

Abstract
Natural and synthetic electrophilic compounds have been shown to activate the antioxidant protective Nrf2 (nuclear factor erythroid 2‐related factor 2)/heme oxygenase‐1 (HO‐1) axis in cells and tissues. Here, we tested the ability of different isoxazoline‐based electrophiles to up‐regulate Nrf2/HO‐1. The potency of activation is dependent on the leaving group at the 3‐position of the isoxazoline nucleus, and an additional ring on the molecule limits the Nrf2/HO‐1 activating properties. Among the synthetized compounds, we identified 3‐bromo‐5‐phenyl‐4,5‐dihydroisoxazole 1 as the derivative with best activating properties in THP‐1 human monocytic cells. We have confirmed that the target of our compounds is the Cys151 of the BTB domain of Keap1 by using mass spectrometry analyses and X‐ray crystallography. Our findings demonstrate that these compounds affect the Nrf2/HO‐1 axis and highlight a positive activity that can be of relevance from a therapeutic perspective in inflammation and infection.
Keywords: 3-bromo-4,5-dihydroisoxazole; crystal structure; cycloaddition; Keap1; mass spectrometry
1. Introduction
The Nrf2 (nuclear factor erythroid 2‐related factor 2) transcription factor plays a key role in cellular stress response mechanisms, controlling the transcription of more than 1000 genes involved in detoxification, anti‐oxidant, metabolic and anti‐inflammatory activities.1, 2, 3 Under unstressed conditions, Nrf2 is retained in the cytoplasm in a silent form by its repressor protein, Keap‐1 (Kelch‐like ECH‐associated protein 1), which contains a subset of 27 highly reactive cysteine (Cys) residues in Human. Oxidative and environmental stimuli modify the cysteine residues of Keap‐1, thus enabling translocation of Nrf2 to the nucleus where it binds to the antioxidant responsive element (ARE) located in the promoter region of detoxifying and protective genes. Keap‐1 also controls Nrf2 by mediating its ubiquitination. Among Nrf2 dependent genes, Heme oxygenase‐1 (HO‐1) is one of the enzymes controlled by Nrf2 that converts the heme to biliverdin, iron and carbon monoxide (CO), important antioxidant and signaling molecules active during stress conditions.4 Therefore, the Nrf2/HO‐1 axis has been shown to be very important for tissue protection and is currently the target of drug discovery approaches.
A successful strategy used to disrupt the Keap1‐Nrf2 interaction consists in using electrophiles from synthetic or natural sources. Bardoxolone methyl (CDDO‐Me, Figure 1) is a potent activator of Nrf2 and like the fumarates is thought to act via reaction with Cys151 of the Keap‐1 protein.5, 6 In addition to these compounds, other electrophilic modulators of the Keap1‐Nrf2 pathway are known including curcumin, 4‐hydroxy‐2‐nonenal, and ebselen (Figure 1). In this context, one of the most studied compounds is sulforaphane, a naturally occurring isothiocyanate isolated from cruciferous vegetables.7–1
Figure 1.
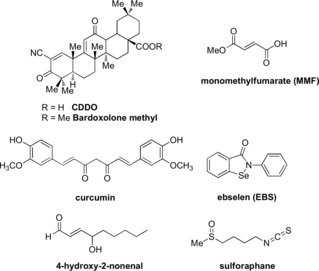
Electrophilic modulators of the Keap1‐Nrf2 pathway.
Numerous electrophiles (e.g., acrylamides and other α,β‐unsaturated groups, boronic acids, and α‐halogen‐substituted ketones) have been widely employed to design enzymatic covalent inhibitors. Some functional groups such as epoxides, aziridines, halomethyl‐ and acyloxymethyl‐ketones are often regarded as too reactive for potential drug applications. Nonetheless, examples of selective and relatively safe inhibitors bearing those functionalities exist (e.g., Fosfomycin).11, 12, 13 The 3‐halo‐4,5‐dihydroisoxazole warhead can be considered a rare electrophile in therapeutics since it reacts solely with cysteine residues activated by surrounding amino acid residues present in the catalytic site of a number of enzymes. Such a peculiarity has already been used to design efficacious inhibitors of different enzymatic targets, ranging from parasitic14, 15, 16, 17 and bacterial enzymes18, 19 to human targets20 involved in the modulation of neuronal metabolic pathways or in tumor cell metabolism.21 In this context, we tested different isoxazoline‐based electrophiles (Figure 2) to examine their effect on the Nrf2/HO‐1 axis.
Figure 2.
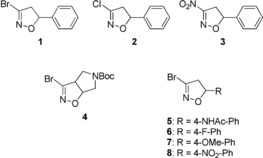
Isoxazoline‐based electrophiles.
