

Abstract
Significance: The long-term hematopoietic stem cell (LT-HSC) demonstrates characteristics of self-renewal and the ability to manage expansion of the hematopoietic compartment while maintaining the capacity for differentiation into hematopoietic stem/progenitor cell (HSPC) and terminal subpopulations. Deregulation of the HSPC redox environment results in loss of signaling that normally controls HSPC fate, leading to a loss of HSPC function and exhaustion. The characteristics of HSPC exhaustion via redox stress closely mirror phenotypic traits of hematopoietic malignancies and the leukemic stem cell (LSC). These facets elucidate the HSC/LSC redox environment as a druggable target and a growing area of cancer research.
Recent Advances: Although myelosuppression and exhaustion of the hematopoietic niche are detrimental side effects of classical chemotherapies, new agents that modify the HSPC/LSC redox environment have demonstrated the potential for protection of normal HSPC function while inducing cytotoxicity within malignant populations.
Critical Issues: New therapies must preserve, or only slightly disturb normal HSPC redox balance and function, while simultaneously altering the malignant cellular redox state. The cascade nature of redox damage makes this a critical and delicate line for the development of a redox-based therapeutic index.
Future Directions: Recent evidence demonstrates the potential for redox-based therapies to impact metabolic and epigenetic factors that could contribute to initial LSC transformation. This is balanced by the development of therapies that protect HSPC function. This pushes toward therapies that may alter the HSC/LSC redox state but lead to initiation cell fate signaling lost in malignant transformation while protecting normal HSPC function. Antioxid. Redox Signal.
Keywords: : HSC, stem cell function, redox-active compound, LSC, hematopoiesis
Introduction
The hematopoietic compartment contains a tightly regulated hierarchy of cell types responsible for the function and maintenance of the bone marrow (BM) and blood systems. All peripheral blood cells, both the myeloid and lymphoid lineage, differentiate from a common pool of hematopoietic stem and progenitor cells (HSPCs) that reside within the BM (54, 138). This pool of HSPCs can be broken down into three subsets of cell types: the long-term hematopoietic stem cell (LT-HSC), short-term hematopoietic stem cell (ST-HSPC), and the multipotent progenitor cell (MPP). Two distinct progenitor cell populations emerge from the MPP pool: the common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs). Differentiation from these progenitor cell populations eventually results in the establishment of peripheral effector cell populations within each lineage, such as B and T cells within the lymphoid lineage or erythrocytes and neutrophils within the myeloid lineage (1, 54, 137). There are several phenotypic and functional facets that define the HSPC populations individually and as a whole. The central defining characteristics of hematopoietic stem cells (HSCs), particularly those within the long-term population, are the ability to self-renew while, at the same time, limit the size of the HSPC population and demonstrate the potential to differentiate into any mature cell type within the hematopoietic system (Fig. 1) (1, 11, 54, 97, 137, 138).
FIG. 1.

The bone marrow compartment contains a complex and highly regulated hierarchy of cell types tasked with maintaining the production and balance of all cells in the blood system. Beginning with the most primitive CD34+ LT-HSCs, the ability to self-renew and differentiate to downstream phenotypes defines the LT-HSC pool with a decreased capacity for self-renewal as differentiation occurs. From the MPP pool, both lymphoid and myeloid cell types develop and eventually differentiate into terminal effector cell types seen in the periphery. CD34, cluster of differentiation protein 34; CLP, common lymphoid progenitor; CMP, common myeloid leukemia; LT-HSC, long-term hematopoietic stem cell; MPP, multipotent progenitor cell; ST-HSC, short-term hematopoietic. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
ST-HSPC and progenitor populations have the capacity to maintain normal hematopoiesis for a period of 6–8 weeks. Serial transplant studies in vivo have identified the LT-HSC as the population and cell type that can sustain normal hematopoiesis throughout an organism's entire lifespan. This fact demonstrates a loss of HSC self-renewal capacity as a function of increased cellular differentiation. For these reasons, normal LT-HSC function must be maintained throughout the lifespan of an organism. This elucidates the LT-HSCs as the only population that holds true characteristics of the HSC. Because self-renewal and differentiation of LT-HSPC and ST-HSPC and MPP populations are essential to normal hematopoietic function, we define this entire population as the HSPCs and reserve the term HSC for the true LT-HSC populations.
Loss of normal LT-HSC and ST-HSPC function is a hallmark of natural stem cell aging and several hematopoietic disorders, most notably the development and progression of hematopoietic malignancies (1, 4, 11, 54, 65, 90, 97, 137, 138, 156, 165, 173). Within these cancers, normal hematopoietic regulation is lost, yet disease still progresses through the differentiation and clonal expansion of progenitor cell pools, eventually leading to a lack of terminal differentiation to functional cell types within the periphery. This observation led to the identification of the cancer stem cell (CSC) or more specifically the leukemic stem cell (LSC) (2, 65, 66, 119, 128, 130, 143, 156, 173). Although we know that hematopoietic neoplasms are driven by LSC populations, developing therapies that treat LSC pools as entities separate from normal HSPCs has been difficult. Thus, little progress has been made in the development of therapies that both eradicate malignant HSPCs while, at the same time, protect or pose no detriment to healthy HSPC populations within a single patient.
There is a heterogeneous and diverse set of cytogenetic abnormalities within various hematopoietic cancers that, in some cases, may lend themselves to personalized treatment plans. However, intrinsic characteristics that separate normal HSPCs from their malignant counterparts are becoming more relevant (7, 12, 13, 16, 77, 105, 121, 150). The identification of these differences will lead to the development of safe therapeutics that have broad implications for treatment of several hematopoietic neoplasms across patient populations. Chief in the differences between normal and malignant HSPCs is the generation of reactive species and the management of the cellular redox environment (5, 22, 67, 75, 82, 106, 107, 119, 128, 129, 143, 150, 155, 159). It has been well established that cancer cells demonstrate elevated levels of reactive species generation and a difference in basal redox environment as compared with their normal counterparts. This difference is heavily rooted in an increased metabolism and production of reactive oxidative species such as superoxide and hydrogen peroxide (H2O2), which, in turn, leans on the cellular antioxidant capacity and thus, enhances the need for reducing species such as glutathione (GSH). The result is an unbalance in equilibria that stresses both sides of cellular oxidoreduction capacity, herein we refer to this stress imbalance simply as redox stress. In fact, the malignant hematopoietic phenotype mirrors the changes in normal hematopoietic architecture brought on by increased production of redox stress, which results in alterations to the HSPC redox environment (66, 69, 70, 117, 129, 155). This fact has recently presented researchers with a druggable target in which a therapeutic index can be defined that exploits the malignant cell redox environment while leaving normal cell populations unharmed (67). This is accomplished by examining the effects of redox-active compounds in both normal and malignant hematopoietic stem and progenitor cell populations. Redox-active compounds have traditionally been defined as those that can undergo single electron transfers acting as either an oxidizing or reducing agent. These compounds include nitroxides such as tempol, flavonoids such as oxorylin or baicalein, and those with transition metal centers, such as Mn (III) meso-tetrakis(N-(n-butoxyethyl)pyridinium-2-yl) porphyrin (MnTnBuOE), capable of cycling through multiple oxidation states acting as active sites for therapeutic efficacy (8–10, 49, 113, 147, 149, 161, 164, 176). Other compounds, such as doxorubicin (DOX), may transition through structural changes as a result of electron exchange via metabolism (34, 36, 50, 86, 100, 101, 172). In addition, we consider those compounds that affect the cellular redox environment by actively modifying concentrations of cellular reductants or those that inhibit normal antioxidant enzyme function, such as buthionine sulfoximine (BSO) or parthenolide (PTL), although actual oxidoreduction reactions do not directly result from the chemistry of the molecules themselves (69, 115). In this review, we offer a concise summary of key studies examining the effects of endogenous mechanisms and redox-active compounds resulting in changes to the cellular redox environment of normal and malignant HSPCs. Furthermore, we present the HSPC cellular redox environment as a druggable target that allows for effective intervention within malignant populations while leaving normal HSPC populations unharmed.
Redox Regulation and Redox-Active Compounds Affect Normal HSC Function
The production and elimination of the initial and ensuing reactive species are the cornerstones of cellular redox environment management. Here, metabolic byproducts such as O2− (superoxide) and H2O2, as well as .OH (hydroxyl radical), and ONOO− (peroxynitrite) are the main reactive species responsible for oxidative damage, including DNA oxidation, lipid peroxidation, and protein oxidation as well as nitration (155). As a result, an increase in metabolic activity and reactive species generation is closely associated with an increase in cellular proliferation and differentiation (70, 117, 138, 153). It should come as no surprise then that various concentrations of the original redox-active compound, molecular oxygen (O2), significantly affect cellular redox status and HSPC proliferation. The BM niche, or extracellular components surrounding HSPCs, and peripheral tissues have oxygen tensions ranging from 1% to 7%. In vitro study of primary normal and malignant HSPCs, as well as various culture lines, has demonstrated the beneficial effects of a hypoxic environment (18, 26, 27, 60, 61, 131, 133). Both normal and malignant HSCs benefit from growth conditions that have oxygen concentrations ranging from 1% to 3%, compared with cultures at normoxic (5%) and atmospheric (21%) O2 concentrations (29, 70, 116, 144, 152). Interestingly, a low oxygen environment leads to a decrease of GSH levels, as well as antioxidant enzyme expression and function (19, 40, 108). Primitive HSC populations have been thought to be refractory to induction of oxidative stress, due to their quiescent state, antioxidant capacity, and local niche environment. However, their environment may be a key factor in keeping HSCs sensitive to redox stress. This facet of the BM environment has potential in allowing for the development of differential sensitivities to redox insult between normal and malignant HSC as well as HSPC populations (Fig. 2).
FIG. 2.
