

Abstract
Background:
In pulmonary arterial hypertension (PAH), pathological changes in pulmonary arterioles progressively raise pulmonary artery pressure and increase pulmonary vascular resistance, leading to right heart failure and high mortality rates. Recently, the first potassium channelopathy in PAH, due to mutations in KCNK3, was identified as a genetic cause and pharmacological target.
Methods:
Exome sequencing was performed to identify novel genes in a cohort of 99 pediatric and 134 adult onset group I pulmonary arterial hypertension patients. Novel rare variants in the gene identified were independently identified in a cohort of 680 adult onset patients. Variants were expressed in COS cells and function assessed by patch-clamp and rubidium flux analysis.
Results:
We identified a de novo novel heterozygous predicted deleterious missense variant c.G2873A (p.R958H) in ABCC8 (ATP-binding cassette, subfamily C, member 8) in a child with idiopathic PAH. We then evaluated all individuals in the original and a second cohort for rare or novel variants in ABCC8 and identified 11 additional heterozygous predicted damaging ABCC8 variants. ABCC8 encodes sulfonylurea receptor 1 (SUR1), a regulatory subunit of the ATP-sensitive potassium channel (KATP). We observed loss of KATP function for all ABCC8 variants evaluated, and pharmacological rescue of all channel currents in vitro by the SUR1 activator, diazoxide.
Conclusions:
Novel and rare missense variants in ABCC8 are associated with pulmonary arterial hypertension. Identified ABCC8 mutations decreased KATP channel function, which was pharmacologically recovered.
Keywords: Pulmonary Hypertension, Ion Channels/Membrane Transport, Genetics, Electrophysiology, Basic Science Research
Keywords: pulmonary hypertension, genetics, human, electrophysiology, ion channel, pharmacology, ABCC8, SUR1
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a rare and often fatal disease characterized by distinctive changes in pulmonary arterioles that lead to elevated pulmonary artery pressure and right sided heart failure. Novel treatment options have decreased mortality, but PAH remains a frequently fatal illness. The heterogeneity in disease etiology and nonspecific patient presentation often delays diagnosis, contributing to poor outcomes.
Genetics are recognized to play an important role in the pathogenesis of PAH in patients with and without a family history of disease. Most genetic studies of the disease have been performed in patients with adult-onset disease. Germline mutations in bone morphogenetic protein receptor type 2 (BMPR2), a member of the transforming growth factor β (TGFβ) superfamily of receptors, have been identified as the major genetic cause, including in 70% of inherited and 10–40% of idiopathic cases.1, 2 Mutations in other TGFβ family members comprise additional rare genetic causes. The prevalence of the disease in children is estimated at 2.2 cases per million, an order of magnitude lower than the estimated prevalence of 15–50 cases per million in adults,3 and there are few genetic studies of individuals with childhood-onset PAH.
We previously used exome sequencing to identify mutations in the KCNK3 potassium channel as a genetic cause of idiopathic and familial PAH.4 Furthermore, we proposed KCNK3 as a novel pharmacological target in PAH, as potassium channel currents of select mutant and wildtype KCNK3 channels were pharmacologically remedied by ONO-RS-082, an experimental compound.4, 5 In the current study, we report a novel association of ABCC8/ SUR1 loss of function mutations in both pediatric and adult onset PAH. ABCC8 encodes sulfonylurea receptor 1 (SUR1), a regulatory subunit of the ATP-sensitive potassium channel (KATP). We have functionally assessed mutant KATP channels, and characterized their pharmacological activation.
METHODS
The sequencing data and methods will be made available to other researchers for purposes of reproducing the results or replicating the procedures. The study was approved by the institutional review board at Columbia University Medical Center. Detailed methods are available as supplemental data.
RESULTS
Inherited and de novo variants in ABCC8
By exome sequencing, we identified a de novo missense variant c.G2873A (p.R958H) in ATP-binding cassette, subfamily C, member 8 (ABCC8) (NM_001287174), which was predicted to be deleterious, in a patient diagnosed with idiopathic PAH at the age of 10 (Table 1). We then examined all CU-PAH patients for rare or novel variants in ABCC8 and identified seven additional rare damaging missense variants, predicted by multiple algorithms to be deleterious in seven unrelated patients with idiopathic, familial, or congenital heart disease associated pulmonary arterial hypertension (Table 1). In one familial case, the p.A240T variant was transmitted from an affected father and also observed in the affected sibling. To replicate the findings in the CU-PAH cohort, we evaluated the UK-PAH cohort and identified three additional rare or novel missense and one splice variant in ABCC8 in three patients with idiopathic and one patient with congenital heart disease associated PAH (Table 1). Five variants – c.A214G (p.N72D), c.G558T (p.E186D), c.G718A (p.A240T), c.G2371C (p.E791Q), and c.T2694+2G -- were novel; four rare variants – c.G331A (p.G111R), c.C403G (p.L135V), c.G2437A (p.D813N), and c.G4414A (p.D1472N) -- have been reported in patients with congenital hyperinsulinism; and two variants – c.C686T (p.T229I) and c.G3941A (p.R1314H) – have been reported in patients with transient or permanent neonatal diabetes mellitus.6–12 Alignment of the ABCC8 sequence revealed that all missense variants occur at amino acid residues conserved across species and in critical domains (Figure 1A and 1B).
Table 1.
Pathogenicity Predictions and Clinical Characteristics of Patients with Pulmonary Arterial Hypertension with ABCC8 Mutations. ExAC frequency lists the allele frequency for all of ExAC followed by the allele frequency in the patient’s ethnic group.
| Nucleotide and AA variant |
c.A214G: p.N72D |
c.G331A: p.G111R |
c.C403G: p.L135V |
c.G558T: p.E186D |
c.C686T: p.T229I |
c.G718A: p.A240T |
c.G2371C: p.E791Q |
c.G2437A: p.D813N |
c.G2873A: p.R958H |
c.G3941A: p.R1314H |
c.G4414A: p.D1472N |
c.T2694+2G: p.NA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ExAC_Freq | 0 | 0.00002/ 0.00001 |
0.00003 /0.00006 |
0 | 0.000008/ 0.00001 |
0 | 0 | 0.0001/ 0.0012 |
0.00002/ 0.00001 |
0.00002/ 0.00001 |
0 | 0 |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Hispanic | Asian | Caucasian | Caucasian | Caucasian | Caucasian |
| MetaSVM_pred | D | D | D | D | D | D | D | D | D | D | D | N/A |
| CADD_phred | 22.8 | 28.4 | 20.5 | 19.46 | 29.4 | 35 | 32 | 35 | 27.9 | 23 | 22.4 | 25.1 |
| Cohort | UK | UK | CU | CU | UK | CU | CU | CU | CU | CU | CU | UK |
| PAH type | CHD associated |
Idiopathic | Idiopathic | Idiopathic | Idiopathic | Familial; Sister and father with PAH |
Familial; two deceased affected siblings not tested |
Idiopathic | Idiopathic | CHD- associated |
Idiopathic | Idiopathic |
|
Inheritance/ segregation |
Unknown | Unknown | Paternal | Unknown | Unknown | Paternal; father and affected sister carry p.A240T |
Maternal; affected siblings unavailable for analysis |
Paternal | De novo | Paternal | Maternal | Unknown |
| Gender | F | M | M | M | F | F | F | M | F | M | F | F |
|
Age of
diagnosis |
35 | 64 | 5 | 79 | 34 | Unknown | 14 | 12 | 10 | <1 | 9 | 60 |
| RAP M (mmHg) | N/A | 11 | 9 | N/A | 9 | N/A | N/A | 25 | 6 | 8 | 7 | 7 |
| PAP M (mmHg) | N/A | 44 | 55 | N/A | 45 | N/A | N/A | 54 | 56 | 54 | 37 | 36 |
| AOP M (mmHg) | N/A | Unknown | 78 | N/A | Unknown | N/A | N/A | 48 | 88 | 57 | 67 | Unknown |
| PVRi (U*m2) | N/A | 19 | 25 | N/A | 13 | N/A | N/A | 29 | 16 | 13.6 | 17 | 19 |
| Art Sat % | N/A | 97 | 91 | N/A | 98 | N/A | N/A | 93 | 91 | 97 | 96 | 97 |
| PCWPm (mmHg) | N/A | 15 | 8 | N/A | 10 | N/A | N/A | N/A | 8 | 8 | N/A | 6 |
| CI (L/min/m2) | N/A | 1.5 | 2.8 | N/A | 2.7 | N/A | N/A | N/A | 2.8 | 3.5 | 2 | 1.6 |
|
Response to acute
vasodilator test |
N/A | Unknown | Yes | Unknown | No | Unknown | Unknown | No | Yes | No | Yes | No |
|
Other genetic
variants in PAH genes |
None | None | None | None | None | None | None | None | None | TBX4 c.1106delC: p.S369fs |
None | None |
|
Cardiac
arrhythmias; other conditions |
Large atrial septal defect |
Type 2 Diabetes Mellitus |
Bigeminy, 1st degree heart block |
None | Hearing loss Hypothyroid Lipodermato- sclerosis ESRD Raynaud syndrome |
None | None | Atrial flutter Nonspecific intra-ventricular block Autism |
None | Ventricular septal defect |
None | None |
Abbreviations: AA= amino acid, M=male, F=female, N/A=not available, RAP M=mean right atrial pressure, PAP M=mean pulmonary arterial pressure, AOP M=mean aortic pressure, PVRi=pulmonary vascular resistance index, Art Sat=arterial oxygen saturation, PCWPm=mean pulmonary capillary wedge pressure, CI=cardiac index. UK=United Kingdom cohort, CU=Columbia University cohort.
Figure 1.
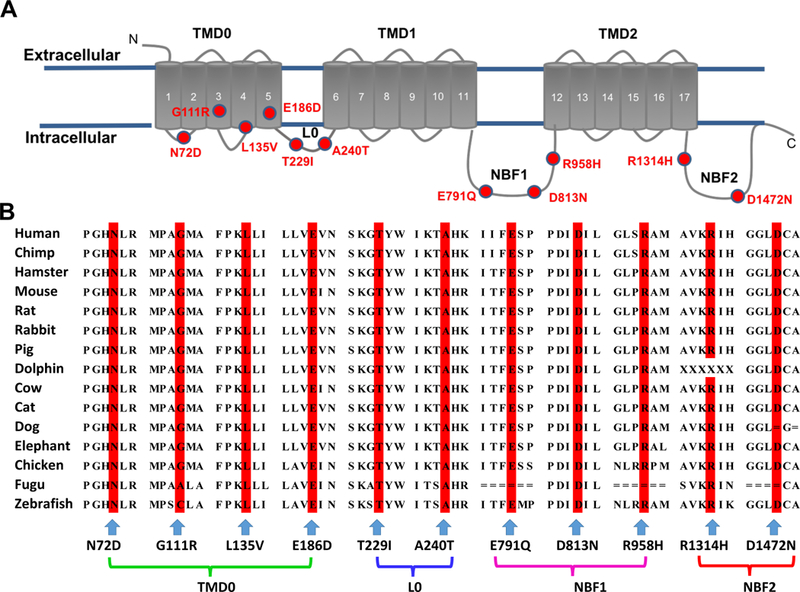
Topologic analysis of the SUR1 protein encoded by ABCC8, and sequence alignment of ABCC8 across species. Panel A shows the topology of the SUR1 protein. The 17 transmembrane segments are grouped into transmembrane domains (TMD): TMD0, TMD1, and TMD2. The two nucleotide-binding fold domains (NBF1 & 2) are indicated. Variants N72D, G111R, L135V, and E186D are located in TMD0; T229I and A240T are located in the cytoplasmic loop, L0; E791Q, D813N, and R958H are located in NBF1; R1314H and D1472N are located in NBF2. The position of each mutation is indicated by a red circle. Panel B shows the alignment of human ABCC8-encoding SUR1 protein with 14 different species, demonstrating conservation across species of each amino acid found mutated in this study.
All individuals were heterozygous for these rare ABCC8 variants. The L135V, D813N, and R1314H variants were inherited from an unaffected father and represented PAH risk variants in childhood-onset PAH, while E791Q and D1472N were inherited from an unaffected mother and represented PAH risk variants in childhood-onset PAH. The ABCC8 p.R1314H carrier also had a TBX4 c.1106delC: p.S369fs mutation. None of the other ABCC8 carriers had mutations in any known PAH genes. Other predicted deleterious variants throughout the genome carried by the ABCC8 discovery cohort are listed in Supplementary Table S1. Seven of the probands were female and five were male. None of the probands had any evidence of hyperinsulinism or transient/permanent neonatal diabetes mellitus although one adult patient had type 2 diabetes. Six of the patients were children at the time of diagnosis. Three patients responded and four patients did not respond to acute vasodilator testing using inhaled nitric oxide during cardiac catheterization. Two patient had evidence of a cardiac arrhythmia (Table 1).
To estimate the genetic effect size of ABCC8 variants, we selected 33,369 European adult subjects from Exome Aggregation Consortium (ExAC) and 49,630 European ancestry subjects from the Regeneron-Geisinger DiscovEHR study independently as controls,13 and tested for an excess of rare (minor allele frequency ≤0.1%) predicted deleterious missense variants in cases compared to controls. There were six rare predicted deleterious alleles in 150 PAH cases of European ancestry in the CU-PAH cohort, while 158 unique deleterious variants were observed a total of 295 times in 33,369 controls with exome sequencing in the ExAC dataset and 165 rare unique deleterious variants were observed 712 times in the DiscovEHR study. With binomial tests, we observed significant excess of rare predicted deleterious missense variants in ABCC8 in CU-PAH cases when comparing to ExAC controls (P-value = 0.0023, Enrichment rate = 4.5) and to DiscovEHR controls (P-value = 0.022, Enrichment rate = 2.8). We tested the association of predicted benign ABCC8 variants and identified two rare synonymous alleles in cases, 223 unique predicted benign missense variants or synonymous variants observed a total of 512 times in controls, and found no significant difference between the CU-PAH group and ExAC (P-value = 1; relative risk = 0.87).
ABCC8 expression in human lung
ABCC8 encodes the sulfonylurea receptor 1 (SUR1) protein, a regulatory subunit of the KATP channel, which associates with the pore-forming Kir6.2 subunit.14 SUR1 controls cell excitability by regulating trafficking and expression of the KATP channel, and confers sensitivity of KATP channels to magnesium-nucleotides and pharmacological modulators. SUR1-dependent KATP channels are prominent in neuronal and pancreatic tissues, but present in many other tissues, including cardiac atria.14–16 We demonstrate that ABCC8 is expressed in lungs of patients with PAH and in healthy individuals (Supplementary Figure S1), providing a potential target for influencing and modulating PAH. We replicated our finding of ABCC8 gene expression in a second set of lung samples from heritable PAH patients with known BMPR2 mutations (HPAH) and healthy controls. We observed a significant increase in ABCC8 expression in lungs of BMPR2-associated HPAH patient samples (Supplementary Figure S2).
To determine the cell types expressing SUR1 protein in lungs of idiopathic PAH (IPAH) patients, confocal microscopic analyses and triple labeling with ABCC8, CD68 and SM22 antibodies were used in paraffin-embedded lungs from six IPAH patient lung samples. Strong staining for SUR1 was found in a population of alveolar macrophages, and moderate staining for SUR1 was observed in proximal pulmonary arteries (Supplementary Figure S3).
Functional characterization of ABCC8 mutations
We examined the consequence of eight of the twelve identified ABCC8 variants putatively associated with PAH on SUR1 function using two complementary measures of KATP activity: (1) patch-clamp electrophysiology provided a direct measurement of whole cell KATP conductance in individual cells across different membrane potentials; and (2) rubidium (86Rb+) flux assays provided quantification of channel activity using 86Rb+ efflux as a measure of macroscopic KATP conductance from a population of intact cells. By co-expressing Kir6.2 with SUR1 in COS cells, functional KATP channels were formed in each assay. All SUR1 variants tested demonstrated loss of function in at least one functional assay.
First, we used patch-clamp experiments to directly measure SUR1-dependent KATP channel activity by applying a voltage ramp in whole-cell conditions (Figure.2A), using an established assay.17 We maximally activated KATP channel currents with diazoxide (100μM), a selective SUR1 activator. Once steady-state diazoxide current activation was achieved, glibenclamide (10μM) – which inhibits KATP channels by binding to the SUR subunit – was co-applied. The glibenclamide-sensitive current was taken as the SUR1-dependent KATP current.17 A series of control experiments for assay validation are shown in Supplementary Figures S4 and S5.
Figure 2.
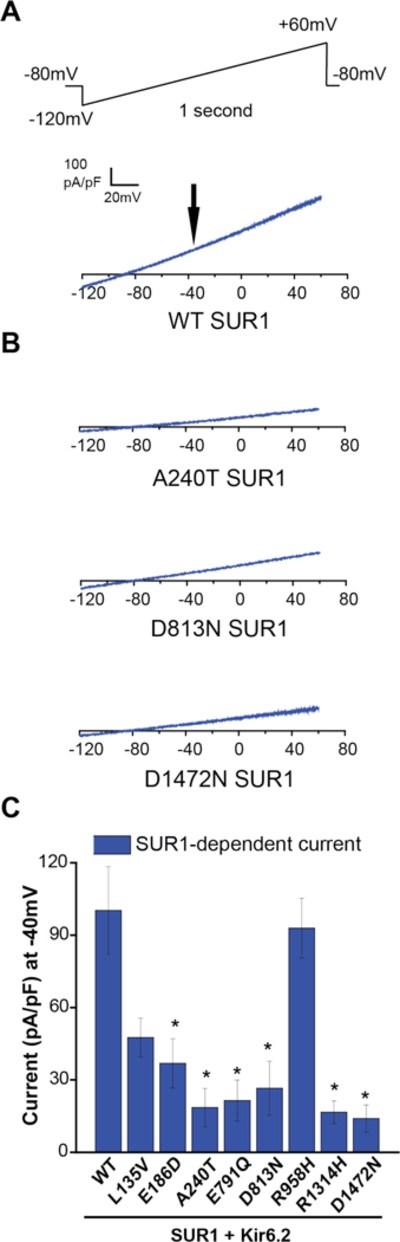
Electrophysiological consequence of SUR1 mutations on KATP (SUR1+Kir6.2) channel function. Whole-cell voltage clamp was used to measure expressed wildtype (WT) versus mutant KATP channel currents containing SUR1+Kir6.2 in COS7 cells. Panel A shows a wildtype SUR1-dependent KATP current trace. A voltage ramp from −120mV to +60mV over 1 second was applied every 3 seconds, from a −80mV holding potential. For all sample current traces, the vertical scale is 100 pA/pF, and the horizontal scale is 20mV. Panel B shows SUR1-dependent current traces of mutant KATP channels containing A240T, D813N, or D1472N SUR1 as indicated. Panel C summarizes SUR1-dependent KATP current densities (pA/pF) for the eight SUR1 mutants evaluated and wildtype, measured at −40mV (indicated by the black arrow in Panel A); 8 to 30 cells were studied per condition. Data are shown as means; T bars indicate standard errors. Asterisks indicate P<0.05 for the comparison between wildtype SUR1 and each mutant, as calculated by a one-way ANOVA and post-hoc Tukey test.
Robust SUR1-dependent KATP currents were measured in cells expressing Kir6.2 and wildtype SUR1 (Figure 2B). By contrast, currents were much smaller in cells expressing A240T SUR1, one of the novel SUR1 variants identified. Similarly small currents were observed in cells expressing D813N or D1472N SUR1, previously reported mutations in congenital hyperinsulinism (Figure 2B). Further analysis demonstrated significantly reduced SUR1-dependent currents in six of eight SUR1 mutants functionally evaluated (E186D, A240T, E791Q, D813N, R1314H, and D1472N), and non-significant reductions in L135V and R958H (Figure 2C).
Next, 86Rb+ efflux rate was recorded as a measure of KATP channel activity in cells expressing wildtype or mutant SUR1 along with Kir6.2 (Figure 3), in basal metabolic conditions and in metabolic inhibition. Compared to wildtype SUR1, basal conditions yielded marked decreases in efflux rate for the A240T, D813N, and D1472N variants, and smaller decreases for L135V, E791Q, R958H, and R1314H [Figure 3A and 3B; Supplementary Table S1]. In metabolic inhibition (extracellular solution supplemented with 2-deoxy-D-glucose and oligomycin to impair ATP synthesis and relieve KATP channels from inhibition by intracellular nucleotides), the flux rates for the L135V, A240T, D813N, R958H and D1472N mutants were markedly lower than wildtype (Figure 3C and 3D; Supplementary Table S2). The E791Q and R1314H mutant fluxes were slightly reduced, while no decrease in flux under basal or metabolic inhibition conditions was observed for E186D.
Figure 3.
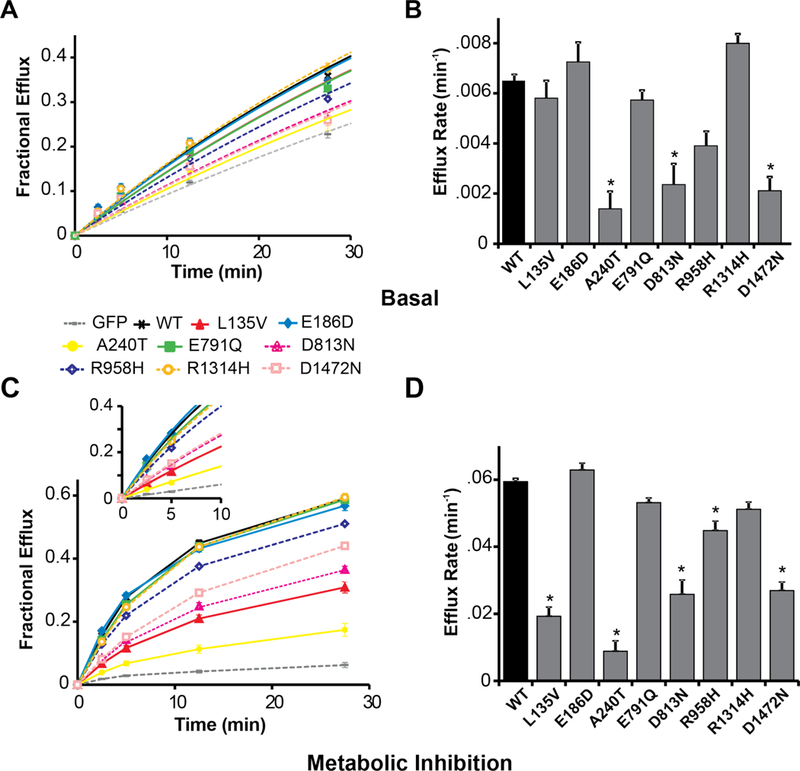
Functional impact of SUR1 mutations on macroscopic KATP (SUR1+Kir6.2) channel activity. 86Rb+ efflux was measured over time from COSm6 cells expressing KATP channels containing SUR1+Kir6.2. Panel A shows basal efflux for wildtype (WT, black curve) versus mutant (colored curves) SUR1-containing KATP channels, and GFP-alone (gray curve). Panel B displays the mean rate constants for KATP-dependent 86Rb+ efflux under basal conditions. Panel C shows efflux from cells exposed to solution containing oligomycin and 2-deoxy-D-glucose to induce metabolic inhibition of cells, thereby relieving KATP channels from intracellular inhibition by ATP. Wildtype versus mutant SUR1-containing KATP channels, and GFP-alone, are compared. The inset shows exponential fits to early time points which were used to derive the efflux rate constants (see Supplementary Table S3). Panel D shows the mean rate constants for KATP-dependent 86Rb+ efflux under metabolic inhibition conditions. For each condition, 7 to 10 cell populations were studied. Data are shown as means; T bars indicate standard errors. Asterisks indicate P<0.05 for the comparison between wildtype SUR1 and each mutant, as calculated by a one-way ANOVA and post-hoc Tukey test.
Thus, when combining results from the whole-cell patch clamp and rubidium flux functional assays, there was a significant decrease in basal and/or maximal channel activity for all SUR1 mutants associated with PAH that were functionally tested. SUR1 and KATP loss of function could result from various mechanisms, but any channel activity present might be augmented by selective potassium channel opening drugs, such as diazoxide. Consistent with this suggestion, all mutants tested were pharmacologically activated by diazoxide (100μM) in rubidium efflux (Figure 4A and 4B; Supplementary Table S3), and whole-cell patch clamp (Figure 4C and 4D; Supplementary Figure S4-F).
Figure 4.
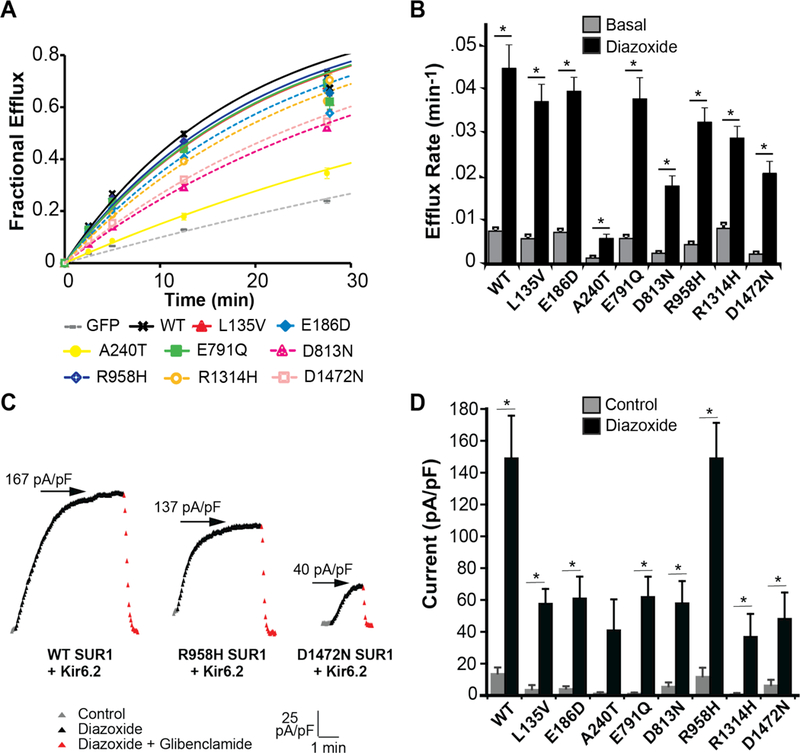
Pharmacological recovery of mutant KATP (SUR1+Kir6.2) channels. Diazoxide restores function of KATP channels (SUR1/Kir6.2) containing mutant SUR1. Panel A shows rubidium efflux in the presence of diazoxide 100μM for wildtype (WT) SUR1 (black curve), mutant SUR1-containing KATP channels (colored curves), and GFP-alone (gray curve). Panel B shows average efflux rates for wildtype and mutant KATP channels in basal (gray) versus diazoxide 100μM (black) conditions. For each condition, 7 to 9 cell populations were studied. Panel C depicts whole-cell drug time courses of wildtype and selected mutant KATP channel currents with varying degrees of pharmacological activation. Time course depicts before drug application (gray, control), during diazoxide 100μM application (black), and during co-application of glibenclamide 10μM with diazoxide 100μM (red). The vertical scale is 25 pA/pF, and the horizontal scale is 1 minute. Arrows indicate the maximal steady-state current-density (pA/pF) achieved during diazoxide 100μM application. Panel D summarizes current density (pA/pF at −40mV) for wildtype and each mutant SUR1-containing KATP channel, in control (gray) and diazoxide 100μM (black) conditions; 6 to 30 cells were studied per condition. Data are shown as means; T bars indicate standard errors. Asterisks denote P<0.05 for the comparison of basal and diazoxide (Panel B), or control and diazoxide (Panel D), calculated by the paired Student’s t-test.
DISCUSSION
Using exome sequencing, we identified de novo and inherited heterozygous mutations in a novel candidate gene, ABCC8, potentially associated with idiopathic, familial, and congenital heart disease associated PAH, in eight independent families, with six of the probands diagnosed as children in the CU-PAH study. We identified an additional four rare or novel predicted damaging missense and splice variants in ABCC8 in a second cohort (UK-PAH) of adult group I PAH patients with idiopathic, familial, congenital heart disease-associated or appetite drug-associated disease. The mutations are incompletely penetrant similar to most other genes for PAH, and penetrance may depend on additional genetic or environmental modifiers. Functional analyses demonstrated reduced ATP-sensitive potassium (KATP) channel activity in all SUR1 mutants tested and pharmacological rescue of KATP activity in vitro by diazoxide.
ABCC8 encodes the sulfonylurea receptor 1 (SUR1) protein, a KATP channel subunit. The eleven identified missense ABCC8 variants are all rare, located at residues highly conserved across species, and reside in intracellular and transmembrane domains of SUR1, including nucleotide binding fold regions (Figure 1). ABCC8 is highly expressed in the human brain and endocrine pancreas, and moderately expressed in human lungs18 (Supplementary Figures S1 and S2). SUR1 expression has been observed in intact rat pulmonary arteries,19 while KATP channel activity was shown to be upregulated by elevated shear stress in pulmonary vascular endothelial cells.20 More recently, SUR1 upregulation by hypoxia was reported in cerebral microvascular endothelial cells.21, 22 As we have observed upregulation of ABCC8 in BMPR2-associated heritable PAH patient samples (Supplementary Figure S2), as well as SUR1 protein in both alveolar macrophages and proximal pulmonary arteries within the lung (Supplementary Figure S3), further studies may elucidate SUR1’s primary physiologic role in the pulmonary vasculature, and how exactly its dysfunction and subsequent reduction in KATP currents contribute to PAH in some patients.
KCNK3, established as the first potassium channelopathy in PAH,4, 5, 23 is also regulated by hypoxia in pulmonary artery smooth muscle cells, and may contribute to hypoxic pulmonary vasoconstriction.24 In the lung, SUR1-dependent KATP channel loss of function alongside KCNK3 channel loss of function represent possible pathogenic mechanisms in PAH, and pharmacologic recovery of channel function a therapeutic avenue.4, 5, 23 Moreover, heteromeric channel assembly of SUR1 and KCNK3 with related channel subunits is well documented for both KATP and KCNK3 in various organs.14, 25, 26 This complementary and redundant potassium channel activity could contribute to the lung-specific phenotype observed clinically in patients with heterozygous ABCC8 or KCNK3 mutation.5
Despite loss of ABCC8 function underlying many cases of congenital hyperinsulinism, the patients in our study have no evidence of hyperinsulinemic hypoglycemia or transient/permanent neonatal diabetes mellitus. This raises the question: why do SUR1-dependent PAH patients not have hyperinsulinism, and vice versa, why do hyperinsulinism patients not have evidence of PAH? Ultimately, a combination of genetic, developmental, and environmental factors may determine which patients with ABCC8 mutations develop PAH.
The mechanism of SUR1 loss of function likely varies based on mutation location with the channel subunit. For instance, G111R and D1472N have been previously shown to decrease SUR1 trafficking to the plasma membrane,7, 12, 27 while nucleotide binding fold mutations, D813N and R958H, may impair magnesium-nucleotide activation. SUR1 mutation severity impacts viability for pharmacological rescue, as previously demonstrated for KCNK3 mutant channels associated with PAH,4, 5 and for SUR1 mutants associated with congenital hyperinsulinism.28 Alongside pharmacological activation of SUR1-containing KATP channels, ascertaining the mechanism of loss of function of all SUR1 variants in our study has important implications for disease pathogenesis and the therapeutic potential of KATP activation in PAH. Understanding mechanism of dysfunction may be accomplished by screening for SUR1 defects in trafficking, gene expression, regulation by nucleotides, and post-translational modifications.
As mainstay treatment in congenital hyperinsulinism, diazoxide administration overcomes disease-causing ABCC8 loss-of-function mutations. We observed variable functional recovery in vitro by diazoxide of each SUR1 mutant tested in our study of PAH patients with ABCC8 mutations. Diazoxide is a SUR1 activator clinically employed as an anti-hypertensive and anti-hyperinsulinism agent. Case reports from many years ago described the successful use of diazoxide to reverse pulmonary hypertension; 29, 30 however, hypoglycemic infants treated with diazoxide have developed pulmonary hypertension.31 This may be secondary to inadequate diuresis with diazoxide treatment, leading to volume overload following systemic blood volume expansion. Until diazoxide is proven to be safe, we do not recommend diazoxide as a treatment for pulmonary hypertension. While SUR1-dependent KATP activation is an intriguing potential basis for pulmonary hypertension therapy,32 ultimately, KATP channel activators with less pulmonary toxicity may prove useful for pulmonary hypertension treatment.33
In conclusion, we have identified mutations in the ABCC8 gene as a potential second potassium channelopathy in PAH, and as a possible therapeutic target.
Supplementary Material
ACKNOWLEDGMENTS:
We thank the patients and their families for their generous contribution. Robyn Barst and Jane Morse were critical members of the team to enroll and clinically characterize patients. Patricia Lanzano provided oversight of the Columbia biorepository. Anthony Marcketta provided bioinformatics support. Jenny Rao assisted with molecular biology and cell culture preparations. Charles Stanley provided helpful discussion. The authors thank Ly Tu and Raphaël Thuillet for their assistance with confocal image processing and analysis. The following research nurses, study coordinators and data managers provided invaluable support to the NIHR BioResource – Rare Diseases PAH study: Amanda Creaser-Myers, Rosa DaCosta, Margaret Day, Natalie Dormand, Dipa Ghedia, Alan Greenhalgh, Mark Grover, Anna Huis in’t Veld, Val Irvine, Fiona Kennedy, Shokri Othman, Alice Parker, Gary Polwarth, Stephen Roney, Gwen Schotte, Debbie Shipley, Della Stokes, Yvonne Tan, Sara Walker, Bath Respiratory Nurse Specialists.
FUNDING SOURCES: Funding support was provided by NHLBI HL060056 (to WKC), NHLBI HL129656 (to MSB), and HL45742 (to CGN). Funding for the Pulmonary Hypertension Breakthrough Initiative (PHBI is provided under an NHLBI R24 grant, #R24HL123767, and by the Cardiovascular Medical Research and Education Fund (CMREF). Additional funding was provided by a Medical Research Council Experimental Challenge Award MR/K020919/1 (to NWM); a British Heart Foundation Special Project Grant SP/12/12/29836 (to NWM); the National Institute for Health Research BioResource – Rare Diseases, and the National Institute for Health Research/Wellcome Trust Imperial Clinical Research Facility, at Imperial College Healthcare NHS Trust, London, UK.
Footnotes
DISCLOSURES: None.
REFERENCES:
- 1.International PPHC et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–4. [DOI] [PubMed] [Google Scholar]
- 2.Thomson JR, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37:741–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peacock AJ, et al. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–9. [DOI] [PubMed] [Google Scholar]
- 4.Ma L, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369:351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohnen MS, et al. The Impact of Heterozygous KCNK3 Mutations Associated With Pulmonary Arterial Hypertension on Channel Function and Pharmacological Recovery. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellanne-Chantelot C, et al. ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. J Med Genet. 2010;47:752–9. [DOI] [PubMed] [Google Scholar]
- 7.Park SE, et al. Characterization of ABCC8 and KCNJ11 gene mutations and phenotypes in Korean patients with congenital hyperinsulinism. Eur J Endocrinol. 2011;164:919–26. [DOI] [PubMed] [Google Scholar]
- 8.Chandran S, et al. Paternally inherited ABCC8 mutation causing diffuse congenital hyperinsulinism. Endocrinol Diabetes Metab Case Rep. 2013;2013:130041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Q, et al. Neonatal diabetes caused by mutations in sulfonylurea receptor 1: interplay between expression and Mg-nucleotide gating defects of ATP-sensitive potassium channels. J Clin Endocrinol Metab. 2010;95:E473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi NQ, et al. Function and distribution of the SUR isoforms and splice variants. J Mol Cell Cardiol. 2005;39:51–60. [DOI] [PubMed] [Google Scholar]
- 11.Ellard S, et al. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am J Hum Genet. 2007;81:375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tornovsky S, et al. Hyperinsulinism of infancy: novel ABCC8 and KCNJ11 mutations and evidence for additional locus heterogeneity. J Clin Endocrinol Metab. 2004;89:6224–34. [DOI] [PubMed] [Google Scholar]
- 13.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–6. [DOI] [PubMed] [Google Scholar]
- 15.Ashcroft FM. From molecule to malady. Nature. 2006;440:440–7. [DOI] [PubMed] [Google Scholar]
- 16.Flagg TP, et al. Arrhythmia susceptibility and premature death in transgenic mice overexpressing both SUR1 and Kir6.2[DeltaN30,K185Q] in the heart. Am J Physiol Heart Circ Physiol. 2007;293:H836–45. [DOI] [PubMed] [Google Scholar]
- 17.Nessa A, et al. Molecular mechanisms of congenital hyperinsulinism due to autosomal dominant mutations in ABCC8. Hum Mol Genet. 2015;24:5142–53. [DOI] [PubMed] [Google Scholar]
- 18.Babenko AP, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–66. [DOI] [PubMed] [Google Scholar]
- 19.Cui Y, et al. The molecular composition of K(ATP) channels in human pulmonary artery smooth muscle cells and their modulation by growth. Am J Respir Cell Mol Biol. 2002;26:135–43. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee S, et al. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am J Physiol Cell Physiol. 2003;285:C959–67. [DOI] [PubMed] [Google Scholar]
- 21.Woo SK, et al. Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J Cereb Blood Flow Metab. 2012;32:525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simard JM, et al. Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest. 2007;117:2105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antigny F, et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation. 2016;133:1371–85. [DOI] [PubMed] [Google Scholar]
- 24.Olschewski A, et al. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res. 2006;98:1072–80. [DOI] [PubMed] [Google Scholar]
- 25.Czirjak G, et al. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J Biol Chem. 2002;277:5426–32. [DOI] [PubMed] [Google Scholar]
- 26.Enyedi P, et al. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559–605. [DOI] [PubMed] [Google Scholar]
- 27.Muzyamba M, et al. Complex ABCC8 DNA variations in congenital hyperinsulinism: lessons from functional studies. Clin Endocrinol (Oxf). 2007;67:115–24. [DOI] [PubMed] [Google Scholar]
- 28.Martin GM, et al. Pharmacological Correction of Trafficking Defects in ATP-sensitive Potassium Channels Caused by Sulfonylurea Receptor 1 Mutations. J Biol Chem. 2016;291:21971–21983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klinke WP, et al. Diazoxide in primary pulmonary hypertension. N Engl J Med. 1980;302:91–2. [DOI] [PubMed] [Google Scholar]
- 30.Chan NS, et al. Reversibility of primary pulmonary hypertension during six years of treatment with oral diazoxide. Br Heart J. 1987;57:207–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yildizdas D, et al. Pulmonary hypertension, heart failure and neutropenia due to diazoxide therapy. Adv Ther. 2008;25:515–9. [DOI] [PubMed] [Google Scholar]
- 32.Adi A, et al. Screening for Mutations in ABCC8 and KCNJ11 Genes in Saudi Persistent Hyperinsulinemic Hypoglycemia of Infancy (PHHI) Patients. Genes (Basel). 2015;6:206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kharade SV, et al. The shifting landscape of KATP channelopathies and the need for ‘sharper’ therapeutics. Future Med Chem. 2016;8:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.