Here the influence of different leaving groups in position 3, i.e., bromine, chlorine and nitro (compounds 1–3, Figure 2) and of the substituents in position 4 and 5 of the isoxazoline ring have been investigated (compounds 4–5, Figure 2). The more interesting derivatives in terms of cytotoxic activity, i.e., compounds 1 and 5, and other three para‐phenyl substituted analogues, i.e., compounds 6–8, have been submitted to a mass spectrometry analysis to get the proof of concept of the covalent addition to Keap1‐BTB domain (Broad complex, Tramtrack, and Bric‐à‐Brac domain). Subsequently, in order to determine the covalent addition of the ligands to Cys151 of Keap1‐BTB domain, the structure of the complex between Keap1‐BTB and 5 was solved by X‐ray crystallography. Cysteine‐151, in fact, represents one of the major sensors in KEAP1 and its modification is sufficient for robust activation of NRF2.22
2. Results and Discussion
2.1. Chemistry
The synthesis of derivatives 1, 2 and 4–8 was based on the 1,3‐dipolar cycloaddition of bromonitrile oxide, generated in situ by dehydrohalogenation of the stable precursor dibromoformaldoxime (DBF)23 to the suitable dipolarophile, affording in all cases the 5‐substituted 3‐bromo‐isoxazoline derivative in very high yield. Derivatives 1, 2, 4 and 7 were previously described by us,24, 25, 26 compound 3 was obtained following a literature procedure27 whereas derivatives 5, 6 and 8 were obtained according to Scheme 1.
Scheme 1.
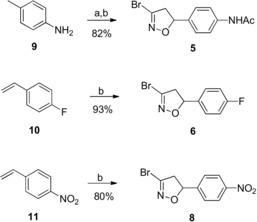
Reagents and conditions: a) NaOAc, Ac2O, AcOH; b) DBF, NaHCO3, EtOAc, room temperature.
2.2. Effects on Nrf2 Activation and HO‐1 Expression
Electrophilic compounds can exert cytotoxic activity and we first examined the viability of human monocytic THP‐1 cells exposed to increasing concentrations of the different compounds. As shown in Figure 3, compounds 1, 2, 4 and 5 did not cause evident cytotoxicity up to 100 μm. Only compound 3 promoted cell toxicity (34 %) at the highest concentration of 100 μm; such a profile could be due to the presence of the nitro group which can be reduced to a toxic hydroxylamine derivative. It is noted that there are many compounds that will induce Nrf2‐HO‐1 at concentrations lower than those used in the present study3, 4 but there are also examples of already approved drugs, such as dimethylfumarate (DMF), that can be used in the higher micromolar range (50–100) to affect Nrf2 due to their low cytotoxicity profile.5 On the contrary, CDDO is very powerful at nanomolar range but it is also very toxic at low micromolar concentrations. Thus, the ability of compounds to induce the Nrf2 system needs to be balanced with their intrinsic cytotoxicity. When tested in Western blot experiments, we observed that compounds 1, 3 and 5 increased Nrf2 and HO‐1 protein expression at different extents with compound 1 being the most efficient inducer of both proteins (Figure 4 a). In contrast, compound 2 increased Nrf2 but not HO‐1 protein (Figure 4 b) while compound 4 did not affect Nrf2 or HO‐1 expression (data not shown). The fact that compound 2 activated Nrf2 but did not induce HO‐1 expression is not understood at present. The activation of Nrf2 by derivatives 1, 2, 3 and 5 is strictly related to the nature of the leaving group appended at the 3 position of the isoxazoline nucleus. As previously observed on different targets,16, 17, 25 the 3‐bromo‐isoxazoline warhead is more active than the 3‐chloro counterparts. Finally, the additional ring present in derivative 4 seems to limit the Nrf2/HO‐1 activating properties.
Figure 3.
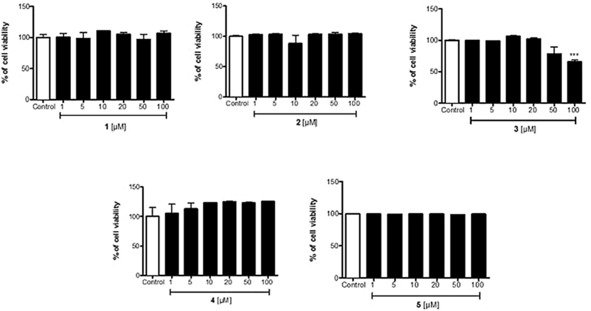
Cytotoxicity data of compounds 1–5. Human monocytic THP‐1 cells were exposed for 24 h to increasing concentrations of the different compounds and assessed for cell viability using the LDH assay, as described in the Experimental Section. Results are mean ± SEM of three independent experiments.
Figure 4.
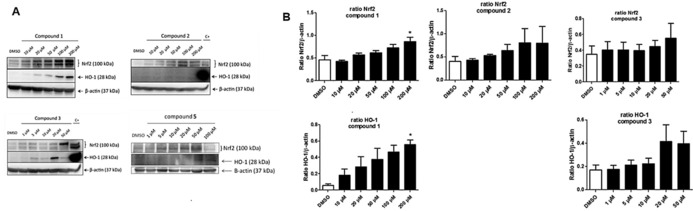
Compound 1 is the most efficient Nrf2 and HO‐1 activator in THP‐1 cells. A) Western blot results of THP‐1 cells treated for 6 h with the different compounds. B) Densitometric analysis of Western blots. C+ is the positive control obtained from THP‐1 cells incubated for 6 h with 100 μm DMF. Results are mean ± SEM of three independent experiments; * p<0.05 vs. control.
2.3. Mass Spectrometry Analysis
To confirm that the target of our compounds is the BTB domain of Keap1, we purified recombinant protein and investigated by mass spectrometry its covalent modification in the absence or presence of 1, 5, 6, 7 or 8. The BTB domain comprises three of the 27 cysteines of Keap1, including Cys151 as the critical stress sensor.22 The mass for the protein incubated with DMSO was determined to be 15,190.9 Da in good agreement (within <30 ppm) to its calculated mass of 15,190.4645 Da (Figure 5 A). Incubation of Keap1 BTB with our compounds resulted in the emergence of a peak with a higher mass confirming covalent modification of the protein by addition of a single compound molecule. For all compounds but 1, the most abundant species corresponds to the derivatized protein with the expected mass (Figure 5 B). For all compounds, however, an additional peak is observed with a mass that fits to a degradation product in which the phenyl substituent in position 5 of the isoxazoline ring was eliminated (Figure 5 C). Variable amounts of the unmodified protein, intact protein adduct and protein modified with degradation product are observed with the different compounds (Figure 5 B).
Figure 5.
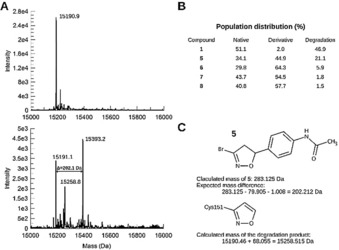
The isoxazoline covalently modifies a single cysteine in the BTB domain of Keap1. A) Deconvoluted intact protein mass spectrum of the native protein (top) and after treatment with 5 (bottom). The masses determined by MS are in good agreement with the theoretical mass for the native protein (15 190.46 Da) and with that of the protein after covalent modification of a cysteine residue of the protein by compounds. B) Population distribution between native unmodified protein, protein modified by the intact compound, and modified protein after elimination of the aryl substituent at position‐5 of the isoxazoline moiety. C) The theoretical and calculated masses are given for the example of the reaction of BTB with 5 and for the degradation product common to all compounds (Figure 5 B).
2.4. Crystallographic Studies
Kelch‐like ECH‐associated protein 1 (Keap1) controls Nrf2 activation by binding Nrf2 in the cytoplasm and inducing its degradation by the proteasome. Keap 1 contains a number of cysteins that react with oxidants and electrophiles, thus inducing modification in the Keap1 protein that prevents the binding to and degradation of Nrf2. As a consequence, Nrf2 accumulates and enters the nucleus to up‐regulate its target genes.28 The Cys151 residue in the Keap1 protein has been shown to be essential for Nrf2 activation by several electrophiles.29 To confirm Cys151 modification and to identify further interactions and mechanistic consequences, we solved the structure of the Keap1‐BTB domain bound to 5 by X‐ray crystallography (Table 1).
Table 1.
Data collection and refinement statistics.[a] Keap1 BTB domain in complex with 5.
| Wavelength [Å] | 0.91814 |
| Resolution range [Å] | 36.8–2.2 (2.28–2.20) |
| Space group | P 65 2 2 |
| Unit cell | a=42.491, b=42.491, c=265.213 |
| Total reflections | 47 296 (4 795) |
| Unique reflections | 7 968 (752) |
| Multiplicity | 5.9 (6.4) |
| Completeness [%] | 98.73 (99.08) |
| Mean I/σ(I) | 9.33 (0.57) |
| Wilson B‐factor [Å2] | 50.25 |
| R‐meas | 0.149 (3.161) |
| CC 1/2 | 1.00 (0.33) |
| Reflections used in refinement | 7 946 (750) |
| R‐work | 0.2506 (0.4153) |
| R‐free | 0.2919 (0.3979) |
| No. non‐hydrogen atoms | 1 085 |
| No. macromolecule atoms | 1 052 |
| No. ligand atoms | 15 |
| No. solvent molecules | 18 |
| No. protein residues | 131 |
| RMS(bonds) [Å] | 0.002 |
| RMS(angles) [°] | 0.40 |
| Ramachandran favored [%] | 96.90 |
| Ramachandran outliers [%] | 0.00 |
| Rotamer outliers [%] | 0.00 |
| Average B‐factor [Å2] | 71.21 |
| Average macromolecules | 71.63 |
| Average ligands | 59.33 |
| Average solvent molecules | 56.54 |
[a] Values for the highest‐resolution shell are shown in parentheses.