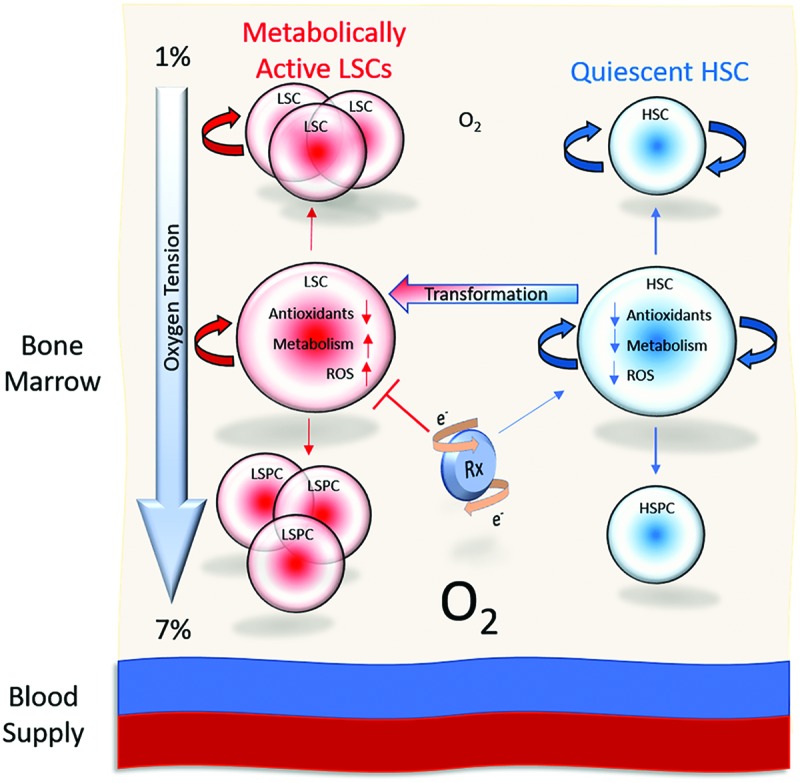
The bone marrow compartment demonstrates oxygen concentrations that range from 1% (furthest for the blood supply) to 7% (closest to the blood supply). Low oxygen concentrations have been referenced as a factor contributing to the protection and quiescence of HSCs. However, upon transformation, LSCs may demonstrate increased levels of metabolism and ROS levels, while maintaining a lower antioxidant capacity than their downstream progeny. This difference, a product of the oxygen environment, may allow for a differential redox insult and targeting of LSCs over normal HSCs driven by the introduction of a redox-active compound. HSC, hematopoietic stem cell; HSPC, hematopoietic stem/progenitor cell; LSC, leukemic stem cell; ROS, reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Cluster of differentiation protein (CD)34+ HSPCs express higher levels of superoxide dismutase (copper-zinc superoxide dismutase [SOD1] and SOD2) and catalase (CAT) relative to other antioxidant enzymes, such as, glutathione peroxidases (GPx), glutathione-S-transferases (GSTs), glutathione reductase (GR), thioredoxin (TXN), and thioredoxin reductase (TXNR) utilized for the removal of H2O2 as well as other radicals and reactive species (Fig. 3) (115). However, the expression level of antioxidant defense enzymes such as SOD and CAT is lower in the primitive LT-HSC compartment than in their downstream differentiated progeny (4, 99, 155). This means that if these defense enzymes fail, a significant buildup of primary and secondary reactive species may occur; thus, quickly and, potentially irreversibly, altering the redox environment within HSCs. In addition, hematopoietic malignancies are generally typified by the uncontrolled clonal expansion of HSPC populations. Therefore, primitive malignant populations may, themselves, express low levels of antioxidant enzyme capacity, such as SOD or CAT. This would then result in higher levels of downstream reducing agents such as the GSH/GSSH redox couple.
FIG. 3.
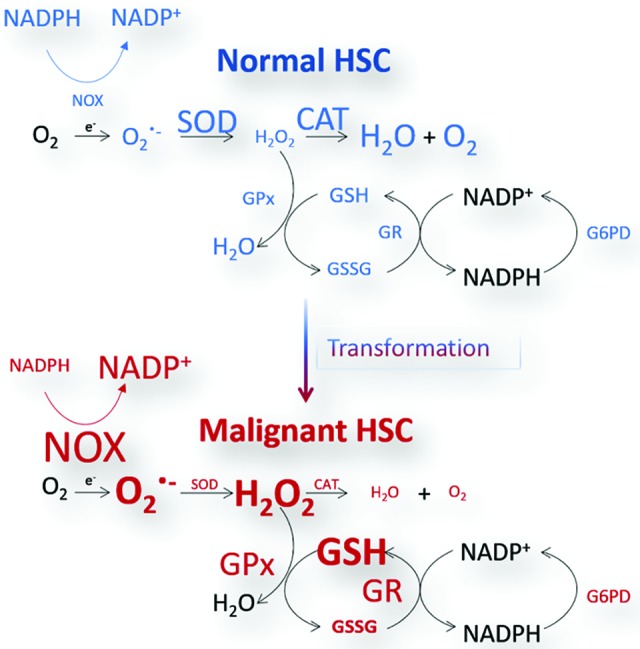
Normal and malignant HSCs demonstrate differences in the regulation and management of the antioxidant system. Normal HSCs express higher levels of primary antioxidant enzymes SOD and CAT. Malignant HSCs are subject to elevated levels of ROS produced by NOX enzyme and rely on higher cellular concentrations of GSH as well as the GSH metabolic system to manage the malignant HSC redox state. CAT, catalase; GPx, glutathione peroxidase; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, nicotinamide adenine dinucleotide phosphate oxidase; SOD, superoxide dismutase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In the past decade, several key studies have identified management of the HSPC redox environment as a crucial factor in the maintenance of the HSPC population. The HSPC compartment itself contains cell populations that can be separated based on their reactive oxygen species (ROS) levels and redox environments. Using 2′-7′-dichlorofluorescence (DCF) diacetate, Jang and Sharkis were able to separate two HSPC populations characterized by high and low levels of ROS (70). These two populations display distinct differences in HSPC function. Limiting dilution transplantation assays showed that HSPCs with low levels of ROS repopulated recipient mice significantly better than HSCs with high levels of ROS. This characteristic was also carried over to secondary and tertiary transplant recipients. In addition, the “ROS low” population contained more primitive long-term culture-initiating cells (LTC-ICs) and resulted in better multilineage reconstitution (70). Interestingly, upon transplantation the “ROS high” population resulted in lineage reconstitution that favored myeloid cells. Conversely, ROS low cells favored lymphoid lineage development. This indicates a potential for the development of myeloid lineage malignancies upon redox deregulation within the normal HSPC populations. The ROS high population demonstrated elevated levels of p38 activity, elevated expression of p16 and lower levels of p53 expression. The LTC-IC capacity of the ROS high population did respond to treatment with either a p38 mitogen-activated protein kinase (p38 MAPK) inhibitor, the GSH precursor N-acetyl-l-cysteine (NAC), or rapamycin (70). This indicates that buffering the HSPC redox environment can improve normal HSPC function. Thus, as one might expect, NAC treatment aids the engraftment of normal human HSPCs isolated from human cord blood into recipient mice. These results are demonstrated even in the highly enriched lineage (Lin)− CD34+ CD38− CD90+ CD45 isoform A (CD45RA)+ CD49f+ Rho GTPase (Rholow) HSC population, indicating that exogenous protection against reactive species challenges and can independently improve HSC function (64).
In 2007, Ito et al. utilized the Atm−/− mouse model to demonstrate that an elevation of ROS activated p38 MAPK signaling, resulting in a loss of normal HSC maintenance as well as quiescence eventually leading to HSPC senescence (69). They found that depletion of cellular GSH via BSO treatment led to an increase in H2O2 within the HSPC Lin−/stem cell antigen 1 (Sca-1)+/c-Kit+ (LSK) population, resulting in an increased expression of cell cycle regulators p15INK4B (cyclin-dependent kinase inhibitor B) p16INK4A cyclin-dependent kinase inhibitor 2A, p18INK4C (cyclin-dependent kinase 4 inhibitor C), and p19ARF (cyclin-dependent kinase 4/6 inhibitor). This led to a loss of HSPC function; yet the upregulation of tumor suppressor proteins was reversed by treatment with NAC or a p38 MAPK inhibitor. These effects were not demonstrated in differentiated cell populations (69). Similarly, ROS accumulation, p15INK4B, p16INK4A, p18INK4C, and p19ARF upregulation, and loss of HSPC function were demonstrated in Atm−/− mice. Within this model, treatment with a p38 MAPK inhibitor both before and after transplant recovered HSC function in Atm−/− mice, compared with wild-type controls. Using NAC and p38 MAPK inhibitor treatments, this study also demonstrated that ROS-mediated p38 activation is responsible for a loss of HSPC quiescence (69). Interestingly, H2O2 oxidation of ataxia-telangiectsia mutated (Atm) results in internal disulfide crosslinking and dimer formation that leads directly to Atm activation independent of DNA double-strand breaks (58). Results found by Ito et al. are very similar to those found by Miyamoto et al. using a Foxo3a−/− mouse model. These studies demonstrate forkhead box O3A (FOXO3A) regulation of HSC function. Importantly FOXO3A operates downstream of phosphatidylinositol-4,5-biphosphate 3-kinase/protein kinase B (PTEN/PI3K/AKT) pathway, and loss of FOXO3A activity results in HSC exhaustion and activation of the p38 MAPK pathway (102).
Just as these phenomena have been demonstrated in mice, human cells subjected to serial transplantation in mice demonstrate similar characteristics. Serial transplantation of human Lin− CD34+ CD38− HSCs loses their ability to repopulate the BM (171). This loss of HSC function in human cells is accompanied by an accumulation of oxidative DNA damage, characterized by an increase in γH2AX foci, as well as an increase in the expression of cell cycle inhibitors p16INK4A, p14ARF (p14 alternate reading frame tumor supressor), and p21CIP (cyclin-dependent kinase inhibitor 1) (171). The formation of these DNA damage foci is recapitulated in normal human HSCs treated with BSO. Upon formation, DNA damage repair enzymes, Atm, p53 binding protein (53BP1), CHK2, and FOXO3A are recruited to the foci. Furthermore, examination of the highly enriched Lin− CD34+ CD38− CD90+ CD45RA+ HSC population showed that although damage is seen in both the stem and progenitor cell (CD34+ CD38+) populations, only the HSC compartment undergoes cell cycle arrest and apoptosis. In addition, HSC populations recover more slowly than progenitor populations after the oxidative insult is removed. Consistent with other studies, addition of NAC as a protecting agent preserves HSC function in correlation with decreased accumulation of DNA damage foci, even in the face of oxidative insult (171). Control of DNA damage response can also be managed via the polycomb repressor (BMI1) signaling via control of INK4A/ARF (CHK2) proteins. Loss of BMI1 function results in mitochondrial dysfunction and accumulation of ROS, and loss of HSPC self-renewal and differentiation, a phenotype that can be partially restored by treatment with NAC or the deletion of the Chk2 gene itself (91).
Clearly the accumulation of oxidative damage incurs a detrimental effect on HSC function, and as one may anticipate, nuclear factor (erythroid-derived 2)-like 2 (NRF2) activity has been identified as a major regulator of the HSC redox environment and HSC function (79, 98, 160). Tsai et al. were able to utilize the Nrf2−/− mouse to demonstrate familiar effects on HSC function. These studies elegantly showed that the loss of NRF2 resulted in an increase in the LSK population, but only within the ST-HSC and MPP compartments, not within the LT-HSC population (153). Furthermore, Nrf2−/− mice demonstrated an increase in both the common myeloid (CMP, granulocyte macrophage progenitor, and megakaryocyte-erythroid progenitor) and lymphoid (lymphoid primed multipotent progenitor, CLP-1, and CLP-2) progenitor cell populations. Although progenitor pools underwent significant expansion, the primitive LT-HSC populations were unchanged. However, MKI67 antigen KI-67 analysis, 5-FU pulse experiments, and culture in OP9-DL1 revealed that NRF2 regulates HSPC proliferation and differentiation resulting in diminished quiescence and increased differentiation within the most primitive LT-HSC populations (153). Importantly, the NRF2-deficient HSPCs also significantly underperformed in colony formation and in vivo transplant assays, indicating that not only does NRF2 regulate proliferation and differentiation but also regulate HSPC self-renewal and function. Proliferation analysis via coculture of deficient cells with WT counterparts demonstrated a proliferative advantage for the Nrf2−/− cells (153). Chimeric studies showed that the proliferative advantage is generated from within the HSPC itself and does not result from signals received from the BM environment. Moreover, the loss of NRF2 results in a decreased BM homing effect, implying that NRF2 even regulates the retention of HSPCs in the BM (153). This homing effect is linked to the loss of chemokine receptor type 4 (CXCR4) expression, resulting in deficient chemotaxis abilities in Nrf2−/− HSPCs (43, 153).
