

Abstract
Purpose
Osteopathy/osteoporosis in Gaucher disease type 1 (GD1) shows variable responses to enzyme replacement therapy (ERT); the pathogenesis is incompletely understood. We aimed to investigate the effects of several gene variants on bone mineral density (BMD) and serum markers of bone metabolism in GD1.
Patients and methods
Fifty adult Caucasian patients with GD1/117 controls were genotyped for gene variants in the osteoprotegerin (TNFRSF11B; OPG), estrogen receptor alpha, calcitonin receptor (CALCR), and vitamin D receptor (VDR) genes. In patients and 50 matched healthy controls, we assessed clinical data, serum markers of bone metabolism, and subclinical inflammation. BMD was measured for the first time before/during ERT (median 6.7 years).
Results
Forty-two percent of patients were splenectomized. ERT led to variable improvements in BMD. Distribution of gene variants was comparable between patients/controls. The AA genotype (c.1024+283G>A gene variant; VDR gene) was associated with lower Z scores before ERT vs GA (P=0.033), was encountered in 82.3% of patients with osteoporosis and was more frequent in patients with pathological fractures. Z score increases during ERT were higher in patients with the CC genotype (c.9C>G variant, TNFRSF11B; OPG gene; P=0.003) compared with GC (P=0.003). The CC genotype (c.1340T>C variant, CALCR gene) was associated with higher Z scores before ERT than the TT genotype (P=0.041) and was absent in osteoporosis. Osteocalcin and OPG were lower in patients vs controls; beta crosslaps, interleukin-6, and ferritin were higher.
Conclusions
We suggest for the first time a protective role against osteoporosis in GD1 patients for the CC genotype of the c.9C>G gene variant in the TNFRSFB11 (OPG) gene and for the CC genotype of the c.1340T>C gene variant (CALCR gene), while the AA genotype of the c.1024+283G>A gene variant in the VDR gene appears as a risk factor for lower BMDs. Serum markers suggest decreased osteosynthesis, reduced inhibition of osteoclast activation, increased bone resorption, and subclinical inflammation during ERT.
Keywords: Gaucher disease, gene variants, osteoporosis, vitamin D receptor, osteoprotegerin, calcitonin receptor
Introduction
Gaucher disease (OMIM#230800) is the most frequent lysosomal storage disorder, caused by mutations in the glucocerebrosidase gene and is inherited in an autosomal recessive manner. The deficiency of acid ß-glucocerebrosidase (GBA) leads to the accumulation of unprocessed substrate (glucosylceramides) in the lysosomes of macrophages and explains the multiorgan damage with splenohepatomegaly, thrombocytopenia, anemia, and bone disease.1 Type I Gaucher Disease (GD1), the most common phenotype, can be distinguished from the more severe types 2 and 3 based on the absence of the typical neurologic manifestations associated with the latter two forms. The prevalence of this rare disease varies, based on literature data, between 1/40,000 and 1/1,00,000.2,3
Bone disease, present in most patients with a variable severity, is due to medular infiltration with glucocerebrosides lowden macrophages, which act directly, by mechanical pressure, and indirectly by cytokine-induced inflammation, influencing osteoblastic and osteoclastic activity.4 This includes abnormal bone remodeling, osteopenia, osteoporosis, lytic lesions, and avascular necrosis. Clinical manifestations are bone pain, bone crises, and pathological fractures, with disability and a progressive reduction in the quality of life.5,6
In a report of the International Collaborative Gaucher Group on 181 untreated European patients, lumbar Z scores between <−1 and >−2.5 (osteopenia) were found in 47.2% and Z scores ≤−2.5 (osteoporosis) in 5.6%.7
Intravenous enzyme replacement therapy (ERT) with recombinant glucocerebrosidase leads to an important improvement in organomegaly, thrombocytopenia, anemia, and bone disease.8,9 However, the bone response to ERT is generally slower compared with the response of other clinical manifestations,9–11 and some complications (eg, avascular necrosis) are irreversible.6,12
Among 1,307 European Gaucher patients being treated with ERT (imiglucerase), 27% had osteopenia and 4.5% had osteoporosis.7 Furthermore, bone mineral density (BMD) increase under ERT was age-dependent, with better responses at young ages.13
Genetic variability may also play a role in the BMD of patients with GD1 and influence its response to treatment. Different gene variants of the vitamin D receptor (VDR) gene, estrogen receptor alpha gene (ESR1), collagen 1A1-gene, calcitonin receptor gene (CALCR), osteoprotegerin gene (TNFRSF11B; OPG), and RANK gene (TNFRSF11A) have been associated with reduced BMD in different populations.14–16
To the best of our knowledge, only four prior studies have addressed the issue of gene variants in GD (VDR, ESR2, and TNFRSF11B), three of them in a Jewish population, suggesting an association between the VDR Bsm1 gene variant (NM_000376.2: c.1024+283G>A; rs1544410) and skeletal involvement as well as no evident effect of TNFRSF11B (OPG) genotype on BMD.17–19 However, the allele distribution of the OPG (TNFRSF11B) gene variants was significantly different in the studied populations compared with Caucasians, and the number of controls was far below the number of patients. There is only one recent European retrospective study reporting the effect of gene variants of the estrogen receptor gene (c.453–397T>C) and of the VDR (c.1024+283G>A) on bone mass. However, the cohort included treated and untreated patients, and the evaluation method for BMD was not the same.20 None of these studies evaluates BMD dynamically under ERT.
Changes in bone mass are accompanied by variable alterations of bone turnover markers,6 requiring a better understanding of bone metabolism and its relation to bone manifestations in GD. According to several studies, osteocalcin (OC), OPG, and type I collagen C-terminal telopeptide had reduced or normal values.21–24
Splenectomy, performed prior to the ERT era in severe forms of the GD1, further aggravates bone disease.25,26
Given these limited and partly controversial literature data, we aimed to analyze the influence of genetic variability on BMD, measured at the moment of diagnosis and at the last follow-up visit, as well as biomarkers of bone metabolism and subclinical inflammation, according to splenic status, in a cohort of Romanian patients with GD1 under ERT.
Patients and methods
Design
The study was a monocentric, cross-sectional, retrospective, observational study.
Patients and controls
Among all the 65 patients with GD1 diagnosed, treated, and followed up in Romania, at the Center of Genetic Diseases, 1st Pediatric Clinic of the Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca, we included 50 adult Caucasian patients (19M/31F), median age 40 (26–51) years, who agreed to participate in the study, as specified in the study protocol. The diagnosis of GD1 was confirmed by demonstration of deficient activity of glucocerebrosidase in leucocytes and genotyping.
There were two control groups. The first control group (control group 1) served for the comparison of the frequencies of five variants in four genes with influence on bone metabolism (see below) between our patients and healthy Romanian subjects. It comprised 117 healthy Romanian Caucasian subjects without history of fractures or known osteoporosis and was built from the general population. The other group (control group 2) was selected for the comparison of biomarkers of bone metabolism and subclinical inflammation between patients and healthy subjects. This group included 50 healthy controls, matched for age, sex, body mass index (BMI), nicotine consumption, calcium dietary intake, and exercise practices, as well as the use of oral contraceptives and for pre- and postmenopausal status in women. No subjects in this control group had a history of fractures, known osteoporosis, or used other chronic medication. No patient/control subject received medication for osteoporosis, glucocorticoids, or supplementation with vitamin D.