The structure comprises residues 48 to 180, which adopt a conformation very similar to the previously reported Keap1‐BTB structures (root mean square deviations on the position of 121 to 131 Cα range from 0.38 to 0.53 Å).6, 30 . The presence of continuous electron density extending from the Sγ atom of Cys 151 shows the presence of covalently bound ligand (Figure 6). Residual difference density extending from Cys151 further suggests that the cysteine side‐chain points in at least two different directions, which could either, indicate that the compound induces a change in orientation or that the ligand induces extreme flexibility and is present in multiple conformations rather than adopting a unique one.
Figure 6.
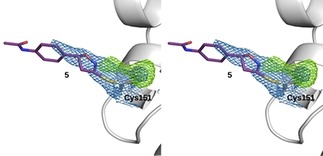
Crystal structure of the Keap1/5 complex (PDB ID 6FFM): Stereoscopic view of the region of Cys151 covalently bound to 5 (purple). The backbone and residue 151 of the BTB domain are shown in white with two types of electron density maps. A feature‐enhanced map of type 2mFo‐DFc in blue is contoured at 1sigma and a Fourier difference omit map (mFo–DFc) of the ligand is presented in green contoured at 3sigma. Both maps support the presence of the covalently bound inhibitor and suggest the existence of 2 to 3 alternative conformations of the cysteine side chain.
The electron density, however, does not cover the ligand completely and the aryl substituent in position 5 of the isoxazoline moiety is only partially defined, the signal fading away from the chiral centre onward. This can be explained by a certain flexibility of this part and/or by the presence of multiple conformations. This is consistent both with the absence of interaction with the protein and with the coexistence of two enantiomers of 5. This limited and rather unspecific interactions between Keap1‐BTB and a covalent inhibitor of Cys151 is, however, not uncommon since it was observed in most other published complex structures.6, 30 The major conformation of 5 that is observed here is consistent with previous Keap1 Cys151 modifier conformations (Figure 7) and, since the isoxazoline substituent is flexible and not significantly involved in binding, is representative of complexes between Keap1‐BTB and our other isooxazoline compounds, including the most potent inhibitor 1 (Sup. Figure 1).
Figure 7.
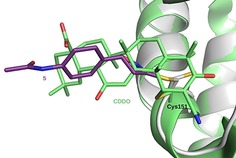
Superposition of the BTB/5 complex to the complex with CDDO. The structure of the BTB/5 complex (white/purple) (PDB ID 6FFM) was superposed to the BTB/CDDO complex structure (light green, PDB ID 4CXT) based on 101 Cα positions (rmsd 0.221 Å).6
3. Conclusions
Our work demonstrates that certain isoxazoline‐based electrophiles, and especially bromo‐based derivatives such as compound 1, are good activators of the Nrf2/HO‐1 protective system. It appears that the potency of activation is dependent on leaving group appended at the 3 position of the isoxazoline nucleus and that an additional ring on the molecule, such as in the case of compound 4, limits the Nrf2/HO‐1 activating properties. The ability of compound 5 to covalently bind the Keap1‐BTB domain at Cys151 indicates the most likely cysteine target that is critical for allowing the modulation of Nrf2 expression in cells. However, we cannot exclude that other cysteine residues of Keap1 are modified by our compound and contribute to functional effects. It is interesting to note that isoxazoline derivatives have been already shown to inactivate glyceraldehydes‐3‐phosphate dehydrogenase from Plasmodium falciparum.25, 26⋅Therefore, our findings that these compounds also affect Nrf2/HO‐1 highlight an additional positive activity for these derivatives that can be of relevance from a therapeutic perspective in inflammation and infection.
Experimental Section
Materials and Methods
All reagents were purchased from Sigma. 1H NMR and 13C NMR spectra were recorded with a Varian Mercury 300 (300 MHz) spectrometer. Chemical shifts (δ) are expressed in ppm, and coupling constants (J) are expressed in Hz. TLC analyses were performed on commercial silica gel 60 F254 aluminium sheets; spots were further evidenced by spraying with a dilute alkaline potassium permanganate solution or ninhydrin. Flash chromatography separations were performed on Büchi Pump Manager C‐615 and C‐601 instruments. Melting points were determined on a model B 540 Büchi apparatus and are uncorrected. Microanalyses (C, H, N) of new compounds were within ±0.4 % of theoretical values.
