

Abstract
Two aldehydes, 2,6-diacetam ido-4-formylpyridine (7) and 1-butyl-6-formyluracil (11), are used to synthesize five pyridyl and four uracyl meso-subsituted porphyrins. With these complementary porphyrin building blocks, it is possible to build various types of multi-porphyrin supramolecules with different spatial relationships in predefined geometries. The formation and properties of self complementary dimers and a closed tetrameric square are presented as a basis of comparison to the latter system in the solid state. An X-ray structure of 5,10-bis(4-tert-butylphenyl)-15,20-bis-(3,5-diacetam ido-4-pyridyl)porphyrin confirm s its molecularstru cture and reve als a hydrogen-bonded supramolecular organization mediated by water molecules.
Introduction
Self-assembly into specifically designed structures involves the spontaneous association of molecules through intermolecular interactions.1 The ability to control the supramolecular organization of molecular entities by nonconvalent interactions remains a challenging goal in materials science,1b because of our still imperfect ability to predictably design supramolecular structures in solution and in the solid state. Due to their key role in many important biological systems, the self-assembly of porphyrin derivatives has attracted considerable attention because of their photoelectric properties, their potential use as components of nanometer scale photonic devices, and as novel functional materials.2 In addition to the free bases, the functionality (luminescence, redox, electronic, etc.) of these large, rigid aromatic macrocycles ma y be altered or fine-tuned by formation of various metal derivatives. For these reasons, porphyrins and metallo-porphyrins are particularly attractive molecular species to incorporate into supramolecular assemblies with special built-in properties or functions. Much effort has been applied to the design and synthesis of covalent porphyrinic arrays analogous to those found in nature for charge separation, electron transport,3 and signal or energy transduction.4 Formation of multiple hydrogen bonds between complementary molecular components is widely used in the fabrication of supramolecular assemblies because of their strength, directionality, specificity, and reversibility. Preparation of monomeric hydrogen bond recognition unit-bearing chromophores allows the self-assembly of both polymers and discrete arrays with various structures in high yields by appropriate combinations of the molecular building blocks.
Designed porphyrin arrays have been formed by hydrogen bonding,5,6 axial metallo-porphyrin coordination,7,8 and coordination of exocyclic ligands.9 Several topologically complex structures such as catenanes and rotaxanes have utilized porphyrins.10,11 Porphyrins have been incorporated into lipid membranes to form photo transis-tors12 and have been used in read-write devices 13 and as receptors.14 Several recent reviews on self-assembly of various molecules, including porphyrins, put the present work into context.15 One conclusion that may be drawn from these numerous studies is that the nature of the chemical linker—covalent, coordination, or hydrogen bond—is just as important to the function of the array as the topology of the chromophores. Thus, porphyrinic squares formed by metal ion coordination have substantially different photophysical properties than porphyrin squares formed via hydrogen bonding.9 For example, if fluorescence is a desired property of the material, say for a ns luminescent switch, then hydrogen bonding is preferred overmetal ion coordination oracetylenic linkers, because these later reduce the quantum yield by heavy atom effects and energy orelectron transfer, respectively. While there is an ever increasing number of discrete porphyrin arrays mediated by metalion coordination, there are few discrete arrays mediated by hydrogen bonding.5,6
The primary objective of this work is to develop synthetic strategies for two sets of porphyrin building blocks bearing rigidly linked peripheral complementary triple hydrogen bonding moieties (Figure 1), and the heretofore unknown aldehydes. The issue of a general synthesis of porphyrins bearing complex heterocyclic substituents is yet unresolved, vide infra. These can be joined into a variety of discrete nanostructures with remarkable control and precision.1 By mixing these complementary synthons in organic solvents with accurately controlled stoichiometry, the directed triple hydrogen bonding moiety makes it possible to form various types of rigid multi-porphyrin arrays with different, designed spatial relationships. Low molecular weight polymers with somewhat tunable size, depending on the thermodynamics of binding, and molecular organization into higher order structures are also possible.16 The use of free base or metalloporphyrin building blocks enables the fabrication of arrays with predetermined metalation states. These building blocks also provide hydrogen-bonded pathways through which energy or electron transfer might be facilitated.17 In addition, an X-ray study of a 5,15 disubstituted pyridyl derivative reveals an un expected new mode of hydrogen-bonded supramolecular aggregation mediated by adventitious water molecules.
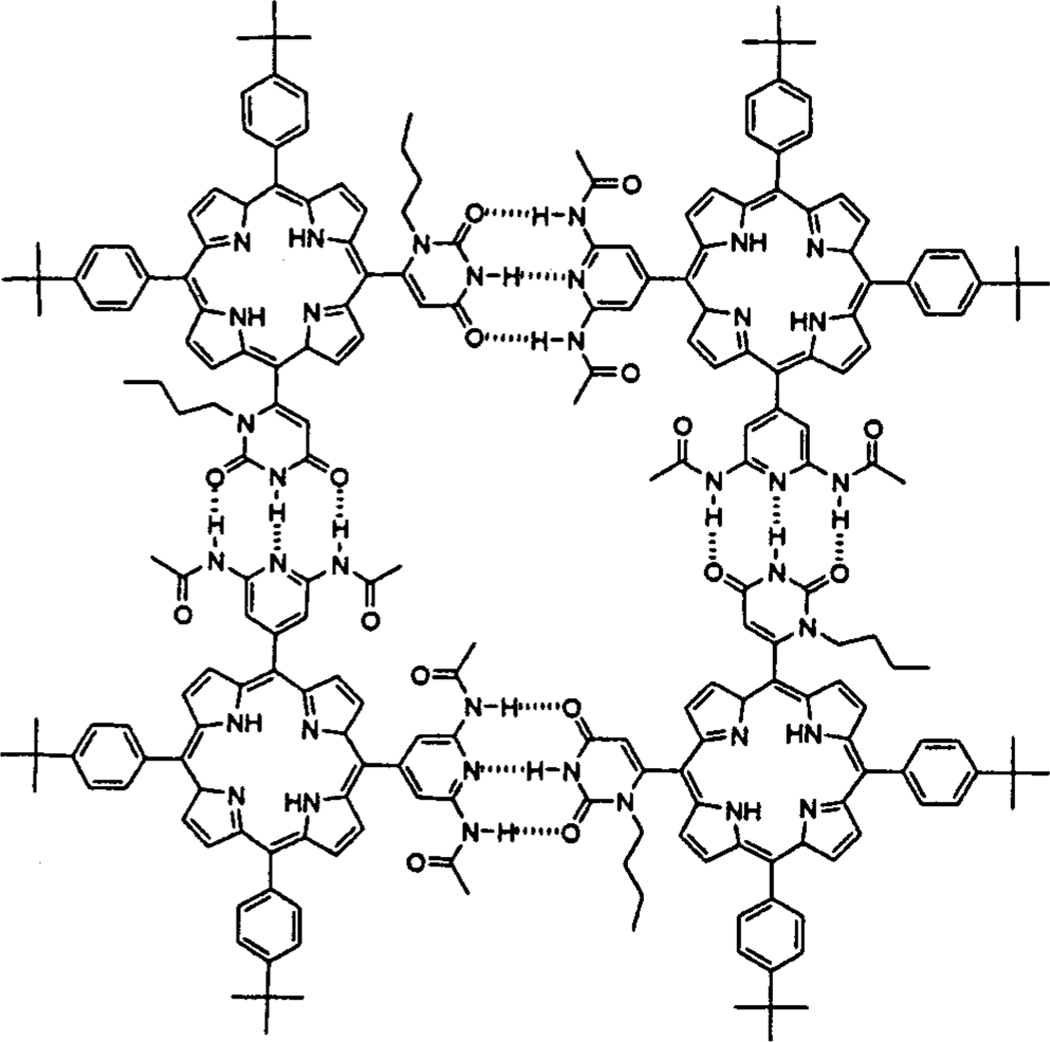
Figure 1.
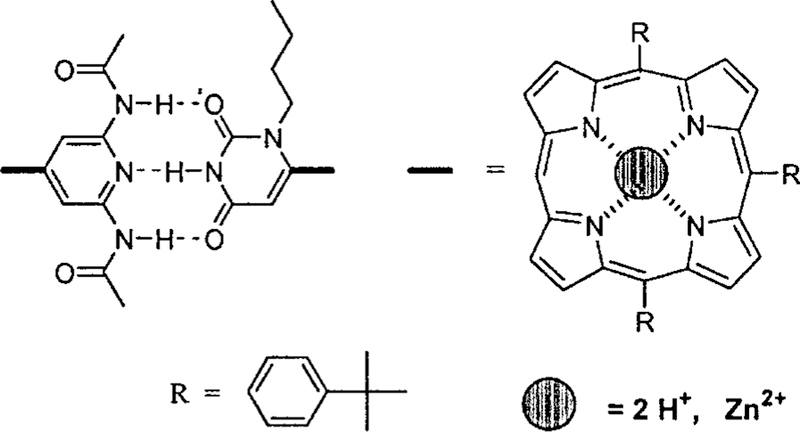
Interactions between two porphyrin building blocks via complementary hydrogen bonding. Assemblies containing more than two uracyl groups will have one or more isomeric forms.