The studies already discussed have laid the groundwork for a clear understanding of how the HSPC redox environment affects normal HSPC regulation and function. Ultimately, dysregulation of HSPC redox balance results in well-defined changes to HSPC populations. As previously noted, the resulting alterations to HSPC populations mimic those demonstrated in several disease states, most notably, the myeloproliferative disorders (MPDs). Clearly, regulating the cellular GSH pool is required for faithful management of ROS and HSPC function. HSPCs that undergo redox deregulation resulting in an increase in ROS seem to demonstrate a natural response that functions through the Jun N-terminal kinase (JNK)/MAPK signaling as well as FOXO and Atm DNA damage response pathways (Fig. 4) (69, 70, 102). This results in loss of HSPC quiescence and function. Despite the perception that HSPCs maintain robust function as a result of their protected location and their highly quiescent state, we now know that HSPCs not only rely on intrinsic signaling but also that HSPCs are more sensitive to redox stress than previously thought (171). These facets are highlighted by improving HSPC function through buffering redox capacity but also by the result of losing major regulatory control over redox environment management via transcription factors such as NRF2 (64, 153). Thus, redox modulators, such as BSO, which induce strong conditions of oxidative stress, work to the detriment of normal HSPC function. Conversely, compounds such as NAC that provide antioxidant aid improve HSPC function.
FIG. 4.

A schematic diagram demonstrates the differential effect and graded response of HSCs to various redox-active compounds and ROS inducers. Normal hematopoiesis is characterized by primitive LT-HSCs that have the ability to self-renew and differentiate into any other cell type. As differentiation occurs, progenitor cell types lose their ability to self-renew. This process is accelerated in the presence of potent ROS induces, such as conventional chemotherapeutic agents and ionizing radiation. Response to these stimuli results in DNA damage response and activation of signaling that results in the upregulation of cellular senescence and apoptosis. New evidence now suggests that mild ROS inducers have an opposite effect wherein generation of a mild ROS milieu results in the activation of antioxidant defense pathways leading to an improvement in HSC function. The result is a graded response to different levels of ROS stimulant that leads to variations of HSC function and either exhaustion or strengthening of the hematopoietic niche. Atm, ataxia-telangiectsia mutated; BSO, buthionin sulfoximine; DOX, doxorubicin; MnP, Mn porphyrin; Nq01, NAD(P)H dehydrogenase quinone 1; Nrf2, nuclear factor (erythroid-derived 2)-like 2. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Chemotherapeutics and Normal HSCs
The work already summarized demonstrates the importance of HSPC redox regulation in maintaining normal HSPC function. Normal HSPC toxicity and functional exhaustion are a significant problem underlying the use of traditional chemotherapeutics for the treatment of hematopoietic malignancies. Most chemotherapies currently utilized for the treatment of the hematopoietic malignancies are not designed to manipulate the cellular redox environment of the HSPC; rather, they target DNA synthesis or immune function. However, we now know that many cancer therapeutics do in fact impact the redox milieu of target and normal tissues. These redox-active therapeutics illicit detrimental effects on HSCs that echo the results previously discussed. Methotrexate (MTX), a long-approved cancer drug within the antifolate class that inhibits endogenous nucleotide synthesis, has been well documented to induce oxidative stress within target tissues (6, 141). MTX treatment results in an increase of superoxide production while concurrently depleting cellular GSH and total thiol levels by 70%. These MTX-mediated changes in the cellular redox environment are accomplished via inhibition of MnSOD, GPx, GR, and γ-glutamylcysteine synthetase (6). Although chemotherapeutics such as MTX target dividing cells, quiescent HSC populations seem to remain refractory to MTX treatment even in the face of cell cycle inducers. However, inhibition of nucleoside transport allows for HSPC sensitization to MTX cytotoxicity, resulting in a loss of HSPC function (3). Although antifolate drugs mainly target DNA synthesis, their effects on redox management cannot be ignored, particularly in the HSC population where GSH metabolism and homeostasis is important. Similarly, cyclophosphamide, an agent that targets dividing cells via induction of DNA cross-linking, causes a significant amount of reactive species production measured by DCF in HSPCs. This leads to hematopoietic suppression demonstrated by loss of HSPC quiescence and function followed by increases in peripheral blood and splenic progenitor populations (104). This is typified by a significant decrease in the GSH/oxidized glutathione (GSSG) ratio. Interestingly, the application of a glutathione disulfide mimetic significantly decreases the production of ROS and partially rescues the changes in cellular GSH (35). This GSSG mimetic also increases the number of HSPCs depleted by cyclophosphamide treatment and partially rescues HSPC function. Treatment also rescues cyclophosphamide suppression of antioxidant gene expression, particularly SOD3 (copper-zinc extracellular superoxide dismutase) and GPX2 (35).
Perhaps one of the most potent and popular chemotherapeutics approved for treatment of leukemias is the anthracyclines. Of this class, doxorubicin/adriamycin (DOX/ADR) is possibly the most well-documented redox stress inducer. DNA intercalation and inhibition of topoisomerase II is the primary mechanism of DOX-induced cytotoxicity in cancer. However, the ability to act as a redox modifier results from nicotinamide adenine dinucleotide phosphate (NADPH)-mediated formation of a semiquinone structure, which allows for the generation of oxygen radicals, H2O2, DNA damage, and alterations in bioenergetics (34, 36, 50, 86, 100, 101, 172, 178). Fenton type chemistry may then exacerbate this process, leading to the generation of hydroxyl radicals. It is no surprise then that the use of anthracyclines such as DOX is applied at the detriment of normal hematopoiesis (14, 28, 31). Traditionally, pharmacological doses of DOX induce myelosuppression, cardiac toxicity, and the enrichment of refractory CSCs through the elevated expression of multidrug-resistant proteins such as MDR1 and MRP1/ABCC1 (multidrug resistant protein 1/ATP-binding cassette subfamily C member 1) (14). DOX treatment specifically suppresses the number of ATP binding cassette subfamily G member 2 (ABCG2)+ HSPCs and BM sphere formation in comparison with vehicle and paclitaxel controls in vitro. Decreases in ABCG2+ HSPCs as well as peripheral white blood cells have also been demonstrated in vivo at doses of 1–5 mg/kg DOX (14). More recently, Mahbub et al. discovered that DOX and etoposide (ETP) can be combined with several different polyphenol compounds to enhance malignant cell death in both lymphoid and myeloid lineage cancer models while at the same time protect normal CD34+ and CD133+ HSPC populations (92, 93). With DOX and ETP in vitro concentrations ranging from 0.1 to 0.4 μM, combinations of five different polyphenol compounds (quercetin, apigenin, emodin, rhein, and cis-stilbene) all recovered levels of adenosine tri-phosphate (ATP) production and caspase 3 activity when compared with DOX and ETP treatment alone. In these studies, cellular GSH levels were identified as a key factor in mediation of protection in combination treatments (92, 93). Interestingly, quercetin and apigenin, compounds recognized as mitocans (which specifically target mitochondria of cancerous cells), can bind adenine nucleotide translocase of the mitochondrial permeability transition pore, inhibiting its function and the efflux of mitochondrial ATP (52, 111).
Redox-Based Therapeutic Protection of the HSPC
A precedent for HSPC protection has been established in the past with the use of granulocyte colony-stimulating factor, erythropoietin, and other hematopoietic stimulants such as pegfilgrastim (Nulasta). These have been used to bolster production of peripheral blood cells in the face of small molecule chemotherapeutics, such as cyclophosphamide, as well as antibody-based therapies such as ublituximab (89). Although the use of antioxidants, such as resveratrol and NAC, in cancer therapeutic regimens has been a subject of debate, the prospect of protecting normal HSPC function while killing malignant cells merits investigation and consideration, as the potential impact of accomplishing both simultaneously cannot be over stated. HSPC protection in the face of oxidative insult has been accomplished through the upregulation of antioxidant enzyme function. Furthermore, this protection is actually supported by the induction of redox stress, (i.e., the generation of oxidoreductive reactive species that upregulate endogenous antioxidant machinery). Liposomal delivery of a vector containing an MnSOD construct protected HSPCs of mice receiving a limiting dose of donor BM in the face of a 10 gray (Gy) total body irradiation (TBI) (99). This protective effect is enhanced when the MnSOD vector is delivered before the TBI. Protection of the HSPC is accompanied by a significant decrease in ROS production. Interestingly, HSCs overexpressing CAT and MnSOD performed better in long-term competitive repopulation assays, with CAT outperforming the MnSOD over expressers. Importantly, the repopulation ability of these subjects was significantly increased after recipients were exposed to a lower 2 Gy dose of radiation. This indicates that upregulation of antioxidant defenses primes HSCs to rebuff subsequent oxidative insult, resulting in an increase in HSC function (99).
The antioxidant protective effect of upregulated MnSOD in HSPCs can also be achieved through the application of manganese-containing porphyrin compounds (8, 10, 147). A significant characteristic of these compounds is the stable Mn3+ center and the ability to accumulate in the mitochondrial fraction (132). Manganese and iron-containing porphyrins mimicking endogenous SOD function have been successfully utilized for the protection of BM, among other tissues, against the oxidative challenges of radiation therapy (10, 21, 147, 149, 166). Mn (III) meso-tetrakis (N-ethylpyridinium-2-yl) porphyrin (MnTE-2-PyP5+ or MnTE) has demonstrated protection of the HSPC in the face of a sublethal TBI dose of 6.5 Gy. Administered at a dose of 6 mg/kg on the day of TBI injury and for 30 days subsequent to TBI injury, MnTE-2-PyP5+ improved hematopoietic recovery (10, 88, 147). This resulted from improved function of both: hematopoietic stem and progenitor cells compared with control-treated mice. Importantly, MnTE-treated HSPCs demonstrated a decrease in senescence induced by TBI through the downregulation of p16Ink4a expression (114, 127, 163). Pharmacological protection of both the splenic and BM HSPC compartments from TBI of 7.5 Gy has also been achieved through the implementation of the flavonoid, baicalein (113). Both in vitro and in vivo treatment with baicalein resulted in an inhibition of phosphatase MKP3, ERK activation, nuclear accumulation of NRF2, and an increase in NRF2 target mRNA (113). These studies indicate that redox-active compounds may offer dual targeting of the NRF2 and MAPK signaling pathways in normal HSPCs, a concept that may aid redox-active therapeutic targeting of malignant HSPC populations, over normal HSPCs.
Our studies demonstrated similar effects of improved number of HSPCs and function generated by overexpression of mitochondrial MnSOD. Our more recent work has further explored this concept utilizing the MnSOD mimetic Mn (III) meso-tetrakis(N-(n-butoxyethyl) pyridinium-2-yl) porphyrin (MnTnBuOE-2-PyP5+ or MnTnBuOE). MnTnBuOE treatment in vitro as well as chronic treatment in vivo results in an increase in the HSPC LSK population, similar to MnSODTgH mice (transgenic mice overexpressing MnSOD) (179). Moreover, LSK populations significantly increase through tertiary BM transplants from donor cells treated with MnTnBuOE for 16 h in vitro as well as donor BM from mice treated for a total duration 60 days at 2 mg/kg, 3 times/week. Not only does MnTnBuOE increase HSPC function evidenced by long-term transplant studies, but MnTnBuOE treatment also significantly improves the recovery of red blood cells (RBCs) and hemoglobin after induced acute blood loss (179). Although MnTnBuOE is traditionally considered to function as an antioxidant, due to its MnSOD-like activity, we found that MnTnBuOE treatment decreases mitochondrial function and ATP-linked oxygen consumption within the HSPC fraction. Concurrently, MnTnBuOE induces mild ROS production, measured by DCF. Here, an increase in DCF identifies an increase in general reactive species. This is presumably due to an increase in H2O2 production resulting from the reduction of superoxide by MnTnBuOE. However, because SOD mimetics are also known to modulate peroxynitrite levels, further study is required to elucidate the precise ROS resulting from MnTnBuOE treatment. Rather than oxidative activation of p38 MAPK signaling resulting in cellular senescence and apoptosis as previously mentioned, MnTnBuOE treatment results in the activation of E26 transformation specific and NRF2 transcriptional machinery. MnTnBuOE treatment results in an increase of NRF2 target transcript levels (CAT, MnSOD, glutatmate-cysteine ligase catalytic subunit [GCLC], GCLC, NAD(P)H dehydrogenase quinone 1 [Nq01], uncoupling proteins 1 and 3 [UPC1/3], and PRDXR1) (179). Western blot analysis of the LSK population confirms the increase of MnSOD, CAT, GSTp1, and UPC3 protein levels after MnTnBuOE treatment. These results indicate that MnTnBuOE initially acts as a transient pro-oxidant, ultimately resulting in activation of an antioxidant phenotype that improves and protects HSPC function. In fact, we found that the HSPC/LSK population has a significantly higher 2GSH/GSSG ratio as well as an increased ability to remove H2O2 in comparison with differentiated myeloid progenitor cells (179).
Protecting or supplementing HSPC function through the upregulation of antioxidant activity via gene delivery or chemotherapies is demonstrated by the application of limiting doses of sublethal and lethal TBI (10, 55, 88, 99, 114, 127, 163, 179). The management of NADPH/NADP+ levels has emerged as a critical factor for the protection of HSPC function. HSPCs express multiple isoforms of the NADPH oxidases (NOXs) that constantly produce ROS (118). TBI causes DNA damage and double strand breaks leading to the upregulation of the INK4A cell cycle regulators and HSPC senescence (127, 163). Inhibition of NOX by treatment with diphenylene iodonium (DPI) resulted in HSPC protection from ROS-induced DNA damage(114). This is similar to the protection achieved by HSPC treatment with MnTE (88). As previously noted, MnTE treatment protects irradiated HSPC function by downregulation of p16INK4A, a key player in p38 MAPK-induced HSPC senescence. MnTnBuOE treatment of nonirradiated HSPCs significantly improves HSPC function through the transient production of ROS and activation of NRF2. This results in upregulation of not only MnSOD and CAT protein levels but also results in an increase of NQO1 transcript levels (179). Together, these results indicate that the redox management of the NADPH/NADP+ system lies squarely at the center of HSPC regulation. This also presents the NOX and NADPH dehydrogenases as potential targets for redox-active compounds that include the Mn porphyrins (MnPs). Other natural extract compounds such as apocynin and pumbagin also target the NOX system as well as modify the cellular redox environment through changing the cellular GSH concentrations (95). Thus, emerging redox-active compounds with the ability to act not only as a mild antioxidant but also as a mild pro-oxidant may not only be successful in eradicating malignant cells but also may protect normal HSPC function.
These studies further suggest that, contrary to the currently accepted dogma of ROS effects on long-term HSC function, modulation of oxidative challenge to illicit a low-to-moderate level of redox stress can actually offer protection by upregulating HSPC function through NRF2 activation. This is accomplished through functional duality of redox-cycling compounds (Fig. 4). For example, MnTnBuOE has the ability to oxidize superoxide to molecular oxygen in one half reaction while maintaining the same proclivity for the reduction of superoxide to H2O2 in another half reaction (8–10, 147, 149). Redox-active compounds can accomplish this by closely mimicking structure–function characteristics of endogenous enzymes. In the case of MnTnBuOE, functional mimic is characterized by the half-wave reduction potential of the manganese center, which is very close to the potential of MnSOD itself (8–10, 147, 149). Other compounds such as polyphenols and flavonoids can have differential effects on normal and malignant cells also by targeting antioxidant machinery such as components of GSH metabolism and NRF2 activation discussed later in this review. Mixed oxidoreductase function of compounds such as MnTnBuOE are contrary to conventional chemotherapeutics, such as DOX, that can act as oxidizing agents resulting from metabolic/bioenergetic alteration or interaction with the cellular redox milieu. With this principal in mind, differences in the basal redox environment between normal and malignant cells now expose cancer cells to redox-based therapies that offer little harm to normal HSPCs (Fig. 5).
FIG. 5.

The prosurvival response of normal HSCs to mild pro-oxidants has led to the concept of differential response to redox cycling compounds between normal and malignant HSCs. Redox-active compounds will use the normal levels of ROS produced by healthy HSCs to activate an antioxidant response leading to increased proliferation and HSC function. Conversely, LSCs perpetually produce elevated levels of superoxide via aberrant constitutive enzyme activity that can be used by redox-active compounds to produce second messenger ROS such as hydrogen peroxide. This may lead to LSC senescence and death. The theoretical result is the protection of normal HSC function with simultaneous cytotoxic, anticancer affects in the LSC population. AP-1, activating protein-1. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Redox-Based Therapies Targeting the LSC Redox Environment
The reactive species generated by malignant HSPCs is closely linked to metabolism. Thus, as malignant HSPCs or LSCs undergo clonal expansion at an unchecked rate, in comparison with their normal counterparts, the HSPC redox environment has emerged as an identifying trait between normal and malignant stem cells (1, 2, 54, 66, 67, 117, 119, 129, 137, 143). Although most cancer drugs were developed to interfere with DNA synthesis, a number of common therapeutic compound classes already discussed do in fact induce ROS generation. Because the compounds such as DOX, MTX, ETP, cyclophosphamide, and cytarabine are all administered to the peril of the normal HSPC, new methods of employment must be considered. Transient, low-dose regimens and/or combination treatments present viable options for safer usage, and several studies within the past 5 years have begun to explore these options and are reviewed here.
As previously discussed, the GSH system has emerged as a key regulator of HSPC function and as such may be targeted by redox-active compounds for the treatment of hematopoietic malignancies. BSO is a potent inhibitor of GSH synthesis and as previously discussed has been used to demonstrate stem cell exhaustion. For this reason, clinical utilization of BSO has some potential downfalls. Recently, however, Tagde et al. have shown the effective combination of melphalan (L-PAM) and BSO in preclinical models of multiple myeloma (MM). High GSH levels are indicative of an aggressive MM relapse phenotype that may be resistant to L-PAM, depletion of GSH with BSO sensitizes nine MM cell lines and primary MM samples to L-PAM treatment (142). This potentiation is blocked by treatment with NAC, which does not change cellular GSH concentrations. The combination of BSO and L-PAM results in DNA strand breaks, mitochondrial dysfunction, and activation of apoptosis. This combination was also successful in depleting GSH and inducing engraftment suppression within three human MM xenograft models (142).
The regulation of antioxidant enzyme expression within LSCs differs versus their normal counterparts, but these defense enzymes also differ between types of hematopoietic malignancies. For example, CD34+ cells isolated from acute myeloid leukemia (AML) patients express low levels of manganese superoxide dismutase (SOD2) and CAT and higher levels of GCLC, glutamate-cysteine ligase regulatory subunit (GCLM), GPX1, and TXN than their normal CD34+ counterparts (115). SOD2 and CAT levels vary among other cancers within the myeloid lineage (151, 177). However, elevated expression of GSH metabolism enzymes highlights, just as in normal HSPCs, the importance of GSH regulation within malignant HSPC populations. Indeed the GSH/GSSG system has successfully been targeted for therapy using several compounds that contain α,β-unsaturated-γ-lactone groups or have strong electrophilic character and have the potential to react with free thiol groups: PTL and piperlongumine (PLM) (115). Both of these compounds preferentially deplete cellular GSH levels in CD34+ malignant cells compared with normal counterparts. In addition, PTL and PLM-treated normal HSPCs demonstrate some degree of GSH/GSSG recovery, whereas the malignant cells do not, offering protection of normal HSPCs and cytotoxic effects in malignant HSPCs. The result of this GSH depletion is an increase in cleaved caspase 3 and cleaved poly (ADP-ribose) polymerase (PARP), leading to a significant induction of LSC cell death compared with normal CD34+ HSPCs. Ultimately, PTL was found to actually bind several enzymes involved with GSH synthesis, including GCLC and GCLM (115). PTL also exhibited a synergistic effect when paired with low doses of cytarabine and idarubicin (59).
PTL and several other natural compounds, including polyphenol and flavonoids, have demonstrated potential therapeutic efficacy in chronic myelogenous leukemia (CML) and lymphocytic leukemia. For example, Wu et al. evaluated the effects of 27 different compounds that include α-β-unsaturated carbonyls, sulfhydryl reactive metals, isothiocyanates, polyphenols, and flavones on NRF2 activation and cytotoxicity in peripheral blood mononuclear cells (PBMCs) isolated from normal and chronic lymphocytic leukemia (CLL) patients (168). Through the analysis of the NRF2 target heme oxygenase-1 (HO-1) and NRF2 protein levels, they first found that NRF2 activity was higher in PBMCs from CLL patients than in their normal controls. In all, six α-β unsaturated carbonyls, three arsenic-containing sulfhydryl reactive metals, two isothiocyanates, two flavones, and five polyphenol compounds were able to activate NRF2 in a FRET β-lactamase-based reporter assay in HepG2 cells (168). Cytotoxicity evaluation via MTT analysis showed that several α-β-unsaturated carbonyls, sulfhydryl reactive metals, isothiocyanates, and flavones were selectively cytotoxic to primary CLL PBMCs compared with their normal counterparts. In the case of PTL and ethacrynic acid, loss of α-β unsaturated carbonyl groups diminished NRF2 activation and CLL-selective cytotoxicity (168).
As discussed previously, Tsai et al. were able to utilize the Nrf2−/− mouse model to demonstrate a downregulation of homing and chemotaxis associated with the loss of CXCR4 (153). Similarly, an increase in CXCR4 expression resulting from an upregulation of CXCL12 results in ADR-resistant K562 CML cells (164). This is characterized by an increase in PI3K activation and nuclear factor-κB (NF-κB) translocation to the nucleus, whereas CXCR4 siRNA downregulates PI3K/AKT, and IKKα activation sensitizing K562/ADM cells to ADR treatment demonstrated by an inhibition of cell growth and increase in apoptosis (164). Both in vitro and in vivo, treatment with oroxylin A, a monoflavonoid, targets leukemic/ADM cells with low off target toxicity by increasing apoptosis via downregulation of CXCR4 expression (164). Taken together, these results suggest that compounds such as polyphenols or flavonoids may be able to target the malignant cell redox environment through the manipulation of the NRF2 pathway. As discussed earlier, the flavone baicalein offers protection of splenic and BM HSPC from TBI through ERK and NRF2 activation (113, 176). However, within malignant K562 CML cells, treatment with both baicalein and baicalin results in an increase in NOTCH-1 activation followed by an increase in Hes-1, cyclin D1, Hey-1, and Hey-2 mRNA levels (161). In addition, treatment of K562 cells with baicalin resulted in an increase of cells in G0/G1 phase as well as a decrease of cells entering S phase and G2/M phase, whereas treatment with baicalein only resulted in a decrease of cells in G2/M phase (161). Interestingly, both compounds inhibited K562 colony formation in soft agar assays (161). Again, this highlights the ability of redox-active compounds to selectively target the malignant cellular redox environment, while, at the same time, offering protection to normal HSPC populations.
GSH homeostasis is also targeted by synergistic combination treatments of lymphoid and myeloid cell lines with polyphenol compounds and DOX or ETP (92, 93, 176). Quercetin, apigenin, emodin, rhein, and cis-stilbene all had synergistic effects in cytotoxic induction of DOX and ETP within CCRF-CEM and Jurkat malignant lymphoid cell lines. Quercetin and apigenin, however, were the only polyphenols that demonstrated synergistic effects with DOX and ETP in THP-1 and KG-1a malignant myeloid cell lines. All synergistic effects were closely associated with a decrease of cellular GSH, a decrease of ATP production, and activation of the apoptosis cascade (92, 93). Of note, the lymphoid lineage cell lines being most sensitive to this redox-based combination treatment actually demonstrated the lowest basal levels of GSH, indicating that lymphoid cancers may be excellent targets for redox-based therapies. Other examples of polyphenol induction of LSC cytotoxicity include the use of Nispex, a polyphenol mixture that mainly comprises quercetin, quercitroside, avicularin, and oleandrin. This mixture has been proven effective against the AML/HL-60 cell line both in vivo and in a xenograft in vivo model (56, 57, 169).
The HL-60 cell line is one of the earliest malignant promyelocytic leukemia cell lines used for the elucidation of chemotherapeutic mechanisms. DOX treatment of HL-60 cells results in the activation of caspases 3, 8, and 9 as well as DNA fragmentation (140). The development of the HL-60/ADR (DOX resistant) cell line has led to the implementation of alternative therapies that include DOX. For example, pretreatment with a sublethal dose of DOX and subsequent combination with TRAIL, a member of the tumor necrosis factor (TNF) family, induces cytotoxicity in the parental and HL-60/ADR cell lines, indicating DOX induced redox signaling even in drug-resistant tumor types (78). This occurs through the depolarization of the mitochondrial membrane and increased levels of caspase 9 and Apaf-1 from the activation of mitochondrial type II apoptosis pathway (78). Importantly, HL-60 cells have demonstrated sensitivity to other more basic redox-active compounds such as exogenous H2O2 and the nitroxide compound, tempol (109). HL-60 cells demonstrate significant sensitivity to H2O2-induced cell death measured by apoptotic markers at concentrations as low as 5 μM, whereas normal BM and mononuclear cells isolated from cord blood remain resistant to cell death. Primitive populations isolated from normal control were not affected by H2O2 treatment, yet CD34+ populations isolated from the HL-60 pool were significantly reduced without differentiation at all H2O2 concentrations evaluated. The authors continued to identify AKT as a potential mediator of H2O2-induced cell death within progenitor populations of HL-60 cells (109). Tempol treatment of HL-60 cells results in a p53-independent upregulation of p21, indicating that peroxide generating and nitroxide compounds may be promising alternative therapies for chemo-resistant tumor types (52, 109).
MnSOD has been widely discussed as a tumor suppressor for many years, and just as the roles of GSH homeostasis and CAT activity play important parts of normal and malignant HSPC treatment, MnSOD has also been recently demonstrated as a potential therapeutic specific to malignant HSCs (110, 180). A phenotypic trait that separates HSPCs from LSCs is the differential expression of various surface markers such as CD33 and CD123 (119). For example, some LSC populations exhibit high levels of CD123 or interleukin-3 receptor (IL-3R) expression, whereas normal HSC populations do not (119). This has led to a targeted approach for the delivery of MnSOD as a tumor suppressor. Much like the liposomal delivery of MnSOD previously discussed in normal HSPCs, Li et al. utilized CD123 for the targeted delivery of adenoviral particles carrying the MnSOD gene. Adenoviral delivery of MnSOD induced dose-dependent cytotoxicity in HL-60 and KG-1 malignant myeloid cell lines (87). Upon infection, cytotoxicity is accompanied by increases in cleaved caspases 7 and 9 as well as increases in cleaved PARP, indicating mitochondrial activation of apoptosis. In addition, delivery of MnSOD to xenografted HL-60 cells prolonged survival in mice (87). Similar results were seen via treatment of murine thymic lymphoma cells. Here, Jaramillo et al. found that elevated expression of MnSOD increased cellular H2O2, sensitizing WEHI7.2 cells to glucocorticoid and H2O2-induced apoptosis. This result was recapitulated with the application of MnTE. Although application of MnTE can in some cases produce peroxide, sensitization of WEHI7.2 cells may occur through another mechanism, such as the oxidation of NF-κB (71). In addition, MnTE combination treatment with cyclophosphamide inhibited cell growth. No sensitization of WEHI7.2 cells was demonstrated in a combination of MnTE and DOX; however, MnTE addition did protect cardiomyocytes from DOX-induced injury (71). These findings have large implications for the future of leukemia drug development: delivery of MnSOD to normal HSPC populations either by genetic overexpression or by treatment with an SOD2 mimetic promotes HSPC function, whereas ectopic MnSOD expression or MnP treatment in leukemia cell populations clearly induces cell death (71, 87, 88, 99, 179). Furthermore, MnP function within malignant populations may be enhanced by the presence of other biologically relevant redox-active compounds and/or proteins. For example, Ferrer-Sueta et al. have shown that MnPs may be reduced by enzymes such as xanthine oxidase and complexes I and II of the mitochondrial electron transport chain (42). This reduction shown in submitochondrial particles allows for catalytic redox cycling and reduction of peroxynitrite, offering mitochondrial protection against peroxynitirite formation and damage (42, 158). In addition, the combination of MnPs with vitamin C has a promising outlook. Vitamin C itself has demonstrated a protective effect against oxidative insult within HL-60 cells (53). However, various concentrations of vitamin C in combination with MnPs may be effective. Although this combination has not been evaluated in HSPC or LSC populations, vitamin C has been shown to sensitize pancreatic and breast cancer cell populations through metabolic alterations to the cellular redox environment and production of species such as peroxides (37, 148, 154). Furthermore, combination of MnP with vitamin C has demonstrated significant effects on the malignant cell redox environment, primarily through the generation of H2O2, within prostate and breast cancer, as well as glioblastoma populations (38, 148, 174, 181). This strategy is supported by the finding that exogenous H2O2 kills primitive LSCs while leaving normal HSPC populations unharmed (109). This indicates that redox-active compounds such as MnSOD mimetics, themselves or in combination with other redox-active drugs, may be able to modulate redox levels that support healthy HSPC function, leading to the safe eradication of LSCs from the hematopoietic compartment.
Perhaps one of the most primitive and aggressive of all hematopoietic cancers are CMLs. CML patients in blast crisis undergo rapid clonal expansion of progenitor cell compartments and are often resistant to tyrosine kinase inhibitor (TKI) therapies due to altered expression of the breakpoint cluster region-Abl kinase fusion protein (BCR-Abl) fusion gene (30, 63). Redox-based therapies, however, offer potential solutions to refractory disease. Treatment of the KBM5- and the KBM5-T315I TKI-resistant CML cell line with ROS inducers such as adaphostin or β-phenylethyl isothiocyanate (PEITC) results in complete inhibition of CML cell growth within cell lines and primary samples (24, 112, 151, 177). The cytoxicity is based on these ROS inducers' ability to modify the redox environment of the CML cell that results in the oxidative modification and subsequent cleavage of the BCR-ABL protein. The modification of the redox milieu is caused by a sharp depletion of cellular GSH. Decreases in GSH, BCR-ABL cleavage, and cytotoxic effects are reversed by the pretreatment of CML cells with NAC. Similar effects of BCR-ABL degradation are caused by treatment with BSO. PEITC-induced cytotoxicity is also observed in primary CML patient samples drawn from individuals who display both imatinib sensitivity and resistance (177).
Similar studies have been conducted in the CD34+ K562 CML cell line. The K562 line is a BCR-ABL+ cell line that demonstrates resistance to imatinib. However, K562 cells are sensitized to imatinib by cotreatment with simvastatin, a known nitric oxide producer (25). The sensitization of K562 cells to this combination treatment is rescued by pretreatment with NAC, implying that the production of reactive species allows for successful treatment. This may very well proceed through the depletion of cellular GSH subsequent to the production of peroxynitrite (25). More recently, a combination treatment of DOX and polyphenol extract from Viscum album rescued K562 cells from G2/M arrest by expression of cyclins B1 and D1. This resulted in the activation of apoptosis and sensitization of K562 cells to DOX treatment (135). Elucidation of redox modifiers that can impact primitive and resistant malignant populations is clearing the way for emerging combination treatments, allowing for the use of subtoxic doses of classic chemotherapeutics that, originally, were detrimental to the patient hematopoietic compartment. These findings highlight the important ability of redox modifiers to provide new solutions for refractory diseases, ultimately improving outcomes for patients with a poor prognosis.
Redox-Based Therapies Targeting LSC Metabolism
Expertly reviewed by Pollyea and Jordan, the LSC redox environment can be specifically targeted through differential metabolic profiles between HSPCs and LSCs (119). Potential targets of LSC mitochondrial-induced apoptosis are IKK2 (IKB kinase 2) and NF-κB (47). Inhibition of NF-κB activation results in mitochondrial dysfunction, rupture, and cell death. Importantly, NF-κB demonstrates constitutive activation in AML and has been used to characterize LSC populations (15, 103). As demonstrated in the combination treatment of TNF-related apoptosis-inducing ligand (TRAIL) and DOX for refractory AML cells, inhibition of NF-κB with small molecules such as JSH-23 or AS602868 also sensitizes LSCs to treatment with TRAIL (39, 47, 146). As mentioned throughout this review, targeting GSH metabolism may also occur in the mitochondria, separating the LSC population apart from normal HSPC populations. Recently, however, more exciting discoveries have come to light. Several recent studies have identified the isocitrate dehydrogenases as redox-active targets for cancer therapies. For example, downregulation of IDH1 may sensitize cells to H2O2 production. Conversely, over production of the oxidative metabolite 2-hydroxyglutarate sensitizes LSCs to B cell lymphoma 2 downregulation (23, 44, 83, 119). The overproduction of 2-hydroxyglutarate, stemming from mutations in the isocitrate dehydrogenase enzymes, may cause epigenetic changes that push leukemogenesis (23, 44, 83, 84, 119, 130). Thus, from these studies, we may learn of redox-driven epigenetic events that initialize HSPC transformation in the BM niche. In addition, we have found that the MnTnBuOE porphyrins are excellent candidates for the alteration of HSPC and LSC mitochondrial function. Treatment of HSPCs with MnTnBuOE results in a decrease of mitochondrial function, an increase in antioxidant defense enzyme levels, and an increase in uncoupling protein expression, eventually offering protection of HSPCs (179). Moreover, as previous evidence has suggested, these same changes may have cytotoxic effects in LSC populations.
Targeting the NADPH Oxidase System
As previously mentioned, normal HSPC irradiation produces a phenotype of HSPC exhaustion resulting from DNA damage and the upregulation of cell cycle inhibitor proteins, leading to HSPC senescence (127, 163). This exhaustion can be protected against by treatment with redox-active compounds (71, 88, 99, 179). The principal mediator of these effects is the ROS produced by the NOX enzymes, implicating NOX enzymes as potential redox-based therapeutic target for leukemias (62, 72, 94, 118, 136). Aggressive subpopulations of AML can be identified by internal tandem duplication of the FMS-like tyrosine kinase 3-internal tandem repeat (FLT3-ITD) (122, 136). This population has elevated levels of DNA damage and ROS production. However, recent studies, particularly those demonstrating that Nox4−/− mice are resistant to a FLT3-ITD-mediated induction of an MPD phenotype, elucidate NOX as a driving factor in the generation or propagation of LSC (72). These studies further suggest that redox-based therapies targeting NOX-produced ROS may simultaneously be effective in the treatment of LSCs. Inhibition of the FLT3-ITD signaling via PCK412 treatment results in decreased STAT5 signaling and the downregulation of the NOX subunit p22phox (167). This and the inhibition of NOX via DPI or VAS2870 treatment led to a decrease in DNA damage as well as a decrease in H2O2 production in the cytosol and nucleus of AML blast cells. Both culture models and primary patient HSCs that demonstrate elevated ROS production by NOX have depleted GSH levels and diminished antioxidant capabilities (136, 167). These facets promote the growth of LSC population over their normal CD34+ counterparts, and this growth advantage is also mediated through loss of response to the p38 MAPK signaling pathway. Taken together, these results show that just as normal HSPCs undergo exhaustion in response to ROS-mediated MAPK signaling, malignant LSC populations are propagated through NOX-induced ROS and loss of their normal p38 MAPK response that limits HSPC pools. In addition to NOX inhibitors, this presents another therapeutic window for the MnPs in leukemia therapeutic regimens, for the redox-active MnTnBuOE and MnTE are able to modulate the cellular redox environment offering control over the p38 MAPK, NRF2, and NOX/NQO1 systems that may protect normal HSPCs while driving malignant cells to activate cell senescence mechanisms (Fig. 5).
Epigenetic Considerations in LSC Redox Treatments
Targeting epigenetic characteristics have also become a successful avenue of treatment for many hematopoietic disorders, including AML, myelodysplastic syndrome (MDS), and CML. Several cytogenetic abnormalities have become very prominent across the leukemias, particularly those involving fusion of mixed lineage leukemia/eleven nineteen leukemia and HOXA and nuclear pore complex protein 98 (NUP98) genes within AML, BCR-ABL in CML and acute lymphoblastic leukemia (ALL), and myelodysplastic 5q deletion syndrome in MDS (12, 13, 17, 25, 77, 85, 126, 130, 135, 145). Aberrant DNA methylation patterns have widely been recognized for the characterization of hematopoietic disorders within both the myeloid and lymphoid lineage (45, 48, 51, 68, 74). As patient populations grow increasingly resistant to therapies, methyl transferase or demethylase inhibitors such as the azacytidines and histone deacetylase inhibitors have been developed (128, 130). Even still, fractions of patient populations demonstrate resistance to epigenetic therapies. As with resistant CMLs however, there does seem to be a role for redox-active compounds in combination treatments that result in sensitization to epigenetic treatments. Recently, inhibition of lysine specific histone demethylase 1A (LSD1) demethylase with trans-2-phenylcyclopropylamine combined with all-trans retinoic acid (ATRA) treatment induced sensitivity in several AML models, demonstrated by decreased AML engraftment in vivo as well as an increase in the expression of differentiation markers such as CD11b (124). This has potential redox implications as ATRA has been known to induce ROS-mediated embryonic stem cell death (20). In addition, ATRA has been identified as an NRF2 inhibitor and has potentiated arsenic trioxide toxicity in human AML cells (157, 162). Combination treatment in MM with the histone deacetylase inhibitor PXD101 and bortezomib induces cell death in primary CD138+ MM cells but not in normal mononuclear populations (41).
These effects are driven by activation of caspases 3, 8, and 9 with coordinate activation of p53 and the p38 MAPK pathway, all of which can be blocked with NAC treatment (33, 41, 139, 175). Finally, the methyltransferase inhibitor 3-deazaneplanocin (DZNep) inhibits the polycomb-repressive complex-2 (PRC2) through the upregulation of thioredoxin interacting protein (TXNIP). TXNIP activity then results in TXN inhibition and a subsequent increase in oxidative stress that may not be a direct result of TXNIP-mediated inhibition of TXN itself. This ultimately results in apoptotic signaling within AML culture lines as well as primary samples (182). This also identifies DZNep as a potential therapy for CML (125, 170). The discovery of TXNIP downregulation in AMLs and the identification of PRC2/EZH2 as a target for redox-based therapies are an exciting advancement (125, 170). Recently the Txnip−/− model has identified TXNIP as a crucial regulator of normal LT-HSC function (73, 76). Txnip−/− mice undergo HSPC exhaustion that is characterized by a loss of repopulation capacity, increased Wnt signaling, a decrease in p21 expression, and a loss of CXCL12 activity within the BM niche (73). Loss of TXNIP, as has been demonstrated in AMLs, results in an advanced aging phenotype in which HSC exhaustion and BM failure occurs through activation of the p53 and p38 MAPK pathway and induction of cell cycle regulators (182). Together, these results demonstrate that the Txnip−/− phenotype closely resembles hematopoietic phenotypes characterized by pharmacological induction of redox stress or those generated by the selection of ROS high populations. These studies highlight the potential redox targeting of pathways of normal HSPCs use for maintenance within malignant populations. This supports the employment of ROS and redox modulation for the successful development of cancer therapies that are highlighted by the protection of normal tissues (Fig. 5). We have summarized the redox-active compounds as they have been cited and discussed throughout this review to highlight potential areas of therapeutic development that may safely and successfully target malignant populations in the future (Fig. 6).
FIG. 6.

The table summarizes the redox-active compounds that have been discussed throughout this review as well as their effects within their indicated disease states and normal HSPC populations where applicable. This dual application highlights the potential for selective targeting of malignant populations while offering protection to normal HSPCs for many redox-active compounds discussed and cited within this review. ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; FLT3-ITD, FMS-like tyrosine kinase 3-internal tandem repeat; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase regulatory subunit; LSD1, lysine specific histone demethylase 1A; MDS, myelodysplastic syndrome; MM, multiple myeloma; NF-κB, nuclear factor-κB; PARP, poly (ADP-ribose) polymerase; PBMC, peripheral blood mononuclear cell; TRAIL, TNF-related apoptosis-inducing ligand; TXNIP, thioredoxin-interacting protein.
Summary and Future Directions
In the past, the limited, primitive, and quiescent state of HSPCs has made it difficult to meaningfully evaluate the impacts of redox modification in normal and malignant HSPCs. In the past 10 years, we have begun to significantly evaluate and realize the important impact that management of the HSPC redox environment has on normal hematopoiesis and overall patient outcome. We now know that the HSPC redox environment can be manipulated by redox-active compounds to protect and improve HSPC function while still creating stress conditions that may differentially target malignant LSCs. This may be accomplished through combination therapies utilizing low-dose chemotherapeutics and the implementation of a single redox cycler. To this purpose, several crucial pathways, such as FOXO, Atm, NRF2, NOX, NF-κB, and p38 MAPKs, have emerged as critical HSPC redox regulators.
Through the years, there has been some debate over the importance of an impact of GSH homeostasis in the management of cellular redox environments and the treatment of cancers (46). However, within the HSPC compartment, the management of GSH/GSSG levels appears to be indispensable. Accurate measurement of cellular GSH and GSSG concentrations obviously provides insight into the mechanistic underpinnings of drug activity as previously discussed, and new methods of quantitation have emerged allowing for GSH measurement in HSPC populations (19, 134). These studies may also aid in the identification of therapeutics not previously thought to be redox active. For example, lenalidomide (LEN) has been used for the treatment of several hematopoietic diseases (96, 126, 145). This compound, previously thought to only act as an immunosuppressive agent, has demonstrated the redox-mediated activation of AP-1 signaling in MM treatment (32). Subsequently, we found that LEN can deplete cellular GSH levels in an MDS model at a level and rate similar to DOX (19). This now opens both LEN and MDS to consideration for redox-based therapies. In addition, our previous work has demonstrated that modulation of MRP1 expression dictates cellular GSH/GSSG concentrations. Most notably, an increase in HSPC GSH concentrations is demonstrated in the Mrp1−/− mouse model, in which loss of MRP1 results in an increase in HSPC function. These results are demonstrated by colony formation assays as well as by in vivo serial transplantation experiments. Improvements in WT HSPC function, compared with the Mrp1−/−, can also be recapitulated with the addition of NAC (120). This indicates that ABC transporter inhibitors may have a role in the development of leukemia therapies that protect the HSPC compartment. Moreover, GSH and GSSG levels appear to be a potential biomarker for the redox-based therapy candidacy. We have previously found that peripheral leukocytes isolated from AML patients contain significantly higher cellular GSH levels, resulting in a significant difference in the 2GSH/GSSG ratio between patients and healthy donors (19). These elevated GSH levels may be the result of an increase in reactive species produced by malignant cells, relative to healthy cells. Redox-active therapies must then be able to overcome the increased redox buffer capacity demonstrated in malignant cells to elicit a desired therapeutic effect. Thus, the 2GSH-GSSG ratio may not only be a marker for disease but also serve as an indicator for redox-based therapies. Because the cellular redox potential can be calculated based on the 2GSH/GSSG ratio, we may be able to establish limits of HSPC/LSC redox potentials in which we know that certain redox-based therapies will be efficacious (81, 123). In the future, evaluation of patient cellular redox potentials across diseases (CML, AML, ALL etc.) may allow for the prediction of response of disease states to specific redox-based therapies.
The focus of these principles should expand to include a better understanding of other factors affecting the HSPC redox environment, such as thioredoxin activity and the function of TXNIP. TXNIP has recently been identified as an important regulator of the HSPC, wherein loss results in p38 activation and LT-HSC exhaustion (73). If this occurs through NRF2 regulation, TXNIP may emerge as a druggable target for the treatment of hematopoietic malignancies. In addition, cytogenetic abnormalities, such as chromosomal rearrangements and deletions, have been well documented throughout the hematopoietic diseases, yet major changes in epigenetic programming remain unclear. For example, TXNIP studies involving DNA methylation status indicate that future efforts must focus on the impact that redox-active compounds have on the epigenetic regulation of the normal HSPC as well as the malignant LSC. Redox evaluation of epigenetic changes in hematopoietic malignancies, such as those demonstrated by the isocitrate dehydrogenase enzymes (IDH1/2), may provide valuable insight into potential initiating events that drive LSC propagation (23, 44, 80, 84, 130). This may provide avenues for new genetic-based therapies that utilize the manipulation of the LSC redox environment.
Finally, redox modifiers and redox cycling drugs should be used to elucidate the impact of signaling pathways contributing to malignant HSPC expansion. For example, subpopulations of aggressive hematopoietic malignancies express several isoforms of the NOX family, as compared with limited NOX expression in normal HSCs (62, 72, 94). However, in normal HSPCs, increased NOX expression and activity results in overproduction of ROS (O2.−, H2O2) leading to the activation p38 MAPKs, which results in the upregulation of cell cycle regulators. It is unclear whether constitutive NOX activity in malignant populations leads to the loss of AKT and p38 MAPK regulation over LSC expansion or whether components within, such as the AP-1 family of transcription factors, may be targeted for redox regulation by redox-active compounds to regain control of progenitor compartments undergoing clonal expansion (Fig. 5). These aspects have the potential to improve redox-based therapies, providing new directions for safe, effective treatments.
Abbreviations Used
- 53BP1
p53 binding protein
- ABCG2
ATP binding cassette subfamily G member 2
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- AP-1
activating protein-1
- Atm
ataxia-telangiectsia mutated
- ATP
adenosine tri-phosphate
- ATRA
all-trans retinoic acid
- BCL-2
B cell lymphoma 2
- BCR-Abl
breakpoint cluster region-Abl kinase fusion protein
- BM
bone marrow
- BSO
buthionine sulfoximine
- CAT
catalase
- CD
cluster of differentiation protein
- CD45RA (PTPRC)
cluster of differentiation protein 45 isofrom A (protein tyrosine phosphatase, receptor type C isoform A)
- CLL
chronic lymphocytic leukemia
- CLP
common lymphoid progenitor
- CML
chronic myelogenous leukemia
- CMP
common myeloid progenitors
- CSC
cancer stem cell
- CXCR4
chemokine receptor type 4
- DCF
2′-7′-dichlorofluorescence
- DOX
doxorubicin
- DPI
diphenylene iodonium
- DZNep
3-deazaneplanocin
- ETP
etoposide
- FLT3-ITD
FMS-like tyrosine kinase 3-internal tandem repeat
- Foxo3a
forkhead box O3a
- GCLC
glutatmate-cysteine ligase catalytic subunit
- GCLM
glutatmate-cysteine ligase regulatory subunit
- GPx
glutathione peroxidase
- GR
glutathione reductase
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- GST
glutathione-S-transferase
- Gy
gray
- H2O2
hydrogen peroxide
- HSC
hematopoietic stem cell
- HSPC
hematopoietic stem/progenitor cell
- IDH1
isocitrate dehydrogenase 1
- LEN
lenalidomide
- Lin
lineage
- L-PAM
melphalan
- LSC
leukemic stem cell
- LSD1
lysine specific histone demethylase 1A
- LSK
Lin−/Sca-1+/c-Kit+
- LTC-IC
long-term culture-initiating cell
- LT-HSC
long-term hematopoietic stem cell
- MDS
myelodysplastic syndrome
- MM
multiple myeloma
- MnPs
Mn porphyrins
- MnTE/MnTE-2-PyP5+
Mn (III) meso-tetrakis (N-ethylpyridinium-2-yl) porphyrin
- MnTnBuOE/MnTnBuOE-2-PyP5+
Mn (III) meso-tetrakis(N-(n-butoxyethyl)pyridinium-2-yl) porphyrin
- MPD
myeloproliferative disorder
- MPP
multipotent progenitor cell
- MRP1/ABCC1
multidrug-resistant protein 1/ATP-binding cassette subfamily C member 1
- MTX
methotrexate
- NAC
N-acetyl-l-cysteine
- NADPH
nicotinamide adenine dinucleotide phosphate
- NF-κB
nuclear factor-κB
- NOX
NADPH oxidase
- Nq01
NAD(P)H dehydrogenase quinone 1
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- NUP98
nuclear pore complex protein 98
- O2
molecular oxygen
- O2.−
superoxide
- p15INK4B
cyclin-dependent kinase 4 inhibitor B
- p16INK4A
cyclin-dependent kinase inhibitor 2A
- p18INK4C
cyclin-dependent kinase 4 inhibitor C
- p19ARF
cyclin-dependent kinase 4/6 inhibitor
- p21CIP
cyclin-dependent kinase inhibitor 1
- p38 MAPK
p38 mitogen-activated protein kinase
- PARP
poly (ADP-ribose) polymerase
- PBMC
peripheral blood mononuclear cell
- PEITC
β-phenylethyl isothiocyanate
- PLM
piperlongumine
- PRC2
polycomb-repressive complex-2
- PTL
parthenolide
- ROS
reactive oxygen species
- Sca-1
stem cell antigen 1
- SOD2
manganese superoxide dismutase
- ST-HSC
short-term hematopoietic stem cell
- TBI
total body irradiation
- TKI
tyrosine kinase inhibitor
- TNF
tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
- TXN
thioredoxin
- TXNIP
thioredoxin-interacting protein
- UPC1/3
uncoupling protein 1/3
Acknowledgments
This work is supported by National Institute of Health training grant T32 ES007266, the Edward P. Evans Foundation, and the NIH grant CA 205400-01.
References
- 1.Adams GB. and Scadden DT. The hematopoietic stem cell in its place. Nat Immunol 7: 333–337, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Al-Hajj M, Becker MW, Wicha M, Weissman I, and Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev 14: 43–47, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Allay JA, Spencer HT, Wilkinson SL, Belt JA, Blakley RL, and Sorrentino BP. Sensitization of hematopoietic stem and progenitor cells to trimetrexate using nucleoside transport inhibitors. Blood 90: 3546–3554, 1997 [PubMed] [Google Scholar]
- 4.Arai F, Hirao A, and Suda T. Regulation of hematopoietic stem cells by the niche. Trends Cardiovasc Med 15: 75–79, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, and Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J 418: 29–37, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babiak RM, Campello AP, Carnieri EG, and Oliveira MB. Methotrexate: pentose cycle and oxidative stress. Cell Biochem Funct 16: 283–293, 1998 [DOI] [PubMed] [Google Scholar]
- 7.Bartholdy B, Christopeit M, Will B, Mo Y, Barreyro L, Yu Y, Bhagat TD, Okoye-Okafor UC, Todorova TI, Greally JM, Levine RL, Melnick A, Verma A, and Steidl U. HSC commitment-associated epigenetic signature is prognostic in acute myeloid leukemia. J Clin Invest 124: 1158–1167, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batinic-Haberle I. and Spasojevic I. Complex chemistry and biology of redox-active compounds, commonly known as SOD mimics, affect their therapeutic effects. Antioxid Redox Signal 20: 2323–2325, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Batinic-Haberle I, Spasojevic I, Tse HM, Tovmasyan A, Rajic Z, St Clair DK, Vujaskovic Z, Dewhirst MW, and Piganelli JD. Design of Mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids 42: 95–113, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batinic-Haberle I, Tovmasyan A, Roberts ER, Vujaskovic Z, Leong KW, and Spasojevic I. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid Redox Signal 20: 2372–2415, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beerman I, Luis TC, Singbrant S, Lo Celso C, and Mendez-Ferrer S. The evolving view of the hematopoietic stem cell niche. Exp Hematol 50: 22–26, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernt KM. and Armstrong SA. Targeting epigenetic programs in MLL-rearranged leukemias. Hematol Am Soc Hematol Educ Program 2011: 354–360, 2011 [DOI] [PubMed] [Google Scholar]
- 13.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL, and Armstrong SA. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20: 66–78, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhinge KN, Gupta V, Hosain SB, Satyanarayanajois SD, Meyer SA, Blaylock B, Zhang QJ, and Liu YY. The opposite effects of doxorubicin on bone marrow stem cells versus breast cancer stem cells depend on glucosylceramide synthase. Int J Biochem Cell Biol 44: 1770–1778, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosman MC, Schuringa JJ, and Vellenga E. Constitutive NF-kappaB activation in AML: causes and treatment strategies. Crit Rev Oncol Hematol 98: 35–44, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, Dohner H, and Pollack JR. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med 350: 1605–1616, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Calvo KR, Sykes DB, Pasillas MP, and Kamps MP. Nup98-HoxA9 immortalizes myeloid progenitors, enforces expression of Hoxa9, Hoxa7 and Meis1, and alters cytokine-specific responses in a manner similar to that induced by retroviral co-expression of Hoxa9 and Meis1. Oncogene 21: 4247–4256, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, and Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15: 1239–1253, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carroll D, Howard D, Zhu H, Paumi CM, Vore M, Bondada S, Liang Y, Wang C, and St Clair DK. Simultaneous quantitation of oxidized and reduced glutathione via LC-MS/MS: an insight into the redox state of hematopoietic stem cells. Free Radic Biol Med 97: 85–94, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castro-Obregon S. and Covarrubias L. Role of retinoic acid and oxidative stress in embryonic stem cell death and neuronal differentiation. FEBS Lett 381: 93–97, 1996 [DOI] [PubMed] [Google Scholar]
- 21.Celic T, Spanjol J, Bobinac M, Tovmasyan A, Vukelic I, Reboucas JS, Batinic-Haberle I, and Bobinac D. Mn porphyrin-based SOD mimic, MnTnHex-2-PyP(5+), and non-SOD mimic, MnTBAP(3-), suppressed rat spinal cord ischemia/reperfusion injury via NF-kappaB pathways. Free Radic Res 48: 1426–1442, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chai X, Li D, Cao X, Zhang Y, Mu J, Lu W, Xiao X, Li C, Meng J, Chen J, Li Q, Wang J, Meng A, and Zhao M. ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci Rep 5: 10181, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, Zhao F, Medeiros BC, Tyvoll DA, and Majeti R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med 21: 178–184, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandra J, Tracy J, Loegering D, Flatten K, Verstovsek S, Beran M, Gorre M, Estrov Z, Donato N, Talpaz M, Sawyers C, Bhalla K, Karp J, Sausville E, and Kaufmann SH. Adaphostin-induced oxidative stress overcomes BCR/ABL mutation-dependent and -independent imatinib resistance. Blood 107: 2501–2506, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen R, Xiao W, Li D, and Mu S. Combination of simvastatin and imatinib sensitizes the CD34+ cells in K562 to cell death. Med Oncol 28: 528–531, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Chow DC, Wenning LA, Miller WM, and Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I. Krogh's model. Biophys J 81: 675–684, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chow DC, Wenning LA, Miller WM, and Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys J 81: 685–696, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chow KU, Boehrer S, Bojunga J, Stieler M, Rummel MJ, Fauth F, Schneider B, Martin H, Hoelzer D, Weidmann E, and Mitrou PS. Induction of apoptosis by cladribine (2-CdA), gemcitabine and other chemotherapeutic drugs on CD34+/CD38+ and CD34+/CD38- hematopoietic progenitor cells: selective effects of doxorubicin and 2-CdA with protection of immature cells. Leuk Lymphoma 43: 377–384, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Cipolleschi MG, Dello Sbarba P, and Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood 82: 2031–2037, 1993 [PubMed] [Google Scholar]
- 30.Clarke CJ. and Holyoake TL. Preclinical approaches in chronic myeloid leukemia: from cells to systems. Exp Hematol 47: 13–23, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colinas RJ, Burkart PT, and Lawrence DA. In vitro effects of hydroquinone, benzoquinone, and doxorubicin on mouse and human bone marrow cells at physiological oxygen partial pressure. Toxicol Appl Pharmacol 129: 95–102, 1994 [DOI] [PubMed] [Google Scholar]
- 32.Colla S, Zhan F, Xiong W, Wu X, Xu H, Stephens O, Yaccoby S, Epstein J, Barlogie B, and Shaughnessy JD., Jr. The oxidative stress response regulates DKK1 expression through the JNK signaling cascade in multiple myeloma plasma cells. Blood 109: 4470–4477, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D'Arena G, Valentini CG, Pietrantuono G, Guariglia R, Martorelli MC, Mansueto G, Villani O, Onofrillo D, Falcone A, Specchia G, Semenzato G, Di Renzo N, Mastrullo L, Venditti A, Ferrara F, Palumbo A, Pagano L, and Musto P. Frontline chemotherapy with bortezomib-containing combinations improves response rate and survival in primary plasma cell leukemia: a retrospective study from GIMEMA Multiple Myeloma Working Party. Ann Oncol 23: 1499–1502, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Davies KJ. and Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 261: 3060–3067, 1986 [PubMed] [Google Scholar]
- 35.Diaz-Montero CM, Wang Y, Shao L, Feng W, Zidan AA, Pazoles CJ, Montero AJ, and Zhou D. The glutathione disulfide mimetic NOV-002 inhibits cyclophosphamide-induced hematopoietic and immune suppression by reducing oxidative stress. Free Radic Biol Med 52: 1560–1568, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doroshow JH. and Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem 261: 3068–3074, 1986 [PubMed] [Google Scholar]
- 37.Du J, Cieslak JA, 3rd, Welsh JL, Sibenaller ZA, Allen BG, Wagner BA, Kalen AL, Doskey CM, Strother RK, Button AM, Mott SL, Smith B, Tsai S, Mezhir J, Goswami PC, Spitz DR, Buettner GR, and Cullen JJ. Pharmacological ascorbate radiosensitizes pancreatic cancer. Cancer Res 75: 3314–3326, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans MK, Tovmasyan A, Batinic-Haberle I, and Devi GR. Mn porphyrin in combination with ascorbate acts as a pro-oxidant and mediates caspase-independent cancer cell death. Free Radic Biol Med 68: 302–314, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fadeev RS, Solovieva ME, Slyadovskiy DA, Zakharov SG, Fadeeva IS, Senotov AS, Golenkov AK, and Akatov VS. [Inhibition of NF-kB activation decreases resistance in acute myeloid leukemia cells to trail-induced apoptosis in multicellular aggregates]. Biofizika 60: 1146–1150, 2015 [PubMed] [Google Scholar]
- 40.Fan J, Cai H, Yang S, Yan L, and Tan W. Comparison between the effects of normoxia and hypoxia on antioxidant enzymes and glutathione redox state in ex vivo culture of CD34(+) cells. Comp Biochem Physiol B Biochem Mol Biol 151: 153–158, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Feng R, Oton A, Mapara MY, Anderson G, Belani C, and Lentzsch S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol 139: 385–397, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Ferrer-Sueta G, Hannibal L, Batinic-Haberle I, and Radi R. Reduction of manganese porphyrins by flavoenzymes and submitochondrial particles: a catalytic cycle for the reduction of peroxynitrite. Free Radic Biol Med 41: 503–512, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Fiegl M, Samudio I, Clise-Dwyer K, Burks JK, Mnjoyan Z, and Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood 113: 1504–1512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, and Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18: 553–567, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, Valk PJ, Lowenberg B, Delwel R, and Melnick A. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17: 13–27, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flohe L. The fairytale of the GSSG/GSH redox potential. Biochim Biophys Acta 1830: 3139–3142, 2013 [DOI] [PubMed] [Google Scholar]
- 47.Frelin C, Imbert V, Griessinger E, Peyron AC, Rochet N, Philip P, Dageville C, Sirvent A, Hummelsberger M, Berard E, Dreano M, Sirvent N, and Peyron JF. Targeting NF-kappaB activation via pharmacologic inhibition of IKK2-induced apoptosis of human acute myeloid leukemia cells. Blood 105: 804–811, 2005 [DOI] [PubMed] [Google Scholar]
- 48.Garcia-Manero G, Daniel J, Smith TL, Kornblau SM, Lee MS, Kantarjian HM, and Issa JP. DNA methylation of multiple promoter-associated CpG islands in adult acute lymphocytic leukemia. Clin Cancer Res 8: 2217–2224, 2002 [PubMed] [Google Scholar]
- 49.Gariboldi MB, Rimoldi V, Supino R, Favini E, and Monti E. The nitroxide tempol induces oxidative stress, p21(WAF1/CIP1), and cell death in HL60 cells. Free Radic Biol Med 29: 633–641, 2000 [DOI] [PubMed] [Google Scholar]
- 50.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 57: 727–741, 1999 [DOI] [PubMed] [Google Scholar]
- 51.Goncalves AC, Cortesao E, Oliveiros B, Alves V, Espadana AI, Rito L, Magalhaes E, Pereira S, Pereira A, Costa JM, Mota-Vieira L, and Sarmento-Ribeiro AB. Oxidative stress levels are correlated with P15 and P16 gene promoter methylation in myelodysplastic syndrome patients. Clin Exp Med 16: 333–343, 2016 [DOI] [PubMed] [Google Scholar]
- 52.Gorlach S, Fichna J, and Lewandowska U. Polyphenols as mitochondria-targeted anticancer drugs. Cancer Lett 366: 141–149, 2015 [DOI] [PubMed] [Google Scholar]
- 53.Guaiquil VH, Vera JC, and Golde DW. Mechanism of vitamin C inhibition of cell death induced by oxidative stress in glutathione-depleted HL-60 cells. J Biol Chem 276: 40955–40961, 2001 [DOI] [PubMed] [Google Scholar]
- 54.Gunsilius E, Gastl G, and Petzer AL. Hematopoietic stem cells. Biomed Pharmacother 55: 186–194, 2001 [DOI] [PubMed] [Google Scholar]
- 55.Guo CY, Luo L, Urata Y, Goto S, Huang WJ, Takamura S, Hayashi F, Doi H, Kitajima Y, Ono Y, Ogi T, and Li TS. Sensitivity and dose dependency of radiation-induced injury in hematopoietic stem/progenitor cells in mice. Sci Rep 5: 8055, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo JF, Zhou JM, Feng GK, Liu ZC, Xiao DJ, Deng SZ, Deng R, and Zhu XF. [Apoptosis of human leukemia HL-60 cells induced by rhabdastrellic acid-A and its mechanisms]. Ai Zheng 26: 809–814, 2007 [PubMed] [Google Scholar]
- 57.Guo JF, Zhou JM, Zhang Y, Deng R, Liu JN, Feng GK, Liu ZC, Xiao DJ, Deng SZ, and Zhu XF. Rhabdastrellic acid-A inhibited PI3K/Akt pathway and induced apoptosis in human leukemia HL-60 cells. Cell Biol Int 32: 48–54, 2008 [DOI] [PubMed] [Google Scholar]
- 58.Guo Z, Kozlov S, Lavin MF, Person MD, and Paull TT. Atm activation by oxidative stress. Science 330: 517–521, 2010 [DOI] [PubMed] [Google Scholar]
- 59.Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, and Jordan CT. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 105: 4163–4169, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harrison DK. Optical measurement of tissue oxygen saturation. Int J Low Extrem Wounds 1: 191–201, 2002 [DOI] [PubMed] [Google Scholar]
- 61.Harrison JS, Rameshwar P, Chang V, and Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood 99: 394, 2002 [DOI] [PubMed] [Google Scholar]
- 62.Hole PS, Zabkiewicz J, Munje C, Newton Z, Pearn L, White P, Marquez N, Hills RK, Burnett AK, Tonks A, and Darley RL. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 122: 3322–3330, 2013 [DOI] [PubMed] [Google Scholar]
- 63.Holyoake TL. and Vetrie D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood 129: 1595–1606, 2017 [DOI] [PubMed] [Google Scholar]
- 64.Hu L, Cheng H, Gao Y, Shi M, Liu Y, Hu Z, Xu J, Qiu L, Yuan W, Leung AY, Yang YG, and Cheng T. Antioxidant N-acetyl-L-cysteine increases engraftment of human hematopoietic stem cells in immune-deficient mice. Blood 124: e45–e48, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huntly BJ. and Gilliland DG. Cancer biology: summing up cancer stem cells. Nature 435: 1169–1170, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Huntly BJ. and Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer 5: 311–321, 2005 [DOI] [PubMed] [Google Scholar]
- 67.Irwin ME, Rivera-Del Valle N, and Chandra J. Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 18: 1349–1383, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Issa JP, Baylin SB, and Herman JG. DNA methylation changes in hematologic malignancies: biologic and clinical implications. Leukemia 11 Suppl 1: S7–S11, 1997 [PubMed] [Google Scholar]
- 69.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, and Suda T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 12: 446–451, 2006 [DOI] [PubMed] [Google Scholar]
- 70.Jang YY. and Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 110: 3056–3063, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jaramillo MC, Frye JB, Crapo JD, Briehl MM, and Tome ME. Increased manganese superoxide dismutase expression or treatment with manganese porphyrin potentiates dexamethasone-induced apoptosis in lymphoma cells. Cancer Res 69: 5450–5457, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jayavelu AK, Muller JP, Bauer R, Bohmer SA, Lassig J, Cerny-Reiterer S, Sperr WR, Valent P, Maurer B, Moriggl R, Schroder K, Shah AM, Fischer M, Scholl S, Barth J, Oellerich T, Berg T, Serve H, Frey S, Fischer T, Heidel FH, and Bohmer FD. NOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cells. Leukemia 30: 473–483, 2016 [DOI] [PubMed] [Google Scholar]
- 73.Jeong M, Piao ZH, Kim MS, Lee SH, Yun S, Sun HN, Yoon SR, Chung JW, Kim TD, Jeon JH, Lee J, Kim HN, Choi JY, and Choi I. Thioredoxin-interacting protein regulates hematopoietic stem cell quiescence and mobilization under stress conditions. J Immunol 183: 2495–2505, 2009 [DOI] [PubMed] [Google Scholar]
- 74.Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O'Keefe C, Sekeres M, Saunthararajah Y, and Maciejewski JP. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 113: 1315–1325, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jorgenson TC, Zhong W, and Oberley TD. Redox imbalance and biochemical changes in cancer. Cancer Res 73: 6118–6123, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jung H, Kim DO, Byun JE, Kim WS, Kim MJ, Song HY, Kim YK, Kang DK, Park YJ, Kim TD, Yoon SR, Lee HG, Choi EJ, Min SH, and Choi I. Thioredoxin-interacting protein regulates haematopoietic stem cell ageing and rejuvenation by inhibiting p38 kinase activity. Nat Commun 7: 13674, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jung N, Dai B, Gentles AJ, Majeti R, and Feinberg AP. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun 6: 8489, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ke H, Wang Y, Ren D, and Wang L. Adriamycin sensitizes ardriamycin resistant HL-60/ADR cells to TRAIL-mediated apoptosis. Chin J Clin Oncol 5: 354–360, 2008 [Google Scholar]
- 79.Kensler TW, Wakabayashi N, and Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47: 89–116, 2007 [DOI] [PubMed] [Google Scholar]
- 80.Kim S, Kim SY, Ku HJ, Jeon YH, Lee HW, Lee J, Kwon TK, Park KM, and Park JW. Suppression of tumorigenesis in mitochondrial NADP(+)-dependent isocitrate dehydrogenase knock-out mice. Biochim Biophys Acta 1842: 135–143, 2014 [DOI] [PubMed] [Google Scholar]
- 81.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, and Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic Biol Med 27: 1208–1218, 1999 [DOI] [PubMed] [Google Scholar]
- 82.Kizaki M, Sakashita A, Karmakar A, Lin CW, and Koeffler HP. Regulation of manganese superoxide dismutase and other antioxidant genes in normal and leukemic hematopoietic cells and their relationship to cytotoxicity by tumor necrosis factor. Blood 82: 1142–1150, 1993 [PubMed] [Google Scholar]
- 83.Ku HJ. and Park JW. