

Abstract
Amplification of the MYCN oncogene predicts treatment resistance in childhood neuroblastoma. We used a MYC target gene signature that predicts poor neuroblastoma prognosis to identify the histone chaperone, FAcilitates Chromatin Transcription (FACT), as a crucial mediator of the MYC signal and a therapeutic target in the disease. FACT and MYCN expression created a forward feedback loop in neuroblastoma cells that was essential for maintaining mutual high expression. FACT inhibition by the small molecule curaxin compound, CBL0137, markedly reduced tumor initiation and progression in vivo. CBL0137 exhibited strong synergy with standard chemotherapy by blocking repair of DNA damage caused by genotoxic drugs, thus creating a synthetic lethal environment in MYCN-amplified neuroblastoma cells and suggesting a treatment strategy for MYCN-driven neuroblastoma.
One Sentence Summary:
Histone chaperone FACT acts in a positive feedback loop with MYCN and is a therapeutic target in neuroblastoma.
Introduction
Neuroblastoma is a pediatric cancer of the sympathetic nervous system, which often presents at a clinically advanced stage with primary or acquired resistance to conventional chemotherapy (1, 2). Amplification of the c-MYC homolog, MYCN, is observed in ~15–20% of patients (3, 4), and this feature strongly correlates with poor clinical outcome (1, 2). Direct evidence of MYCN as an oncogene is highlighted by the TH-MYCN mouse model, in which a MYCN transgene driven by the tyrosine hydroxylase (TH) promoter is sufficient to recapitulate many of the features of human neuroblastoma (5). In addition to MYCN amplification being an effective biomarker for poor prognosis in high risk neuroblastoma patients, it has also been reported that increased activation of MYC transcriptional target genes predicts poor prognosis in clinical stage 4 patients, independently of MYCN amplification (6). Paradoxically, these patients had very low expression of MYCN, suggesting that c-MYC, the expression of which is inversely correlated to MYCN, acts in a functionally redundant manner for MYC-directed transcription (6). Thus, targeting genes downstream of MYC proteins may be an alternative therapeutic strategy for high risk neuroblastoma patients.
Here we used a prognostic MYC target gene signature to identify the histone chaperone, FAcilitates Chromatin Transcription (FACT), as a promising therapeutic target in neuroblastoma. We found that FACT and MYCN expression act in a positive feedback regulatory loop in neuroblastoma cells, are strongly correlated during tumor initiation and result in a remarkable susceptibility of neuroblastoma cells to the FACT inhibitor, CBL0137. Moreover, due to the role of FACT in DNA repair, CBL0137 created a synthetic lethal environment when combined with genotoxic chemotherapy, suggesting a promising treatment approach for this aggressive childhood malignancy.
Results
A MYC target gene signature identifies histone chaperone FACT as a prognostic marker in neuroblastoma
To identify MYC target genes as therapeutic targets in neuroblastoma, we examined the expression of a previously reported MYC core target gene signature (7) in neuroblastoma tumors derived from patients at primary diagnosis. We conducted unsupervised hierarchical clustering on 649 neuroblastoma patients (4) and as expected, this identified marked upregulation in a cluster that corresponded to MYCN-amplified patients (MYCN-amplified cluster), but also high expression in some MYCN non-amplified tumors (MYC activation cluster) (Fig. 1A). The MYC activation cluster was associated with indicators of poor prognosis, including older patient age (>18 months) or advanced clinical stage 3 or 4, and the MYC target signature as a whole strongly predicted poor overall and event free survival (Fig. 1A-B, fig. S1A). Next we examined each gene member of the MYC activation signature by Cox proportional hazards modeling for prognostic significance, either by i) univariate analysis, ii) multivariate analysis considering MYCN amplification status, or iii) multivariate analysis considering MYCN amplification status, patient age, and stage (table S1). Eleven of 51 genes were found to be significant predictors of poor prognosis in all three analyses (table S1). Among these 11 candidates, we sought a MYC-regulated target with a clinically relevant and strong chemical inhibitor and pre-clinical/clinical evidence for a role in cancer (table S2). Suppressor of Ty 16 (SUPT16H, hereafter referred to as SPT16) was the only gene that conformed to these criteria. Together with Structure Specific Recognition Protein 1 (SSRP1), SPT16 comprises the histone chaperone heterodimer complex FACT (8, 9), which has been identified as a potential therapeutic target in a number of cancer models (10–12). Curaxin class compounds, including lead compound, CBL0137, are potent inhibitors of FACT that deplete free FACT from the soluble fraction of the nucleus, functionally reducing FACT regulation of chromatin (12). CBL0137 exhibits potent anti-tumor efficacy and minimal toxicity in experimental cancer models (12) and is currently being tested in early phase clinical trials (http://clinicaltrials.gov/show/NCT01905228).
Fig. 1: FACT predicts poor prognosis in neuroblastoma patients and is associated with MYC signaling.
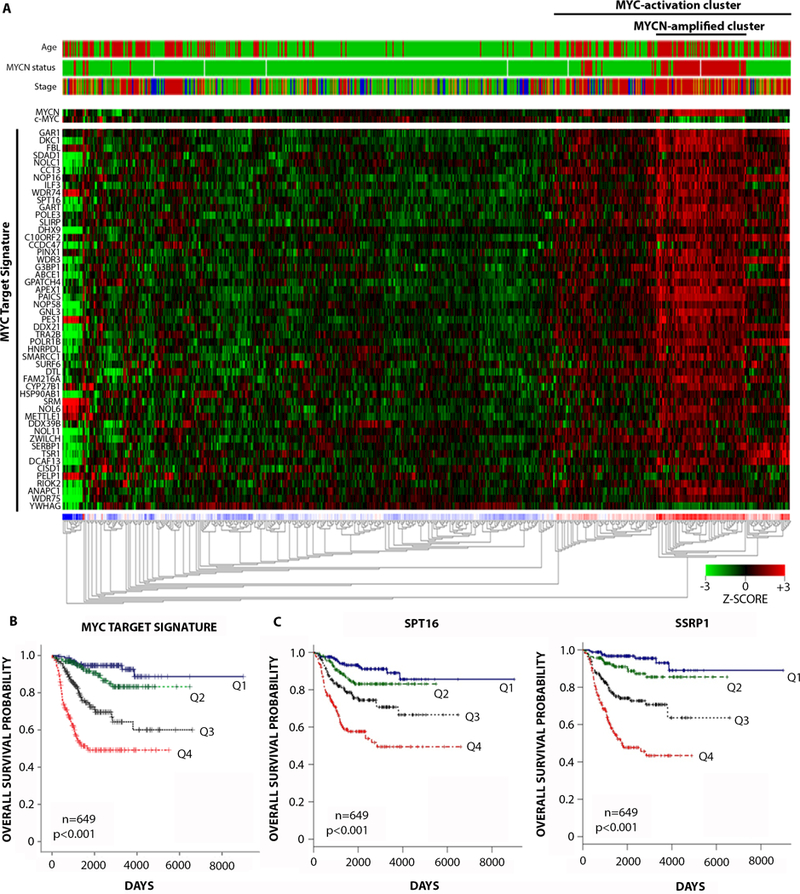
(A) Unsupervised hierarchical clustering on 649 neuroblastoma patients according to a 51-gene MYC target signature (7). Clinical parameters (top) were included as follows: patient age (>18 months: red, <18 months: green), MYCN amplification status (amplified: red, non-amplified: green), International Neuroblastoma Staging System (INSS) (Stage 1+2: green, Stage 3: orange, Stage 4: red, Stage 4S: blue). Clustering was performed according to Euclidean distance using the R2 Microarray Analysis and visualization platform (http://r2.amc.nl). (B) Kaplan Meier plots for overall survival of 649 neuroblastoma patients stratified by expression quartiles of the MYC target signature. MYC target signature expression quartiles were generated by averaging the expression of all 51 gene members of the MYC signature per patient and ranking lowest to highest: Q1, 0–25th patient percentile, Q2, 25th-50th patient percentile, Q3, 50th-75th patient percentile, Q4, 75th-100th patient’s percentile. P-value: pairwise log-rank tests on Q1 vs Q4, Q2 vs Q4, or Q3 vs Q4. See table S7 for individual p-values. (C) Kaplan Meier plots for overall survival of 649 neuroblastoma patients stratified by expression quartiles of SPT16 (left) and SSRP1 (right) as for (B) above.
SPT16 and SSRP1 are highly dependent on each other for regulation of expression and function (13), so next we examined the expression of SSRP1 as well as SPT16 in neuroblastoma tumors. Both FACT subunits were associated with poor overall and event-free survival in two cohorts of primary neuroblastoma patients (4, 14)(Fig. 1C, fig. S1B-C). Using Cox proportional hazards modeling, high mRNA expression of SSRP1 and SPT16 were found to be strong and independent predictors of poor prognosis by univariate analysis or when considering MYCN amplification status alone or combined with patient age and stage (table S3A-B). Moreover, SPT16 and SSRP1 expression in the 649 patient tumor cohort showed a strong association with clinical parameters of poor prognosis (fig. S1D-E), as well as a correlation with MYCN expression or average MYC target gene expression (fig. S1F-G).
Next we examined whether high SSRP1 protein expression was similarly associated with poor clinical outcome. We used a tissue microarray of 47 primary untreated neuroblastomas with representative prognostic indicators for neuroblastoma (fig. S1H), and we found that high SSRP1 protein expression was a strong predictor of poor prognosis (fig. S1I). Using semi-quantitative scoring, high SSRP1 expression was associated with MYCN amplification, but this comparison did not reach statistical significance (fig. S1J-K). To confirm that SSRP1 protein expression was higher in MYCN-amplified tumors, we used quantitative western blotting on 12 primary neuroblastoma tumors and showed markedly higher amounts of SSRP1 in the MYCN-amplified tumors (fig. S1L-M). Moreover, high amounts of SSRP1 and SPT16 protein correlated with high MYCN or c-MYC protein expression in cultured neuroblastoma cell lines (fig. S1N-Q).
FACT and MYCN expression are controlled in a forward feedback loop
Because SPT16 is a direct transcriptional target of c-MYC in fibrosarcoma cell lines (7), we hypothesized that MYCN transcriptionally regulates FACT expression in neuroblastoma cells. Consistent with this prediction, siRNA knockdown of MYCN decreased SPT16 and SSRP1 mRNA and protein expression in MYCN-amplified BE(2)C and KELLY neuroblastoma cell lines (Fig. 2A-B). Both SPT16 and SSRP1 gene promoters contain a MYC E-box transactivation motif ~500 base pairs upstream of their transcriptional start sites (TSS). We used chromatin immunoprecipitation assays to demonstrate enrichment of MYCN at the E-box motif of both the SPT16 and SSRP1 core promoters (fig. S2A-C). Moreover, siRNA knockdown of MYCN reduced binding at these sites, suggesting that MYCN can transcriptionally upregulate both FACT components. Additionally, we found that this was a general feature of MYC proteins, because c-MYC siRNAs also resulted in markedly reduced protein expression of both FACT subunits in MYCN non-amplified neuroblastoma cells (fig. S2D).
Fig. 2: FACT and MYCN act in a transcriptional positive feedback loop.
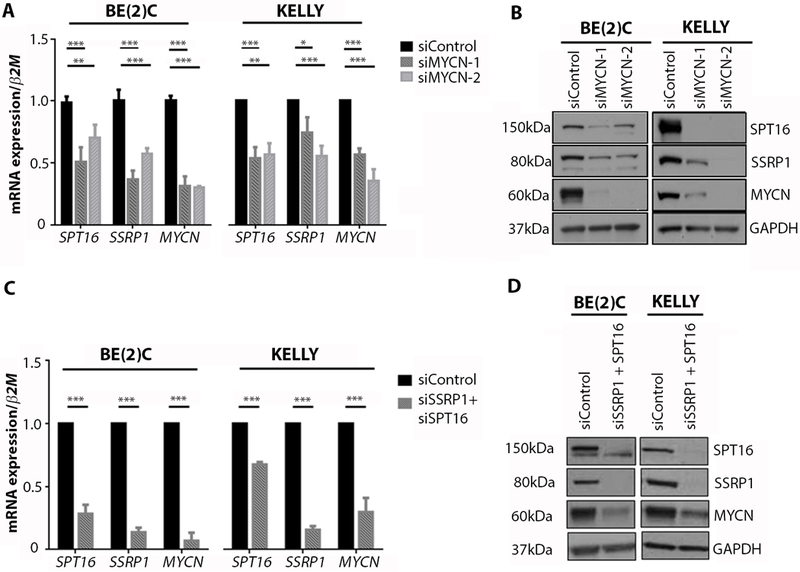
(A) Mean mRNA expression for SPT16, SSRP1, or MYCN in BE(2)C and KELLY cells treated with control, MYCN-1 or MYCN-2 siRNA. Data displayed were obtained 48 hours after transfection and were normalized to mRNA expression in a control siRNA sample. β2-Microglobulin (β2M) was used as a reference gene. *, p<0.05; **, p<0.01; ***, p<0.001. See table S7 for individual p-values. (B) Western blots for SPT16, SSRP1, and MYCN protein expression in BE(2)C cells and KELLY cells treated with control, MYCN-1, or MYCN-2 siRNA. Data displayed were obtained 48 hours after transfection. GAPDH was used as a loading control. (C) Mean mRNA expression for SPT16, SSRP1, or MYCN in BE(2)C and KELLY cells treated with control or combined SSRP1 and SPT16 siRNA. Data displayed were obtained 72 hours after transfection and normalized to mRNA expression in a control siRNA sample. β2M was used as a reference gene. ***, p<0.001. See table S7 for individual p-values. (D) Western blots for SPT16, SSRP1, and MYCN protein expression in BE(2)C cells and KELLY cells treated with control or combined SSRP1 and SPT16 siRNA. Data displayed were obtained 72 hours after transfection. GAPDH was used as a loading control.
FACT regulates nucleosome structure to promote transcriptional initiation and elongation (8, 9). Because FACT is enriched at the c-MYC promoter in HT1080 fibrocarcinoma cells (10), we hypothesized that FACT may also enhance the transcription of MYCN in neuroblastoma cells, completing a forward feedback loop. Indeed, we found that siRNA knockdown of SSRP1 and SPT16 decreased MYCN mRNA and protein expression (Fig. 2C-D, fig. S2E), whereas overexpression of SSRP1 and/or SPT16 increased MYCN mRNA and protein concentrations in neuroblastoma cells (fig. S2F-G). In addition, cycloheximide assays showed that SSRP1 or SPT16 knockdown markedly reduced MYCN protein half-life while conversely, SPT16/SSRP1 overexpression prolonged the half-life of MYCN (fig. S2H-I), suggesting that FACT regulates MYCN protein expression by two discrete mechanisms, one at the transcriptional level and the other post-translational. This was further supported by gene set enrichment analysis, which showed a strong correlation between the pattern of altered gene expression after SSRP1 knockdown and MYCN target gene signatures (fig. S2J). Taken together our data indicate a positive regulatory expression loop involving FACT and MYCN, which drives MYCN transcription and protein stability by independent mechanisms.
FACT is highly expressed in neuroblastoma precursor cells in TH-MYCN+/+ mice
We have previously shown that high MYCN expression caused postnatal persistence and hyperplasia of neuroblasts as precancerous lesions in the developing sympathetic ganglia of TH-MYCN mice (15). Immunohistochemical analyses showed that in wildtype ganglia, SSRP1 protein expression was rapidly down-regulated after birth, whereas in contrast, TH-MYCN+/+ ganglia displayed high SSRP1 expression up until tumor formation at six weeks of age (Fig. 3A, fig. S3A). SSRP1 expression was highly correlated with MYCN expression (Fig. 3A, fig. S3A-C), and the degree of MYCN and SSRP1 expression was highly correlated with the extent of neuroblast hyperplasia in ganglia (Fig. 3B, fig. S3A,D-F). These results suggest that FACT is upregulated in pre-cancerous TH-MYCN+/+ neuroblasts.
Fig. 3: CBL0137 overcomes developmental signals that drive neuroblast persistence in TH-MYCN mice.
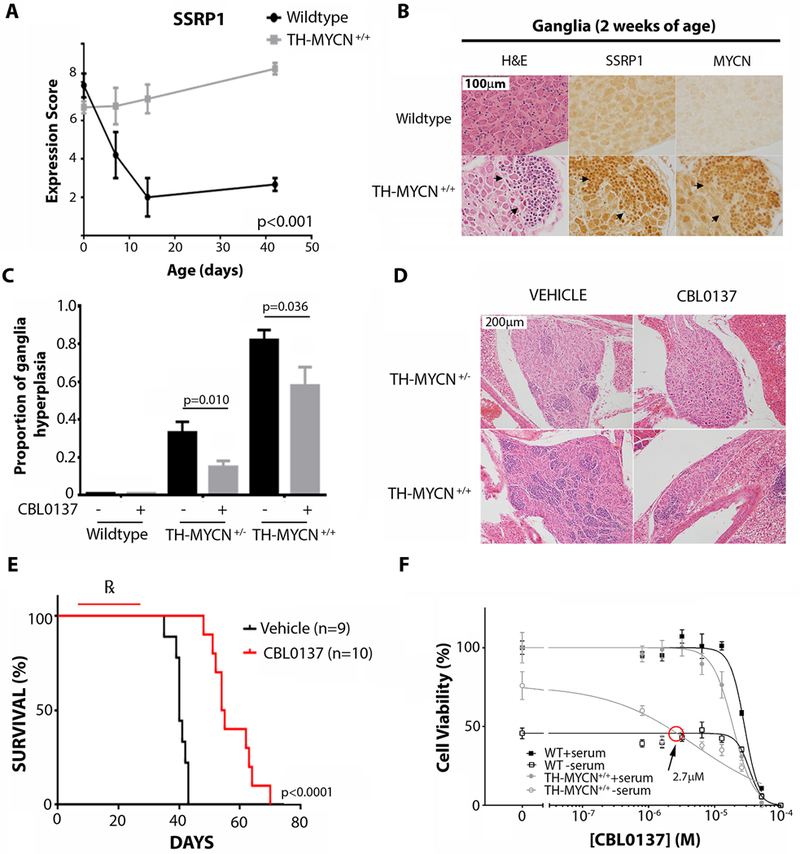
(A) Semi-quantitative histology scoring was used to determine the relative expression of SSRP1 in wildtype and TH-MYCN+/+ mice through perinatal time points as indicated. P-value was calculated using ANOVA, testing the significance of genotype as the source of variation. At least 3 independent mice had one or more ganglia present for analysis for each time point/genotype. (B) Representative immunohistochemistry images comparing serial ganglion sections of two-week old wildtype and TH-MYCN+/+ mice. Hematoxylin and eosin (H&E) staining and immunoblotting for SSRP1 and MYCN is shown. Arrowheads indicate areas of neuroblast hyperplasia. (C) 6 day old wildtype, TH-MYCN+/−, and TH-MYCN+/+ mice were treated with intraperitoneal vehicle control (0.2% methylcellulose) or CBL0137 for 5 days (10 mg/kg/day), and the proportion of ganglia with hyperplastic neuroblasts was evaluated by histological examination. Ganglia were considered to be hyperplastic if >30 focal neuroblasts were present per ganglion. Data displayed indicate the average proportion of hyperplastic ganglia per mouse ± standard error. Samples sizes were as follows: wildtype/vehicle=6, wildtype/CBL0137=4, TH-MYCN+/−/vehicle=13, TH-MYCN+/−/CBL0137=13, TH-MYCN+/+/vehicle=9, TH-MYCN+/+/CBL0137=9. (D) Representative H&E images of ganglion histology from mice treated as in (C). (E) 6 day old TH-MYCN+/+ mice were treated with intraperitoneal vehicle control (0.2% methylcellulose x 5 days/week) or CBL0137 for 3 weeks (10 mg/kg/day x 5 days/week) and thereafter assessed for tumor growth. A Kaplan Meier plot is displayed for vehicle or CBL0137 treated mice considering time to maximum tumor burden (when tumors were 10 mm in diameter by palpation). ℞ indicates treatment period. Log rank test was used to calculate statistical significance between CBL0137 and vehicle treated tumors. (F) Primary sympathetic ganglia were isolated and cultured from 2 week old TH-MYCN+/+ or wildtype age-matched controls. Ganglia cultures were treated with serum-rich or serum-deprived medium (1/30 normal concentration of serum) for 48 hours, as well as with various concentrations of CBL0137 for 24 hours, and viability was calculated by cell counts on βIII-tubulin positive cells for each of the TH-MYCN+/+ or wildtype cultures. Plot shows the non-linear regression line of percentage cell viability as determined by counts for βIII-tubulin positive immunofluorescence normalized to untreated/serum-rich samples for each of the TH-MYCN+/+ or wildtype cultures. Data displayed represent average cell viability ± standard error from the combined data of 3 independent biological replicates. Red circle indicates the concentration of CBL0137 that causes serum-deprived TH-MYCN+/+ cultures to have equal cell viability to serum-deprived wildtype cultures.
FACT inhibitor CBL0137 restores developmental signals in postnatally persistent neuroblasts from TH-MYCN mice
To evaluate whether FACT is required for TH-MYCN neuroblast persistence and hyperplasia, we treated perinatal TH-MYCN mice with the FACT inhibitor, CBL0137. Low dose intraperitoneal administration of CBL0137 from six days of age for a total of five days reduced the proportion of ganglia with neuroblast hyperplasia in both hemi- (TH-MYCN+/−) and homozygote (TH-MYCN+/+) mice at two weeks of age (Fig. 3C-D). Furthermore, low dose CBL0137 prophylaxis from six days of age until four weeks of age delayed subsequent tumor growth in TH-MYCN+/+ mice (Fig. 3E). Histological analysis of tumors from mice given CBL0137 prophylaxis revealed an increase in ganglion-like cells within tumors (fig. S3G), suggesting that CBL0137 promoted the differentiation of neuroblasts that had pathologically persisted postnatally. Consistent with this observation, we also demonstrated that all-trans-retinoic acid treatment of human neuroblastoma cell line, SH-SY5Y, rapidly downregulated expression of both SSRP1 and SPT16 (fig. S3H) at a time that coincided with terminal neuritic differentiation in vitro (16, 17).
We have previously shown that MYCN blocked developmental cell deletion signals in pre-malignant TH-MYCN+/+ ganglia cells after nerve growth factor (NGF) or serum withdrawal in vitro, which mirrored postnatal neuroblast persistence and hyperplasia in vivo (15, 18, 19). To evaluate the role of FACT in neuroblast death responses, we cultured primary ganglia from 2-week old TH-MYCN+/+ mice with CBL0137. TH-MYCN+/+ ganglia were more sensitive to the cytopathic effects of CBL0137 in cultures deprived of serum, compared with wildtype ganglia (Fig. 3F, fig. S3I). Moreover, when 1 week old TH-MYCN+/+ mice were treated with low dose CBL0137 for five days, primary ganglia cultures derived from treated mice also exhibited sensitivity to trophic factor withdrawal compared with vehicle-treated mice (fig. S3J). Together this suggested that CBL0137 restored normal cell deletion responses to trophic factor withdrawal, despite high MYCN expression, blocking the very earliest steps of this MYCN-driven embryonal cancer.
We have previously shown that p53 stress responses to trophic factor withdrawal are impaired in a MYCN-dependent manner in TH-MYCN+/+ ganglia, compared with wildtype ganglia (19). Consistent with this observation, gene set enrichment analysis identified suppression of gene sets related to p53 target genes in TH-MYCN+/+ mice compared with age matched wildtype mice (table S4). CBL0137 has previously been shown to activate p53, thus we evaluated whether CBL0137 could restore p53 stress responses in serum-deprived TH-MYCN+/+ ganglia. Rescue experiments with the p53 inhibitor, Pifithrin-α (PFTα) (20) and pan-caspase inhibitor OPH-Q-VD, demonstrated that CBL0137 sensitized TH-MYCN+/+ ganglia to trophic factor withdrawal in a p53-dependent manner (fig. S3K) and required activation of caspase signaling (fig. S3L). Inactivation of p53 as the mechanism of resistance to trophic withdrawal in TH-MYCN+/+ ganglia was further supported by the finding that the p53 signal activator, Nutlin3a (21), also restored the death responses to trophic factor withdrawal in a p53-dependent, pro-apoptotic manner (fig. S3M-P).
FACT inhibition is an effective therapeutic strategy for MYCN-driven neuroblastoma
Next we examined the impact of FACT inhibition on neuroblastoma cell viability. FACT siRNAs reduced cell viability of BE(2)C and SH-SY5Y cells, neuroblastoma cells with high expression of MYCN or c-MYC respectively (fig. S4A-B, fig. S1N). Moreover, FACT siRNAs inhibited MYCN protein expression, as well as colony number increases in SHEP.MYCN3 cells, a doxycycline-inducible neuroblastoma cell model of MYCN overexpression (fig. S4C-D). CBL0137 was highly toxic to a panel of seven human neuroblastoma cell lines compared with normal fibroblasts and was particularly potent against neuroblastoma cell lines with high c-MYC or MYCN protein expression (Fig. 4A, fig. S4E, fig. S1N). Consistent with this observation, CBL0137 markedly decreased colony-forming capacity of doxycycline-treated SHEP.MYCN3 cells, as well as MYCN mRNA, MYCN protein, and MYCN binding to the promoter of FACT subunits in BE(2)C and KELLY cells (Fig. 4B-D, fig. S4F).
Fig. 4: High MYCN expression sensitizes neuroblastoma cells to CBL0137.
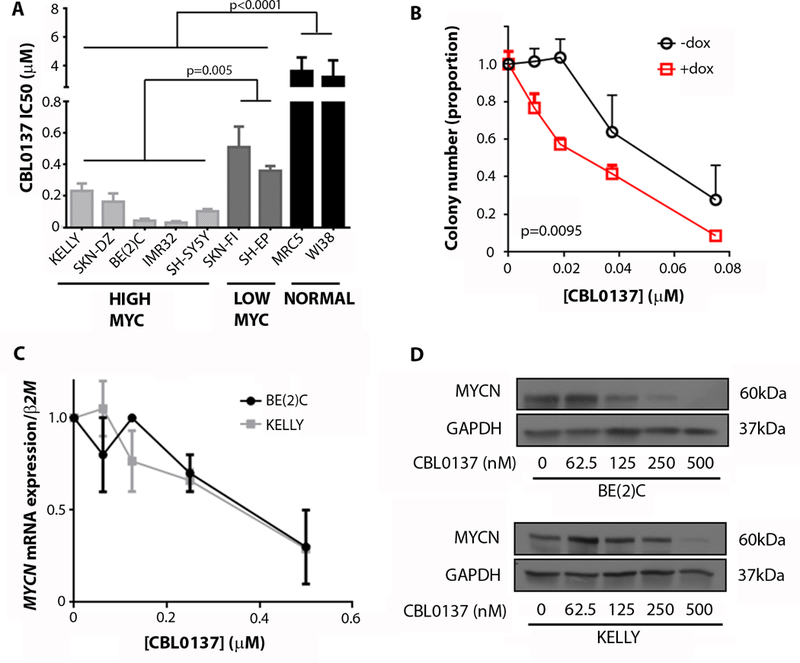
(A) CBL0137 inhibitory concentration 50 (IC50) was determined from resazurin reduction assays on a panel of neuroblastoma cell lines and primary lung fibroblast controls. Cells were classified as high MYC expressing and low MYC expressing cell lines on the basis of their MYCN or c-MYC expression as displayed in fig. S1N. Data displayed are 72 hours after treatment. IC50 was determined using non-linear regression analysis from at least 3 independent biological replicates. Average IC50 ± standard error is shown. (B) Colony assays were conducted for doxycycline (dox) inducible MYCN overexpressing SHEP.MYCN3 cells (51) treated with an escalating dose of CBL0137. The graph shows the average number of colonies at each concentration of drug from 3 independent biological replicates + standard error. Extra sum of squares F-test was used to determine statistical significance between non-linear regression of +dox (+MYCN) vs –dox (-MYCN) cells. (C) MYCN mRNA expression in BE(2)C and KELLY cells treated with increasing concentrations of CBL0137. Data displayed are mean normalized mRNA expression from 3 replicate experiments ± standard error. Data obtained 48 hours after treatment. β2-Microglobulin (β2M) was used as a reference gene. (D) Western blot for MYCN protein expression in BE(2)C and KELLY cells treated with increasing concentrations of CBL0137. Data displayed are 48 hours after treatment. GAPDH was used as a loading control.
We have previously shown that CBL0137 exhibited promising anti-tumor efficacy in multiple pre-clinical cancer models (12), so next we examined CBL0137 in vivo against established neuroblastoma in TH-MYCN+/+ mice. Intravenous (i.v) administration of CBL0137 (60 mg/kg every 4 days for 8 doses) to 6-week old TH-MYCN+/+ mice with advanced neuroblastoma delayed tumor growth, with the majority of mice exhibiting long term tumor regression (Fig. 5A, fig. S5A). Oral CBL0137 (20 mg/kg, 5 doses weekly for 4 weeks) and low dose i.v CBL0137 (40 mg/kg every 4 days for 8 doses) were less effective but still prolonged time until maximum tumor burden (Fig. 5A, fig. S5A). We next examined the histology of tumors derived from TH-MYCN+/+ mice after short term CBL0137 treatment either as an oral (20 mg/kg for 5 days) or i.v (20, 40, or 60 mg/kg for 1 day) formulation. We found that tumor tissue demonstrated a marked treatment effect, particularly for the 60 mg/kg i.v. treated group, characterized by extensive necrosis, hemorrhage, and apoptosis compared to controls (Fig. 5B, fig. S5B, table S5–6). Moreover, i.v 60 mg/kg CBL0137 treated mice exhibited evidence of metastatic regression with no mice demonstrating pulmonary metastases, whereas vehicle and low dose CBL0137 treated mice demonstrated marked metastatic spread to the lungs (fig. S5C). There was a substantial accumulation of CBL0137 in treated tumors compared to other organs/body fluids, with 60 mg/kg i.v CBL0137-treated animals showing the highest concentrations of intratumoral CBL0137 (fig. S5D). Tumor concentrations of CBL0137 also showed a strong inverse correlation with tumor viability (Fig. 5C, fig. S5E). SSRP1, SPT16, and MYCN protein expression was decreased in i.v 60 mg/kg CBL0137-treated tumors compared to both oral CBL0137 and vehicle treatment (Fig. 5D). Moreover, i.v administration of CBL0137 to BALB/c nude mice bearing flank xenografts of the human neuroblastoma cell line, BE(2)C, resulted in delayed tumor growth and decreased MYCN protein expression (fig. S5F-G). Altogether this suggests that the inhibitory effect of CBL0137 on FACT has a downstream negative impact on MYCN expression and results in a marked therapeutic selectivity for MYCN-driven neuroblastoma tissues in vitro and in vivo.
Fig. 5: CBL0137 cytotoxicity in TH-MYCN+/+ tumors is associated with inhibition of the FACT/MYCN positive feedback loop.
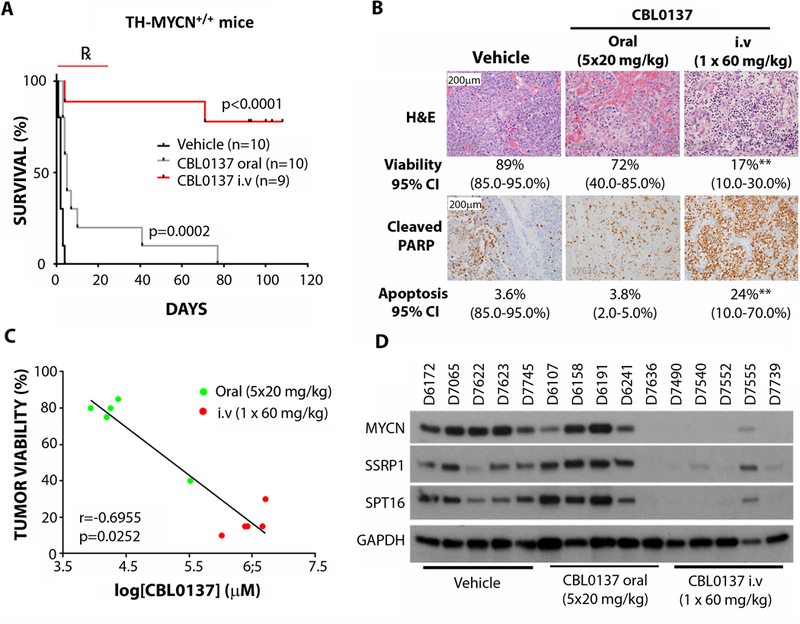
(A) Kaplan Meier plot for survival of 6 week old tumor-bearing TH-MYCN+/+ mice treated with vehicle (5% dextrose given intravenously), oral CBL0137 (20 mg/kg, 5 days/week for 4 weeks) or intravenous CBL0137 (60 mg/kg, once every 4 days for 4 weeks). Log-rank test was used to determine statistical significance between both CBL0137-treated groups, compared to vehicle. Mice began treatment when medium sized (~5 mm diameter) palpable tumors were detected. Endpoint was when tumors reached 10 mm by palpation. ℞ indicates treatment period. (B) Representative images for H&E staining or cleaved PARP (cPARP) in TH-MYCN+/+ tumors treated with oral CBL0137 (5 days at 20 mg/kg/day) or CBL0137 i.v. (1day at 60 mg/kg/day). Quantitation of tumor viability or cPARP positive cells and 95% confidence interval (CI) is displayed for each treatment group. Statistical significance was determined using Mann-Whitney test. **; p<0.01. See table S7 for individual p-values. (C) Correlation of CBL0137 tissue concentration and tumor viability in CBL0137-treated tumors treated as in (B). P-value and R-value determined using Spearman’s rank correlation coefficient. (D) Western blot for MYCN, SSRP1, and SPT16 protein expression in individual TH-MYCN+/+ tumors treated as in (B). GAPDH was used as a loading control. Tumor identifiers are indicated above each lane.
CBL0137 exhibits synergy with DNA-damaging cytotoxic chemotherapy
Knockdown of SSRP1 has been previously identified to sensitize ovarian and breast cancer cells to cytotoxic chemotherapy (22), suggesting that CBL0137 may enhance neuroblastoma chemotherapy. We used colony formation and cell viability assays in neuroblastoma cells to demonstrate that CBL0137 synergizes with mafosfamide (the active metabolite of cyclophosphamide) and that this synergy was exclusive to neuroblastoma cells in comparison with normal primary fibroblasts (Fig. 6A, fig. S6A). The combination of CBL0137 and mafosfamide was far more toxic in neuroblastoma cells compared to normal controls, indicating that a wide therapeutic index exists for this combination treatment (fig. S6B). CBL0137 combination synergy extended to several other chemotherapeutics used in neuroblastoma therapy, albeit with some exceptions (fig. S6C-D). Consistent with these findings, cyclophosphamide, etoposide, cisplatin, and vincristine each potently increased CBL0137 efficacy in vivo against established TH-MYCN+/+ tumors (Fig. 6B). Moreover, when i.v CBL0137 was combined with clinically relevant chemotherapy regimens used to treat high-risk/refractory neuroblastoma patients, namely cyclophosphamide/topotecan and irinotecan/temozolomide, strong tumor delay was observed in both the TH-MYCN+/+ transgenic mice (Fig. 6C) and BALB/c nude mice harboring BE(2)C xenografts (fig. S6E). Combination therapies were mostly well tolerated in recipient mice, with CBL0137/vincristine a notable exception. Some occasions of weight loss were observed and mice were removed from study as follows: cisplatin (2/10 mice), cisplatin/CBL0137 (2/10 mice), vincristine/CBL0137 (5/10 mice) and CBL0137/irinotecan/temozolomide (2/10 mice).
Fig. 6: CBL0137 synergizes with neuroblastoma chemotherapy.
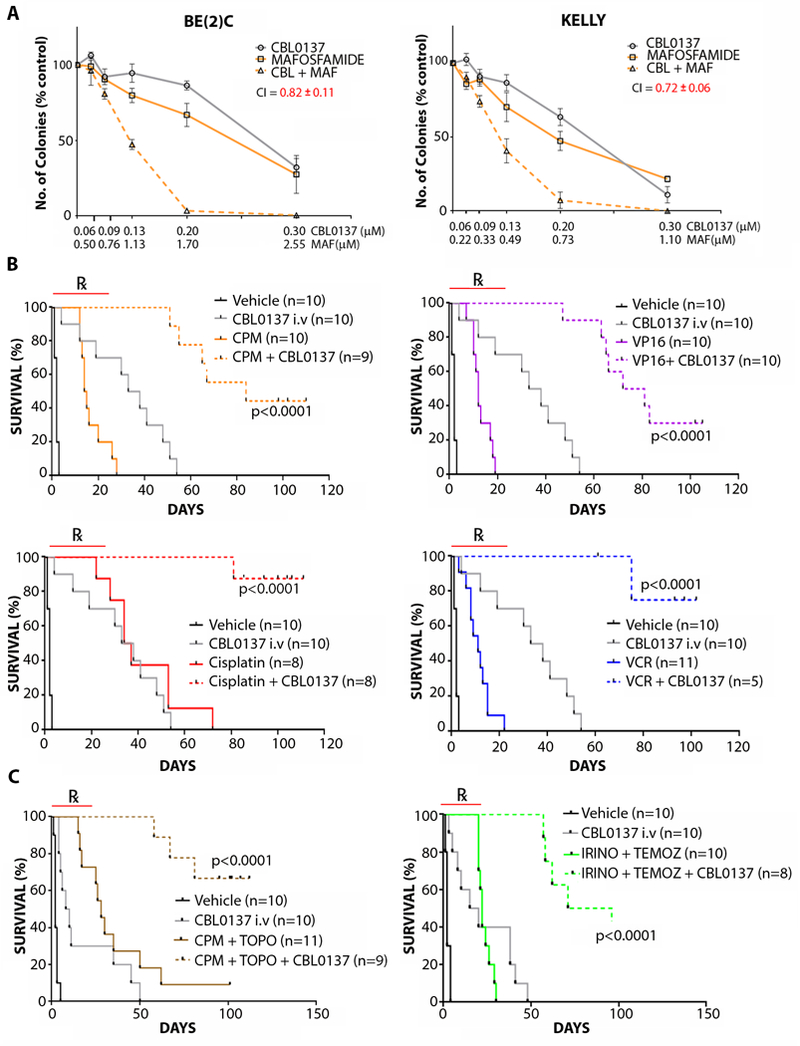
(A) Colony-forming assays were performed for BE(2)C and KELLY cells treated with CBL0137 (CBL) in combination with mafosfamide (MAF), the active metabolite of cyclophosphamide. Combination Index (CI) at a fractional cell kill of 0.80 is shown using the Chou and Talalay method (52). CI<0.9 = synergistic. Data are displayed as the average number of colonies at each concentration of drugs from 3 independent biological replicates ± standard error. (B) Kaplan Meier plots of tumor-bearing TH-MYCN+/+ mice treated with vehicle (5% dextrose) or i.v. 40 mg/kg CBL0137 (once every 4 days for 4 weeks) combined with 15 mg/kg/day cyclophosphamide (CPM), 6 mg/kg/day etoposide (VP16), 2 mg/kg/day cisplatin, or 0.2 mg/kg/day vincristine (VCR). Mice were treated starting at 6 weeks of age, when 5 mm palpable tumors were detected. End point was at 10 mm tumor diameter by palpation. CPM, VP16, cisplatin, and VCR were all administered intraperitoneally for 5 consecutive days when treatment began. P-value was determined using pairwise log-rank test for CBL0137/chemotherapy combinations vs their respective single treatment controls. ℞ indicates treatment period. (C) Kaplan Meier plots of tumor-bearing TH-MYCN+/+ mice treated with vehicle (5% dextrose) or intravenous 40 mg/kg CBL0137 (once every 4 days for 4 weeks) combined with 10 mg/kg/day CPM and 0.5 mg/kg/day topotecan (topo) (left) or 2 mg/kg/day irinotecan (irino) and 5 mg/kg/day temolozomide (temoz) (right). Mice were treated at 6 weeks of age, starting when 5 mm palpable tumors were detected. End point was at 10 mm tumor diameter by palpation. CPM, topo, irino, and temoz were all administered intraperitoneally for 5 consecutive days when treatment began. P-value was determined using pairwise log-rank test for CBL0137/chemotherapy combinations vs their respective treatment controls. ℞ indicates treatment period.
FACT was previously implicated in cisplatin resistance as part of a DNA repair complex containing DNA-PK (22), and FACT is well established to have roles in DNA repair (8, 23, 24). Thus, we reasoned that the synergy exhibited by CBL0137 in combination with genotoxic chemotherapy may be dependent on inhibiting FACT during DNA repair. Thus, we next examined the extent of DNA repair when CBL0137 was added to cells after they were treated with the genotoxic agents, cisplatin or etoposide. CBL0137 inhibited DNA double strand break repair, which otherwise occurred after withdrawal of either cisplatin or etoposide, despite the fact that CBL0137 exhibited no genotoxicity alone (Fig. 7A). Moreover, CBL0137 resulted in a marked increase in DNA damage markers, γH2AX and 53BP1, in the presence of etoposide (Fig. 7B-C). These findings were recapitulated when inhibition of DNA repair by CBL0137 was evaluated after hydroxyurea treatment, but not when CBL0137 was added after vincristine (fig. S7A-B), suggesting that the synergistic action of CBL0137 was specific to direct DNA-damaging agents and not microtubule poisons.
Fig. 7: CBL0137 prevents DNA damage repair after genotoxic chemotherapy.
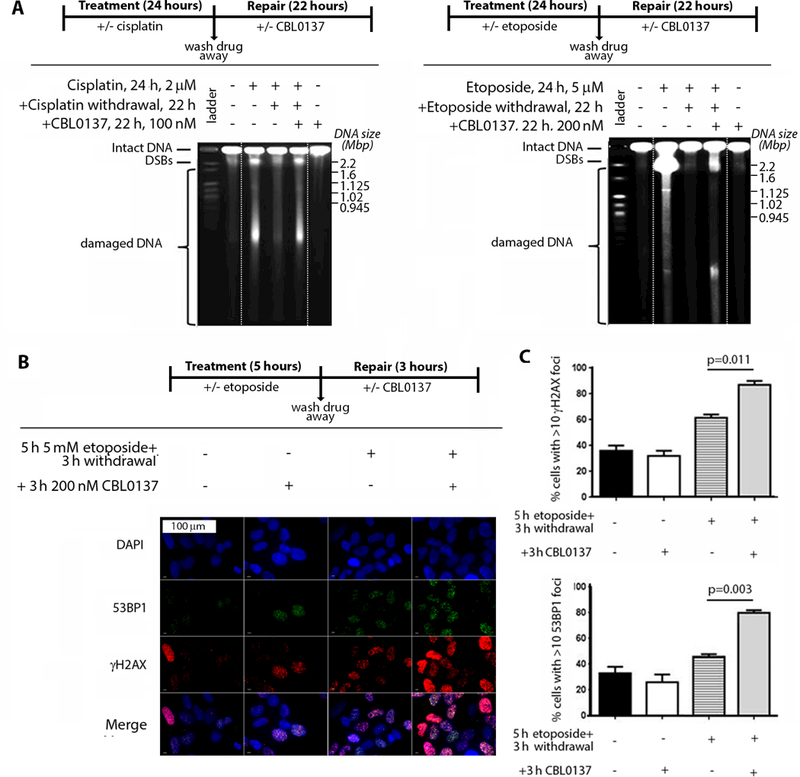
(A) BE(2)C cells were treated for 24 hours with or without the following chemotherapeutics: 2 μM cisplatin (left) or 5 μM etoposide (right). Chemotherapeutics were subsequently removed, and medium was supplemented with or without 100 nM CBL0137 for 22 hours (refer to schematic above gel for the administration schedule). The repair of chemotherapy-induced DNA double-stranded breaks (DSBs) in the presence or absence of CBL0137 was determined using pulsed-field gel electrophoresis. Data displayed are representative of 2 independent biological replicates. Dashed white lines indicate areas where gel was cropped for presentation. (B) BE(2)C cells were treated with or without 5 μM of etoposide for 5 hours. Etoposide was subsequently removed and medium was supplemented with or without 200 nM of CBL0137 for 3 hours (refer to schematic above the images for the administration schedule). Cells were then fixed and processed for immunofluorescence with anti-γH2AX (red) and anti-53BP1 (green), as well as DAPI DNA stain (blue). (C) From (B), quantitation of the percentage of cells with >10 positive immunofluorescent foci for DNA damage markers γH2AX (top) or 53BP1 (bottom). Data are displayed as the average percentage of positive cells ± standard error from 2 independent biological replicates.
Discussion
Pathologic epigenetic regulation of transcription by abnormal chemical modification of histone tails and DNA methylation has been extensively studied in human cancer (25). However, less is known about the role of nucleosome remodelers, such as the histone chaperone proteins in cancer (26). Here we used a MYC target gene expression signature to identify the H2A/H2B histone chaperone FACT as a key driver and treatment target in the MYCN-driven pediatric cancer, neuroblastoma. FACT and MYCN expression acted in a forward feedback loop, and chemical inhibition of FACT impaired neuroblastoma initiation, suggesting that FACT and MYCN may together inhibit developmentally-timed maturation signals in TH-MYCN+/+ mice. This oncogenic cooperation between MYCN and FACT created a synthetic lethality when conventional DNA-damaging chemotherapy was administered with the FACT inhibitor CBL0137, providing the mechanistic basis for a combination anti-cancer therapy approach.
Aberrant activity of nucleosome remodeling proteins has been observed in pediatric cancer. Most evidence suggests that these proteins more frequently act as tumor suppressors, as evidenced by the loss-of-function mutations in SWI/SNF complexes in malignant rhabdoid tumors (27) and genomic alterations of the H3.3 chaperone, ATRX, in neuroblastoma (14) and pediatric glioma (28). In contrast, our data support a gain-of-function model whereby FACT is present in pathologically high amounts in cancer tissues. Although there are other examples of similar “oncogenic” histone chaperones such as DEK (29) and CHAF1A (30), this study identifies an oncogenic histone chaperone that has a potent and effective chemical inhibitor available for clinical use.
FACT is normally expressed in embryonal or primitive tissues and is thereafter down-regulated during tissue differentiation and in mature tissues, in a manner similar to MYCN (11, 13, 31). We showed a strong correlation between MYCN and FACT expression in normal sympathetic ganglia, including high expression in normal developmental neuroblasts, the putative cell of origin of neuroblastoma (15, 18). SSRP1 knockout mice are embryonic lethal before E5.5 (32), so later developmental roles are unknown, but this expression pattern suggests that FACT may function in physiological regulation of sympathoadrenal development. Our data show that FACT expression is also correlated with MYCN at tumor initiation and chemical inhibition of FACT delayed neuroblastoma onset. This suggests that FACT may have functions in neuroblastoma tumor initiation and progression. This is consistent with the observation that aberrant expression or activity of developmental regulators are key determinants of neuroblastoma tumorigenesis (18). Animal models with conditional knockout of SSRP1 or SPT16 will reveal more specific roles of FACT in sympathoadrenal development and by association, MYCN-driven tumorigenesis.
FACT controls nucleosome ‘eviction’ and ‘reassembly’ of histones, thus promoting the smooth passage of RNA polymerase II during gene transcription (8, 9, 33). Our previous data have demonstrated that SSRP1 is enriched at the promoter regions of oncogene transcription factors and their respective transcriptional targets (10). We show here that FACT maintains high concentrations of MYCN protein in neuroblastoma cells by driving MYCN transcription and protein stability. Moreover, FACT is a functional co-factor to oncogenic mutant H-RasV12 in malignant transformation of mammary epithelial cells (10). These observations support a model whereby FACT drives both malignant transcription and oncogenic transcription factor activity, yet the mechanism that governs FACT transcriptional specificity in cancer cells remains unclear. It may simply be that inappropriate expression of FACT in normal cell types is sufficient to collaborate with other oncogenic transcription factors to drive a gene program that favors malignant transformation and transcription.
Our results demonstrate that FACT can enhance MYCN transcription and protein stability in neuroblastoma cells, even those with multiple copies of MYCN and an already high MYCN expression. Although MYCN amplification is a common feature of human neuroblastoma, recent studies have indicated that mechanisms in addition to gene amplification are required to maintain the very high MYCN protein concentrations necessary for the fully malignant phenotype (34–37). Collectively, these findings are consistent with observations made in tumorigenic murine Myc models showing that different thresholds of MYC expression result in different components of the malignant phenotype, and suggest that MYCN expression and protein stability are important treatment targets (38).
For neuroblastoma patients with advanced disease, prognosis is still poor (1, 2). Many of these patients’ tumors are characterized by MYCN amplification or over-activation of MYC target genes (6). We show here that CBL0137 was highly effective against established primary tumors and pulmonary metastases in TH-MYCN+/+ mice and that CBL0137 treatment was accompanied by marked MYCN depletion in tumors after only a single intravenous dose, suggesting that targeting FACT may be an important strategy to decrease MYCN expression and overcome primary and metastatic disease (37, 39–41). This treatment approach is particularly promising because the expression of MYCN and FACT are markedly higher in malignant compared with normal tissues (10, 11, 15). Accordingly, our data showed that CBL0137 inhibition of FACT and MYCN was associated with extensive cytotoxicity in advanced TH-MYCN+/+ tumors but exhibited minimal toxicity, consistent with findings in other animal models of cancer (12). Moreover, the previously described inhibitory activity of CBL0137 against NFκB transcriptional targeting and activation of p53 by casein kinase dependent serine 32 phosphorylation (12) makes CBL0137 a potential multi-modal cancer therapeutic for a variety of malignancies characterized by defects within these pathways.
FACT has been shown to promote DNA damage repair in the face of DNA-damaging chemotherapy (22), suggesting that it may act as a chemo-resistance factor. Here we showed that CBL0137 was particularly effective as a combination therapy with chemotherapeutic agents. In addition, combining CBL0137 with chemotherapy regimens currently used after relapse potentiated its activity, highlighting a potential therapeutic strategy for high risk neuroblastoma patients. Although our data and those of others have shown that CBL0137 alone is not genotoxic (12), inhibition of FACT created a synthetic lethal environment in the presence of cisplatin and etoposide but not vincristine. Moreover, combinations of CBL0137 with DNA-damaging agents such as cisplatin, cyclophosphamide, and etoposide were better tolerated in our mouse models than the non-genotoxic microtubule inhibitor vincristine, suggesting that the most effective role for CBL0137 in anti-cancer therapy will be co-administration with primary DNA-damaging agents. We also observed that p53 status was not a determinant of CBL0137 combination therapy sensitivity, consistent with previous findings for CBL0137 as a single agent in adult cancer types (12). Considering that genotoxic chemotherapy is associated with marked non-specific toxicity in patients, targeting FACT with the non-genotoxic CBL0137 promises to enhance cancer cell specificity of these agents, which are the current mainstay of neuroblastoma therapy.
Future studies will need to address some of the limitations of this study. First, it will be necessary to unveil the specific mechanism by which FACT modifies MYCN expression at both a transcriptional and post-translational level. Second, we still lack in-depth understanding of the mechanism and consequences of CBL0137-induced changes in chromatin, in particular how this relates to inhibition of FACT-dependent transcriptional programs and to specific DNA-repair pathways that are affected. Detailed analyses of CBL0137 effects on DNA-repair pathways such as homologous repair and non-homologous end joining will be required. Third, the mechanism by which CBL0137 accumulates at high concentration in neuroblastoma tumors and whether this occurs in other cancer types is unknown. In particular, mechanisms of CBL0137 uptake, metabolism, and efflux will need to be elucidated. Finally, it remains to be explored whether CBL0137 can be used as a therapy for other cancers, and whether MYC deregulation can serve as a biomarker for CBL0137 sensitivity. With these additional studies, CBL0137 or newer FACT inhibitors may serve as a therapy for neuroblastoma and potentially other MYC-driven cancers.
Materials and Methods
Detailed methods are available in the supplementary materials.
Supplementary Material
Data Supporting Figure 1
Data Supporting Figure 2
Data Supporting Figure 3
Data Supporting Figure 4
Data Supporting Figure 5
Data Supporting Figure 6
Data Supporting Figure 7
Fluorescence in situ hybridization for MYCN amplification in tissue microarray.
Cox proportional hazards modeling of MYC target signature members in 649 neuroblastoma patients.
Literature analysis of lead candidates that favor poor outcome in neuroblastoma.
Cox proportional hazards modeling of FACT subunits, SSRP1 and SPT16 in 649 neuroblastoma patients.
Gene set enrichment analysis on 2 week old TH-MYCN+/+ ganglia compared with age matched wild type mice
Histology of TH-MYCN+/+ mice treated with oral or i.v CBL0137.
Histology of TH-MYCN+/+ mice treated with a dose escalating regimen of i.v CBL0137.
P-values not shown in figures
Acknowledgments
We thank Robyn Lukeis and Mary Suter at the Department of Cytogenetics (SydPath), St Vincents Hospital, Darlinghurst, NSW, Australia for histological assistance with the tissue microarray. We also thank Dr Jason Shohet (Baylor College of Medicine) for kindly providing the SHEP.MYCN3 cells used in this study.
Funding: This work was supported by Program Grants from the NHMRC Australia (grant numbers APP1016699 and APP1085411); Cancer Institute NSW (grant number 10/TPG/1–03); Cancer Council NSW (grant numbers PG-11–06), Australian Rotary Health / Rotary Club of Adelaide, Fund for Scientific Research Flanders (FWO), German Cancer Aid (grant no. 110122), German Ministry of Science and Education (BMBF) as part of the e:Med initiative (grant nos. 01ZX1303A and 01ZX1307D), and Fördergesellschaft Kinderkrebs-Neuroblastom-Forschung e.V.
Competing interests: This work was funded in part by grants to K.V.G., A.V.G., M.D.N. and M.H from Incuron, LLC. K.V.G. and A.V.G. are co-inventors on patents describing CBL0137. M.D.N. and M.H. own some stock in Cleveland Biolabs, which is entitled to a portion of the royalties for sale of Incuron’s product, CBL0137.
Footnotes
The other authors declare that they have no competing interests.
Data and materials availability: Gene expression data for neuroblastoma tumors (n=649) are available at the Gene Expression Omnibus under accession #GSE45547. All other gene expression data are available at the Gene Expression Omnibus under accession #GSE71062. CBL0137 is available under MTA from Incuron, LLC.
References and Notes:
- 1.Brodeur GM, Neuroblastoma: Biological insights into a clinical enigma. Nature Reviews Cancer 3, 203–216 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Maris JM, Recent advances in neuroblastoma. The New England journal of medicine 362, 2202–2211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M, Trent J, Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 305, 245–248 (1983). [DOI] [PubMed] [Google Scholar]
- 4.Kocak H, Ackermann S, Hero B, Kahlert Y, Oberthuer A, Juraeva D, Roels F, Theissen J, Westermann F, Deubzer H, Ehemann V, Brors B, Odenthal M, Berthold F, Fischer M, Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell death & disease 4, e586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM, Targeted expression of MYCN causes neuroblastoma in transgenic mice. Embo J.. 16, 2985–2995 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westermann F, Muth D, Benner A, Bauer T, Henrich KO, Oberthuer A, Brors B, Beissbarth T, Vandesompele J, Pattyn F, Hero B, Konig R, Fischer M, Schwab M, Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol 9, R150 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, Cleveland JL, Dang CV, Zeller KI, Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One 6, e26057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Formosa T, The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta 1819, 247–255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reinberg D, Sims RJ 3rd, de FACTo nucleosome dynamics. The Journal of biological chemistry 281, 23297–23301 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Garcia H, Miecznikowski JC, Safina A, Commane M, Ruusulehto A, Kilpinen S, Leach RW, Attwood K, Li Y, Degan S, Omilian AR, Guryanova O, Papantonopoulou O, Wang J, Buck M, Liu S, Morrison C, Gurova KV, Facilitates chromatin transcription complex is an “accelerator” of tumor transformation and potential marker and target of aggressive cancers. Cell reports 4, 159–173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia H, Fleyshman D, Kolesnikova K, Safina A, Commane M, Paszkiewicz G, Omelian A, Morrison C, Gurova K, Expression of FACT in mammalian tissues suggests its role in maintaining of undifferentiated state of cells. Oncotarget 2, 783–796 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA, Pal S, Safina A, Sviridov S, Koman IE, Veith J, Komar AA, Gudkov AV, Gurova KV, Curaxins: anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci Transl Med 3, 95–74 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Safina A, Garcia H, Commane M, Guryanova O, Degan S, Kolesnikova K, Gurova KV, Complex mutual regulation of facilitates chromatin transcription (FACT) subunits on both mRNA and protein levels in human cells. Cell cycle 12, 2423–2434 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, Schild L, Molenaar P, Stroeken P, van Noesel MM, Ora I, Santo EE, Caron HN, Westerhout EM, Versteeg R, Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–U107 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Hansford LM, Thomas WD, Keating JM, Burkhart CA, Peaston AE, Norris MD, Haber M, Armati PJ, Weiss WA, Marshall GM, Mechanisms of embryonal tumor initiation: Distinct roles for MycN expression and MYCN amplification. Proc. Natl. Acad. Sci. U. S. A 101, 12664–12669 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishida Y, Adati N, Ozawa R, Maeda A, Sakaki Y, Takeda T, Identification and classification of genes regulated by phosphatidylinositol 3-kinase- and TRKB-mediated signalling pathways during neuronal differentiation in two subtypes of the human neuroblastoma cell line SH-SY5Y. BMC research notes 1, 95 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marshall GM, Bell JL, Koach J, Tan O, Kim P, Malyukova A, Thomas W, Sekyere EO, Liu T, Cunningham AM, Tobias V, Norris MD, Haber M, Kavallaris M, Cheung BB, TRIM16 acts as a tumour suppressor by inhibitory effects on cytoplasmic vimentin and nuclear E2F1 in neuroblastoma cells. Oncogene 29, 6172–6183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marshall GM, Carter DR, Cheung BB, Liu T, Mateos MK, Meyerowitz JG, Weiss WA, The prenatal origins of cancer. Nat Rev Cancer 14, 277–289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calao M, Sekyere EO, Cui HJ, Cheung BB, Thomas WD, Keating J, Chen JB, Raif A, Jankowski K, Davies NP, Bekkum MV, Chen B, Tan O, Ellis T, Norris MD, Haber M, Kim ES, Shohet JM, Trahair TN, Liu T, Wainwright BJ, Ding HF, Marshall GM, Direct effects of Bmi1 on p53 protein stability inactivates oncoprotein stress responses in embryonal cancer precursor cells at tumor initiation. Oncogene 32, 3616–3626 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV, A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285, 1733–1737 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Sand-Dejmek J, Adelmant G, Sobhian B, Calkins AS, Marto J, Iglehart DJ, Lazaro JB, Concordant and opposite roles of DNA-PK and the “facilitator of chromatin transcription” (FACT) in DNA repair, apoptosis and necrosis after cisplatin. Mol Cancer 10, 74 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kari V, Shchebet A, Neumann H, Johnsen SA, The H2B ubiquitin ligase RNF40 cooperates with SUPT16H to induce dynamic changes in chromatin structure during DNA double-strand break repair. Cell cycle 10, 3495–3504 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, Goodman R, Lozano G, Zhao Y, Lu H, A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Molecular cell 7, 283–292 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Baylin SB, Jones PA, A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11, 726–734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgess RJ, Zhang Z, Histone chaperones in nucleosome assembly and human disease. Nature structural & molecular biology 20, 14–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson BG, Roberts CW, SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11, 481–492 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N, Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Wise-Draper TM, Mintz-Cole RA, Morris TA, Simpson DS, Wikenheiser-Brokamp KA, Currier MA, Cripe TP, Grosveld GC, Wells SI, Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer research 69, 1792–1799 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barbieri E, De Preter K, Capasso M, Chen Z, Hsu DM, Tonini GP, Lefever S, Hicks J, Versteeg R, Pession A, Speleman F, Kim ES, Shohet JM, Histone chaperone CHAF1A inhibits differentiation and promotes aggressive neuroblastoma. Cancer research 74, 765–774 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Zimmerman KA, Yancopoulos GD, Collum RG, Smith RK, Kohl NE, Denis KA, Nau MM, Witte ON, Toran-Allerand D, Gee CE, et al. , Differential expression of myc family genes during murine development. Nature 319, 780–783 (1986). [DOI] [PubMed] [Google Scholar]
- 32.Cao S, Bendall H, Hicks GG, Nashabi A, Sakano H, Shinkai Y, Gariglio M, Oltz EM, Ruley HE, The high-mobility-group box protein SSRP1/T160 is essential for cell viability in day 3.5 mouse embryos. Molecular and cellular biology 23, 5301–5307 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D, FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92, 105–116 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Marshall GM, Liu PY, Gherardi S, Scarlett CJ, Bedalov A, Xu N, Iraci N, Valli E, Ling D, Thomas W, van Bekkum M, Sekyere E, Jankowski K, Trahair T, Mackenzie KL, Haber M, Norris MD, Biankin AV, Perini G, Liu T, SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS genetics 7, e1002135 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, Ling D, Su SP, Nelson C, Chang DK, Koach J, Tee AE, Haber M, Norris MD, Toon C, Rooman I, Xue C, Cheung BB, Kumar S, Marshall GM, Biankin AV, Liu T, The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell death and differentiation 20, 503–514 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, Berwanger B, Eilers M, Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell 15, 67–78 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, Matthay KK, Rowitch D, Weiss WA, Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer research 66, 8139–8146 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI, Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 14, 447–457 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chanthery YH, Gustafson WC, Itsara M, Persson A, Hackett CS, Grimmer M, Charron E, Yakovenko S, Kim G, Matthay KK, Weiss WA, Paracrine signaling through MYCN enhances tumor-vascular interactions in neuroblastoma. Sci Transl Med 4, 115–113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS, BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, Stegmaier K, Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer discovery 3, 308–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chesler L, Weiss WA, Genetically engineered murine models--contribution to our understanding of the genetics, molecular pathology and therapeutic targeting of neuroblastoma. Seminars in cancer biology 21, 245–255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, Osborne CK, Paik S, Perlmutter J, Rhodes A, Sasano H, Schwartz JN, Sweep FC, Taube S, Torlakovic EE, Valenstein P, Viale G, Visscher D, Wheeler T, Williams RB, Wittliff JL, Wolff AC, American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 28, 2784–2795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC, PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burkhart CA, Cheng AJ, Madafiglio J, Kavallaris M, Mili M, Marshall GM, Weiss WA, Khachigian LM, Norris MD, Haber M , Effects of MYCN antisense oligonucleotide administration on tumorigenesis in a murine model of neuroblastoma. J Natl Cancer Inst 95, 1394–1403 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Henderson MJ, Haber M, Porro A, Munoz MA, Iraci N, Xue C, Murray J, Flemming CL, Smith J, Fletcher JI, Gherardi S, Kwek CK, Russell AJ, Valli E, London WB, Buxton AB, Ashton LJ, Sartorelli AC, Cohn SL, Schwab M, Marshall GM, Perini G, Norris MD, ABCC multidrug transporters in childhood neuroblastoma: clinical and biological effects independent of cytotoxic drug efflux. J Natl Cancer Inst 103, 1236–1251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer 86, 349–363 (1999). [PubMed] [Google Scholar]
- 49.Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, Stram DO, Gerbing RB, Lukens JN, Matthay KK, Castleberry RP, The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 86, 364–372 (1999). [PubMed] [Google Scholar]
- 50.Geran R, Greenberg N, Macdonald M, Schumacher A, Abbott B, Protocols For Screening Chemical Agents and Natural Products Against Tumors and Other Biological Systems. Cancer Chemotherapy Reports 3, 47–51 (1972). [Google Scholar]
- 51.Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A, Shohet JM, The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc Natl Acad Sci U S A 102, 731–736 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chou TC, Talalay P, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation 22, 27–55 (1984). [DOI] [PubMed] [Google Scholar]
- 53.Chou TC, Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research 70, 440–446 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Issaeva N, Thomas HD, Djureinovic T, Jaspers JE, Stoimenov I, Kyle S, Pedley N, Gottipati P, Zur R, Sleeth K, Chatzakos V, Mulligan EA, Lundin C, Gubanova E, Kersbergen A, Harris AL, Sharma RA, Rottenberg S, Curtin NJ, Helleday T, 6-thioguanine selectively kills BRCA2-defective tumors and overcomes PARP inhibitor resistance. Cancer research 70, 6268–6276 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marcel V, Ghayad SE, Belin S, Therizols G, Morel AP, Solano-Gonzalez E, Vendrell JA, Hacot S, Mertani HC, Albaret MA, Bourdon JC, Jordan L, Thompson A, Tafer Y, Cong R, Bouvet P, Saurin JC, Catez F, Prats AC, Puisieux A, Diaz JJ, p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 24, 318–330 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang YC, Lu TY, Huang DY, Kuo YS, Kao CF, Yeh NH, Wu HC, Lin CT, NOLC1, an enhancer of nasopharyngeal carcinoma progression, is essential for TP53 to regulate MDM2 expression. Am J Pathol 175, 342–354 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang C, Yin C, Wang L, Zhang S, Qian Y, Ma J, Zhang Z, Xu Y, Liu S, HSPC111 governs breast cancer growth by regulating ribosomal biogenesis. Mol Cancer Res 12, 583–594 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Shamanna RA, Hoque M, Pe’ery T, Mathews MB, Induction of p53, p21 and apoptosis by silencing the NF90/NF45 complex in human papilloma virus-transformed cervical carcinoma cells. Oncogene 32, 5176–5185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou XZ, Lu KP, The Pin2/TRF1-interacting protein PinX1 is a potent telomerase inhibitor. Cell 107, 347–359 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supporting Figure 1
Data Supporting Figure 2
Data Supporting Figure 3
Data Supporting Figure 4
Data Supporting Figure 5
Data Supporting Figure 6
Data Supporting Figure 7
Fluorescence in situ hybridization for MYCN amplification in tissue microarray.
Cox proportional hazards modeling of MYC target signature members in 649 neuroblastoma patients.
Literature analysis of lead candidates that favor poor outcome in neuroblastoma.
Cox proportional hazards modeling of FACT subunits, SSRP1 and SPT16 in 649 neuroblastoma patients.
Gene set enrichment analysis on 2 week old TH-MYCN+/+ ganglia compared with age matched wild type mice
Histology of TH-MYCN+/+ mice treated with oral or i.v CBL0137.
Histology of TH-MYCN+/+ mice treated with a dose escalating regimen of i.v CBL0137.
P-values not shown in figures