Results and Discussion
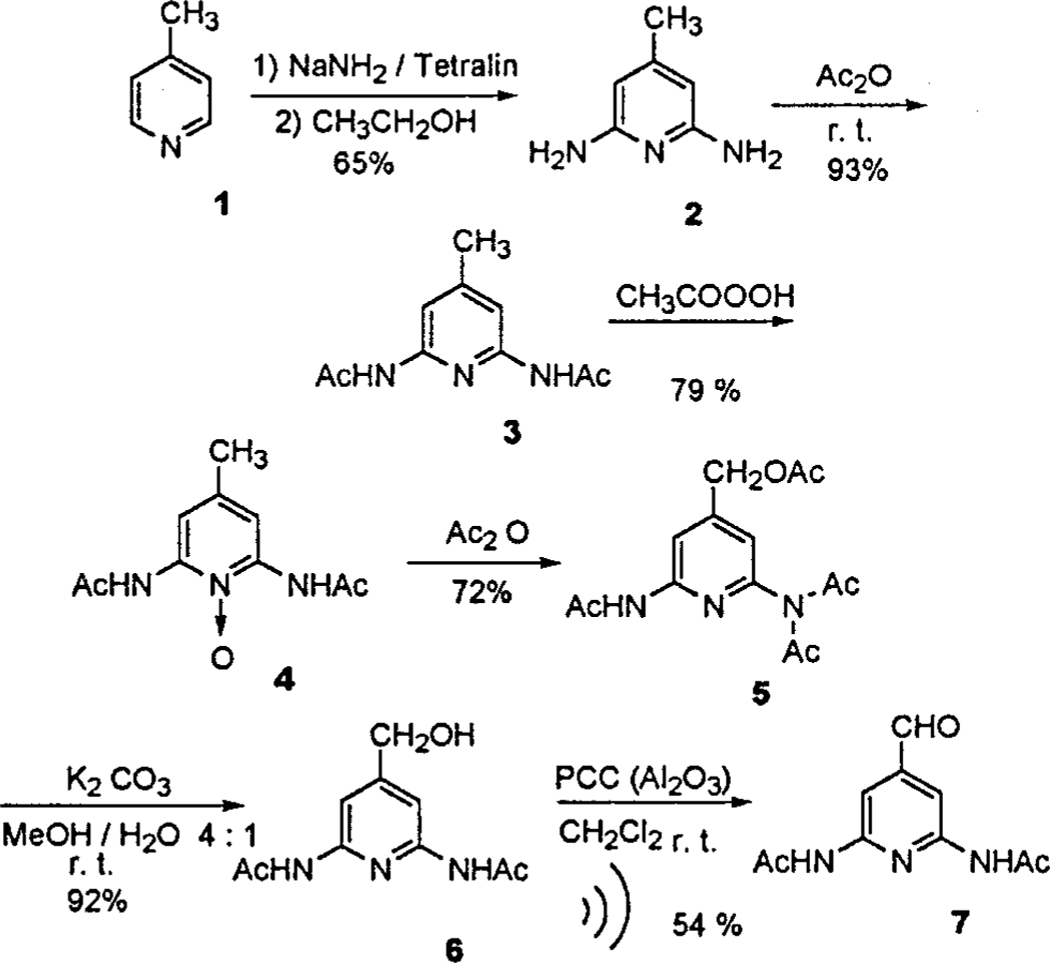
The syntheses of two aldehydes 717c and 11 are shown in Schemes 1 and 2. The direct amination of 4-methylpy-ridine, 1, with sodium amide in a tetralin solution affords 2,6-diamino-4-methylpyridine, 2, in 65% yield. This factor of 2 increase in yield over previous methods 18 is largely due to milder reaction conditions (slowly heating by stages) and workup (hydrolysis with ethanol). 2,6-Diac-etam ido-4-methylpyridine, 3, is made through acylation of 2 with acetic anhydride. The oxidation of compound 3 with peracetic acid yields 2,6-diacetam ido-4-methylpy-ridine-1-oxide, 4. Refluxing 4 in acetic anhydride results in the oxidation of the methyl group by a transfer of the intermediate 1-acetoxy group19 to yield 2-diacetimido-6-acetam ido-4-acetoxy methylpyridine 5. Selective hydrolysis of one acetate of the imide and the ester group of 5 with K2CO3 in a methanol/water solution affords the 2,6-diacetam ido-4-hydroxymethylpyridine, 6. The alcohol 6 is oxidized with PCC (on alumina) under ultrasound conditions to yield 2,6-diacetam ido-4-formylpyridine, 7, where yields as high as 65% are observed in small-scale oxidation reactions. Starting from 2, the 26–32% overall yield of this synthetic route (Scheme 1) is reasonable because of the inexpensive starting materials and the easy scale-up. Other routes to this aldehyde are not successful because of the substantial electronic effects at the 4 position. For example, the 2,6-diamino-3-formylpy-ridine isomer can be synthesized by the formylation of 2,6-diaminopyridine by a Vilsmeyer reaction.20 Completely optimized structure calculations using Gaussian 98 (B3LYP/6–311**) on aldehyde 7 indicate that both the acetamido and aldehyde groups are coplanar with the pyridine ring, with the amide oxygen atoms pointing toward the ring. These calculations also suggest that there is some hydrogen bonding between the amide oxygen atoms and the pyridyl hydrogens, and one of the pyridyl hydrogens is near enough to hydrogen bond with the aldehyde oxygen. The difference in the chemical shifts for the two pyridyl protons is calculated (GIAO) to be 0.220 ppm in the gas phase, and that measured in chloroform-d is <0.07 ppm. Interestingly, the pyridyl protons sense the formation of the self-complementary hydrogen bond dimer of II, manifested as a small chemical shift of ∼0.1 ppm, which is consistent with the expected inductive and geometric effects, Supporting Information.
Scheme 1
Scheme 2
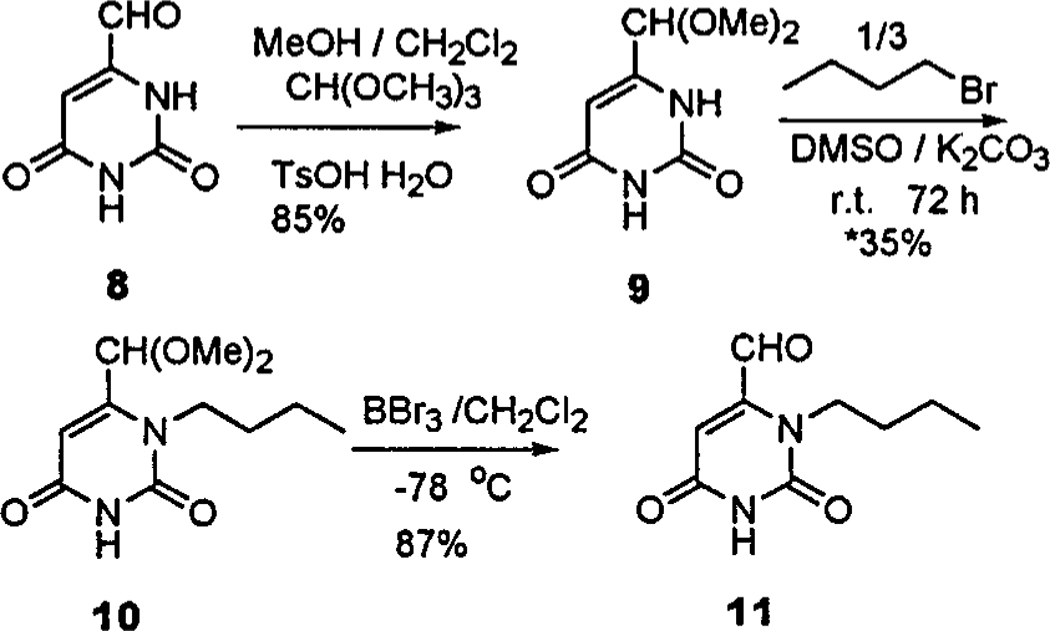
The synthesis of the complementary 1-butyl-6-formyl-uracil, 11, from 6-form ylura cil, 8, is substantially different than that of the known 1-butyl-5-formyluracil5a (Scheme 2). Unlike the 5-formyl derivative, 21a the 6-formyluracil21b does notalkylate preferentially at the 1-position so must be protected as the acetal, alkylated, and deprotected. Trimethyl orthoformate in methanol/ methylene chloride and 8 were refluxed for10 h in the presence of the p-toluenesulfonic acid to form the 6-(dimethoxymethyl)uracilin 86% yield.22a The dimethyl acetal was alkylated with n-butylbromide catalyzed by K2CO3 in DMSO at room temperature for 72 h to form the 1-butyl-6-(dimethoxymethyl)uracil (37% based on n-butylbromide)23 and two byproducts. The 1-butyl-6-(dimethoxymethyl)uracil could not be converted to its corresponding aldehyde under a variety of aqueous acid conditions, so a Lewis acid, BBr3, in CH2Cl2 was used to cleave the dimethyl acetal22b at-78 °C in 87% yield. In dry solvents the uracil vinyl proton can be observed as two peaks, possibly as a consequence of self-complementary hydrogen bonding of 11.