Patients and controls were included in this exploratory study after getting written informed consent from all participants. The study followed the ethical guidelines of the Declaration of Helsinki and the study protocol was approved by the Medical Ethics Committee of the University of Medicine and Pharmacy, Cluj-Napoca, Romania.
Methods
The following data were registered from medical records: age (at diagnosis, at start of ERT, and at the last visit), duration of ERT up to the last visit, genotype, splenic status, clinical data on bone disease (fractures and bone crises), BMD at the start of ERT, and presence/absence of avascular necrosis. Severity of bone disease was estimated according to the disease severity scoring system.26
All patients received a complete physical examination at the last visit, with calculation of BMI (kg/m2, Seca 702, Seca GmBH & Co, Hamburg, Germany), assessment of BMD, and collection of blood samples for the analysis of gene variants with possible influences on bone metabolism and measurement of serum markers of bone metabolism and subclinical inflammation.
Control subjects received a physical examination. Additionally, blood samples for analysis of gene variants were taken from subjects in control group 1. For control group 2, age, sex, and BMI were registered and blood samples were collected for the measurement of serum markers of bone metabolism and subclinical inflammation.
BMD assessment
Values of BMD were recorded in patients before the start of ERT and at the last visit. BMD (g/cm2) was measured at the lumbar spine (L1–L4) with a dual X-ray absorptiometer (Lunar 8743, DXA Prodigy, Schick Healthcare, Long Island City, NY, US). The instrument was calibrated on a daily basis according to the manufacturer’s instructions. Reproducibility was calculated as coefficient of variation, obtained by weekly measurements of a standard phantom with the instrument. Conversion to Z scores was done in relationship to the mean of an age- and sex-matched reference group provided by the manufacturer. Z scores ≥−1 were considered normal. Z scores <−1 and >−2.5 were considered moderately reduced (osteopenia), while values ≤−2.5 SD were severely reduced (osteoporosis), as accepted in the literature.27 The Z score was registered before the start of ERT, measured during treatment (at the last visit), and the Z-increase under ERT was calculated (delta Z).
Genotyping
The following gene variants were analyzed in patients and controls from the general population (control group 1):
c.9C>G; rs2073618 (G1181C) in the osteoprotegerin gene (TNFRSF11B; OPG), NM_002546.3)
c.453-351A>G; rs9340799 (A351G) and c.453–397T>C; rs2234693 (C397T) in the estrogen receptor gene alpha (ESR1, NM_000125.3)
c.1340T>C; rs1801197 (C1377T) in the calcitonin receptor gene (CALCR, NM_001742.3)
c.1024+283G>A; rs1544410 (G283A) in the vitamin D receptor gene (VDR, NM_000376.2)
Genotyping analysis was performed by using the polymerase chain reaction-restriction fragment length polymorphism technique. Genomic DNA was extracted for all subjects from 300 µL of blood using a commercial kit (Wizard Genomic DNA Purification kit, Promega, Cambridge, MA, USA). After DNA extraction, genotyping of the OPG gene was performed according to Langdahl et al.15 The protocols described by Boroumand et al were used for estrogen receptor gene variants.28 For the CALCR gene c.1340T>C; rs1801197 variant, the genotypes were determined as previously described.29 For the VDR c.1024+283G>A; rs1544410 gene variant, we used a protocol described by Kaya et al.30 The PCR reactions were prepared in a 25 µL reaction volume, containing 12.5 µL 2× PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA), 1 µL BSA (Thermo Fisher Scientific) solution 5 mg/mL; 8 pM of each primer, forward and reverse (Eurogentec, Seraing, Belgium); and 2 µL of genomic DNA and DNase/RNase free water to complete the final volume. After specific amplifications, 12 µL from the amplification products were digested with the appropriate restriction enzymes (SmoI for TNFRSF11B [OPG] gene, FastDigestXbaI and PvuII, respectively, for ESR1 gene variants, FastDigestAluI for the CALCR gene variant, and FastDigest Mva1269I for the VDR gene variant; Thermo Fisher Scientific). The digestion products were resolved in 3% high-resolution agarose gel electrophoresis (MetaPhor, Lonza, Basel, Switzerland) and stained with RedSafe (Chembio Ltd, Rickmansworth, UK) for visualization on a UV transilluminator.
Biomarkers of bone metabolism and subclinical inflammation
Blood samples were taken in a fasting state at 8:00–9:00 am in all patients and controls. Serum was used for all analyses except for OC measurements (EDTA plasma). The following parameters were measured at the Institute for Clinical Chemistry and Laboratory Medicine: calcium (mmol/L), phosphorus (mg/dL), creatinine (mg/dL; Jaffe reaction), alkaline phosphatase (U/L), high-sensitive C-reactive protein (mg/L), iron (µg/dL), and transferrin (ng/mL) were measured on an Abbott ARCHITECT c8000 Clinical Chemistry Analyzer (Abbott, Wiesbaden, Germany) using Abbott reagents. Transferrin saturation (%) was calculated from transferrin and iron serum values. Parathyroid hormone (intact PTH), ferritin, and 25-hydroxy vitamin D (ng/mL) were obtained by chemiluminescent microparticle immunoassay carried out on an Abbott ARCHITECT i2000 immunoanalyzer using Abbott reagents. Serum interleukin-6 (IL-6; pg/mL), C-terminal telopeptide of type 1 collagen (β-CrossLaps; β-CTX; pg/mL), OC (N-MID OC; ng/mL), and N-terminal propeptide of type 1 collagen (total P1NP; µg/L) were measured by electrochemiluminescence immunoassay on a Cobas e 411 Analyzer (Roche Diagnostics GmbH, Mannheim, Germany).
The following parameters were obtained by ELISA and were carried out manually: OPG (pmol/L; Osteoprotegerin ELISA Kit, Immundiagnostik AG, Bensheim, Germany), total soluble RANKL (OPG ligand; pg/mL; total sRANKL [human] ELISA Kit, Immundiagnostik AG), and sclerostin (pmol/L; sclerostin ELISA, Biomedica Medizinprodukte GmbH, Wien, Austria).
Statistical analysis
Quantitative variables were expressed as mean and standard deviations if they followed a normal distribution or as medians and quartiles otherwise. The distribution of continuous variables was assessed with the Shapiro Wilk test and quantile–quantile plots. If the data were normally distributed, we analyzed the equality of variances with the Levene’s test and compared the independent samples using the unpaired t-test. Otherwise, the nonparametric Mann– Whitney U test was used. For comparisons of matched, dependent groups of continuous data, the paired t-test was used for data following the normal distribution; otherwise, the Wilcoxon signed-rank test was used. When performing multiple pairwise comparisons for subgroups regarding quantitative data, we used permutation tests from the coin R package – Conditional Inference Procedures in a Permutation Test Framework with post hoc tests with corrections for multiple testing (Nemenyi–Damico–Wolfe–Dunn post hoc test). Where appropriate, we added a Bonferroni correction for the statistically significant results. The Spearman correlation coefficient was employed to measure the relationships between parameters of bone metabolism and Z scores within the different genotype subgroups. Each gene variant was tested for Hardy–Weinberg equilibrium. Frequencies of gene variants in patients and controls were compared using the Fisher’s exact test. Two-tailed P-values <0.05 were considered statistically significant. Statistical evaluation was performed with the statistical package R environment for statistical computing and graphics, version 3.2.3.31 The statistical methodology was prespecified in the protocol.
Ethics statement
The work followed the ethical standards of national research committee and the 1964 Helsinki declaration and its later amendments or comparable ethical standards. It was approved by the Ethics Committee of the University of Medicine and Pharmacy, Cluj-Napoca, Romania, no 353/8.10.2014. Written informed consent was obtained from all participants included in the study.
Results
The characteristics of patients and controls are shown in Table 1.
Table 1.
Patients’ characteristics, compared to controls
| Characteristics | Patients | Controls (group 2) | P-value |
|---|---|---|---|
| Age (years) | 40 (26–51) | 39 (28–48) | 0.429 |
| Sex (M/F) | 19/31 | 19/31 | 1 |
| Body mass index (kg/m2) | 24.47±3.72 | 24.09±4.23 | 0.196 |
| Age at diagnosis (years) | 32.2 (20.4–40.4) | ||
| Duration of treatment (years) | 6.7 (1.27–8.75) |
The median duration of treatment was 6.7 (1.27–8.75) years. After diagnosis, ERT was started with human recombinant glucocerebrosidase (imiglucerase, Cerezyme, Genzyme Europe B.V., North Holland, the Netherlands) with 30–60 U/kg, according to disease severity, as intravenous infusions every 2 weeks.
Table 2 describes the patients’ characteristics, according to splenic status, genotype of the glucocerebrosidase gene, and specific bone parameters. Bone density values (Z scores) before the start of ERT were −1.81 (−2.62; −0.84) for the whole group. Z score values at the last follow-up visit (under ERT) were −0.5 (−1.1; −0.1), while the mean increase under treatment (delta Z) was 1.3 (0.73; 2.12).
Table 2.
Patients’ characteristics, according to splenic status, genotype of the glucocerebrosidase gene, and specific bone parameters
| Splenectomy: n (%) | 21/50 (42) | With splenectomy (a) (n=21) | Without splenectomy (b) (n=29) | P (a,b) |
|---|---|---|---|---|
|
| ||||
| Genotype: n (%) | ||||
| N409S/N409S | 13 (26) | 2/21 (9.5) | 11/29 (37.9) | 0.047 |
| N409S/L444P | 10 (20) | 5/21 (23.8) | 5/29 (17.2) | 0.723 |
| N409/other alleles | 27 (54) | 14/21 (66.6) | 13/29 (44.4) | 0.157 |
|
| ||||
| Pathological fractures: n (%) | 14/50 (28) | 11/21 (52.3) | 3/29 (10.3) | 0.001 |
|
| ||||
| Bone crises: n (%) | 4/50 (8) | 4/21 (19.0) | 0/29 | 0.025 |
|
| ||||
| BMD (Z score L1–L4) | ||||
| Before ERT | −1.81 (−2.62; −0.84) | −2.07 (−2.87; −1.20) | −1.69 (−2.5; −0.69) | 0.14 |
| Last visit | −0.5 (−1.1; −0.1) | −0.8 (−1; −0.1) | −0.5 (−1.1; −0.4) | 0.46 |
| Delta Z | 1.3 (0.73; 2.12) | 1.43 (0.78; 2.21) | 1.2 (0.6; 2.1) | 0.65 |
|
| ||||
| Avascular necrosis: n (%) | 14/50 (28) | 11/21 (52.3) | 3/29 (10.3) | 0.001 |
|
| ||||
| Severity score for bone disease25 | 4.44±1.86 | 5.64±1.91 | 3.39±1.20 | 0.011 |
Note: Quantitative variables were expressed as means and standard deviations if they followed a normal distribution or as medians and quartiles otherwise. Bold values indicate statistically significant values, P<0.05.
Abbreviations: BMD, bone mineral density; ERT, enzyme replacement therapy.
Before the start of ERT, osteoporosis was registered in 17, osteopenia in 18, and normal BMD values in 15 patients. Osteoporosis disappeared during treatment (34% before ERT vs 0% under ERT, P<0.001), osteopenia decreased from 18 (36%) to 15 (30%; P=0.830), and the percentage of patients with normal BMD significantly increased from 30% (n=15) to 70% (n=35; P<0.001; Figure 1).
Figure 1.
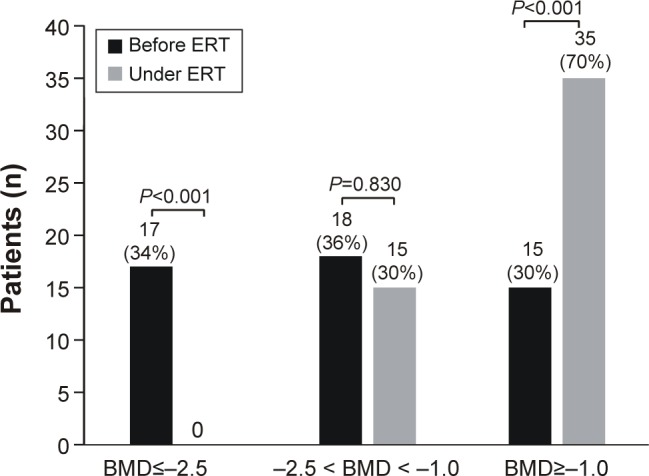
Improvement of BMD under ERT in patients with Gaucher disease type 1.
Abbreviations: BMD, bone mineral density; ERT, enzyme replacement therapy.
The Z score increases under ERT did not correlate with the ERT doses (P=0.090) and did not differ between patients receiving lower doses (<45 UI/kg) compared to those treated with higher doses (≥45 U/kg): 1.38±1.22 vs 1.84±1.38 (P=0.628). Z score increases during ERT showed a negative correlation with age (P=0.029). Delta Z was higher in patients <40 years of age (1.97±1.40) than in patients >40 years (1.13±0.99; P=0.027). Delta Z was independent of sex: 1.23±1.10 in women and 1.84±1.39 in men (P=0.107).
Splenectomy was performed in 42% of our patients. We observed a significantly higher proportion of pathological fractures (P=0.001) and avascular necrosis (P=0.001), as well as significantly higher severity scores for bone disease (P=0.011) in splenectomized patients, compared with those without splenectomy. Bone crises were present only in splenectomized patients (Table 2). Even if Z scores before ERT were lower in patients with splenectomy than in those without, the difference was not statistically relevant (P=0.14).
Glucocerebrosidase genotypes (GBA gene) were N409S (c.[1226A>G])/other alleles, N409S/N409S, and N409S/L444P (c.[1226A>G]; c.[1448T>C]), in 54%, 26%, and 20% of patients. The N409S/N409S genotype was more frequent in patients without splenectomy (P=0.047). In patients with Z scores≤−2.5 before the start of treatment, BMDs were lower in patients with the genotype N409S/L444P compared with those with N409S/N409S (−3.35 [−3.31; −2.63] vs −2.70 [−2.70, −2.63]; P≤0.001). No differences related to the glucocerebrosidase genotype were found in patients with BMDs >−2.5.
Table 3 shows the distribution of the analyzed gene variants in the 50 patients compared to 117 controls from the general population (control group 1). The distribution of the analyzed gene variants was in Hardy–Weinberg equilibrium, except the ESR1 gene variant c.453-351A>G, and did not differ between patients and controls. There were no differences in allele distribution according to splenic status.
Table 3.
Distribution of gene variants (alleles) with implications in bone metabolism in patients, stratified according to splenic status and controls
| Gene | Gene variant/rs identification | Patients | Control group 1, (n=117, 234 alleles) (B) | P A,B OR (95% CI) | HWE (P) | |||
|---|---|---|---|---|---|---|---|---|
| Total (n=50, 100 alleles) (A) | With splenectomy (a) (n=21, 42 alleles) | Without splenectomy (b) (n=29, 58 alleles) | P (a,b) OR (95% CI) | |||||
| TNFRSF11B (OPG) |
c.9C>G rs2073618 |
53G/47C | 21G/21C | 32G/26C | 1 0.96 (0.4–2.29) |
141G/93C | 0.228 0.81 (0.49–1.33) |
0.095 |
| ESR1 | c.453-351A>G rs9340799 |
44G/56A | 19G/23A | 25G/33A | 0.841 0.92 (0.38–2.21) |
107G/127A | 0.810 1.07 (0.65–1.77) |
0.0002* |
| c.453–397T>C rs2234693 |
51C/49T | 17C/25T | 34C/24T | 0.104 0.48 (0.2–1.16) |
106C/128T | 0.282 1.31 (0.8–2.15) |
1 | |
| CALCR | c.1340T>C rs1801197 |
26C/74T | 12C/30T | 14C/44T | 0.649 1.25 (0.46–3.39) |
71C/163T | 0.510 0.81 (0.46–1.4) |
0.064 |
| VDR | c.1024+283G>A rs1544410 |
66A/34G | 28A/14G | 38A/20G | 1 0.95 (0.37–2.38) |
140A/94G | 0.223 0.73 (0.43–1.23) |
1 |
Notes: Bold values indicate statistically significant values, P<0.05.
Bonferroni correction for statistically significant results, corrected for the analyses in the two groups, separately for each gene variant.
Abbreviations: CALCR, calcitonin receptor gene; ESR1, estrogen receptor-α gene; HWE, Hardy–Weinberg equilibrium; OPG, osteoprotegerin gene; OR, odds ratio; rs, reference single nucleotide polymorphisms cluster; VDR, vitamin D receptor gene.
Table 4 presents the bone density values expressed as Z scores before the start of ERT (Z pre-ERT), at the last follow-up visit (Z under ERT), and the difference between these two values (delta Z), according to the analyzed gene variants, and the P-values resulting from the pairwise comparison of the subgroups.
Table 4.
Comparison between the bone densities (Z score) of patients before start of ERT (Z pre-ERT), at the moment of evaluation (Z under ERT), and the Z score increase under ERT (delta Z), depending on the analyzed gene variants of the osteoprotegerin, estrogen receptor 1, calcitonin receptor, and vitamin D receptor genes
| TNFRSF11B (OPG) (c.9C>G; rs2073618) | ||||||
|---|---|---|---|---|---|---|
| GG (n=17) | GC (n=19) | CC (n=14) | P(GG,GC) | P(GG,CC) | P(GC,CC) | |
| Z pre-ERT | −1.8 (−2.2; −0.73) | −1.9 (−2.48; −0.59) | −2.69 (−3.09; −2.29) | 0.98 | 0.32 | 0.35 |
| Z under ERT | −0.1 (−0.95; 0.03) | −0.8 (−1.1; 0.1) | −0.3 (−0,4; 0.3) | 0.88 | 0.99 | 0.18 |
| Delta Z | 1.11 (1.95; 2.70) | 1.10 (0.6; 1.7) | 2.89 (2.54; 3.04) | 0.991 | 0.093 | 0.003* |
| ESR1 (c.453–351A>G; rs 9340799) | ||||||
| AA (n=9) | AG (n=38) | GG (n=3) | P(AA,AG) | P(AA,GG) | P(AG,GG) | |
| Z pre-ERT | −2.10 (−2.65; −1.75) | −1.86 (−2.57; −0.85) | −3.28 (−3.56; −2.99) | 0.88 | 0.30 | 0.17 |
| Z under ERT | 0.0 (−1.00; 0.05) | −0.75 (−1.10; 0.12) | −0.40 (−0.45; −0.35) | 0.98 | 0.6 | 0.38 |
| Delta Z | 1.70 (0.74; 2.00) | 1.18 (0.76; 1.78) | 2.88 (2.54; 3.21) | 0.95 | 0.31 | 0.15 |
| ESR1 (c.453–397T>C; rs 2234693) | ||||||
| CC (n=13) | CT (n=25) | TT (n=12) | P(CC,CT) | P(CC,TT) | P(CT,TT) | |
| Z pre-ERT | −1.81 (−2.62; −0.27) | −2.08 (−2.57; −1.59) | −1.40 (−2.71; −0.28) | 0.90 | 1.00 | 0.81 |
| Z under ERT | −0.10 (−0.95; 0.25) | −0.65 (−1.02; 0.18) | −0.90 (−1.50; −0.35) | 0.89 | 0.41 | 0.60 |
| Delta Z | 1.27 (0.67; 1.75) | 1.42 (0.97; 2.52) | 0.85 (−0.35; 1.58) | 0.68 | 0.53 | 0.30 |
| CALCR (c.1340T>C; rs 1801197) | ||||||
| CC (n=6) | CT (n=14) | TT (n=30) | P(CC,CT) | P(CC,TT) | P(CT,TT) | |
| Z pre-ERT | −0.89 (−1.38; −0.59) | −1.7 (−2.83; −1.4) | −2.16 (−2.65; −1.50) | 0.240 | 0.041* | 0.998 |
| Z under ERT | −0.25 (−0.73; 1.12) | −0.30 (−0.90; 0.20) | −0.80 (−1.15; 0.05) | 0.89 | 0.40 | 0.62 |
| Delta Z | 0.88 (0.18; 1.83) | 1.60 (1.10; 2.59) | 1.16 (0.73; 2.03) | 0.67 | 0.93 | 0.60 |
| VDR (c.1024+283G>A; rs1544410) | ||||||
| GG (n=6) | GA (n=22) | AA (n=22) | P(GG,GA) | P(GG,AA) | P(GA,AA) | |
| Z pre-ERT | −1.85 (−2.61; −1.05) | −1.47 (−1.81; −0.45) | −2.60 (−2.83; −2.07) | 0.734 | 0.689 | 0.033* |
| Z under ERT | −0.65 (−0.8; −0.12) | −0.40 (−1.10; 0.28) | −0.60 (−1.30; 0.00) | 0.970 | 0.870 | 0.750 |
| Delta Z | 1.94 (1.05; 2.95) | 1.13 (0.50; 1.54) | 1.70 (0.80; 2.63) | 0.580 | 0.990 | 0.120 |
Notes: Values are given as medians and interquartile ranges. Bold values indicate statistically significant values, P<0.05. All P-values for each genotype were corrected for multiple testing;
Bonferroni correction was added for statistically significant P-values (corrected for the three variables that were assessed per each genotype).
Abbreviations: CALCR, calcitonin receptor gene; delta Z, difference between Z score at present and Z score before the start of ERT; ERT, enzyme replacement therapy; ESR1, estrogen receptor α gene; TNFRSF11B (OPG), osteoprotegerin gene; VDR, vitamin D receptor gene.
For the TNFRSF11B (OPG) gene variant c.9C>G (rs2073618), patients with the CC genotype had lower Z score values before the start of ERT compared with the GC and GG genotypes; however, the differences were not significant (median −2.6 vs −1.9 and −1.8, respectively, P=0.35 and P=0.32, respectively). However, patients with the CC genotype showed a higher increase in bone density under ERT (delta Z) compared with patients with the GC genotype (P=0.003). The distribution of the CC genotypes was comparable among patients <40 years (4/19) and patients .40 years (9/27; P=0.509).
Regarding the CALCR gene variant c.1340T>C (rs 1801197), patients with the CC genotype showed higher Z scores before ERT compared with the TT genotype (P=0.041). The CC genotype of this gene variant was absent in all patients with osteoporosis.
Patients with AA genotype of the c.1024+283G>A (rs1544410) gene variant of VDR had lower Z scores before the start of ERT compared with patients with the GA genotype (P=0.033).
When stratifying the patients according to BMD before ERT, we observed that the AA genotype of the c.1024+283G>A (rs1544410) gene variant of VDR, encountered in 82.3% of patients with osteoporosis, was seen in only 27.8% of patients with osteopenia and in only 20% of patients with normal BMD (P=0.001). Compared to the AA genotype, the GG and the GA genotypes were present in only 11.7% and 5.8% of patients with osteoporosis, respectively (P=0.006). This difference holds true independent of the splenic status (Table 5). The severity score for bone disease in patients with the AA genotype (4.86±1.87) was tendentially higher than the score of the GA genotype (3.87±1.53); however, the difference was not significant (P=0.06, data not shown).
Table 5.
Distribution of the genotypes of the c.1024+283 G>A variant of VDR gene in patients with Gaucher disease, stratified according to BMD before ERT and to splenic status
| BMD | n | Genotypes: n (%) | P-value | ||||
|---|---|---|---|---|---|---|---|
| GG | GA | AA | |||||
| All patients (n=50) | |||||||
| OP | 17 | 2/17 (11.7) | 1/17 (5.8) | 14/17 (82.3) | 0.006* | }0.001 | |
| O | 18 | 2/18 (11.1) | 11/18 (61.1) | 5/18 (27.8) | |||
| N | 15 | 2/15 (13.3) | 10/1 (66.6) | 3/15 (20) | |||
| Total | 50 | 6 (12) | 22 (44) | 22 (44) | |||
| Patients with splenectomy (n=2l) | |||||||
| OP | 8 | 2/8 (25) | 0/8 (0) | 6/8 (75) | 0.011* | }0.06# | |
| O | 8 | 1/8 (12.5) | 5/8 (62.5) | 2/8 (25) | |||
| N | 5 | 0/5 (0) | 3/5 (60) | 2/5 (40) | |||
| Total | 21 | 3 (14.2) | 8 (38.1) | 10 (47.6) | |||
| Patients without splenectomy (n=29) | |||||||
| OP | 9 | 0/9 (0) | 1/9 (11.1) | 8/9 (88.8) | 0.03* | }0.006# | |
| O | 10 | 1/10 (10) | 6/10 (60) | 3/10 (30) | |||
| N | 10 | 2/10 (20) | 7/10 (70) | 1/10 (10) | |||
| Total | 29 | 3 (10.3) | 14 (48.3) | 12 (41.3) | |||
Notes: Bold values indicate statistically significant values, P<0.05.
Bonferroni-corrected P-value for BMD subgroup analysis (P × 3);
Bonferroni-corrected P-value for splenectomy subgroup analysis (P × 2).
Abbreviations: BMD, bone mineral density; ERT, enzyme replacement therapy; N, normal BMD; O, osteopenia; OP, osteoporosis.
Furthermore, we analyzed the distribution of genotypes according to bone crises, pathological fractures, avascular necrosis, and severity score for bone disease. We found that the AA genotype of the VDR gene was significantly more often encountered in patients with pathological fractures than in those without (P=0.004). Among patients with pathological fractures, 9 had an AA genotype vs 2 and 3 with the G/A and the G/G genotypes, respectively, while in patients without pathological fractures the genotype distribution was 3, 20, and 13 for the AA, GA, and GG genotypes, respectively. No further statistically relevant relationships were found between the analyzed gene variants and these clinical parameters of bone disease.
Table 6 presents the values of different markers of bone metabolism and subclinical inflammation for comparison between patients and controls. We observed significantly lower values for OC and OPG and higher values of ßcrosslaps, IL-6, and ferritin in patients compared with controls. There were no differences according to splenic status, except for higher concentrations of alkaline phosphatase and transferrin saturation in splenectomized patients.
Table 6.
Biomarkers of bone metabolism and subclinical inflammation
| Patients (A) | Control group 2 (B) | P-value (A,B) | Patients with splenectomy (a) | Patients without splenectomy (b) | P-value (a,b) | |
|---|---|---|---|---|---|---|
| Calcium (mmol/L) | 2.46 (2.36; 2.53) | 2.43 (2.34; 2.50) | 0.085 | 2.49 (2.36–2.56) | 2.45 (2.38–2.49) | 0.36 |
| Phosphate (mg/dL) | 3.8 (3.6; 4.2) | 3.9 (3.6; 4.1) | 0.897 | 4 (3.7–4.4) | 3.8 (3.5–4.2) | 0.447 |
| Total alkaline phosphatase (U/L) | 59 (49–68) | 63 (52; 80) | 0.056 | 69 (55–79) | 54 (45–62) | 0.002 |
| Parathormone intact (pg/mL) | 44.2 (35.8; 55.7) | 44.3 (36.9; 58.2) | 0.942 | 48.1 (33.9–60.1) | 44.2 (38.1–53.1) | 0.569 |
| 25-hydroxy-vitamin D (ng/mL) | 24.6 (18.8; 28.3) | 19.8 (17.6; 28.4) | 0.208 | 22.3 (18.4–26.2) | 24.6 (20.4–29.5) | 0.219 |
| OC (ng/mL) | 16.8 (13.1; 21.9) | 20.6 (17.3; 24.7) | 0.011 | 16.8 (13.8–24) | 16.8 (13.1–21.5) | 0.609 |
| P1NP (µL/L) | 59 (42; 73) | 56 (42; 76) | 0.886 | 65 (46–79) | 52 (42–63) | 0.135 |
| OPG (pmol/L) | 4.7±2.8 | 6.2±2.7 | 0.009 | 5.6±2.9 | 4.1±2.5 | 0.068 |
| Total soluble RANKL (pg/mL) | 49,250 (20,317; 1,06,000) | 54,910 (14,510–79,125) | 0.976 | 65,590 (23,200–99,500) | 46,950 (19,700–1,34,000) | 0.953 |
| C-terminal telopeptide of type 1 collagen (ß-CTX, ß-CrossLaps) (pg/mL) | 390 (273; 518) | 318 (256; 364) | 0.009 | 423 (240–518) | 390 (276–555) | 0.922 |
| Sclerostin (pmol/L) | 35 (29; 43) | 32 (27; 47) | 0.956 | 36 (32–46) | 34 (29–39) | 0.201 |
| hsCRP (mg/L) | 1.34 (0.49; 2.53) | 1.2 (0.5; 1.52) | 0.134 | 1.59 (0.74–2.53) | 1.09 (0.45–2.44) | 0.345 |
| IL-6 (pg/mL) | 2 (2–3) | 2 (2–2) | 0.008 | 2 (2–3) | 2 (2–2) | 0.7 |
| Iron (µg/dL) | 107.1±43.5 | 109.1±32.5 | 0.607 | 115 (90–147) | 93 (70–130) | 0.075 |
| Ferritin (ng/mL) | 251 (72; 476) | 36 (20; 106) | <0.001 | 252 (165–689) | 181 (44–383) | 0.13 |
| Transferrin (g/L) | 2.52 (2.27; 2.8) | 2.82 (2.49; 2.91) | 0.140 | 2.48 (2.31–2.79) | 2.55 (2.26–2.83) | 0.665 |
| Transferrin saturation (%) | 26.6 (22.5; 42.1) | 26.3 (23.3; 40.2) | 0.802 | 32.9 (24.9–44) | 23.9 (19.9–36.4) | 0.046 |
Note: Quantitative variables were expressed as means and standard deviations if they followed a normal distribution or as medians and quartiles otherwise. Bold values indicate statistically significant values, P<0.05.
Abbreviations: hsCRP, high-sensitive C-reactive protein; IL-6, interleukin-6; OC, osteocalcin; OPG, osteoprotegerin; P1NP, N-terminal propeptide of type 1 collagen.
For OPG, OC, ßcrosslaps, IL-6, and ferritin, no specific relationships with bone density and the analyzed genotypes could be detected. No significant correlation between the individual values of OC, OPG, ßcrosslaps, ferritin, and IL-6 could be found (data not shown).
Discussion
Splenectomy, performed in 42% of our patients, was indicated in the past, when ERT was not available and worsened bone disease, as previously reported.25,26 The GBA N409S/L444P genotype was associated with the lowest BMD values before ERT in patients with osteoporosis (P<0.001), while the N409S/N409S was more frequently encountered in nonsplenectomized patients (P=0.047), possibly due to the less severe resulting phenotype. Furthermore, younger ages were associated with a better BMD increase under ERT, as previously described.13
Beyond these factors, genetic variability plays a role in bone mass determination and the response of bone disease to ERT. Gene variants in the VDR gene and the ESR1 gene account for 1%–18.7% of changes in BMD in Caucasian post-menopausal women under hormone replacement therapy.32 Gene variants of the CALCR are associated with BMD at the femoral neck in Spanish postmenopausal women.14 Regarding the c.9C>G (rs2073618) gene variant of TNFRSF11B (OPG), Langdahl et al showed that the CC genotype was less common among fracture patients (26.3%) compared with normal controls (36.7%).15 A meta-analysis showed that the GG genotype of this variant was associated with lower lumbar BMD in Europeans and Asians, while lower femoral neck and total hip BMDs were found in Europeans only.33
Few data exist regarding the effect of genetic variability on bone mass in patients with GD. Most of them were not obtained from European populations and the number of controls was lower than the number of patients.17–19 As shown above, the only study on Caucasian patients, reporting an impact of gene variants on BMD, comes from Spain. In this study, BMD was evaluated by two different methods and the group was not homogenous regarding treatment.20
In the present study, we analyzed a group of 50 Romanian patients with GD1, who were also Caucasians, but with a different genetic background than Spanish patients. All patients received ERT. BMD was measured by the same method in all patients before and while under ERT and the controls had the same ethnicity. We evaluated five gene variants from four genes, which had previously been associated with bone metabolism,14,15,32,33 and analyzed our findings in relationship to BMD before and during ERT and according to the splenic status.
We found significant results for three of the five gene variants: c.1024+283G>A in the VDR gene; c.9C>G in the TNFRSF11B (OPG) gene, and c.1340T>C in the CALCR gene.
VDR belongs to the superfamily of nuclear receptors, which regulate gene expression in a ligand-dependent manner and herewith plays a central role in the metabolism of vitamin D. Binding of the active vitamin D (1α,25-dihydroxy-vitamin D3) to its receptor leads to facilitation of intestinal calcium absorption and osteoblast differentiation, allowing a normal bone mineralization and remodeling.34
The gene variant VDR c.1024+283C>A has been evaluated previously in two studies. Greenwood et al found in a group of Ashkenazi Jewish patients with GD a weak association of this gene variant with BMD (P=0.084).17 The second study on Caucasian patients from Spain described a protective role of the AA genotype against osteoporosis.20
In contrast to these findings, the AA genotype of the c1024+283 gene variant in the VDR gene presented as a risk factor for bone disease in our patients. The AA genotype was associated with the lowest BMD values before ERT (P[AA, GA]=0.033; Table 4), displayed the highest frequency among patients with osteoporosis (P=0.006), was less frequently encountered in those with osteopenia and normal BMD (P=0.001, Table 5), and was more frequently identified in patients with pathological fractures (P=0.004). Patients with the AA genotype had tendentially higher severity scores for bone disease.
The TNFRSFB11 (OPG) codes for OPG, a member of the TNF receptors superfamily. It inhibits osteoclastogenesis and protects the bone against excessive resorption.35
In our patients, the CC genotype of the c.9C>G gene variant in TNFRSFB11 was associated with the highest bone mass increase under ERT (delta Z; P(CC,GC)=0.003; Table 4), suggesting a protective role of the CC genotype concerning bone mass, independent of age. This finding is consistent with previous findings in Caucasian patients with GD1, in whom the same genotype has been found to be associated with a less severe or absent bone disease on MRI.20
The CALCR gene codes for the calcitonin receptor, which is implicated in the calcium homeostasis and the regulation of osteoclast mediated bone resorption. To the best of our knowledge, there are no previous reports on the effect of gene variants in CALCR on bone mass in GD1.
The CC genotype of the c.1340T>C variant of the CALCR gene was associated in our patients with the highest BMD values before ERT (pCC, TT=0.041; Table 4) and was missing in patients with osteoporosis. This finding suggests a protective role of the CC genotype in CALCR against osteoporosis in these patients.
The analysis of the c.453–397T>C of ESR1 in our patients did not confirm a protective effect of the TT genotype on bone mass, as previously reported by another study.20
As an expression of incomplete skeletal response under treatment, we observed significantly lower values of OC and OPG in patients compared with controls, suggesting a reduced osteoblastic bone formation and a reduced inhibition of osteoclast activation. Increased bone resorption is suggested also by the elevated serum concentration of ßcrosslaps. These findings are placed in the context of a relevant subclinical inflammation with increased concentrations for IL-6 and ferritin, as an expression of macrophage dysfunction in GD1 (Table 6). Here, we performed many assessments, and thus some of the statistically significant results in the analysis of biochemical parameters might have been spurious due to a multiple comparison issue.
The interpretation of these findings based on literature is difficult, since other studies, analyzing markers of bone turnover in GD1, included a smaller number of patients, who were untreated or were not separately analyzed according to treatment status, and thus the published results are discordant. Our results are in line with some previous data, reporting lower values for OC,21,22 for OPG,23 and increased concentrations for beta-CTX.21–24 However, other studies described normal values for OC,24 OPG,19 and for beta-CTX.22
The markers of bone metabolism did not correlate with BMD in our patients, as previously reported by Magal et al for OPG.19
The storage of glucosylceramide in macrophages produces an inflammatory response with iron recycling deregulation and release of cytokines. Gaucher mesenchymal stromal cells have a marked increase in COX-2, prostaglandin E2, interleukin-8, and CCL2 production compared with normal controls.36 Iron homeostasis is controlled by the circulating peptide hepcidin, and its production is influenced by inflammatory cytokines. Serum ferritin was increased in 54 patients before ERT, with a significant decrease and partial normalization under ERT, while iron, transferrin, and transferrin saturation were normal.37 In our study, ferritin values remained higher than those found in healthy controls even under ERT, and IL-6 levels were significantly increased, while iron, transferrin, and transferrin saturation did not differ from controls. Further evidence of subclinical inflammation in GD comes from recent data describing increased values of osteopontin, a protein identified in bone cells and produced by T-cells and macrophages in untreated GD patients, with normalization under ERT.38
An important limitation of our study concerning the markers of bone metabolism is that values before the start of ERT were not available. However, the serological changes observed in our patients, after a median of 6.7 years of ERT, are in accordance with previous data, which describe a slower and often incomplete response of bone disease under ERT, compared with the improvement of anemia, thrombocytopenia, and visceromegaly.9,10,26,39 A further limitation of the study is the lack of external or internal validation, based on the small number of patients. Due to the low prevalence of this rare disease, a larger cohort was not available in our country at the time of the study. To clarify the significance of markers of bone turnover for bone response under ERT in GD1 and for validation of the results from this exploratory work, further prospective studies on a larger number of patients in different countries are needed.
Conclusion
We provide the first evidence about genetic variability of bone density in a representative cohort of Romanian patients with GD1. This is the second study on this topic in Caucasian patients with this disease and the first in which BMD has been measured before and while under ERT.
We suggest for the first time a protective role against osteoporosis in GD1 patients for the CC genotype of the c.9C>G gene variant in the TNFRSFB11 (OPG) gene and for the CC genotype of the c.1340T>C gene variant in the CALCR gene, while the AA genotype of the c.1024+283G>A gene variant in the VDR gene seems to be a risk factor for lower BMD values and not a protective one, as previously reported.
Even if we registered a good skeletal outcome after a median of 6.7 years of ERT, with disappearance of osteoporosis, the cluster of modified biomarkers suggests a reduced bone formation, a reduced inhibition of osteoclast activation, and an increased bone resorption, in the context of subclinical inflammation. Splenectomy worsened bone disease.
These data add to the knowledge about the relationship between genetic variability, bone density, and serum biomarkers of bone metabolism in Caucasian patients with GD1 treated with ERT, with new insights regarding the role of gene variants in the VDR, the TNFRSFB11 (OPG), and the CALCR genes.
Acknowledgments
The manuscript has been checked for spelling and grammar by a professional English editing service. We thank Mrs Adriana Degreif for excellent technical assistance. This work was supported by a research grant from SC Sanofi-Aventis Romania to the Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania. The grant covered purchasing chemicals for genotyping and measurement of serum markers of bone metabolism. The project was designed as a scientific cooperation between the Center of Genetic Diseases, 1st Pediatric Clinic of the Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania (PG-S) and the 1st Medical Clinic and Polyclinic, Department of Endocrinology and Metabolic Diseases of the University of Mainz (AZ). The funding source was not involved in study design, data collection, conduct of research, analysis and interpretation of data, writing of the manuscript, and in the decision to submit the article for publication.
Footnotes
Author contributions
AZ designed the study and prepared the first draft of the paper. RAP, HR, SB, IN, and MMW contributed to data acquisition. DL was responsible for statistical analysis of the data. PG-S was also responsible for conception and design and is the guarantor. All authors revised the paper critically for important intellectual content and approved the final version. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors confirm independence from the sponsors. The content of the article has not been influenced by the sponsors. PG-S discloses that she received a research grant designed as cooperation between the University of Medicine and Pharmacy Cluj and the University of Mainz and speaker fees at National Symposia on Lysosomal Diseases sponsored by Sanofi-Genzyme. There is no relationship to any own or company-associated patents and no nonfinancial competing interest. The authors report no other conflicts of interest in this work.
References
- 1.Beutler E, Grabowski G. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Vaile D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3635–3668. [Google Scholar]
- 2.Mehta A. Epidemiology and natural history of Gaucher’s disease. Eur J Intern Med. 2006;17(Suppl):S2–S5. doi: 10.1016/j.ejim.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Belmatoug N, Stirnemann J. Gaucher disease type 1. Orphanet. 2012. [Accessed October 4, 2018]. Available from: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=11102&Disease_Disease_Search_diseaseGroup=Gaucher-disease-type-1.
- 4.Masi L, Brandi ML. Gaucher disease: the role of the specialist on metabolic bone diseases. Clin Cases Miner Bone Metab. 2015;12(2):165–169. doi: 10.11138/ccmbm/2015.12.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.vom Dahl S, Poll L, di Rocco M, et al. Evidence-based recommendations for monitoring bone disease and the response to enzyme replacement therapy in Gaucher patients. Curr Med Res Opin. 2006;22(6):1045–1064. doi: 10.1185/030079906X104623. [DOI] [PubMed] [Google Scholar]
- 6.Giuffrida G, Cingari MR, Parrinello N, et al. Bone turnover markers in patients with type 1 Gaucher disease. Hematol Rep. 2012;4(4):e21. doi: 10.4081/hr.2012.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.I.C.G.G. (International Collaborative Gaucher Group) [homepage on the Internet] Gaucher Registry. Annual Report. 2014. [Accessed July 15, 2017]. Available from: http://www.registrynxt.com.
- 8.Charrow J, Scott CR. Long-term treatment outcomes in Gaucher disease. Am J Hematol. 2015;90(Suppl 1):S19–S24. doi: 10.1002/ajh.24056. [DOI] [PubMed] [Google Scholar]
- 9.Serratrice C, Carballo S, Serratrice J, Stirnemann J. Imiglucerase in the management of Gaucher disease type 1: an evidence-based review of its place in therapy. Core Evid. 2016;11:37–47. doi: 10.2147/CE.S93717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wenstrup RJ, Kacena KA, Kaplan P, et al. Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res. 2007;22(1):119–126. doi: 10.1359/jbmr.061004. [DOI] [PubMed] [Google Scholar]
- 11.Grigorescu Sido P, Drugan C, Creţ V, et al. Outcome of enzyme replacement therapy in patients with Gaucher disease type I. The Romanian experience. J Inherit Metab Dis. 2007;30(5):783–789. doi: 10.1007/s10545-007-0621-z. [DOI] [PubMed] [Google Scholar]
- 12.Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine. 2011;90(1):52–60. doi: 10.1097/MD.0b013e3182057be4. [DOI] [PubMed] [Google Scholar]
- 13.Mistry PK, Weinreb NJ, Kaplan P, Cole JA, Gwosdow AR, Hangartner T. Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol Dis. 2011;46(1):66–72. doi: 10.1016/j.bcmd.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandrés E, Pombo I, González-Huarriz M, Rebollo A, López G, García-Foncillas J. Association between bone mineral density and polymorphisms of the VDR, ERalpha, COL1A1 and CTR genes in Spanish postmenopausal women. J Endocrinol Invest. 2005;28(4):312–321. doi: 10.1007/BF03347196. [DOI] [PubMed] [Google Scholar]
- 15.Langdahl BL, Carstens M, Stenkjaer L, Eriksen EF. Polymorphisms in the osteoprotegerin gene are associated with osteoporotic fractures. J Bone Miner Res. 2002;17(7):1245–1255. doi: 10.1359/jbmr.2002.17.7.1245. [DOI] [PubMed] [Google Scholar]
- 16.Roshandel D, Holliday KL, EMAS Study Group Genetic variation in the RANKL/RANK/OPG signaling pathway is associated with bone turnover and bone mineral density in men. J Bone Miner Res. 2010;25(8):1830–1838. doi: 10.1002/jbmr.78. [DOI] [PubMed] [Google Scholar]
- 17.Greenwood A, Elstein D, Zimran A, Altarescu G. Effect of vitamin D receptor (VDR) genotypes on the risk for osteoporosis in type 1 Gaucher disease. Clin Rheumatol. 2010;29(9):1037–1041. doi: 10.1007/s10067-010-1464-9. [DOI] [PubMed] [Google Scholar]
- 18.Arnheim E, Chicco G, Phillips M, et al. Molecular aspects of osteopathy in type 1 Gaucher disease: correlation between genetics and bone density. Rheumatol Int. 2008;28(9):873–877. doi: 10.1007/s00296-008-0550-7. [DOI] [PubMed] [Google Scholar]
- 19.Magal I, Lebel E, Altarescu G, et al. Serum levels of osteoprotegerin and osteoprotegerin polymorphisms in Gaucher disease. Br J Haematol. 2006;133(1):93–97. doi: 10.1111/j.1365-2141.2006.05978.x. [DOI] [PubMed] [Google Scholar]
- 20.Gervas-Arruga J, Cebolla JJ, de Blas I, Roca M, Pocovi M, Giraldo P. The influence of genetic variability and proinflammatory status on the development of bone disease in patients with Gaucher disease. PLoS One. 2015;10(5):e0126153. doi: 10.1371/journal.pone.0126153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drugan C, Jebeleanu G, Grigorescu-Sido P, Caillaud C, Craciun AM. Biochemical markers of bone turnover as tools in the evaluation of skeletal involvement in patients with type 1 Gaucher disease. Blood Cells Mol Dis. 2002;28(1):13–20. doi: 10.1006/bcmd.2001.0479. [DOI] [PubMed] [Google Scholar]
- 22.van Dussen L, Lips P, Everts VE, et al. Markers of bone turnover in Gaucher disease: modeling the evolution of bone disease. J Clin Endocrinol Metab. 2011;96(7):2194–2205. doi: 10.1210/jc.2011-0162. [DOI] [PubMed] [Google Scholar]
- 23.Vargiami E, Dimitradou M, Economou M, Cristoforidis A, Zaferiou DI. Long-term response in biochemical markers of bone turnover during enzyme replacement therapy in a case-series of patients with Gaucher disease type 1 from Northern Greece. Hyppokratia. 2016;20(2):153–159. [PMC free article] [PubMed] [Google Scholar]
- 24.Ciana G, Martini C, Leopaldi A, et al. Bone marker alterations in patients with type 1 Gaucher disease. Calcif Tissue Int. 2003;72(3):185–189. doi: 10.1007/s00223-001-2072-0. [DOI] [PubMed] [Google Scholar]
- 25.Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis. 2008;31(3):319–336. doi: 10.1007/s10545-008-0779-z. [DOI] [PubMed] [Google Scholar]
- 26.Weinreb NJ, Cappellini MD, Cox TM, et al. A validated disease severity scoring system for adults with type 1 Gaucher disease. Genet Med. 2010;12(1):44–51. doi: 10.1097/GIM.0b013e3181c39194. [DOI] [PubMed] [Google Scholar]
- 27.I.C.G.G. (International Collaborative Gaucher Group) Gaucher Registry. Annual Report. 2006. [Accessed July 15, 2017]. Available from: http://www.lsdregystry.net/gaucherregistry.
- 28.Boroumand M, Ghasemi Y, Shirani S, et al. Association between estrogen receptor-α PvuII and XbaI gene polymorphisms with extracranial carotid stenosis. Lab Med. 2011;42(11):663–667. [Google Scholar]
- 29.Mittal RD, Bid HK, Manchanda PK, Kapoor R. Predisposition of genetic polymorphism with the risk of urolithiasis. Indian J Clin Biochem. 2008;23(2):106–116. doi: 10.1007/s12291-008-0027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaya TI, Erdal ME, Tursen U, et al. Association between vitamin D receptor gene polymorphism and psoriasis among the Turkish population. Arch Dermatol Res. 2002;294(6):286–289. doi: 10.1007/s00403-002-0326-y. [DOI] [PubMed] [Google Scholar]
- 31.R Core Team [homepage on the Internet] R: A language and environment for statistical computing. Vienna, Austria: 2016. [Accessed on June 15, 2017]. Available from: http://www.r-project.org. [Google Scholar]
- 32.Deng HW, Li J, Li JL, et al. Change of bone mass in postmenopausal Caucasian women with and without hormone replacement therapy is associated with vitamin D receptor and estrogen receptor genotypes. Hum Genet. 1998;103(5):576–585. doi: 10.1007/s004390050872. [DOI] [PubMed] [Google Scholar]
- 33.Lee YH, Woo JH, Choi SJ, Ji JD, Song GG. Associations between osteoprotegerin polymorphisms and bone mineral density: A meta-analysis. Mol Biol Rep. 2010;31(1):227–234. doi: 10.1007/s11033-009-9637-9. [DOI] [PubMed] [Google Scholar]
- 34.Haussler MR, Whitfield GK, Haussler CA, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13(3):325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 35.Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 36.Campeau PM, Rafei M, Boivin MN, Sun Y, Grabowski GA, Galipeau J. Characterization of Gaucher disease bone marrow mesenchymal stromal cells reveals an altered inflammatory secretome. Blood. 2009;114(15):3181–3190. doi: 10.1182/blood-2009-02-205708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mekinian A, Stirnemann J, Belmatoug N, et al. Ferritinemia during type 1 Gaucher disease: mechanisms and progression under treatment. Blood Cells Mol Dis. 2012;49(1):53–57. doi: 10.1016/j.bcmd.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 38.Vairo F, Sperb-Ludwig F, Wilke M, et al. Osteopontin: a potential biomarker of Gaucher disease. Ann Hematol. 2015;94(7):1119–1125. doi: 10.1007/s00277-015-2354-7. [DOI] [PubMed] [Google Scholar]
- 39.Giraldo P, Pérez-López J, Núñez R, et al. Patients with type 1 Gaucher disease in Spain: a cross-sectional evaluation of health status. Blood Cells Mol Dis. 2016;56(1):23–30. doi: 10.1016/j.bcmd.2015.10.001. [DOI] [PubMed] [Google Scholar]