, white; n, sample number
Reference genes are employed as internal controls for normalizing real-time quantitative reverse transcription PCR (qRT-PCR) data during analysis of target gene expression, and this approach can capture all non-biological variation [17]. At present, β-actin, 18S rRNA, 5S rRNA and glycer-aldehyde-3-phosphate dehydrogenase (GAPDH) are most widely used as candidate reference genes for mRNA expression [18]. In typical miRNA expression studies, 18S rRNA and small nuclear RNAs such as U6 snRNA are commonly used as reference genes [19, 20]. The selection of appropriate reference genes is not trivial, and studies indicate that a single gene is unlikely to behave consistently in all tissues and/or in all physiological and pathological situations [21, 22]. Therefore, internal genes should be verified and selected prudently. However, to date, few reports have analyzed the expression stability of miRNAs used as reference in teleosts.
Herein, we investigated the potential functions of miRNAs in post-transcriptional regulation of pigmentation by constructing Koi carp miRNA libraries from white, red, and black fish skin, identified hundreds of conserved miRNAs and candidate novel miRNAs, and revealed evidence of differential expression of miRNAs in different colors. We examined candidate reference miRNAs in 15 tissues (blood, brain, muscle, liver, kidney, spleen, intestine, heart, gill, fin, eye, ovary, and white/red/black skin) to minimize the risk of co-regulation artifacts. The results provide fundamental knowledge for unraveling the genetic mechanisms underlying color trait variation, and could assist the exploitation of gene regulation linked to skin color determination.
Materials and methods
Sample collection
The Koi carp used in this study were obtained from the aquaculture base of Freshwater Fisheries Research Center (FFRC) affiliated with the Chinese Academy of Fishery Sciences. Animals (average weight: 1000 ± 80 g) were raised at 24 ± 1 °C in 256 L tanks in a circulation water system for 1 week before experiments and fed twice daily (in the morning and late afternoon). Aeration was supplied constantly and a 12 h light / 12 h dark photoperiod was employed.
All of the surviving fish were tranquilized in 20–30 mg/L MS-222 buffered to pH 7.0–7.5, and three sets of skin tissues were collected from nine Koi carp (black, white and red spots in one fish synchronous); three black skin (BS), white skin (WS), and red skin (RS) samples per group, and another 12 tissue samples (blood, brain, muscle, liver, kidney, spleen, intestine, heart, gill, fin, eye, and ovary) were also collected. All fresh tissue samples were immediately snap-frozen in liquid nitrogen and stored at − 80 °C until use.
RNA isolation, library preparation and sequencing
Total RNA was obtained from Koi carp samples using TRIzol (Invitrogen, UK) according to the manufacturer’s protocol, and genomic DNA was removed using DNase I (New England Biolabs). RNA purity was assessed using the Nanodrop-2000 instrument (Thermo Scientific, USA), and all RNA samples had an A260:A280 ratio > 1.9. An equal amount of total RNA from three fish individuals from each color group (black, white, and red spots) was pooled, and a total of nine RNA pools were prepared and used for library construction. A pair of Illunima adapters was ligated to the RNA 5′ and 3′ termini of RNAs, and the resultant products were reverse-transcribed and amplified through 15 PCR cycles. Fragments ranging in size from 140 to 150 nt were retained and purified to generate the sequencing libraries. Each library was loaded into a single Illunima Hiseq2500 lane for 1× 50 base pair (bp) single-end (SE) sequencing. All raw transcriptome data have been deposited in the NCBI Short Read Archive (SRA) with the accession numbers SRR7476960, SRR7476961, SRR7476962, SRR7476963, SRR7476964, SRR7476965, SRR7476966, SRR7476967, and SRR7476968.
Analysis of sequencing data and miRNA identification
FasQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to control the data quality. An initial filtering step was performed to exclude poor quality reads, 3′ and 5′ adaptor reads, and reads shorter than 18 nt. The remaining sequences were mapped to the C. carpio genome using the SOAP program (http://soap.genomics.org.cn) with a tolerance of one mismatch. Sequences matched to the C. carpio genome were subsequently analyzed to filter out rRNAs, tRNAs, snRNAs, snoRNAs, and non-coding RNA reads by BLAST searching against the Rfam (11.0, http://rfam.xfam.org/) and GenBank (https://www.ncbi.nlm.nih.gov/) databases. To ensure each unique small RNA (sRNA) was mapped to only one annotation, we followed the rule rRNAetc (Genbank> Rfam) > known miRNA >repeat >exon >intron3, as previously described [23]. Subsequently, the remaining reads were identified as conserved miRNAs by BLAST searching against miRBase 21.0, allowing no more than two mismatches [24]. Sequences that did not match existing or conserved miRNAs were used to identify potentially novel miRNA candidates [25, 26]. The enrichment degree of each miRNA was identified by counting the number of reads in each sample, and miRNAs were family clustered in each library through miRBase 21.0 database.
Differential expression of miRNAs
To compare the expression levels of miRNAs among the black, white, and red skin libraries, the frequency of miRNA counts was normalized as transcripts per million (TPM). Calculated as follows: normalized expression (TPM) = (actual miRNA count/number of total clean reads) × 1,000,000. Only miRNAs with altered by more than two-fold relative to controls were considered significantly differentially expressed (p-value < 0.05) [27] and classified as DEMs. A positive value indicated up-regulation, while a negative value indicated down-regulation.
Fold-change was calculated as follows: Fold-change = log2 (treatment/control).
The p-value was calculated as follows:
Prediction and analysis of miRNA target genes
Target genes of DEMs were identified using the miRanda [28], RNAhybrid [29] and TargetScan [30], and overlapping DEMs from all three programs provided the final results. The rules used for target prediction were based on those suggested previously by Allen et al... [31] and Schwab et al [32], as follows: (1) No more than four mismatches between sRNA and target (G-U bases count as 0.5 mismatches); (2) No more than two adjacent mismatches in miRNA/target duplex; (3) No adjacent mismatches in positions 2–12 of the miRNA/target duplexes (5′ ends of miRNA); (4) No mismatches in positions 10–11 of miRNA/target duplex; (5) No more than 2.5 mismatches in positions 1–12 of the of miRNA/target duplexes (5′ ends of miRNAs); (6) Minimum free energy (MFE) of miRNA/target duplexes≥75% of MFEs of miRNAs bound to perfect complementary sequences.
Functions strongly associated with the predicted target genes of miRNAs were determined using Gene Ontology (GO; http://www.geneontology.org) [33] biological process categories, and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was conducted using pathway database (http://www.genome.jp/kegg/pathway.html) [34] to statistically test DEMs.
QRT-PCR and analysis of reference miRNAs
Total RNA was extracted as described above. For reverse-transcription of miRNAs, the PrimeScript RT Reagent Kit (Takara Bio, Dalian, China) were used. qRT-PCR was performed on a CFX-96 Real-time PCR System (Bio-Rad, CA, USA) in 25 μL reactions containing 12.5 μL SYBR Advantage Premix (2×) reagent (Takara Bio), 0.5 μL miRNA-specific forward primer (10 μM), 0.5 μL miScript universal primer (10 μM), and 2 μL PCR template (cDNA). Amplification was performed with an initial denaturation at 95 °C for 10s, followed by 40 cycles at 95 °C for 5 s and 60 °C for 20s, and a final cycle from 95 °C to 65 °C. All reactions were conducted in triplicate, which six biological replicates. The relative expression levels of DEMs were measured in terms of threshold cycle value (Ct) and were normalized to U6 snRNA using the eq. 2-ΔΔCt method [35]. Candidate reference miRNAs used for evaluation included let-7a, miR-140-3p, miR-21, miR-15c, miR-26b, miR-92-5p, and miR-145a-5p due to their low expression differences according to the Illumina deep sequencing results (data not shown). Additionally, 5 s rRNA, 18 s rRNA and U6 snRNA were also selected for evaluation as reference genes due to their broad use in the literature. QRT-PCR methods were the same as described above for DEMs, and the stability of reference miRNAs was tested using geNorm [36], NormFinder [37] and Bestkeeper [38] programs. All primers used in qPCR experiments (Additional file 1: Table S1) were synthesized by Sangon Biotech. (Shanghai, China). Data were analyzed statistically with SPSS 20 (IBM, Chicago, IL, USA) by t-test. Thresholds for statistical significance were set at P < 0.05 (significant) and P < 0.01 (highly significant).
Results
Identification of Koi carp miRNAs via sRNA sequencing
To identify miRNAs expressed in the skin of Koi carp, BS, RS, and WS sRNA libraries were determined and analyzed by Illumina deep sequencing. A Total of 20,502,959, 16,916,198, and 19,839,224 reads were obtained from the BS, RS, and WS libraries, respectively. After filtering out low-quality reads, insert-null, reads < 18 nt, and removing adaptor sequences, 19,649,441 (96.01%), 16,193,916 (95.91%), and 18,981,507 (95.87%) clean reads were retrieved for further analysis (Additional file 2: Table S2). In order to annotate the different classes of sRNAs, clean reads were compared with GenBank, Rfam, miRbase, exon and intron, and repeat-associated RNA databases. Representations of different types of RNA sequences, including rRNAs, tRNAs, snRNAs, snoRNAs, and scRNAs are shown in Table 1.
Table 1.
Distribution of sequenced reads from raw data to cleaned sequences
| Type | Black skin | Red skin | White skin | |||
|---|---|---|---|---|---|---|
| Total reads | % | Total reads | % | Total reads | % | |
| Raw reads | 20,502,959 | 100 | 16,916,198 | 100 | 19,839,224 | 100 |
| tRNA | 173,761 | 0.88 | 247,404 | 1.53 | 221,565 | 1.17 |
| snoRNA | 4455 | 0.023 | 4818 | 0.029 | 5169 | 0.027 |
| snRNA | 3143 | 0.016 | 4346 | 0.027 | 4199 | 0.022 |
| rRNA | 175,928 | 0.90 | 150,346 | 0.93 | 180,280 | 0.95 |
| scRNA | 6980 | 0.036 | 7887 | 0.048 | 7890 | 0.042 |
| Intro_antisense | 503,101 | 2.56 | 437,439 | 2.70 | 558,702 | 2.94 |
| Exon_antisense | 197,485 | 1.01 | 186,048 | 1.14 | 168,655 | 0.889 |
| Other | 18,584,128 | 94.58 | 15,155,625 | 93.59 | 17,835,047 | 93.96 |
The length distribution of sRNA reads is shown in Fig. 2. The results were similar among the three BS, RS, and WS groups, with 22- or 23- nt sRNAs accounting for 73.82, 74.20, and 73.10% of total sequences, respectively (Fig. 2).
Fig. 2.

The length distribution of cleaned sequencing reads
Characterization of miRNAs in different skin tissues
To further identify conserved miRNAs and predict novel miRNAs in the three skin color tissues, all sRNA sequences were mapped to known miRNAs in miRBase 21.0. After alignment and additional sequence analysis, 330, 397, and 335 conserved miRNAs were identified from BS, RS, and WS libraries, respectively, that are highly conserved in other species (Additional file 3). These miRNAs exhibited a broad range of expression levels, ranging from 32,428,443 counts for the most abundant, to a single count. The results including known carp miRNAs in the miRBase database for common carp, including 137, 144, and 138 miRNAs for BS, RS, and WS groups, respectively, of which miR-199-5p, miR-199-3p, let-7a, miR-21, miR-26a, miR-99, miR-100, miR-125b, miR-22a, miR-126-3p, and miR-146a were the most abundant (Additional file 4). To obtain a clearer perspective of the most abundant known carp miRNAs, we compared those with the 10 highest read numbers (Table 2) and ~ 81 miRNA families were identified in each library. Several miRNA families were expressed at levels > 100,000 reads, such as let-7, miR-199, miR-126, miR-27, miR-21, etc. The degree of sequence conservation of miRNAs was assessed to investigate phylogenetic evolution, and 20 conserved families were clustered into three groups based on phylogenetic distribution (Fig. 3). Five miRNA families (let-7, miR-124, miR-184, miR-375, and miR-31) are shared in protostomes and deuterostomes, 13 families are presented only in vertebrate, and the other two families (miR-135 and miR-727) have only been identified in fish.
Table 2.
Ten most abundant known carp miRNAs identified in black, white, and red Koi carp skin samples
| miRNA | Sequence | BS Count | RS Count | WS Count |
|---|---|---|---|---|
| ccr-miR-199-5p | CCCAGTGTTCAGACTACCTGTTC | 4,100,063 | 3,122,326 | 3,587,226 |
| ccr-miR-199-3p | ACAGTAGTCTGCACATTGGTT | 1,730,490 | 1,389,612 | 1,653,539 |
| ccr-let-7a | TGAGGTAGTAGGTTGTATAGTT | 1,297,246 | 1,133,283 | 1,272,622 |
| ccr-miR-21 | TAGCTTATCAGACTGGTGTTGGC | 1,309,364 | 1,138,255 | 1,255,244 |
| ccr-miR-26a | TTCAAGTAATCCAGGATAGGCT | 1,086,517 | 902,538 | 1,092,863 |
| ccr-miR-99 | AACCCGTAGATCCGATCTTGT | 809,551 | 750,404 | 781,225 |
| ccr-miR-100 | AACCCGTAGATCCGAACTTGT | 877,228 | 697,664 | 706,497 |
| ccr-miR-125b | TCCCTGAGACCCTAACTTGTGA | 742,182 | 506,300 | 590,501 |
| ccr-miR-22a | AAGCTGCCAGCTGAAGAACTGT | 529,973 | 402,752 | 535,663 |
| ccr-miR-126-3p | CTCGTACCGTGAGTAATAATGC | 485,109 | 394,630 | 476,118 |
Fig. 3.

Phylogenetic tree of 20 conserved miRNA families in Koi carp. Note: The presence and absence of miRNAs is indicated by plus (+) and minus (−) symbols, respectively. Abbreviations: ccr, Cyprinus carpio; dre, Danio rerio; fru, Takifugu rubripes; ssa, Salmo salar; aca, Anolis carolinensis; hsa, Homo sapiens; mmu, Mus musculus; gga, Gallus gallus; tgu, Taeniopygia guttata; bbe, Branchiostoma belcheri; csa, Ciona savignyi; cel, Caenorhabditis elegans; dme, Drosophila melanogaster; P, Protostomia; D, Deutostomia; V, Vertebrata
We also found 1563 miRNAs not previously identified in Koi carp, comprising 340, 353, and 351 in BS, RS, and WS groups, respectively (Additional file 5). These were considered specific candidate miRNAs for further investigation. The seven most abundant miRNAs (novel-miRn0378, novel-miRn0924, novel-miRn1323, novel-miRn0321, novel-miRn0902, novel-miRn0547, and novel-miRn1422) all had > 300 reads (Table 3). We also investigated the most abundant novel miRNAs in each skin tissue, and novel-miRn0022 and novel-miRn0032 were dominantly expressed in white skin rather than black or red skin, with 1165 and 1108 reads, respectively (Table 4).
Table 3.
Seven most abundant novel miRNAs identified in black, white, and red Koi carp skin samples
| miRNA | Sequence | BS Count | RS Count | WS Count |
|---|---|---|---|---|
| novel-miRn0378 | CAAGCTTGTTTCTATGGGTCTC | 1720 | 1693 | 1940 |
| novel-miRn0924 | TAGATCAGTGAACTTGCCTTTA | 2056 | 1388 | 1664 |
| novel-miRn1323 | TCAGTCACCGTTCACTTACATT | 1414 | 1129 | 1356 |
| novel-miRn0321 | TGGAAACATTCTACACTCTCAGA | 1242 | 931 | 1163 |
| novel-miRn0902 | CAAGCTCGATTCTGTGGGTCT | 1027 | 746 | 792 |
| novel-miRn0547 | AATGCAAGAACACATCCTGAGT | 832 | 662 | 805 |
| novel-miRn1422 | ATTATGAACATGATATTGAAT | 415 | 370 | 424 |
Table 4.
Ten most abundant novel miRNAs differentially expressed in black, white, and red skin samples
| miRNA | Sequence | BS Count | RS Count | WS Count |
|---|---|---|---|---|
| novel-miRn0022 | TAAAATGGACCATTGACACTCT | 750 | 531 | 1165 |
| novel-miRn0032 | TCTGCAACACGAAACTGTCTTA | 522 | 47 | 1108 |
| novel-miRn0678 | AAGTTCTGTGGTCCACTCTGGCT | 1328 | 337 | 569 |
| novel-miRn1387 | ACTGATTTCCTCTGGTGCTTGGA | 1099 | 779 | 577 |
| novel-miRn1534 | TCACGCTGCGGATCAGATGCTC | 804 | 629 | 501 |
| novel-miRn0032 | TAAAGAGAACCGCCGCAAACGC | 307 | 53 | 170 |
| novel-miRn0824 | AACGATCTTTAAACATTAATCT | 266 | 77 | 510 |
| novel-miRn0849 | TTCAAACGGACCATTGACATTC | 97 | 131 | 238 |
| novel-miRn1070 | GATCGTGATGAAACTTTAACC | 376 | 138 | 581 |
| novel-miRn1199 | TTCAGTGAAGATAATCTGTCC | 139 | 55 | 84 |
Expression profiling of miRNAs in different skin patterns
In the high-throughput sequencing analysis, many miRNAs exhibited dissimilar expression levels among the three skin color samples. Using the criteria |log2(Fold-change)| ≥ 1 and q-value ≤0.001, volcano plots of the three pairwise comparisons (BS vs. WS, RS vs. BS, and RS vs. WS) revealed the expression trends for each pair (Fig. 4a). We also constructed a histogram of DEMs in the three skin tissues (Fig. 4b). Compared with black skin, 53 miRNAs were up-regulated in white skin, including miR-200b and miR-135c, while 36 miRNAs were down-regulated. A total of 50 miRNAs were down-regulated in white skin compared with red skin, including miR-217, while 45 miRNAs were up-regulated. There were 47 miRNAs displaying greater abundance in black skin compared with red skin, including miR-196a, while 34 miRNAs were down-regulated (Additional file 6). The proportion of DEMs reflects their specific functions and related biological mechanisms in different skin tissues. A Venn diagram (Fig. 4c) was generated to visually compare the expression of miRNAs in Koi carp. Among the significant DEMs, 164 miRNAs (including 14 overlapping DEMs) were identified, and 30 miRNAs (including 17 conserve and 13 novel DEMs) were significant DEMs in WS and RS group compared with the BS group, suggesting that they likely play a key role in the color variation process (Fig. 4c). The heatmap of these 17 known DEMs (Fig. 5) indicates that eight miRNAs including miR-203b-3p, miR-26-3p, miR-205-5p, etc. were up-regulated in WS and RS groups compared with the BS group, while six miRNAs, including aca-miR-210-3p, miR-125c, miR-206, etc. were down-regulated in WS and RS groups compared with BS group. The remaining miRNAs (ccr-miR-196a, miR-202, and ipu-miR-196a) exhibited different expression trends (Fig. 5).
Fig. 4.

Differentially expressed miRNAs (DEMs) in white, red and black Koi carp skin. a, MA plot of differential miRNA expression levels among the three pairwise comparisons, black, red, and green dots represent non-significant, up-regulated, and down-regulated miRNAs, respectively; b, Number of DEMs among the three pairwise comparisons, red and blue indicate the number of up- and down-regulated miRNAs, respectively; c, Venn diagram comparing the expression distribution of miRNAs
Fig. 5.

Heatmap of 17 known DEMs in WS, BS and RS groups. Note: The 17 rows and three columns correspond to each miRNA and skin tissue, respectively. The heatmap was drawn based on log10 normalized expression values for each miRNA in relation to expression in WS, BS, and RS skin groups
In order to validate the reliability of the Illumina sequencing results, expression in different skin tissues was examined by qRT-PCR for 18 miRNAs, including conserved and specific miRNAs, selected randomly from the dataset. The results showed that qRT-PCR expression patterns of all selected miRNAs were absolutely in agreement with the results of RNA-seq analysis, indicating that the RNA-seq data was reliable (Fig. 6). For example, based on the deep sequencing result, the expression level of let-7f-5p in RS was almost 10 times higher than in WS, compared with an 8-fold difference in the qRT-PCR results.
Fig. 6.

Comparison of fold change in expression between qRT-PCR and RNA-seq data
Target genes prediction and signaling pathways analysis
Target genes were predicted based on the common carp (C. carpio) genome sequence (http://www.fishbrowser.org/database/Commoncarp_genome/) using a combination of TargetScan, RNAhybrid, and miRandato refine and improve the prediction results. A total of 2495 putative target genes were predicted, and some unique miRNAs, such as miR-206, miR-125c, miR-217, miR-205-5p, and novel-miRn1185, appear to target hundreds of genes. Moreover, our results indicate that some genes may be regulated by more than one miRNAs, such as cytoglobin-1-like (No: DN51764) that is targeted by nine miRNAs. Target genes were subjected to GO analysis for functional classification (Additional file 7). The result showed that many miRNAs were assigned to pigmentation-related terms such as melanocyte migration (GO: 0097324), retinal pigment epithelium development (GO: 0010669), and melanosome transport (GO: 0032402). These genes enriched in pigmentation-related processes are informative and worthy a further study.
KEGG pathway analysis was also performed to classify putative miRNA targets, and 275 annotated signaling pathways were found to be involved in a wide range of biological functions. For instance, at least 10 pathways involved in pigmentation-related biology were identified, including Wnt/β-catenin signaling, MAPK signaling, melanogenesis, oxidative phosphorylation, cell cycle, TGF-beta signaling, melanoma, cAMP signaling, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor resistance, and Notch signaling (Fig. 7). Herein, we mainly focused on melanogenesis, Wnt signaling, and MAPK signaling pathways, and putative gene pathways involved in different colored skin pigmentation process are shown in Fig. 8. Many genes in the melanogenesis pathway have been linked to skin color in Koi carp, including Mc1r, Msh, Mitf, kit oncogene (KIT), and Tyr [39]. We identified Mc1r as a target of miR-200b, miR-206, miR-196a, and novel-miRn1185. Meanwhile, Msh is a target of miR-200a, miR-206, miR-142b-3p, and miR-26a-3p. Wnt and MAPK signaling pathway are also involved in the pigmentation process in fish, and Sox10, Fgf, Mitf, and Silv are indispensable [40, 41]. These results showed that miR-133c, miR-26a-3p, miR-196a, and miR-181a-5p all target the Mitf gene. Foxd3 is one of the earliest molecular makers in the Guanine pathway, and altering the expression level of this gene can control lineage choice between neural or glial and pigment cells, as demonstrated by repressing Mitf during the early phase of neural crest migration [42]. Herein, Foxd3 was found to be targeted by miR-429b, miR-26a-3p, miR-200b, miR-10b-5p, and miR-181a-5p.
Fig. 7.

Predicted pigmentation-related pathways and target genes based on Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Gene number, number of target genes in each pathway; Rich factor; ratio of the number of target genes divided by the total number of genes in each pathway
Fig. 8.
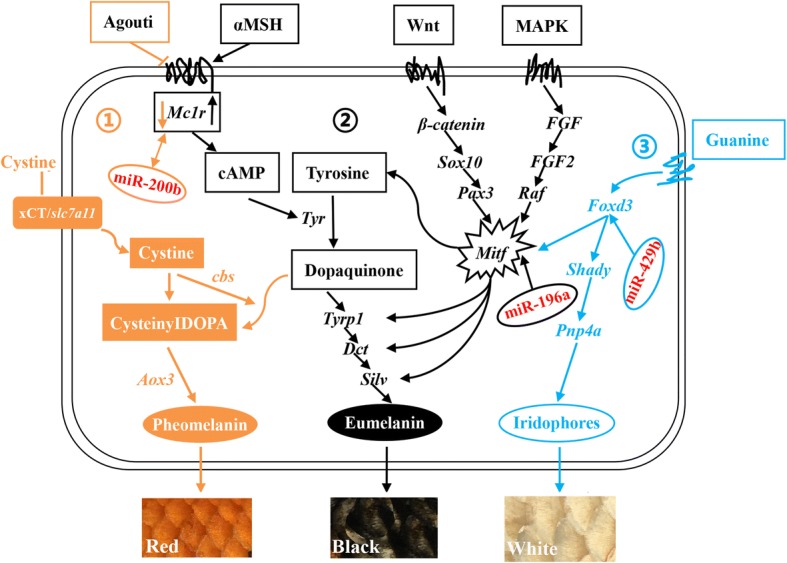
Schematic diagram of putative gene pathways related to the skin pigmentation process in Koi carp which drawn referred to reference literature [54, 55, 57]. (1) represents the pheomelanin biosynthesis pathway; (2) represents the eumelanin pathway. (3) represents the guanine pathway
Identification of optimal reference genes
As shown in Fig. 9, the Ct values of 10 genes (let-7a, 140-3p, 21, 15c, 26b, 92-5p, 145a-5p, 5 s rRNA, 18 s rRNA, and U6) ranged from 15.67 to 31.12 according to the qRT-PCR results, indicating that these 10 genes are expressed at normal levels in different tissues in Koi carp, and are therefore suitable as internal reference genes for screening purposes. Let-7a was the most highly expressed miRNA with the lowest Ct value, ranging from 17.65 to 19.89; and 18 s rRNA had the lowest Ct value, while miR-92a-5p had the largest. The largest variation range was observed for 5 s rRNA (6.28), while miR-26b (1.86) exhibited the least variation.
Fig. 9.

Box plot of the Ct values of the candidate reference genes. Note: Each box represents an internal candidate gene, with horizontal lines representing the median, the top of the box representing the third quartile, the bottom representing the first quartile, and lines crossing the box representing maximum and minimum values
Analysis of miRNA expression stability (M) values using the geNorm algorithm was applied to each reference gene based on average pairwise variation between all candidate genes (M value is negatively correlated with miRNA stability; the lower the value, the higher the stability, and vice versa). The results showed that comparison of let-7a and miR-26b gave an M value of 0.661, which indicates high stability. The stability of the 10 reference genes in different tissues was ranked let-7a and miR-26b > U6 > miR-145a-5p > miR-21 > miR-92a-5p > miR-15c > miR-140-3p > 5 s rRNA > 18 s rRNA (Table 5). The NormFinder algorithm is similar to that used by geNorm, and the lower the expression of internal reference genes, the higher the stability. As shown in Fig. 10, the NormFinder reference gene stability in different Koi carp tissues was ordered let-7a > U6 > miR-26b > miR-15c > miR-145a-5p > miR-92a-5p > miR-21 > 5 s rRNA > miR-140-3p > 18 s rRNA, hence let-7a was the most stable gene. In BestKeeper analysis, based on average Ct values, the stability of internal reference genes should have a low standard deviation (SD) and coefficient of variation (CV), as well as a high Pearson correlation coefficient (R). The BestKeeper results showed that the best-performing internal reference gene was let-7a, followed by miR-26b, while the most unstable gene was miR-140-3p, followed by 18 s rRNA (Table 6). Taking the results of these three analyses together, let-7a was the most stable single reference gene, let-7a and miR-26b was the most stable reference combination in different tissues, while 18 s rRNA was the most unstable gene.
Table 5.
Matrix of different reference gene combinations in different tissues
| Gene name | let-7a | miR-140-3p | miR-21 | miR-15c | miR-26b | miR-92-5p | miR-145a-5p | 5 s rRNA | U6 |
|---|---|---|---|---|---|---|---|---|---|
| let-7a | – | – | – | – | – | – | – | – | – |
| miR-140-3p | 2.762 | – | – | – | – | – | – | – | – |
| miR-21 | 1.577 | 2.311 | – | – | – | – | – | – | – |
| miR-15c | 2.332 | 1.298 | 2.562 | – | – | – | – | – | – |
| miR-26b | 0.661 | 2.864 | 1.824 | 2.218 | – | – | – | – | – |
| miR-92-5p | 1.811 | 1.783 | 2.042 | 1.726 | 1.756 | – | – | – | – |
| miR-145a-5p | 1.328 | 2.465 | 2.036 | 2.78 | 1.124 | 2.119 | – | – | – |
| 5 s rRNA | 3.44 | 2.745 | 3.07 | 3.002 | 3.228 | 3.064 | 3.136 | – | – |
| U6 | 0.985 | 3.228 | 1.756 | 3.11 | 0.867 | 2.198 | 1.033 | 3.176 | – |
| 18 s rRNA | 3.541 | 3.186 | 3.426 | 3.464 | 3.398 | 3.345 | 3.422 | 2.033 | 3.348 |
Note: The left diagonal represents stability in different tissues, and — line represents no values
Fig. 10.

The stability of candidate reference genes analyzed by NormFinder. Values with a different superscript letter denote significant differences between genes (P < 0.05)
Table 6.
Reference gene stability in different tissues based on BestKeeper analysis
| Parameter | let-7a | miR-140-3p | miR-21 | miR-15c | miR-26b | miR-92-5p | miR-145a-5p | U6 | 5 s | 18 s |
|---|---|---|---|---|---|---|---|---|---|---|
| R | 0.936 | 0.243 | 0.52 | 0.769 | 0.904 | 0.582 | 0.468 | 0.821 | 0.419 | 0.412 |
| SD | 0.866 | 0.83 | 1.426 | 1.377 | 0.824 | 1.124 | 1.59 | 1.12 | 1.396 | 2.114 |
| CV | 6.835 | 10.923 | 9.923 | 9.8 | 7.164 | 7.63 | 9.78 | 8.832 | 9.182 | 10.48 |
Discussion
Body color and coloration patterns are prominent features related to intraspecific communication, interspecific interactions, photoprotection, photoreception, thermoregulation, and market value in many animals, including Koi carp. Thus, in the present work we explored the molecular mechanisms of pigmentation regulation and evolution to facilitate improving coloration and ornamental value [43]. Increasing evidence suggests that miRNAs play a vital role in skin pigmentation by controlling the expression of a vast number of genes [44, 45]. Herein, we generated miRNA transcriptome data from black, white, and red skin samples of Koi carp using a deep sequencing approach, and the findings provide a foundation for future skin pigmentation studies.
The BS, WS, RS miRNA libraries constructed from different colored skin tissue displayed a similar read length distribution of 22-23 nt miRNAs, equivalent in size to typical Dicer-derived products. The most abundant miRNAs were let-7a, miR-21, miR-26a, miR-15c, etc. with > 10,000 read each. These miRNAs are also highly expressed in other species; let-7 family was first characterized in Caenorhabditis elegans, and plays a key role in regulating late developmental events by down-regulating lin-41 and possibly other genes [46]; miR-21 is an abundantly expressed miRNA in mammalian cells, and is evolutionarily conserved across a wide range of vertebrate species. Additionally, it is significantly up-regulated in gastric cancer, and it targets 15-PGDH, suggesting the PGE2/PI3K/Akt/Wnt/β-catenin axis might be a novel pathway for gastric cancer treatment [47]. Thus, these miRNAs are considered to be housekeeping molecules involved in the maintenance of basic cellular activities.
To gain insight into the possible significance of skin miRNAs in Koi Carp, we determined expression profile of miRNAs in different colored (red, black, and white) skin tissues. We found that 30 miRNAs (17 conserved and 13 novel) were significant DEMs in WS and RS groups compared with the BS group, and 14 overlapping DEMs were shared by all three skin tissues, including miR-196a, miR-125b, miR-202, miR-125c, miR-205-5p, miR-200b, and etc. implying that these DEMs may therefore be involved in skin pigmentation. Knowledge of tissue- and cell-specific expression patterns of miRNAs could directly inform functional studies [48]. A previous study has revealed miR-200b and miR-196a as candidates that may be important for hair follicle (HF) development because their expression was reduced in the Dkk1 transgenic skin [49]. Meanwhile, miR-125b serves as a rheostat that controls stem cell proliferation, fate commitment, and differentiation [50]. Serum levels of miR-205-5p were determined by real-time PCR in 11 patients with metastatic melanoma and 16 patients without metastasis, and differences indicated that the miRNA network may be involved in the pathogenesis of melanoma metastasis [51]. Also, a significant correlation was observed between miR-9-5p and miR-205-5p, and between miR-203a-3p and miR-205-5p, suggesting that expression levels of these miRNA were not independently regulated in melanoma patients [51]. Herein, we found that miR-196a, miR-202, miR-125b andmiR-125c, etc. were highly expressed in white and red skin compared with black skin, indicating specific functions in difference tissues. Thus, specific DEMs may regulate skin pigmentation in fish.
MiRNA target identification is important for predicting their functions. Although computational approaches have been widely used to predict targets, most methods suffer from a high false-positive rate [52]. Therefore, we used three software packages for analysis of target genes to refine and improve the results. We found that some miRNAs such as miR-206, miR-125c, miR-217, etc. targeted hundreds of genes, and some genes were regulated by more than one miRNA, indicating a complicated regulatory network between miRNAs and their targets gene. Skin pigmentation in fish is a complex process which involves numerous physiological, cellular and genetic factors [53]. Previous studies found that signaling pathway such as the Wnt, MAPK, cAMP, melanogenesis, Notch, and Fgf pathways strongly influence skin pigmentation process [41]. Herein, we constructed the putative gene pathways with some miRNAs involved in red, black, and white color skins pigmentation process. The molecular mechanism of melanin biosynthesis was extensively studied due to its biomedical significance, in mammals and birds, the ratio of eumelanin and pheomelanin largely determines an animal’s overall color: darker (black to brown) phenotypes result from the increased deposition of eumelanin, while lighter (red to yellow) phenotypes result from the increased deposition of pheomelanin [54]. Xu (2014) also elucidated the pigmentation mechanisms of Hebao and Songpu carp, in the melanogenesis pathway, tyrosine is oxidized to form dopaquinone, and then intracellularly catalyzed to become eumelanin; However, cystine and dopaquinone can switch off the eumelanin synthesis pathway and promote the synthesis of pheomelanin; And slc7a11 encodes the transmembrane cystine/glutamate exchanger (xCT), which transports cystine into melanocytes to synthesize pheomelanin [55]. Both miR-200 and miR-205 are highly expressed in normal hair follicles compared epithelia tumors in human [56]. The Mc1r gene is a key factor involved in the production of pheomelanin and eumelanin in melanocytes, and we found that it is targeted by miR-200a, miR-200b, and miR-206, and miR-206 presented up-regulated in black skins compared with red and white skins suggested that it played important roles in pigmentation in Koi carp. Wnt and MAPK are implicated in numerous development and physiological process. Sun reported that both Wnt and MAPK signaling pathways are highly likely to be involved in melanin biosynthesis in common carp [57]. Mitf is a member of the Myc-related family of basic helix-loop-helix leucine zipper (bHLH-Zip) transcription factors, and Mitf was found in cells related to the retinal pigment epithelium (RPE), as well as cells behind the optic cup that were probably derived from neural crest tissue and could develop into iridophore pigment cells [58]. Mutations of the Mitf gene causes a variety of phenotypes, most notably in pigmented cells [59]. Foxd3 can affect the lineage between neural or glial and pigment cells by repressing Mitf during the early phase of neural crest migration [42]. Kevin et al... reported a model which melanophores and iridophores descend from a common precursor whose fate is regulated by the interplay transcription factors regulating Mitf and Foxd3, and loss of Mitfa resulted in supernumerary ectopic iridophores, while loss of Foxd3, a Mitfa repressor, resulted in fewer iridophores [60]. Double mutants displayed restoration of iridophores, suggesting that one of Foxd3 is to suppress Mitfa to promote iridophore development [60]. In our present study, we also observed miR-429b, miR-26a-3p, and miR-181a-5p can target the Foxd3 gene, and miR-133c, miR-196a act on the Mitf gene. Therefore, we have carried out a partial analysis of the regulatory network of the three color skins, while the specific mechanisms and functional need to be deeply excavated.
A number of reference genes for mRNA qRT-PCR analysis have been established, but miRNA qRT-PCR studies are fewer in number. Thus, we attempted to select suitable reference genes for normalization to eliminate or decrease non-biological variation between test samples. The two most commonly used reference genes for normalization in miRNA analyses are U6 and 18S rRNA, but there has been some debate as to the validity since they are expressed at levels greater than target genes [61, 62]. Xu et al (2014). found that miR-101a was the most stable miRNA when all tissue types were considered separately [63]. In Siniperca chuatsi, all six miRNAs (miR-101a, miR-146a, miR-22a, miR-23a, miR-26a and let-7a) exhibited better expression consistency than U6 in most of the conditions examined, and the best combination of reference genes was miR-22a and miR-23a [64]. Herein, we systematically evaluated the reliability of seven miRNA genes across 15 tissues. Our results suggest that let-7a was the most stable single reference gene, and the best combination of reference genes was let-7a and miR-26b, followed by U6, while 18 s rRNA was the most unstable gene.
Conclusions
Our findings provide fundamental information on the expression of conserved and potential novel miRNAs in Koi carp skin, and identified DEMs and their respective target genes that may be related to skin color. We identified 137, 144, and 138 miRNAs for BS, RS, and WS groups which already present in the C.carpio miRBase database, and miR-203b-3p, miR-125c, ccr-miR-196a, miR-202, and etc. were presented differences in expression in the three different skin color tissue samples. The best combination of reference genes was let-7a and miR-26b. Validation of the functional role of DEMs and reference genes will provide a better understanding of miRNA-mediated regulatory mechanisms influencing skin color in fish.
Additional files
Table S1. Primers for skin DEMs and reference miRNAs in Koi carp. (DOCX 14 kb)
Table S2. Read statistics of the obtained small RNAs. (DOCX 13 kb)
Conserved miRNAs identified in black, white, and red skin samples from Koi carp. (XLSX 91 kb)
Known carp miRNAs identified in black, white, and red skin samples from Koi carp. (XLSX 45 kb)
Novel miRNAs identified in black, white, and red skin samples from Koi carp. (XLSX 228 kb)
Differentially expressed miRNAs (DEMs) identified in black, white, and red skin samples from Koi carp. (XLSX 40 kb)
Predicted target genes of DEMs and target genes associated with significantly enriched Gene Ontology (GO) terms in Koi carp. (XLSX 208 kb)
Acknowledgments
We would like to thank Nanjing Decode Genomics Biotechnology Co., Ltd. for assistance with the bioinformatics analysis.
Funding
This study was supported by grants from the “333 project” training fund project of Jiangsu province (BRA2017083), and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX18_0738). The funders had no role in the study design, data collection, analysis, interpretation of data, or manuscript preparation.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files, as well as in the NCBI Short Read Archive (SRA), under accession numbers SRR7476960, SRR7476961, SRR7476962, SRR7476963, SRR7476964, SRR7476965, SRR7476966, SRR7476967, and SRR7476968.
Abbreviations
- Akt
Protein kinase B
- cAMP
Cyclic Adenosine Monophosphate
- cDNA
Complementary DNA
- CV
Coefficient of variation
- DEMs
Differentially expressed miRNAs
- Dkk1
Dickkopf-related protein 1
- EGFR
Epidermal growth factor receptor
- FFRC
Freshwater Fisheries Research Center
- Fgf
Fibroblast growth factor
- Foxd3
Forkhead box D3
- GAPDH
Glycer-aldehyde-3-phosphate dehydrogenase
- GO
Gene ontology
- HF
Hair follicles
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- Kit
Kit oncogene
- MAPK
Mitogen-activated protein kinase
- Mc1r
Melanocortin receptor 1
- MFE
Minimum free-energy
- Mitf
Microphthalmia-associated transcription factor
- mRNA
Messenger RNA
- Msh
Melanocyte-stimulating hormone
- NCBI
National Center for Biotechnology Information
- Notch
Notch signaling pathway
- Nt
Nucleotides
- PGDH
Phosphoglycerate dehydrogenase
- PGE2
Prostaglandin E2
- PI3K
Phosphoinositide 3-kinase
- qRT-PCR
Real-time quantitative reverse transcription PCR
- R
Pearson correlation coefficient
- RISC
RNA-inducing silencing complex
- RPE
Retinal pigment epithelium
- SD
Standard deviation
- Silv
Silver locus protein homolog
- Sox
SRY-related HMG-box
- TGF-beta
Transforming growth factor beta
- TPM
Transcripts per million
- Tyr
Tyrosinase
- Tyrp1
Tyrosine related protein-1
- Wnt
Wingless and INT-1
Authors’ contributions
The authors’ responsibilities were as follows: ZD designed and supervised the study. ML performed experiments and analyzed the transcriptome data. LW and WZ analyzed the transcriptome data. JF prepared tables and figures; FS, MF, and JD performed qRT-PCR. ML wrote the manuscript. All authors read, reviewed, and approved the manuscript for submission.
Ethics approval and consent to participate
Efforts were made to minimize suffering as much as possible. All animals were handled and experimental procedures were carried out in accordance with the guidelines for the Care and Use of Animals for Scientific Purposes set by the Ministry of Science and Technology, Beijing, China (No. 398, 2006). This research was approved by the Animal Care and Use committee of the Centre for Applied Aquatic Genomics at the Chinese Academy of Fishery Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mingkun Luo, Email: mingkunluo@163.com.
Lanmei Wang, Email: wanglm@ffrc.cn.
Wenbin Zhu, Email: zhuwb@ffrc.cn.
Jianjun Fu, Email: fujj@ffrc.cn.
Feibiao Song, Email: songfb1014@126.com.
Min Fang, Email: fmptr@qq.com.
Juanjuan Dong, Email: m18114862625@163.com.
Zaijie Dong, Phone: +8651085558831, Email: dongzj@ffrc.cn.
References
- 1.Eugene KB. Origin and domestication of the wild carp, Cyprinus carpio: from Roman gourmets to the swimming flowers. Aquaculture. 1995;129:3–48. doi: 10.1016/0044-8486(94)00227-F. [DOI] [Google Scholar]
- 2.Cieslak M, Reissmann M, Hofreiter M, et al. Colors of domestication. Biol Rev. 2011;86:885–899. doi: 10.1111/j.1469-185X.2011.00177.x. [DOI] [PubMed] [Google Scholar]
- 3.Aspengren S, Skold HN, Wallin M. Different strategies for color change. Cell Mol Life Sci. 2009;66:187–191. doi: 10.1007/s00018-008-8541-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bar I, Kaddar E, Velan A, et al. Melanocortin receptor 1 and black pigmentation in the Japanese ornamental carp (Cyprinus carpio var. Koi) Front Genet. 2013;4:6. doi: 10.3389/fgene.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu JH, Wen S, Xiao YM, et al. Involvement of the Mitfa gene in the development of pigment cell in Japanese ornamental (Koi) carp (Cyprinus carpio L.) Genet Mol Res. 2015;14(1):2775–2784. doi: 10.4238/2015.March.31.7. [DOI] [PubMed] [Google Scholar]
- 6.Hubbard JK, Hauber ME, Safran RJ, et al. Vertebrate pigmentation: from underlying genes to adaptive function. Trends Genet. 2010;26:231–239. doi: 10.1016/j.tig.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNAs biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 9.Xu P, Vemooy SY, Guo M, et al. The Drosophila microRNA mir-14 suppresses cell death and is required for normal fat metabolism. Curr Biol. 2003;13:790–795. doi: 10.1016/S0960-9822(03)00250-1. [DOI] [PubMed] [Google Scholar]
- 10.Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumor-genesis. Br J Cancer. 2006;94:776–780. doi: 10.1038/sj.bjc.6603023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang F, Jia YF, Chang ZJ, et al. Identification and profiling of Cyprinus carpio microRNAs during ovary differentiation by deep sequencing. BMC Genomics. 2017;18(1):333. doi: 10.1186/s12864-017-3701-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan B, Liu B, Wang CH, et al. MicroRNAs regulation of skin pigmentation in fish. J Cell Sci. 2013;126:3401–3408. doi: 10.1242/jcs.125831. [DOI] [PubMed] [Google Scholar]
- 13.Yi R, Poy MN, Stoffel M, et al. A skin microRNA promotes differentiation by repressing ‘stemness’. Nature. 2008;452:225–229. doi: 10.1038/nature06642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennell JA, Cadigan KM, Waldron EJ, et al. The microRNA miR-8 is a positive regulator of pigmentation and eclosion in Drosophila. Development Dynamics. 2012;241:161–168. doi: 10.1002/dvdy.23705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong C, Wang H, Smith GW, et al. Coat color determination by miR-137 mediated down-regulation of microphthalmia-associated transcription factor in a mouse model. RNA. 2012;18:1679–1686. doi: 10.1261/rna.033977.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian X, Jiang JB, Dong CS, et al. Identification and characterization of microRNAs in white and brown alpaca skin. BMC Genomics. 2012;13:555. doi: 10.1186/1471-2164-13-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng D, Zhang Z, Liang G, et al. Validation of reference genes in Solenopsis invicta in different developmental stages, castes and tissues. PLoS One. 2013;8:e57718. doi: 10.1371/journal.pone.0057718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su JG, Zhang RF, Yang CR, et al. Evaluation of internal control genes for qRT-PCR normalization in tissues and cell culture for antiviral studies of grass carp (Ctenopharyngodon idella) Fish & Shellfish Immunology. 2011;30:830–835. doi: 10.1016/j.fsi.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Yan B, Zhao LH, Zhao JL, et al. miR-429 regulation of osmotic stress transcription factor 1 (OSTF1) in tilapia during osmotic stress. Biochem Biophys Res Commun. 2012;426:294–298. doi: 10.1016/j.bbrc.2012.08.029. [DOI] [PubMed] [Google Scholar]
- 20.Yan B, Zhao JL. miR-1228 prevents cellular apoptosis through targeting of MOAP1 protein. Apoptosis. 2012;17:717–724. doi: 10.1007/s10495-012-0710-9. [DOI] [PubMed] [Google Scholar]
- 21.Thellin O, Zorzi W, Hennen G, et al. Housekeeping genes as internal standards: use and limits. J Biotechnol. 1999;75:291–295. doi: 10.1016/S0168-1656(99)00163-7. [DOI] [PubMed] [Google Scholar]
- 22.Vandesompele J, Pattyn F, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):1–11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calabrese JM, Seila AC, Yeo GW, et al. RNA sequence analysis defines Dicer’s role in mouse embryonic stem cells. Proc Natl Acad Sci. 2007;104(46):18097–18102. doi: 10.1073/pnas.0709193104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffiths-Jones S, Grocock RJ, Enright AJ, et al. MiRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearson WR. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the smith-waterman and FASTA algorithms. Genomics. 1991;11:635–650. doi: 10.1016/0888-7543(91)90071-L. [DOI] [PubMed] [Google Scholar]
- 27.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis BP, Shih IH, Burge CB, et al. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 29.Rehmsmeier M, Steffen P, Giegerich R, et al. Fast and effective prediction of microRNA target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Enright AJ, John B, Sander C, et al. MicroRNA targets in Drosophila. Genome Biol. 2004;5(1):R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allen E, Xie Z, et al. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121(2):207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 32.Schwab R, Palatnik JF, et al. Specific effects of microRNAs on the plant transcriptome. Dev Cell. 2005;8(4):517–527. doi: 10.1016/j.devcel.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 33.Da Wei H, Brad TS, Richard AL. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 34.Kanehisa M, Araki M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lardizabal MN, Nocito AL, Daniele SM, et al. Reference genes for real-time PCR quantification of microRNAs and messenger RNAs in rat models of hepatotoxicity. PLoS One. 2012;7:e36323. doi: 10.1371/journal.pone.0036323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vandesompele J, Preter DK, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:7. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andersen CL, Jens LJ, Torben FR. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64(15):5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 38.Michael WP, Ales T, Christian P, et al. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: best keeper-excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26(6):509–515. doi: 10.1023/B:BILE.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 39.Li XM, Song YN, Li JT, et al. Gene expression variation of red-white skin coloration in common carp (Cyprinus carpio) Int J Mol Sci. 2015;16:21310–21319. doi: 10.3390/ijms160921310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denecker G, Vandamme N, Brochez L, et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014;21(8):1–12. doi: 10.1038/cdd.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelsh RN. Genetics and evolution of pigment patterns in fish. Pigment Cell Res. 2004;17:326–336. doi: 10.1111/j.1600-0749.2004.00174.x. [DOI] [PubMed] [Google Scholar]
- 42.Thomas AJ, Erickson CA. Foxd3 regulates the lineage switch between neural crest-derived glial cells and pigment cells by repressing Mitf through a non-canonical mechanism. Development. 2009;136:1849–1858. doi: 10.1242/dev.031989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bercovich D, Korem S, Shauder L, et al. Genetic diversity of color phenotypes in the Koi (Cyprinus carpio L.) as identified by molecular markers. J Biophys Chem. 2012;3:249–255. doi: 10.4236/jbpc.2012.33029. [DOI] [Google Scholar]
- 44.Wang LM, Zhu WB, Dong ZJ, et al. Comparative microRNA-seq analysis depicts candidate miRNAs involved in skin color differentiation in red tilapia. Int J Mol Sci. 2018;19:1209. doi: 10.3390/ijms19041209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi R, Fuchs E. MicroRNA-mediated control in the skin. Cell Death Differ. 2010;17:229–235. doi: 10.1038/cdd.2009.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pasquinelli AE, Reinhart BJ, Kuroda MI, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 47.Li LH, Wang XJ, Shou T, et al. miR-21 modulates prostaglandin signaling and promotes gastric tumorigenesis by targeting 15-PGDH. Biochem Biophys Res Commun. 2018;495(1):928–934. doi: 10.1016/j.bbrc.2017.09.137. [DOI] [PubMed] [Google Scholar]
- 48.Aboobaker AA, Tomancak P, Lai EC, et al. Drosophila microRNAs exhibit diverse spatial expression patterns during embryonic development. PNAS. 2005;102(50):18017–18022. doi: 10.1073/pnas.0508823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andl T, Murchison EP, Tobias JW, et al. The miRNA processing enzyme Dicer is essential for the morphogenesis and maintenance of hair follicles. Curr Biol. 2006;16(10):1041–1049. doi: 10.1016/j.cub.2006.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Stokes N, Fuchs E, et al. Specific microRNAs are preferentially expressed by skin stem cells to balance self-renewal and early lineage commitment. Cell Stem Cell. 2011;8(3):294–308. doi: 10.1016/j.stem.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masatoshi J. Recent progress in studies of miRNA and skin diseases. J Dermatol. 2015;42:551–558. doi: 10.1111/1346-8138.12904. [DOI] [PubMed] [Google Scholar]
- 52.Bandyopadhyay S, Mitra R. Target miner: microRNA target prediction with systematic identification of tissue-specific negative examples. Bioinformatics. 2009;25(20):2625–2631. doi: 10.1093/bioinformatics/btp503. [DOI] [PubMed] [Google Scholar]
- 53.Colihueque N. Genetics of salmonid skin pigmentation: clues and prospects for improving the external appearance of farmed salmonids. Rev Fish Biol Fish. 2010;20:71–86. doi: 10.1007/s11160-009-9121-6. [DOI] [Google Scholar]
- 54.Hoekstra HE. Genetics, development and evolution of adaptive pigmentation in vertebrates. Heredity. 2006;97(3):222–234. doi: 10.1038/sj.hdy.6800861. [DOI] [PubMed] [Google Scholar]
- 55.Xu P, Zhang X, Sun XW, et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat Genet. 2014;46(11):1212–1219. doi: 10.1038/ng.3098. [DOI] [PubMed] [Google Scholar]
- 56.Yi R, Pasolli H, Fuchs E, et al. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat Genet. 2006;38(3):356–362. doi: 10.1038/ng1744. [DOI] [PubMed] [Google Scholar]
- 57.Jiang YL, Zhang SH, Sun XW, et al. Comparative transcriptome analysis reveals the genetic basis of skin color variation in common carp. PLoS One. 2014;9(9):e108200. doi: 10.1371/journal.pone.0108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakayama A, Nguyen MT, Arnheiter H, et al. Mutations in microphthalmia, the mouse homolog of the human deafness gene Mitf, affect neuroepithelial and neural crest-derived melanocytes differently. Mech Dev. 1998;70(1–2):155–166. doi: 10.1016/S0925-4773(97)00188-3. [DOI] [PubMed] [Google Scholar]
- 59.Levy C, Chen PH, Beermann F, et al. Lineage-specific transcriptional regulation of DICER by Mitf in melanocytes. Cell. 2010;141:994–1005. doi: 10.1016/j.cell.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kevin C, James AL, David WR, et al. Interplay between Foxd3 and Mitf regulates cell fate plasticity in the zebrafish neural crest. Dev Biol. 2010;344:107–118. doi: 10.1016/j.ydbio.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jenny MJ, Aluru N, Hahn ME. Effects of short-term exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on microRNA expression in zebrafish embryos. Toxicol Appl Pharmacol. 2012;264(2):262–273. doi: 10.1016/j.taap.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 2000;25(2):169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 63.Xu XY, Shen YB, Li JL, et al. Determination of reference microRNAs for relative quantification in grass carp (Ctenopharyngodon idella) Fish Shellfish Immunol. 2014;36(2):374–382. doi: 10.1016/j.fsi.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 64.Zhu X, Li YL, Chen DX, et al. Selection of reference genes for microRNA quantitative expression analysis in Chinese perch, Siniperca chuatsi. Int J Mol Sci. 2015;16(4):8310–8323. doi: 10.3390/ijms16048310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers for skin DEMs and reference miRNAs in Koi carp. (DOCX 14 kb)
Table S2. Read statistics of the obtained small RNAs. (DOCX 13 kb)
Conserved miRNAs identified in black, white, and red skin samples from Koi carp. (XLSX 91 kb)
Known carp miRNAs identified in black, white, and red skin samples from Koi carp. (XLSX 45 kb)
Novel miRNAs identified in black, white, and red skin samples from Koi carp. (XLSX 228 kb)
Differentially expressed miRNAs (DEMs) identified in black, white, and red skin samples from Koi carp. (XLSX 40 kb)
Predicted target genes of DEMs and target genes associated with significantly enriched Gene Ontology (GO) terms in Koi carp. (XLSX 208 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional files, as well as in the NCBI Short Read Archive (SRA), under accession numbers SRR7476960, SRR7476961, SRR7476962, SRR7476963, SRR7476964, SRR7476965, SRR7476966, SRR7476967, and SRR7476968.