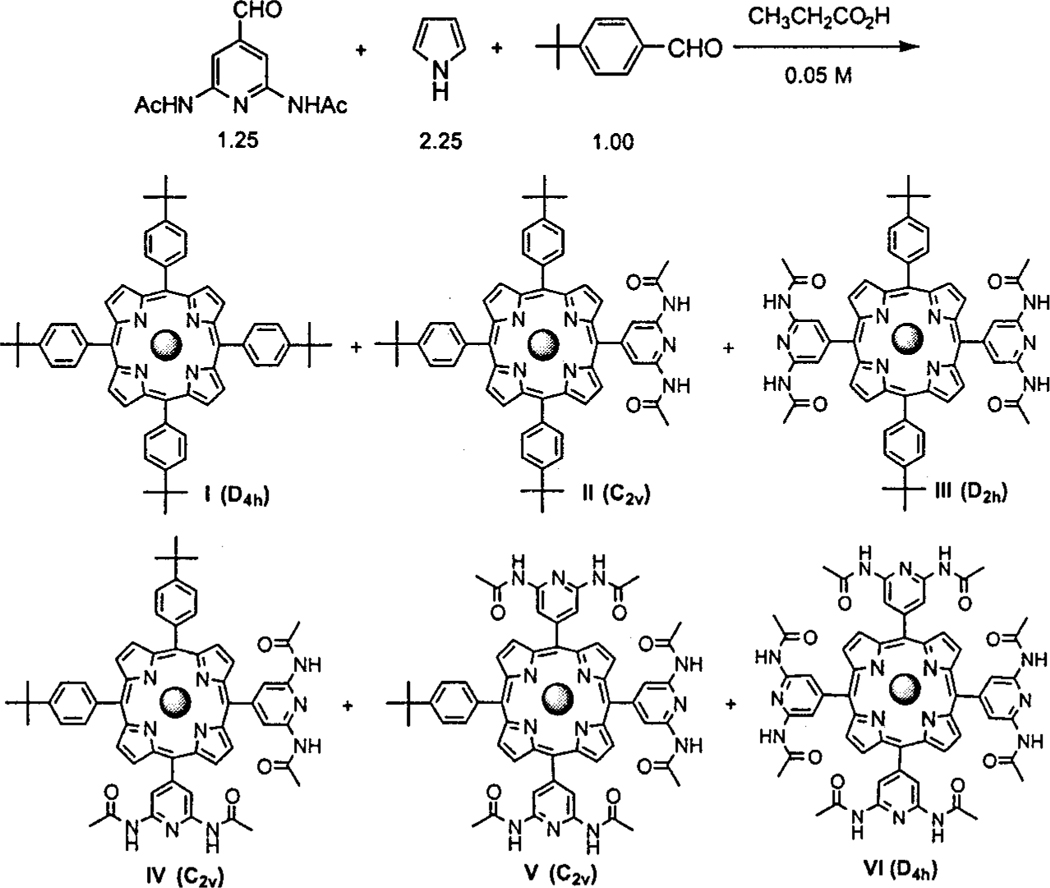
Both sets of porphyrin building blocks were prepared by the Adler24 synthesis (Schemes 3 and 4) and modifications thereof.25 Using a mixture of two different aldehydes results in a mixture of six porphyrins in yields that are weighted by the reactivity, the solubility, and the relative amounts of the aldehydes.8a,9a,25 All three of these can be exploited to obtain maximum yields of the desired porphyrins, vide infra. Since the polarities of the six porphyrins are very different, they are readily separated by column chromatography. The mixed aldehyde condensation8a,25 was chosen for several reasons. (1) Heterocyclic aldehydes and/or those bearing Lewis bases are generally not tolerated or have poor yields using the Lindsey porphyrin syntheses 26 as well as most of the [n + (4 -n)] coupling reactions. (2) The five compounds containing the heterocycles are all needed as building blocks for the self-assembled arrays. (3) The Adler24 synthesis and its modifications25 are quite amenable to large scale, multigram reactions without significant sacrifice in yields or complications in purification. (4) All the porphyrins and isomers can be well characterized by the 1H NMR spectra in the aromatic region, especially by the pyrrole β-H. Thus, with ample quantities of the two aldehydes in hand, a number of porphyrin syntheses were explored: three different reaction solvents, acetic acid, propionic acid, and a mixture of nitrobenzene/acetic acid were tried each with and without zinc acetate as a possible tem-plating agent. All these conditions give good results with7, but11 was more problematic.
Scheme 3
Scheme 4
To increase the yield of the four porphyrin building blocks bearing both heterocyclic and alkylphenyl groups, we adopted three measures: (i) increasing the relative amount of the heterocyclic aldehydes, (ii) decreasing the concentration of the reaction mixture (0.05 M), (iii) for the uracyl porphyrin building blocks, changing the reactant addition order whereby the 4-tert-butylbenzal-dehyde was added 10 min after adding the pyrrole rather than adding two aldehydes together. During the course of this investigation only one method for the synthesis of the tetrauracylporphyrin was discovered, namely using acetic acid and nitrobenzene as reaction solvents25d with zinc acetate (Scheme 4.). The 5.1% yield of XII is consistent with previous Adler synthesis 25a considering that it bears a nonaromatic heterocycle with a sterically hindering substituent next to the aldehyde.
The one striking aspect of the physical properties of both the diheterocyclic porphyrin isomers is the abnormally low solubility of the 5,15-isomercompared to that of the 5,10-isomer. In nonpolar organic solvents the solubility of the 5,10-disubstituted porphyrins is usually expected to be less than that of the 5,15-isomers due to greater polarity; however, on the contrary, these 5,10-porphyrins are found to have much greater solubility than the 5,15-isomers. As discussed below, this anomalous solubility is explained by the self-complementary hydrogen-bond self-assembly of linear polymeric tapes vs closed tetrameric squares.
While the characterization of the 3,5-diacetam ido-4-pyridylporphyrins is straightforward, the characterization of meso-1-butyl-6-uracylporphyrins merits comment. The four1-butyl-6-uracyl groups are rigidly linked to the meso-positions of the porphyrin and rotation about the connecting bond is hindered by the butyl substituents. Thus the 1-butyl-6-uracyl group is oriented nearly normal to the porphyrin plane, projecting the butyl substituents above or below the macrocycle. The 1H NMR spectra, see Supporting Information, of the tetrauracyl compound XII reveals that: (1) all the chemical shifts of the butyl group are shifted up field due to ring current effects; (2) the methylene group next to the 1-N atom showed four sets of multiplets rather than one triplet and the methyl group showed four sets of triplets rather than one triplet. This proves that the butyl groups are on the 1-positions of the uracyl, and that there are four rotameric forms27 in a ratio of 1:3:2:1 from low to high field. The statistical ratio for the αααα,αααβ, ααββ, and αβαβ rotamers would be 1:4:2:1, respectively (α above and β below the porphyrin plane).28 The integral areas of the terminal methyl group are consistent with the N-methyle ne, but may be in a different order. The observed deviations from the ideal ratios may be due to losses during the purification process. The rotameric forms of VIII-XII result in self-assembled supramolecular species with different isomeric forms, but the isomers are not observed. There are also no detectable consequences on the energetics and kinetics of the self-assembly process since the uracyl ADA (A = hydrogen bond acceptor, D = hydrogen bond donor) recognition function(s) are directed along the porphyrin plane.29
The 1H NMR spectra of the various other ura cyl porphyrin derivatives, VIII-XI, are similarly complex due to the presence of both4-tert-butylphenyl and 1-bu-tyl-6-uracyl groups and the resulting rotamers. Each porphyrin face is different in the monouracyl porphyrin, VIII. This results in an AB, A′B′ system, where A′, B′ represents the phenyl protons on the opposite side of the macrocycle relative to the butyl group on the uracil. Thus for VIII, the phenyl protons are observed as multiplets rather than a simple AB quartet. This spectrum of VIII shows the expected diagnostic pattern in the β-pyrrole region for this type of derivative, where there are four different chemical environments that should result in two AB quartets, but only the quartet from the pyrroles nearest the heterocycle is observed, while those on the opposite side of the heterocycle are observed as a singlet even on the 500 MHz instrument.
For analytical purposes both rotamers of IX and X, were isolated. Both the 5,15-and 5,10-compounds can existas αα or αβ rotamers, each with a different symmetry, with respect to the relative orientation of the two 1-butyl-6-uracyl groups. Thus, the chemical environments of the porphyrin faces are the same for the αβ rotamers and different for the αα rotamers for both IX and X. As in the mono substituted derivative, these rotameric forms are readily observed in the 1H NMR in the region of the two 4-tert-butylphenyl groups. A simple doublet of doublets for the phenyl groups of the αβ rotamers of both compounds is observed, while multiplets for the same groups are observed for the αα rotamers. Because the pyrrole β-H are in the porphyrin plane, the rotameric forms of IX and X are not observed in this region, so there are the expected four different pyrrole resonances in X and two for IX.
Structural Studies
As an additional, un expected example of the intrinsic tendency of these compounds to aggregate by hydrogen bonding, we also present an X-ray study of IV. Atom names, displacements from planarity and an edge-on view of the macrocycle are given in Figure 2. The crystallographic determination provides unambiguous identification of IV. Complete experimental details, atomic coordinates and molecular geometry are included in the Supporting Information. The C5-,C10-, and C20 -meso substituents are completely ordered, but the tert-butylphenyl group atC15 exists in two discrete orientations with75/25% occupancies. (Only the orientation of the major component is shown in Figure 2.)
Figure 2.
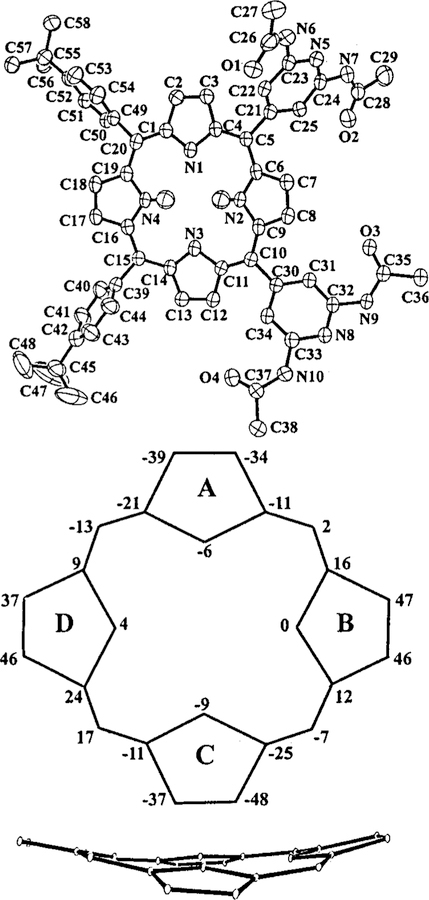
Top: Molecular structure and atom names for IV. Thermal ellipsoids are drawn at the 50% probability level. Peripheral hydrogens are omitted for clarity. Middle : Dis-placements of the 24 atoms that comprise the macrocycle from the 24 atom mean porphyrin plane in units of 0.01 Å. Bottom: Edge-on view of the porphyrin core. Thermal ellipsoids enclose 1% probability forclarity.
The skeleton of IV is mainly saddled with some degree of ruffling. The Cb atoms in each of the four pyrrole rings exhibit substantial unequal displacements from the mean porphyrin plane that range between 0.34 and 0.48 Å (see Figure 2). The deviations of the meso carbons are also unequal; they are smaller at C5 and C10, 0.02 Å and −0.07 Å, than atC15 and C20, 0.17 Å and-0.13 Å. The edge-on view in Figure 2 further illustrates the distorted conformation of the macrocycle, with rings B and D above the mean plane and rings A and C below it. Note that such out-of-plane distortions of the porphyrin macrocycle have been shown to have significant effects on the optical, redox and excited-state properties of the chromophores.30 Biological systems containing porphyrinic proteins such as those used in photosynthesis and electron transport exploit these effects to fine-tune their reactivity and photophysical properties.31
As observed in other saddled porphyrins,30,31 the dihedral angles between the meso-substituents and the porphyrin plane are unusually acute. They assume values of 62° atC5, 36° at C10, 60° and 72° at C15, and 62° at C20. The unusual dihedral angle of 36° of the C10 substitutent, compared to the nearly perpendicular angles of 80–90° normally observed in planararyl-substituted porphyrins, is most likely related to its participation in hydrogen bonds (vide infra).
The sample was crystallized from a mixture of ethyl acetate and hexane, but the crystals do not incorporate either of these solvents. Instead, the lattice contains 2.5 molecules of adventitious water per porphyrin. The unexpected water molecules (O5 and O6) mediate a more extensive supramolecular array of IV via multiple hydrogen bonds (Figure 3).
Figure 3.
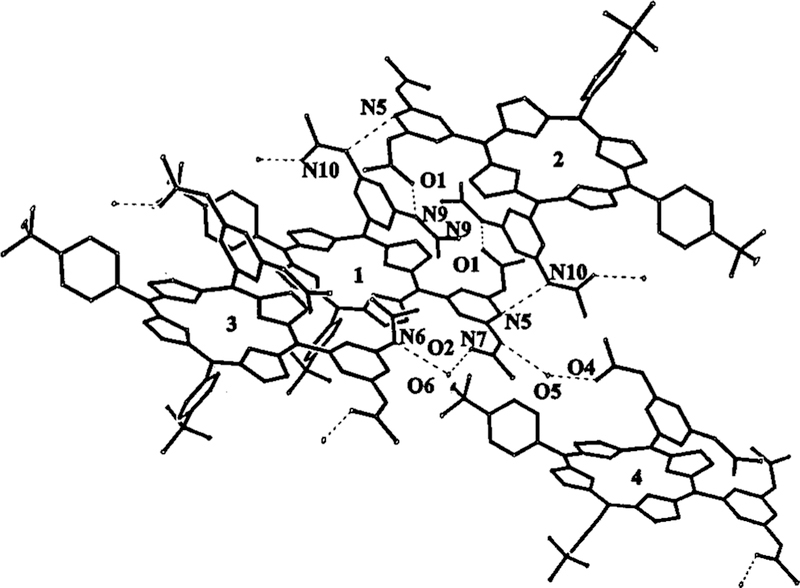
Hydrogen-bonded supramolecular assembly of porphyrin IV mediated by water. (Hydrogen bonds are shown as dotted lines.)
For simplicity, the basic porphyrin building blocks of this array are designated as 1, 2, 3, and 4. Only molecule I comprises the crystallographic asymmetric unit; the restare generated by space group operators. Molecules 1 and 2 are joined by two reciprocal O1···N9 and two reciprocal N5···N10 hydrogen bonds at3.02 Å and 3.05 Å. This dimeric unit is further incorporated into a complex multi-porphyrin array. O2 of molecule 1 associates withO6 (a water molecule of solvation) with an O2···O6 distance of 2.84 Å. O6 then hydrogen bonds to N6 of molecule 3 which is 2.99 Å from it. N7 of molecule 1 associates withO5 (another water molecule of solvation) with N7···O5 = 3.00 Å. O5 sits 2.82 Å away from O4 of molecule 4. (There are additional interactions involving O7, a third water of crystallization that is present only 50% of the time.)
All five possible hydrogen-bond donors and acceptors (O1, N6, N5, N7, and O2) of the C5-meso substituent participate in the network formation, whereas only N9, N10 and O4 of the C10-meso substituent participate, and may thus contribute to the differences in the orientations of the peripheral rings. The closest approach of pyrrole ring centers (A and D) is 3.87 Å involving 1 and another molecule not involved in the hydrogen bonding with1. Since the center to center distance of 1 and 2 is 12.70 Å, they interact only at the periphery. The structure reveals that there are no substantial π-π interactions in this arrangement of macrocycles in the solid state. Therefore, the degree of porphyrin distortion caused solely by hydrogen bonding in this single-component system is unprecedented.31 These results also indicate that the intentional addition of water to molecules designed to form networks or arrays mediated by hydrogen bonding may well lead to novel aggregates, as evidenced by this structure.
Self-Assembled Squares in Solution.
The preponderance of evidence from 1H NMR, UV-visible, ESI-MS, light scattering, and osmometry suggests that IV self- assembles into tetrameric squares in solvents with low hydrogen-bonding potential by self-complementary hydrogen bonds.32 This is well demonstrated by fits of the chemical shift of the amide proton versus concentration to yield an apparent equilibrium constant, K, and the number of molecules in the final assembly, n (Figure 4A). If there are several products or dynamic processes, such as the breaking apart of one or more sets of hydrogen bonds occurring on the NMR time scale, these results will only give rough estimates of K, but this serves as a basis of comparison for the square formed by complementary H-bonds, below. Analysis of the NMR data32 for the self-complementary self-assembly of the square tetramer of IV yields K = 2.1 ± 1 × 109 M−3, and n = 3.9 ± 0.3 in chloroform. In toluene the equilibrium constants are greater, as expected, where K is ∼2 ( 1 × 1010 M−3). ESI-MS and vapor phase osmometry data also indicate a tetrameric species.
Figure 4.
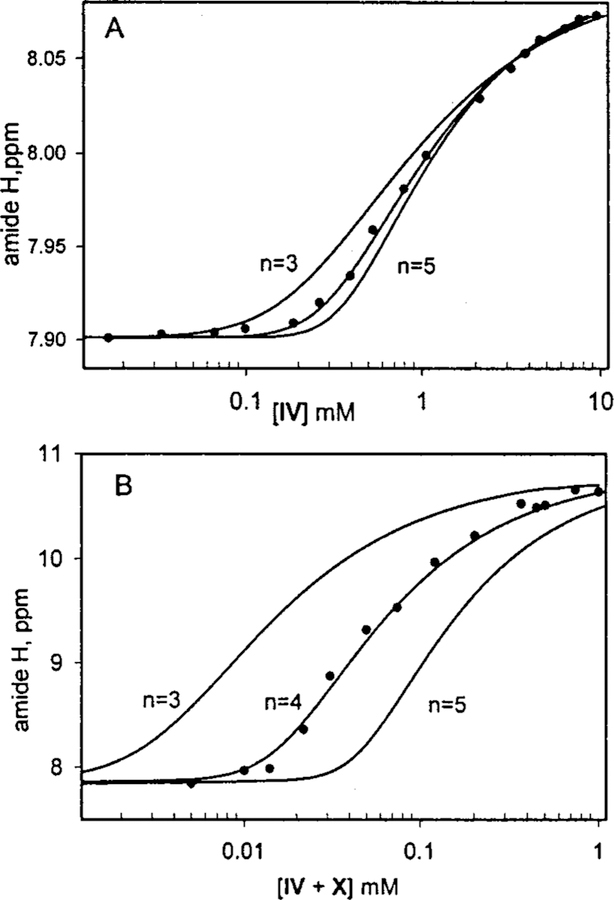
Formation of closed tetrameric squares. (A) The self-complementary association of IV in CDCl3 is compared to (B) the complementary association of a 1:1 mixture of IV and X in THFd8.
The thermodynamics of these self-complementary systems in solution are discussed herein. The association for the diacetamidopyridyl moiety is usually considered weak,29 ∆Gdimerranges from 4 to 13 k J mol-1, and the double hydrogen bond considered the dominant interaction. However, the strengths of these recognition units are substantially altered by the presence of the directly linked porphyrin. Thus there is likely an equilibrium between the various self-complementary interactions of the diacetamidopyridyl groups - double, triple, qua-druple hydrogen bonds. Van’tHoff plots of the 1H NMR data for the self-complementary dimerization of II indicate ∆Hf of the dimeris ∼21 ± 5 k J mol-1 in toluene versus the expected ∼13 kJ mol−1 for the alkyl substituted derivative in the same solvent.29 AM1 calculations indicate that the presence of a phenyl group on the 4 position of the 2,6-diacetamidopyridyl moiety increases the self-complementary H-bond enthalpy by∼4 kJ mol-1. Typically, the electronic effects of the porphyrin are greater than the phenyl group, so a further increase in the ∆H of the self-complementary interaction(s) is expected and calculated to be ∼6 k J mol-1 greater than the parent recognition unit. Thus the experimental and calculated results are qualitatively consistent. The 90° geometry of the rigidly linked recognition groups on the porphyrins, and the thermodynamic stability of the self-assembled H-bond squares compared to other structures,29d ensure that the squares are formed in ∼75% in chloroform, and ∼90% in toluene.32,33 Both experimental and computational results show that the presence of the phenyl has little effect on the self-complementary 1-butyl-6-uracyl system. Calculations indicated there should be a modest increase of ∼3 k J mol-1 for hetero-complementary 2,6-diacetamido-4-phenylpyridine and 1-butyl-6-phenyluracil. The H-bond strengths between the complementary porphyrins II, and VIII, are calculated to be 45 kJ mol-1, ∼7 k J mol-1 greater than for the isolated recognition units bearing neither porphyrin norphenyl substituent.29,33 Thus the porphyrin ring is expected and observed to enhance the basicity of the pyridyl nitrogen and therefore the H-bond strength. This is a well-documented phenomenon in the coordination chemistry of pyridylporphyrins.9
The formation of a tetrameric square from two equivalents each of IV and X (Scheme 5) in THF, used because of the limited solubility of X, is indicated by similar experiments as described above. The overall shape of the NMR concentration vs amide chemical shift(Figure 4B) clearly indicates a tetrameric species with n = 4 and an apparent equilibrium constant, see caveats vide supra, of 6 = 3 × 1012 M−3 which is somewhat greater than that estimated solely by equilibrium considerations from the C1/2 data.29e Note that the C1/2 for the complementary square is about2-fold less than that of the self-complementary species, and the ∆ppm for the amide protons is a factor of 5 greater. These are all indicative that the complementary interactions between the uracyl and diacetamidopyridyl moieties, calculated by various means to be ∼45 k J mol-1, are much stronger than either self-complementary interaction even in a solvent known to be less favorable to H-bonding than chloroform. Dynamic light scattering detects only species with∼ 4 nm hydrodynamic radius between 0.04 and 0.1 mM. Vapor phase osmometry experiments using concentrations between 0.1 and 1 mM yields an average molecular weight of 3500 ± 300 da. ESI-MS using a mixture of IV, Co(III)IV, and X (1:1:2) shows the doubly charged tetramer as well as other multimers and the monomers. Taken together, these data indicate that the complementary tetrameric square forms in solution. Experimental and computational work on these latter assemblies is in progress.
Scheme 5
Conclusion
These two sets of complementary porphyrin building blocks can be used for constructing dive se multi-porphyrin arrays in defined geometries through self-complementary or hetero-complementary assembly. The enthalpy of the latter is stronger by more than 2-fold, and this ensures that the self-complementary products are not complicating factors in the hetero self-assembly process. The 1-butyl-6-uracyl and 3,5-diacetamido-4-pyridyl groups can be used to form linear, square, and junction structures in a predefined metalation state. The ability to self-assemble nanoscaled building blocks with precisely controlled size and composition, and to incorporate them into larger electronics components—which may entail secondary self-assembly steps to form hier-archical structures—with unique properties and functions may have significant implications for the incorporation of these types of molecules into photonic devices. These studies also demonstrate that water can mediate the self-aggregation of H-bonding species and dictate the supramolecular arrangement of subunits, as it does in many protein structures.
Experimental Section
Materials and Methods.
Melting points were determined on a Thomas-Hoover UniMelt capillary apparatus and are uncorrected. Flash column chromatography was performed using 230–400 mesh ASTM Merck silica gel-60. 1H and 13C NMRspectra were recorded on J EOL 400 MHz, a Varian VXR-300 MHz, ora Varian 500 MHz instrument. Chemical shifts are reported in ppm relative to TMS for1 H and 13C spectra, and coupling constants in Hz. NMR assignments are consistent witht hose published previously. Agilent Technologies HP 1100 LC/MSD, and a Cary Bio-3 were used. Typical Electrospray Ionization Mass Spectroscopy (ESI-MS) method: ∼0.05 mM solutions in acetonitrile/water(50:50) containing 1% trifluo-roacetic acid, positive ion mode, and the fragment or voltage between 100 and 350 V. Elemental analysis by Schwarzkopf Microanalytical Laboratory, Inc.
2,6-Diamino-4-methylpyridine (2).
A solution of 4-methylpyridine 1 (9.31 g, 9.73 mL, 0.10 mole) in 50 mL tetralin was added to a solution of sodium amide (9.33 g, 0.24 mole) in 100 mL tetralin at130∼140 °C over6 h, afterward heated to 195 °C and kept at this temperature for10 h.18d After cooling, the reaction mixture was filtered, and the remaining black solid material worked up by the dropwise addition of 150 mL ethanol. 10 g of silica gel was added, and the ethanol was removed under reduced pressure. The resultant dark solid was added to the top of a 45 g column of silica gel and eluted with ethyl acetate to afford 8.02 g (65%) of 2 as a white powder. Recrystallized from chloroform, mp 85–86 °C (lit.18 a 87–88 °C, sublimed crystal, 109–111 °C). 1H NMR(CDCl 3) δ 5.74 (s, 2H), 4.06 (br, s, 4H), 2.11 (s, 3H); 13C NMR(CDCl 3) δ 158.3, 151.3, 99.5, 21.8; MS (ESI) m/z (MH+, 124).
2,6-Diacetamido-4-methylpyridine (3).
16.0 mL of acetic anhydride was added to 1.5 g (12.2 mmol) of the diamine 2. The resultant solution was stirred for1 hand 2.35 g (93%) of a white crystalline solid 3 was collected by filtration, mp 199–201 °C. 1H NMR(CDCl 3) δ 7.72 (s, 2H), 7.64 (br, m, 2H), 2.35 (s, 3H), 2.17 (s, 6H); 13 C NMR(CDCl 3) δ 168.9, 153.4, 149.9, 110.9, 25.5, 22.5; MS (ESI) m/z (MH+, 208). Anal. Calcd forC10 H13 N3O2: C, 57.97; H, 6.32; N, 20.27. Foun d: C, 57.84; H, 6.50; N 20.10.
2,6-Diacetamido-4-methylpyridine-1-oxide (4).
To a solution of the diamide 3 (2.07 g, 10.0 mmol) in 10 mL acetic acid, 3.0 mL of 32% peracetic acid was added carefully at room temperature. The resulting solution was heated for3 h at50–60 °C and 4 h at65–70 °C. After cooling to room temperature, adding FeSO4 solid to destroy residual oxidant, and removing the acetic acid under reduced pressure, the residue was recrystallized from water to yield 1.76 g (79%) of the oxide 4 as a white powder: mp 210–211 °C. 1 H NMR(CDCl 3) δ 9.83 (s, 2H), 7.92 (s, 2H), 2.35 (s, 3H), 2.27 (s, 6H); 13C NMR(CDCl 3) δ 164.4, 137.4, 137.0, 104.1, 21.0, 17.8; MS (ESI) m/z (MH+, 224). Ana l. Calcd for C10 H13 N3O3: C, 53.80; H, 5.87; N, 18.82. Found: C, 53.94; H, 5.96; N, 18.90.
2-Diacetimido-6-acetamido-4-acetoxymethylpyri-dine (5).
A solution of the oxide 4 (1.50 g, 6.73 mmol) in acetic acid (3.0 mL) was added to stirred, refluxing acetic anhydride (20 mL) over a 30-min. period. The reaction mixture was stirred at reflux temperature for 30 h, cooled, 5.0 g silica gel was added to the reaction mixture, and the solvent removed under reduced pressure. The resultant dark material was added to the top of a 20 g column of silica gel and eluted with ethyl acetate/petroleum ether(1:1) to afford 1.49 g (72%) of 5 as a white powder, mp: 177–178 °C. 1H NMR(CDCl 3) δ 8.24 (s, 1H), 7.89 (br, s, 1H), 6.93 (s, 1H), 5.17 (s, 2H), 2.29 (s, 6H), 2.21 (s, 3H), 2.20 (s, 3H); 13C NMR(CDCl 3) δ 172.9, 170.9, 169.2, 152.3, 151.7, 151.6, 118.0, 112.3, 64.7, 27.3, 25.5, 21.5; MS (ESI) m/z (MH+, 308). Anal. Calcd forC14 H17 N3O5 C, 54.72; H, 5.57; N, 13.67. Found: C, 54.66; H, 5.64; N, 13.71.
2,6-Diacetamido-4-hydroxymethylpyridine (6).
A solution of the acetate 5 (1.20 g, 3.9 mmol) and K2CO3 (0.59 g) in methanol/water(10 mL, 4:1) was stirred at room temperature for8 h. The reaction mixture was loaded on 3.0 g silica gel. The resultant yellow material was added to the top of a 25 g column of silica gel and eluted with ethyl acetate/ethanol (9: 1) to afford 0.80 g (92%) of the alcohol 6 as a white powder, mp 194–195 °C. 1H NMR(DMSO-d6) δ 9.93 (s, 2H), 7.65 (s, 2H), 5.34 (t, 1H), 4.43 (d, 2H), 2.05 (s, 6H); 13C NMR(DMSO -d6) δ 170.1, 156.8, 151.2, 107.5, 63.4, 25.2; MS (ESI) m/z (MH+, 224). Ana l. Calcd for C10 H13 N3O3: C, 53.80; H, 5.87; N, 18.82. Found: C, 53.72; H, 6.04; N 18.86.
2,6-Diacetamido-4-formylpyridine (7).
A solution of the alcohol 6 (0.80 g, 3.58 mmol) in CH2Cl2 (15 mL) was treated with NaOAc (2.85 g, 14.32 mmol) followed by portion wise addition of pyridinium chlorochromate (PCC/Alumina, 1.15 g, 5.38 mmol). The reaction mixture was placed in a sonication bath22c at room temperature for1 h, at which time additional PCC/Alumina (0.43 g, 2.01 mmol) was added. After being sonicated an additional 3 h, the reaction mixture was loaded on 3.0 g silica gel, added to the top of a 25 g column of silica gel, and eluted with ethyl acetate/ethanol (9:1) to afford 0.43 g (54%) of the aldehyde 7, mp 223–224 °C. 1H NMR(DMSO -d6) δ 10.33 (s, 2H), 9.96 (s, 1H), 8.11 (s, 2H), 2.10 (s, 6H); 13C NMR(DMSO-d6) δ 194.1, 170.7, 152.6, 146.5, 108.9, 25.3; MS (ESI) m/z (MH+, 222). Anal. Calcd for[C10 H12 N3O3]+: C, 54.05; H, 5.44; N, 18.91. Found: C, 53.63; H, 5.27; N, 18.26.
6-(Dimethoxymethyl)uracil(9).
A solution of orotaldehyde (8) (3.0 g, 21.42 mmol) and trimethyl orthoformate (9.08 g, 9.4 mL, 85.68 mmol) in MeO H (50 mL) and CH2Cl2 (100 mL) was refluxed for10 h in the presence of p-toluenes ulfonic acid (0.3 g, 1.58 mmol). After the reaction mixture cooled to room temperature, triethylamine was added to the reaction mixture until it turned weakly basic. 5.0 g silica gel was added to the reaction mixture and the solvent removed under reduced pressure. The resultant yellow material was added to the top of a 25 g column of silica gel and eluted with ethyl acetate/ ethanol (9:1) to afford 3.46 g (87%) of the acetal 9 as a white powder, mp 185–186 °C, (lit.:22a 185–186 °C). 1H NMR(CDCl 3) δ 11.02 (br, s, 1H), 10.83 (br, s, 1H), 5.45 (s, 1H), 4.99 (s, 1H), 3.25, (s, 6H); 13C NMR(CDCl 3) δ 164.9, 152.4, 151.8, 99.5, 99.1, 54.8; MS (ESI) m/z (M-H+, 185).
1-Buty l-6-(dimethoxymethyl)uracil (10).
To a solution of the acetal (9) (3.20 g, 17.20 mmol) in DMSO (200 mL) were added 1-bromobutane (0.7850 g, 0.62 mL, 5.73 mmol) and anhydrous K2CO3 (2.61 g, 18.92 mmol). The suspension was stirred for72 h at room temperature after which it was filtered and the DMSO was removed in vacuo. The solid was absorbed on 5 g silica gel, loaded on the top of a 25 g column of silica gel, and eluted with hexane to afford 0.140 g (8.2%) of 1,3-dibutyl-6-(dimethoxymethyl)uracil as an oil. Further elution with ethyl acetate/hexane (1:9) afforded 0.222 g (16%) 3-butyl-6-(dimethoxymethyl)uracil and 0.513 g (37%) of 10. Yields reported for this compound are based on 1-bromobutane, mp: 91–92 °C. 1 H NMR(CDCl 3) δ 9.28 (br, s, 1H), 5.95 (s, 1H), 5.05 (s, 1H), 3.87 (t, 2H, J = 7.7 Hz), 1.67–1.57, (m, 2H), 1.42–1.30, (m, 2H), 0.95, (t, 3H, J = 6.9 Hz); 13C NMR(CDCl 3) δ163.4, 152.2, 151.3, 102.7, 99.9, 54.5, 45.1, 31.7, 20.8, 14.5; MS (ESI) m/z (MH+, 243).
1-Butyl-6-formyluracil (11).
A solution of the butylacetal 10 (0.50 g, 2.07 mmol) in 20 mL of CH2Cl2 was cooled to-78 °C and treated with1.6 mL of BBr3 and stirred for2 h at that temperature. The cold bath was removed and the solution was allowed to reach room temperature. Saturated NaH CO3 (15 mL) was added until the p H reached 7–7.5. The organic layer was removed and the aqueous layer was extracted with CH2-Cl2. The extracts were combined and dried (with Na 2SO4), and the solvent was removed to afford 0.315 g of the aldehyde 11 as a white solid (87%), mp 146–147 °C. 1H NMR(CDCl 3), δ 9.57 (s, 1H), 9.18 (br, s, 1H), 6.25 (s, 1H), 4.19 (t, 2H, J =7.3 Hz), 1.63–1.53, (m, 2H), 1.43–1.33, (m, 2H), 0.95, (t, 3H, J = 7.3 Hz); 13C NMR(CDCl 3) δ 186.0, 162.6, 151.5, 147.6, 115.0, 44.5, 32.3, 20.5, 14.4; MS (ESI) m/z (M-H+, 195). Anal. Calcd forC11 H18 N2O4: C, 55.09; H, 6.16; N, 14.28. Found: C, 55.49; H, 6.13; N, 14.25.
5,10,15,20-Tetrakis (3,5-diacetamido-4-pyridyl)porphy-rin (VI).
Pyrrole (69.5 µL, 1.0 mmol) and 2,6-diacetamido-4-formylpyridine 7 (221 mg, 1.0 mmol) were added to boiling propionic acid (10.0 mL). The reaction mixture was refluxed for2 h and then taken to dryness under vacuum. The resulting solid was purified by chromatography on silica gel eluting with ethanol/ethyl acetate (1:1) to yield 77.1 mg (29%) of VI. 1H NMR(DMSO-d6) δ 10.52 (s, 8H), 8.99 (s, 8H), 8.60 (s, 8H), 2.16 (s, 24 H),−3.10, (s, 2H); 13C NMR(DMSO-d6) δ 170.7, 153.2, 150.1, 132.7, 119.4, 116.5, 25.4; MS (ESI) m/z (MH+, 1075 ). Ana l. Calcd for[C56 H50 N16 O8 plus 2 H2O] C, 60.45; H, 4.89; N, 20.16. Foun d: C, 60.92; H, 4.95; N, 20.01.
Diacetamidopyridyl/tert-butylphenyl Porphyrins.
2,6-diacetam ido-4-formylpyridine 7 (2.763 g, 12.5 mmol) and 4-tert-butylbenzaldehyde (1.63 g, 1.68 mL, 10.0 mmol) were added to 450 mL of boiling propionic acid and then 1.56 mL (22.6 mmol) of pyrrole was added. The reaction mixture was refluxed for2 h at which time a 23.8% yield of the mixture of porphyrins was detected spectroscopically. The solvent was removed under vacuum, and the resulting solid was purified by chromatography eluting with hexane (I), to chloroform (II), chloroform/ ethyl acetate (1:1) (III, IV), and ethyl acetate (V, VI) to get the four porphyrins with two different motifs at the meso positions. A second column using 10% v/v dioxane in chloroform is used to purify the fractions containing both III and IV, and typically the tetra substituted derivatives are not isolated as they can be made directly. Based on starting pyrrole, the isolated yield is 20.6%, and the isolated amounts are I, 0.153 g (3.5%); II, 0.261 g (5.14%); III, 0.222 g (4.11%); IV, 0.290 g (5.36%); V, 0.149 g (2.60%); VI, 0.052 g (0.86%). The relative amounts are II (24%), III (20%), IV (26%), and V (11%), and the two homo-substituted porphyrins I (14.5%) and VI (4.5%).
5-(3,5-Diacetamido-4-pyridyl)-10,15,20-tris (4-tert-bu-tylphenyl)porphyrin (II).
1H NMR(CDCl 3) [500 MHz NMR, 50 °C] δ 9.11, 9.05 (d, 2H, J = 5.0 Hz), 8.92 (s, 4H), 8.81 (s, 2H), 8.89 (s, 2H), 8.175, 7.785 (d, 12 H, J = 7.5 Hz), 2.298 (s, 6H), 1.637 (s, 27 H),−2.761 (s, 2H); 13C NMR(DMSO-d6) δ 169.0, 156.1, 151.1, 148.4, 139.7, 135.1, 131.7–131.9, 124.2, 121.6, 121.2, 117.0, 35.7, 32.5, 25.6; MS (ESI) m/z (MH+, 898).
5,15-Bis(3,5-diacetamido-4-py ri dy l)-10,20-bis(4-tert-bu-tylp heny l) po rphy ri n (III).
1H NMR(CDCl 3) δ 9.27, 9.23 (dd, 8H, J = 4.4), 9.15 (s, 4H), 8.49, 8.10 (d, 8H, J = 8.1), 8.35 (s, 4H), 2.62 (s, 12 H), 1.95 (s, 18 H),−2.49 (s, 2H); MS (ESI) m/z (MH+, 957).
5,10-Bis(3,5-diacetamido-4-pyridyl)-15,20-bis(4-tert-bu-tylphenyl) porphyrin (IV).
1H NMR(CDCl 3) [500 MHz NMR, 50 °C] δ 8.98 (s, 2H), 8,95, 8.91 (dd, 4H, J = 5.0 Hz), 8.89 (s, 2H), 8.77 (s, 2H), 7.90 (s, 2H), 8.17, 7.79 (d, 8H, J = 7.5 Hz), 2.29 (s, 6H), 1.64 (s, 27 H),−2.72 (s, 2H); 13 C NMR(DMSO-d6) δ 170.3, 150.9, 149.6, 138.6, 134.7,131 2∼132.6, 124.3, 121.5, 117.9, 115.8, 35.1, 31.9, 24.7; MS (ESI) m/z (MH+, 957).
5,10,15-Tris(3,5-diacetamido-4-pyridyl)-20-(4-tert-bu-tylphenyl) porphyrin (V).
1H NMR(DMSO-d6) δ 10.51 (s, 6H), 8.81∼8.97 (m, 8H), 8.59 (s, 6H), 8.12, 7.77 (dd, 4H, J = 8.1), 2.16 (s, 18 H), 1.51 (s, 18 H),−3.10, (s, 2 H); 13 CNMR(DMSO-d6) δ 170.8, 153.5, 151.4, 150.1, 138.9, 135.2, 132.4∼132.5, 129.9, 129.2, 126.3, 124.7, 122.3, 119.2, 116.4, 35.7, 32.5, 25.4; MS (ESI) m/z (MH+, 1016 ).
5,10,15,20-Tetrakis(1′-butyl-6′-uracyl)porphyrinato-(Zn)(II) (XII).
Pyrrole (69.5µL, 1.0 mmol), 1-butyl-6-formyl-uracil (196 mg, 1.0 mmol) and zinc acetate (109.8 mg, 0.50 mmol) were added to a boiling mixture of acetic acid (7.5 mL) and nitrobenzene (5.0 mL). The reaction mixture was refluxed for10 h while monitoring the yields spectroscopically, and then taken to dryness under vacuum. The resulting solid was purified by chromatography, eluting with ethyl acetate to afford ∼5.1% 12. 1H NMR(DMSO-d6) δ 11.83 (br, s, 4H), 9.50 (m, 8H), 6.26 (s, 4H), 3.56∼3.08 (m, 8H), 1.10∼0.91 (m, 8H), 0.30∼0.08 (m, 8H), 0.00∼−0.60 (m, 12 H); 13 C NMR(CDCl 3) δ 185.0, 163.4, 150.6, 142.3, 110.4, 41.9, 30.3, 20.9, 14.5; MS (ESI) m/z (MH+, 1037 forZnXII). Ana l. Calcd forC52 H52 N12 O8-Zn: C, 60.15; H, 5.05; N, 16.18, forC52 H52 N12 O8Zn with 1 H2O: C, 59.12; H, 5.16; N, 15.90. Foun d: C, 60.04; H, 5.24; N 15.92.
Uracyl/tert-Butylphenyl Porphyrins.
Pyrrole (0.83 mL, 12.0 mmol), 1-butyl-6-formyluracil(1.568 g, 8.0 mmol) and zinc acetate (2.90 g, 13.2 mmol) were added to a boiling mixture of acetic acid (90.0 mL) and nitrobenzene (60.0 mL). After10 min. 4-tert-butylbenzaldehyde (0.652 g, 0.67 mL, 4.0 mmol) was added to the reaction mixture and reflux continued for10 h. A 22.4% overall yield of the mixture of porphyrins was detected spectroscopically in the reaction mixture. The solvent was removed under vacuum. The resulting solid was purified by chromatography using a solvent gradient(isolated yiel d, % yield based on starting pyrrole): starting with hexane I (0.075 g, 2.37%), then chloroform VIII (0.225 g, 6.44%), then chloroform/ethyl acetate (1:1) IX (0.08 g, 2.21%), and X (0.10 g, 2.76%), and ethyl acetate XI (0.02 g, 0.51%), and XII (5 mg, 0.12%) to get the foururacyl/phenyl porphyrins. A second column using a 15% v/v dioxan e/chloroform solution as eluent can be used to separate IX, X, and their rotameric forms. An ∼18%, 0.5 g, isolated yield is obtained for the collection of porphyrins with relative yields of I, 15%; VIII, 45%; IX, 15%; X, 20%; XI, 4%; XII, 1%. Typically the tetra substituted derivatives are not isolated as they can be made directly.
5-(1′-Butyl-6′-u racyl)-10,15,20-tris (4-tert-butylp henyl)-porphyrin (VIII).
1H NMR(CDCl 3) [500 MHz NMR] δ 9.11, 9.05 (dd, 4H, J = 4.5 Hz), 8.92 (s, 4H), 8.81 (s, 1H), 8.12∼8.22, 7.81∼7.84 (m, 12 H), 6.60 (s, 1H), 3.47 (t, 2H, J = 8.0 Hz), 1.20 (m, 2H), 0.41(m, 2H), 0.08(t, 3H, J= 7.5 Hz); 13C NMR(CDCl 3) δ 162.5, 156.9, 151.6, 151.7, 139.4, 135.1, 129.2∼132.6, 124.5, 124.3, 123.56, 122.16, 110.31, 106.81, 47.62, 35.74, 32.48, 31.82, 19.94, 13.65; MS (ESI) m/z (MH+, 873).
5,15-Bis(1′-butyl-6′-uracyl)-10,20-bis(4-tert-butylphenyl)-porphyrinato(Zn)(II) (IX).
1H NMR(DMSO-d6): (IXa) R/3 rotam er(CDCl 3) 11.84 (s, 4H), 9.32, 8.83 (dd, 8H, J = 4.8 Hz), 8.08, 7.81 (2d, 8H, J = 8.2 Hz), 6.21 (s, 2H), 3.20∼3.32 (m, 4H), 1.56 (s, 18 H), 1.05∼1.10 (m, 4H) 0.21∼0.23 (m, 4H),−0.20 (t, 6H, J = 7.33 Hz); MS (ESI) m/z (MH+, 907).
(IXb) RRrotamer(CDCl 3) 11.84(s, 4H), 9.32, 8.82 (dd, 8H, J = 4.76), 8.09, 7.77 (2d, 8H, J = 7.3 Hz), 6.23 (s, 2H), 3.21∼3.34 (m, 4H), 1.54 (s, 18 H), 1.03∼1.08 (m, 4H) 0.31∼0.35 (m, 4H),−0.11 (t, 6H, J = 7.32); MS (ESI) m/z (MH+, 907).
5,10-Bis(1′-butyl-6′-uracyl)-15,20-bis(4-tert-butylphenyl)-porphyrinato (Zn)(II) (X).
1H NMR(DMSO-d6) 11.81 (s, 1H), 9.26, 8.79 (d, 4H, J = 4.8 Hz), 8.75 (s, 4H), 8.12∼7.77 (m, 12 H), 6.27 (s, 2H), 3.22∼3.34 (m, 2H), 1.55 (s, 27 H), 1.01∼1.08 (m, 2H), 0.19∼0.26 (m, 2H),−0.18 (t, 3H, J = 7.32 Hz); 13 C NMR (CDCl3) δ 162.00, 156.10, 151.13, 148.42, 139.75, 135.14, 131.70∼131.88, 124.21, 121.64, 121.15, 116.97, 35.68, 32.48, 25.58; MS (ESI) m/z (MH+, 907).
Supplementary Material
Acknowledgment.
This work was funded by N.S.F. CHE-9732950, PSC-CUN Y-30 grants to C.M.D. We gratefully acknowledge the help of Prof. Joseph Dannenberg for Gaussian 98 calculations, Dr. Michael Blumenstein for high field NMR, Dr. Clifford Soll for MS, and Diana Samaroo for careful reading of the manuscript. The chemistry department infrastructure is partially supported by NIH RCMI program GM3037, and by the NSF for the ESI-MS. The Division of Chemical Sciences, Geosciences and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy, under Contract DE-AC02-98CH10886, supported the work at Brookhaven. We thank Dr. Jonathan C. Hanson for assistance with the crystallographic data collection at beamline X7B of the National Synchrotron Light Source.
References
- (1).(a) Lehn J-M Angew. Chem., Int. Ed. Engl 1990, 29, 1304–1319. [Google Scholar]; (b) Dagan i R Chem., & Eng. News 1998, 76, 35–46. [Google Scholar]
- (2).Lindsey JS New J. Chem 1991, 15, 153–180. [Google Scholar]
- (3).Forreviews on reaction centermodels see: (a) Wasielew ski MR In The Photosynthetic Reaction Center; Deise nhofer J, Norris J, Eds.; Academic Press: New York, 1993. ; Vol. 2, pp 465–511. [Google Scholar]; (b) Gust D; Moore TA In The Porphyrin Handbook; Kadish K, Smith K, Guilard R, Eds.; Academic Press: New York, 2000. ; Vol. 8, pp 153–190. [Google Scholar]
- (4).For examples of various types of discrete multiporphyrinic covalent arrays: (a) Lin VS-Y; DiMagno SG; Therien MJ Science 1994, 264, 1105–1111. [DOI] [PubMed] [Google Scholar]; (b) Wagner RW; Seth J; Yan g SI; Kim D; Bocian DF; Holten D.; Lindsey JS J. Org. Chem 1998, 63, 5042–5049. [Google Scholar]; (c) Lindsey JS; Gust D; Moore TA; Moore AL; Weg horn SJ; John son TE; Lin S; Liddell PA; Kuciau sk as D J. Am. Chem. Soc 1999, 121, 8604–8614. [Google Scholar]; (d) Aratan i N; Os uka A; Kim YH; Jeong DH; Kim D Angew. Chem., Int. Ed 2000, 39, 1458–1462 [DOI] [PubMed] [Google Scholar]; (e) Khoury R; J aquinod L; Nurco DJ; Pandey RK; Senge MO; Smith KM Angew. Chem, Int. Ed. Engl 1996, 35, 2496–2499. [Google Scholar]; (f) Burrell AK; Officer DL Synlett 1998, 1297–1307. [Google Scholar]; (g) Osuka A; Yamazaki I; Nakan o A.; Yama zaki T; Nishimura Y Angew. Chem., Int. Ed 1998, 37, 3023–3027. [DOI] [PubMed] [Google Scholar]; (h) Li J; Lindsey JS J. Org. Chem 1999, 64, 9101–9108. [Google Scholar]; (i) Anderson S; Anderson HL; Sanders JKMJ Chem Soc, Perkin Trans 1 1995, 2247–2254 and references therein. [Google Scholar]
- (5).For examples of discrete multiporphyrinic arrays formed by hydrogen bonding: (a) Dra in CM; Fischer R; Nolen EG; Lehn J-M J. Chem. Soc., Chem. Comm un 1993, 243–245.; (b) Drain CM; Russell KC; Lehn J-M J. Chem. Soc., Chem. Commun 1996, 337–338; (c) Iked a C; Nagahara N; Motegi E; Yoshioka N; Inoue H Chem. Comm un 1999, 1759–1760.; (d) Balaban TS; Eichhoffer A; Lehn J-M Eur. J. Org. Chem 2000, 4047–4057.
- (6).For examples of porphyrin hydrogen bonding in solid-state networks: (a) Daha l S; Goldberg IJ Phys. Org. Chem 2000, 13, 1–6. [Google Scholar]; (b) Bhyrappa P; Wilson SR; Suslick KS J. Am. Chem. Soc 1997, 119, 8492–8502. [Google Scholar]; (c) Diskin-Posner Y; Dahal S; Goldberg I Angew. Chem., Int. Ed. Engl 2000, 39, 1288–1291. [DOI] [PubMed] [Google Scholar]
- (7).For examples of discrete axial coordination arrays: (a) Stibrany RT; Vasudevan J; Knapp S; Potenza JA.; Emge T.; Schugar HJ. J. Am. Chem. Soc 1996, 118, 3980–3981. [Google Scholar]; (b) Knapp S; Vasudevan J; Emge T; Arison BH; Potenza JA; Schugar HJ Angew. Chem., Int. Ed. Engl 1998, 37, 2368–2370. [DOI] [PubMed] [Google Scholar]; (c) Sanders JKM; Bampos N; Mak CC Chem. Commun . 1999, 1086–1086. [Google Scholar]; (d) Chi X; Guerin AJ; Haycock RA; Hunter CA; Sarson LD Chem. Commun 1995, 2563–2565. [Google Scholar]
- (8).For examples of axial coordination polymers or network s: (a) Fleischer EB.; Shachter AM. Inorg. Chem 1991, 30, 3763–3769. [Google Scholar]; (b) Abrahams BF; Hoskins BF; Robson RJ Am. Chem. Soc 1991, 113, 3606–3607. [Google Scholar]; (c) Kumar RK; Balasubramanian S; Goldbe rg I Mol. Cryst. Liq. Cryst 1998, 313, 105–114. [Google Scholar]; (d) Goldberg I Chem. Eur. J 2000, 6, 3863–3870. [DOI] [PubMed] [Google Scholar]
- (9).For examples of discrete arrays formed by exocyclic ligand coordination: (a) Drain CM; Lehn J-M J. Chem. Soc., Chem. Comm un 1994, 2313–2315. [Google Scholar]; (b) Drain CM; Nifiatis F; Vasenko A; Batteas J Angew. Chem., Int. Ed 1998, 37, 2344–2347. [DOI] [PubMed] [Google Scholar]; (c) Yuan H; Thoma s, L.; Woo LK Inorg. Chem 1995, 36, 2808–2817. [Google Scholar]; (d) Slone RV; Hu pp JT Inorg. Chem 1997, 36, 5422–5423. [Google Scholar]; (e) Stang PJ; Fan J; Olenyuk B Chem. Commun 1997, 1453–1454. [Google Scholar]; (f) Fan J; Whiteford JA; Olenyuk B; Levi n MD; Stang PJ; Fleischer EB J. Am. Chem. Soc 1999, 121, 2741–2752. [Google Scholar]
- (10).For examples of hybrid arrays: (a) Burrell AK; Jones BM; Hall SB; Officer DL; Reid DCW; Wild KYJ Incl. Phenom. Macrocycl. Chem 1999, 35, 185–190. [Google Scholar]; (b) Anderson S; Anderson HL; Sanders JK M. Acc. Chem. Res 1993, 26, 469–475. [Google Scholar]; (c) Anderson S; Anderson HL; Bashall A; McPartin M; Sanders JKM Angew. Chem., Int. Ed. Engl 1995, 34, 1096–1099. [Google Scholar]; (d) Funatsu K; Kimura A; Imamura T; Ichimura A; Sasaki Y Inorg. Chem 1997, 36, 1626–1653. [DOI] [PubMed] [Google Scholar]
- (11).For example: (a) Hungerford G; Auweraer MV; Chambron J-C; Heitz V; Sauvage J-P; Pierre J-L; Zurita D Chem. Eur. J 1999, 5(7), 2089–2100. [Google Scholar]; (b) Feiters MC; Fyfe MCT; Martinez-Diaz M-V; Menzer S; Nolte RJM; Stoddart JF; van Kan PJM; Williams DJ J. Am. Chem. Soc 1997, 119, 8119–8120. [Google Scholar]
- (12).(a) Drain CM; Mauzerall D Bioelectrochem. Bioenerg 1990, 24, 263–268. [Google Scholar]; (b) Drain CM; Mauzerall D Biophys. J 1992, 63, 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Drain CM; Christensen B; Mauzerall D Proc. Natl. Acad. Sci., USA 1989, 86, 6959–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Fox MA; Jones WE; Watkins DM Chem. Eng. News 1993, 38–48. [Google Scholar]; (b) Liu C; Pan H; Fox MA; Bard AJ Science 1993, 261, 897–899. [DOI] [PubMed] [Google Scholar]
- (14).Hayashi T; Miyahara T; Koide N; Kato Y; Masuda H; Ogoshi H J. Am. Chem. Soc 1995, 119, 7281–7290. [Google Scholar]
- (15).(a) Stang PJ; Olenyuk B Acc. Chem. Res 1997, 30, 502–518. [Google Scholar]; (b) Burrell AK; Wasielewski MR J. Porph. Phthal 2000, 4, 401–406. [Google Scholar]; (c) Suslick KS; Rakow NA; Kosal ME; Chou J-HJ Porph. Phthal 2000, 4, 407–413. [Google Scholar]; (d) Ph ilp D; Stodd art JF Angew. Chem., Int. Ed. Engl 1996, 35, 1154–1196. [Google Scholar]; (e) Linton B; Hamilton AD Chem. Rev 1997, 97, 1669–1680. [DOI] [PubMed] [Google Scholar]
- (16).(a) Lehn J-M Chem. Eur. J 2000, 6, 2097–2102. [DOI] [PubMed] [Google Scholar]; (b) Berl V; Krische MJ; Huc I; Lehn J-M; Schmutz M Chem. Eur. J 2000, 6, 1938–1946. [DOI] [PubMed] [Google Scholar]
- (17).For example: (a) Sessler JL; Wang B; Harriman A J. Am. Chem. Soc 1995, 117, 704–714. [Google Scholar]; (b) Sessler JL; Wang B; Harriman A J. Am. Chem. Soc 1993, 115, 10418–10419. [Google Scholar]; (c) The synthesis of aldehyde similarto 7 from 2,6-dihydroxyisonicotinic acid in lesseryields, and an deca substituted porphyrin with one diacetamidopyridyl group was heuristically described in: Osuka A; Yoneshima R; Shiratroi H; Okada T; Taniguchi S; Mataga N J. Chem. Soc., Chem. Comm un 1998, 1567–1568. [Google Scholar]; (d) Vollmer MS; Wurthner F; Effenberger F; Emele P; Meyer DU; Stumpfig T; Port H; Wolf HC Chem. Eur. J 1998, 4(2), 260–269. [Google Scholar]; (e) de Rege PJF; Williams SA; Therien MJ Science 1995, 269, 1409–1413. [DOI] [PubMed] [Google Scholar]; (f) Smeets S; Asokan CV; Motman s F; Deha en W J. Org. Chem 2000, 65, 5882–5885. [DOI] [PubMed] [Google Scholar]
- (18).(a) There are two different melting points reported in this reference: one for the recrystallize d material and one for sublimed material, which reverted to the lower melting point on standing. Bernstein, J.; Stearn s B.; Shaw E.; Lott WA. J. Am. Chem. Soc 1947, 69, 9, 1151–1158.20240506 [Google Scholar]; (b) Leffer MT In Organic Reactions; Adams R, Bachmann WE, Fieser LE, Johnson JR, Snyder HR, Eds.; John Wiley and Sons: New York, 1941. ; Vol. 1, pp 91–104. [Google Scholar]; (c) Shreve RN; Riechers EH; Rubenkoenig H; Goodman AH Ind. Eng. Chem 1940, 32, 173–178. [Google Scholar]; (d) 2,6-diamino-4-isoropylpyridine is made in 53% using 4-isopropylpyridine and sodium amide in tetralin K. K. Kogyo, J apanese Patent: Kokai. Tokyo Koho 80 76, 861, 10, June 1980. (CA 93–204, 467 n). [Google Scholar]
- (19).Adams R; Miyano SJ Am. Chem. Soc 1954, 76, 2785–2786. [Google Scholar]
- (20).Fenlon EE; Murray TJ; Baloga MH; Zimmerman SC J. Org. Chem 1993, 58, 6625–6628. [Google Scholar]
- (21).(a) Brossmer R; Ziegler D. Tetrahedron Lett 1966, 5253–5256. [Google Scholar]; (b) Zee-Cheng K-Y; Cheng CC J. Heterocycl. Chem 1967, 4 (1) 163–165. [Google Scholar]; (c) Demuynck M; DeClercq P; Vandewalle MJ Org. Chem 1979, 26, 4863–4866. [Google Scholar]
- (22).(a) Botta M.; DeAngeli s F.; Corelli F.; Menichincheri M.; Nicoletti R.; Marongiu ME.; Pani A.; Colla PL. Arch. Pharm 1991, 324, 203–207. [DOI] [PubMed] [Google Scholar]; (b) Demuynck M; DeClercq P; Vande Walle MJ Org. Chem 1979, 44, 4863–4866. [Google Scholar]; (c) Adams LL; Luzzio FA J. Org. Chem 1989, 54, 5387–5390. [Google Scholar]
- (23).Brown DT; Eisinger J; Leonard NJ J. Am. Chem. Soc 1968, 90, 7302–7306. [DOI] [PubMed] [Google Scholar]
- (24).Adler AD; Longo FR; Finarelli JD; Goldmacher J; Assour J; Korsakoff LJ Org. Chem 1967, 32, 476–480. [Google Scholar]
- (25).(a) Little RG.; Anton JA.; Loach PA; Ibers JA J. Heterocycl. Chem 1975, 12, 343–349. [Google Scholar]; (b) Walker FA; Balke VL; McDermott GA Inorg. Chem 1982, 21, 3342–3348. [Google Scholar]; (c) Milgrom LRJ Chem. Soc., Perkin Trans 1 1984, 1483–1487. [Google Scholar]; (d) John stone RAW; Nunes MLPG; Pereira MM; Gonsalves A. M. d’A R.; Serra AC Heterocycles 1996, 43, 1423–1436. [Google Scholar]; (e) Drain CM; Gong X Chem. Commun 1997, 2117–2118. [Google Scholar]
- (26).(aLindsey JS; Schreiman IC.; Hsu HC.; Kearn ey PC.; Margueretta z AM. J. Org. Chem 1987, 52, 827–836. [Google Scholar]; (b) Gryko D; Lindsey JS J. Org. Chem 2000, 65, 2249–2252. [DOI] [PubMed] [Google Scholar]; (c) Rao PD; Dhanalekshmi S; Littler BJ; Lindsey JS J. Org. Chem 2000, 65, 7323–7344. [DOI] [PubMed] [Google Scholar]
- (27).Collman JP; Gagne RR; Reed CA; Ha lbe rt TR; Lan g G; Robinson WT J. Am. Chem. Soc 1982, 104, 4500–4502. [Google Scholar]
- (28).Lindsey JS In The Porphyrin Handbook; Kadish K, Smith KM, Guilard R, Eds.; Academic Press: San Diego, CA, 2000. ; Vol. 1, pp 45–118 and references therein. [Google Scholar]
- (29).(a) Beijer FH; Sijbe sma RP; Vekemans JAJM; Meijer EW; Kooijman H; Spek AL J. Org. Chem 1996, 61, 6371–6380.(b) Beijer FH; Sijbesma RP; Kooijman H; Spek AL; Meijer EW J. Am. Chem. Soc 2000, 120, 6761–6769.(c) Beijer FH; Kooijman H.; Spek AH.; Sijbesma RP.; Meijer EW. Angew. Chem., Int. Ed. Engl 1998, 37, 75–78.(d) Ercolani GJ Phys. Chem. 1998, 91, 5699–5703.(e) Ercolani G Chem. Commun 2001, June advanced web addition.Note that a ∆G of-7 to-12 k J mol-1 corresponds to 2.8 to 4.9 kBT, thus dynamic processes are expected at room temperature. Though common practice, equilibrium constants for weakly H-bonding systems determined by NMRdata are probably internally comparable, but should be treated with caution unless the dynamics of the system(s) are specifically addressed. Theoretical treatments of equilibrium constants based on simple dimer interactions to predict product distributions in larger systems that neglect cooperativity29d, 29e—especially in aromatic systems such as these—should also be used with caution.
- (30).(a) Drain CM; Gentemann S; Roberts JA; Nelson NY; Medforth CJ; Jia S; Simpson MC; Smith KM; Fajer J; Shelnutt JA; Holten D. J. Am. Chem. Soc 1998, 120, 3781–3791. [Google Scholar]; (b) Drain CM; Kirmaier C; Medforth CJ; Nurco DJ; Smith KM; Holten DJ Phys. Chem 1996, 100, 11984–11993. [Google Scholar]
- (31).Shelnutt JA; Song X-Z; Ma JG; Jia S-L; Jentzen W; Medforth CJ Chem. Soc. Rev 1998, 27, 31–41. [Google Scholar]
- (32).Drain CM; Shi X; Milic T; Nifiatis F Chem. Commun 2001, 287–288.
- (33).(a) DeGrado WF; Lear JD J. Am. Chem. Soc 1985, 107, 7684–7689. [Google Scholar]; (b) Deranleau DA; J. Am. Chem. Soc 1969, 91, 4044–4054. [Google Scholar]; (c) Whitlock BJ; Whitlock HW J. Am. Chem. Soc 1990, 112, 3910–3915. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.