General Procedure for the Cycloaddition Reaction
Solid NaHCO3 (417 mg, 4.96 mmol) and DBF (402 mg, 1.98 mmol) were added to a solution of the appropriate alkene (0.99 mmol) in CH2Cl2 (4.2 mL). The reaction mixture was stirred at room temperature for 48 h and the progress of the reaction was monitored by TLC. Water was added and the organic layer was separated, dried over anhydrous Na2SO4 and evaporated under reduced pressure. Purification of the crude material was performed by column chromatography.
N‐(4‐(3‐Bromo‐4,5‐dihydroisoxazol‐5‐yl)phenyl)acetamide (5)
a) 4‐Vinylaniline (200 μL, 1.71 mmol) was dissolved in AcOH (0.5 mL) and NaOAc (34 mg, 0.51 mmol) and Ac2O (157 μL, 1.71 mmol) were added. The reaction was stirred at room temperature for 30 min. Water (5 mL) was added and the solution was extracted with EtOAc (3×3 mL). The organic phase was dried over anhydrous Na2SO4 and evaporated under reduced pressure. Purification of the crude material by column chromatography (cyclohexane/EtOAc 7:3) afforded the protected aniline (160 mg, 58 % yield). b) Compound 5 was synthesized following the general procedure for the cycloaddition reaction reported above using the acetamide prepared in the previous step.
Yield: 82 %; 1H NMR (300 MHz, CDCl3): 2.18 (s, 3 H), 3.18 (dd, J=9.2, 17.3, 1 H), 3.59 (dd, J=10.7, 17.3, 1 H), 5.63 (dd, J=9.2, 10.7, 1 H), 7.20 (bs, 1 H), 7.30 (d, J=8.5, 2 H), 7.52 (d, J=8.5, 2 H); 13C NMR (75 MHz, CDCl3): 24.8, 42.2, 83.2, 120.4, 127.0, 135.0, 137.1, 138.7, 168.8; Anal. calcd for C11H11BrN2O2: C, 46.66; H, 3.92; N, 9.89; found: C, 47.00; H, 3.99; N, 9.80.
1. ‐Bromo‐5‐(4‐fluorophenyl)‐4,5‐dihydroisoxazole (6)
Compound 6 was synthesized following the general procedure for the cycloaddition reaction reported above.
Yield: 93 %; white solid; crystallized as white needles from n‐hexane; mp=59.2–59.9 °C; R f=0.37 (cyclohexane/EtOAc, 9:1); 1H NMR (300 MHz, CDCl3): δ=3.18 (dd, J=9.1, 17.3, 1 H) 3.61 (dd, J=10.7, 17.3, 1 H), 5.66 (dd, J=9.1, 10.7, 1 H), 7.05–7.15 (m, 2 H), 7.30–7.40 (m, 2 H); 13C NMR (75 MHz, CDCl3): δ=163.1 (d, JC‐F=247.9), 136.9, 135.3 (d, JC‐F=3.1), 128.1 (d, JC‐F=8.3), 116.1 (d, JC‐F=21.7), 82.8, 49.4; MS(ESI): 243.9 [M+H]+. Anal. calcd for C9H7BrFNO: C, 44.29; H, 2.89; N, 5.74; found: C, 44.50; H, 2.99; N, 5.60.
2. ‐Bromo‐5‐(4‐nitrophenyl)‐4,5‐dihydroisoxazole (8)
Compound 8 was synthesized following the general procedure for the cycloaddition reaction reported above.
Yield: 80 %; solid; crystallized as white needles; mp=101.1 −101.5 °C; R f=0.41 (cyclohexane/EtOAc, 8:2); 1H NMR (300 MHz, CDCl3): δ=3.17 (dd, J=8.2, 17.3, 1 H), 3.74 (dd, J=11.1, 17.3, 1 H), 5.78 (dd, J=8.2, 11.1, 1 H), 7.50–7.58 (m, 2 H), 8.22–8.30 (m, 2 H); 13C NMR (75 MHz, CDCl3): δ=148.2, 146.8, 136.9, 126.9, 124.4, 81.9, 49.6; MS(ESI): 270.8 [M+H]+. Anal. calcd for C9H7BrN2O3: C, 39.88; H, 2.60; N, 10.33; found: C, 40.00; H, 2.71; N, 9.98.
Cell culture and Determination of Cell Viability
The human monocytic cell line THP‐1 was cultured under standard conditions in RPMI medium (Gibco) supplemented with 10 % fetal bovine serum, 1 % sodium pyruvate (Gibco) and 1 % penicillin/streptomycin (Gibco). One million cells/well were used for viability assay studies. THP‐1 cells were incubated for 24 h with increasing concentrations (1–100 μm) of the compounds 1, 2, 3, 4 and 5 and solvent control (0.1 % DMSO). At the end of the incubation the cell supernatant was collected and lactate dehydrogenase (LDH) activity was measured as an index of cell toxicity according to the manufacturer's instructions (Roche). Only damaged cells release this enzyme in the supernatant and the level of LDH activity correlates with the extent of cellular damage. Triton 2 % was used as a positive control. Results were expressed as the percentage of cell viability.
Western Blot Analysis for Determination of Nrf2 and HO‐1 Protein Expression
Based on the toxicity results, the ability of 1, 2, 4 and 5 to induce Nrf2/HO‐1 was assessed at concentrations up to 100 or 200 μm. Compound 3, which exhibited significant toxicity at 100 μm, was tested only up to 50 μm. THP‐1 cells were incubated with the compounds for 6 h after which induction of Nrf2 and HO‐1 proteins by Western blot was assessed using specific antibodies. Briefly, cells were washed in cold PBS before lysis in lysis buffer (20 mm Tris pH 7.4, 137 mm NaCl, 2 mm EDTA pH 7.4, 1 % Triton, 25 mm β‐glycerophophate, 1 mm Na3VO4, 2 mm sodium pyrophosphate, 10 % glycerol, 1 mm PMSF, 1 % mammalian protease inhibitor). The homogenates were centrifugated at 15,000 rpm for 20 min at 4 °C. Equal amounts of denaturated protein were loaded onto 10 % SDS‐PAGE gel and transferred on PVDF membrane (Amersham Biosciences, Les Ulis, France). Membranes were then incubated with antibodies directed against Nrf2 (H‐300, Santa Cruz biotechnology, Santa Cruz, USA, 1:1000 dilution in Tris buffer saline Tween 0.2 %, overnight incubation at 4 °C) or HO‐1 (Abcam, UK, 1:1000 dilution in 3 % bovine serum albumin solution prepared with Tris buffer saline‐Tween 0.2 %, 2 h incubation at room temperature). Secondary antibodies were anti‐rabbit‐HRP (7074S, Cell Signaling, 1:2500 dilution in 3 % bovine serum albumin solution prepared with Tris buffer saline‐Tween 0.2 %) for Nrf2 detection and anti‐mouse‐HRP (7076S, Cell Signaling, 1:5000 dilution in 3 % bovine serum albumin solution prepared with Tris buffer saline‐Tween 0.2 %) for HO‐1, incubated for 1 h at room temperature. The wash buffer was Tris buffer saline‐Tween 0.2 %. Immunoreactive bands were detected by chemiluminescence (ECL solution, Amersham Biosciences, Les Ulis, France). β‐actin (Santa Cruz biotechnology, Santa Cruz, USA) was used as a loading control. Images were captured using a G:Box F3 imagery station (Syngene, Cambridge, UK).
Recombinant Expression, Purification and Crystal Structure Determination of the BTB Domain of Keap1
The gene encoding residues 48–180 of human Keap1 (Uniprot Q14 145) were subcloned in a modified pET19 vector to incorporate a TEV cleavable N‐terminal hexahistidine tag. As previously reported, a single point mutation was also introduced at position 172 (S172A), which enhanced protein stability and crystallizability.6 The protein was expressed in E. coli BL21 (DE3) and purified to homogeneity as previously described.6 Prior to crystallization, the BTB domain at 10 mg mL−1 concentration was incubated for 1 h on ice in the presence of 3 mm 5 (10 % final DMSO concentration). Crystallization was carried out by hanging drop vapour diffusion by mixing equal amounts of protein solution with a crystallization solution composed of 25–28 % PEG 4000, 0.1 m Tris‐HCl pH 8.0 and 0.2 m lithium sulfate. For cryoprotection, the crystals were briefly soaked in a solution composed of the original crystallization mother liquor supplemented with 25 % glycerol. Diffraction data have been collected on BL14.1 operated by the Helmholtz‐Zentrum Berlin (HZB) at the BESSY II electron storage ring (Berlin‐Adlershof, Germany)31 and processed using the XDS package.32 The structure was solved by molecular replacement using Phaser33 and the previously published structure of the BTB domain of Keap1 as a model (PDB code 4CXI). Refinement of the structure was performed using phenix.refine34 until convergence of the R factors. The statistics of the diffraction data as well as of the final model are presented in Table 1. The coordinates of the model as well as the structure factors were deposited to the PDB under accession code 6FFM.
Mass Spectrometry Measurement of the Intact Protein Masses
To analyze covalent modification(s) of the BTB domain, 10 μm BTB 48–180 (S172A) were incubated with 3 mm 1, 5, 6, 7 or 8 (or 3 % DMSO final concentration for control) for 1 h on ice in 20 mm Tris‐HCl pH 8.0 and 150 mm NaCl. Intact protein masses were determined through HPLC‐coupled electrospray ionization (ESI)‐MS on an AB sciex TripleTOF 5600+ mass spectrometer. Intact proteins were first concentrated and washed on a Piccolo Proto 200 C4 5‐μm 2.5×0.5‐mm trap column (Higgins Analytical) and subsequently switched in line with, and separated on, a Jupiter C4 5‐μm 300‐Å 150×1‐mm analytical column (Phenomenex) mounted onto a Shimadzu Prominence UFLC (Shimadzu) at a 70 μL min−1 flow rate with the following buffers: A−5 % ACN, 5 % DMSO, and 0.1 % FA; B−90 % ACN, 5 % DMSO, and 0.1 % FA. Proteins were then eluted over with a gradient of 3 min of 1 % B to 55 % B followed by 1 min of 55 % B to 90 % B. Mass analysis was performed by ESI‐TOF‐MS on an AB Sciex TripleTOF 5600+ mass spectrometer (Sciex) with a DuoSpray Ion Source with the following settings: floating voltage of 5,500 V, temperature of 350 °C, declustering potential of 120 with four separate TOF experiments, each, respectively with 4, 12, 20, and 40 time bins summed. Data analysis was performed as follows: spectra were integrated over a retention time period, and the summed TOF experiment with the greatest resolution selected. The raw data were then converted and deconvoluted using the MaxEnt I algorithm (Waters) at a resolution of 0.1 Da.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
A. Pinto, Z. El Ali, S. Moniot, L. Tamborini, C. Steegborn, R. Foresti, C. De Micheli, ChemistryOpen 2018, 7, 858.
References
- 1. Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y., Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [DOI] [PubMed] [Google Scholar]
- 2. Motohashi H., Yamamoto M., Trends Mol. Med. 2004, 10, 549–557. [DOI] [PubMed] [Google Scholar]
- 3. Foresti R., Bains S. K., Pitchumony T. S., de Castro Brás E. L., Drago F., Dubois-Randé J., Bucolo C., Motterlini R., Pharm. Res. 2013, 76, 132–148. [DOI] [PubMed] [Google Scholar]
- 4. Nikam A., Olliver A., Rivard M., Wilson J. L., Mebarki K., Martens T., Dubois-Randé J. L., Motterlini R., Foresti R., J. Med. Chem. 2016, 59, 756–762. [DOI] [PubMed] [Google Scholar]
- 5. Haas B., Chrusciel S., Fayad-Kobeissi S., Dubois-Randé J. L., Azuaje F., Boczkowski J., Motterlini R., Foresti R., J. Cell. Physiol. 2015, 230, 1128–1138. [DOI] [PubMed] [Google Scholar]
- 6. Cleasby A., Yon J., Day P. J., Richardson C., Tickle I. J., Williams P. A., Callahan J. F., Carr R., Concha N., Kerns J. K., Qi H., Sweitzer T., Ward P., Davies T. G., PLOS 2014, 9, e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilson A. J., Kerns J. K., Callahan J. F., Moody C. J., J. Med. Chem. 2013, 56, 7463–7476. [DOI] [PubMed] [Google Scholar]
- 8. Zheng S., Santosh Laxmi Y. R., David E., Dinkova-Kostova A. T., Shiavoni K. H., Ren Y., Zheng Y., Trevino I., Bumeister R., Ojima I., Wigley W. C., Bliska J. B., Mierke D. F., Honda T., J. Med. Chem. 2012, 55, 4837–4846. [DOI] [PubMed] [Google Scholar]
- 9. Satoh T., McKercher S. R., Lipton S. A., Free Radical Biol. Med. 2014, 66, 45–57. [DOI] [PubMed] [Google Scholar]
- 10. Li W., Zheng S., Higgins M., R. P. Morra, Jr. , Mendis A. T., Chien C.-W., Ojima I., Mierke D. F., Dinkova-Kostova A. T., Honda T., J. Med. Chem. 2015, 58, 4738–4748. [DOI] [PubMed] [Google Scholar]
- 11. Hayes J. D., Dinkova-Kostova A. T., Trends Biochem. Sci. 2014, 39, 199–218. [DOI] [PubMed] [Google Scholar]
- 12. De Cesco S., Kurian J., Dufresne C., Mittermaier A. K., Moitessier N., Eur. J. Med. Chem. 2017, 138, 96e114. [DOI] [PubMed] [Google Scholar]
- 13. Kwak E. L., Sordella R., Bell D. W., Godin-Heymann N., Okimoto R. A., Brannigan B. W., Harris P. L., Driscoll D. R., Fidias P., Lynch T. J., Rabindran S. K., McGinnis J. P., Wissner A., Sharma S. V., Isselbacher K. J., Settleman J., Haber D. A., Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ettari R., Pinto A., Previti S., Tamborini L., Angelo I. C., La Pietra V., Marinelli L., Novellino E., Schirmeister T., Zappalà M., Grasso S., De Micheli C., Conti P., Bioorg. Med. Chem. 2015, 23, 7053–7060. [DOI] [PubMed] [Google Scholar]
- 15. Ettari R., Tamborini L., Angelo I. C., Grasso S., Schirmeister T., Lo Presti L., De Micheli C., Pinto A., Conti P., ChemMedChem 2013, 8, 2070–2076. [DOI] [PubMed] [Google Scholar]
- 16. Tamborini L., Pinto A., Smith T. K., Major L. L., Iannuzzi M. C., Cosconati S., Marinelli L., Novellino E., Lo Presti L., Wong P. E., Barrett M. P., De Micheli C., Conti P., ChemMedChem 2012, 7, 1623–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conti P., Pinto A., Wong P. E., Major L. L., Tamborini L., Iannuzzi M. C., De Micheli C., Barrett M. P., Smith T. K., ChemMedChem 2011, 6, 329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jçst C., Nitsche C., Scholz T., Roux L., Klein C. D., J. Med. Chem. 2014, 57, 7590–7599. [DOI] [PubMed] [Google Scholar]
- 19. Orth R., Bçttcher T., Sieber S. A., Chem. Commun. 2010, 46, 8475–8477. [DOI] [PubMed] [Google Scholar]
- 20. Pinto A., Tamborini L., Pennacchietti E., Coluccia A., Silvestri R., Cullia G., De Micheli C., Conti P., De Biase D., J. Enzyme Inhib. Med. Chem. 2016, 31, 295–301. [DOI] [PubMed] [Google Scholar]
- 21. Pinto A., Tamborini L., Cullia G., Conti P., De Micheli C., ChemMedChem 2016, 11, 10–14. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Quinti L., Naidu S. D., Träger U., Chen X., Kegel-Gleason K., Llères D., Connolly C., Chopra V., Low C., Moniot S., Sapp E., Tousley A. R., Vodicka P., Van Kanegan M. J., Kaltenbach L. S., Crawford L. A., Fuszard M., Higgins M., Miller J. R. C., Farmer R. E., Potluri V., Samajdar S., Meisel L., Zhang N., Snyder A., Stein R., Hersch S. M., Ellerby L. M., Weerapana E., Schwarzschild M. A., Steegborn C., Leavitt B. R., Degterev A., Tabrizi S. J., Lo D. C., DiFiglia M., Thompson L. M., Dinkova-Kostova A. T., Kazantsev A. G., Proc. Natl. Acad. Sci. USA 2017, E4676–E4685; [Google Scholar]
- 22b. McMahon M., Lamont D. J., Beattie K. A., Hayes J. D., Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pinto A., Conti P., De Amici M., Tamborini L., Madsen U., Nielsen B., Christesen T., Brauner-Osborne H., De Micheli C., J. Med. Chem. 2008, 51, 2311–2315. [DOI] [PubMed] [Google Scholar]
- 24. Conti P., De Amici M., Pinto A., Tamborini L., Grazioso G., Frølund B., Nielsen B., Thomsen C., Ebert B., De Micheli C., Eur. J. Org. Chem. 2006, 24, 5533–5542. [Google Scholar]
- 25. Bruno S., Pinto A., Paredi G., Tamborini L., De Micheli C., La Pietra V., Marinelli L., Novellino E., Conti P., Mozzarelli A., J. Med. Chem. 2014, 57, 7465–7471. [DOI] [PubMed] [Google Scholar]
- 26. Bruno S., Margiotta M., Pinto A., Cullia G., Conti P., De Micheli C., Mozzarelli A., Bioorg. Med. Chem. 2016, 24, 2654–2659. [DOI] [PubMed] [Google Scholar]
- 27. Ivanova O. A., Budynina E. M., Averina E. B., Kuznetsova T. S., Zefirov N. S., Synthesis 2006, 4, 706–710. [Google Scholar]
- 28. Suzuki T., Yamamoto M., J. Biol. Chem. 2017, 292, 16817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chartoumpekis D. V., Wakabayashi N., Kensler T. W., Biochem. Soc. Trans. 2015, 43, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huerta C., Jiang X., Trevino I., Bender C. F., Ferguson D. A., Probst B., Swinger K. K., Stoll V. S., Thomas P. J., Dulubova I., Visnick M., Wigley W. C., Biochimica et Biophysica Acta 2016, 1860, 2537–2552. [DOI] [PubMed] [Google Scholar]
- 31. Mueller U., Förster R., Hellmig M., Huschmann F. U., Kastner A., Malecki P., Pühringer S., Röwer M., Sparta K., Steffien M., Ühlein M., Wilk P., Weiss M. S., Eur. Phys. J. Plus 2015, 130, 1–10. [Google Scholar]
- 32. Kabsch W., Acta Crystallogr. 2010, D66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bunkoczi G., Echols N., McCoy A. J., Oeffner R. D., Adams P. D., Read R. J., Acta Crystallogr. 2013, D69, 2276–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., Acta Crystallogr. 2012, D68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary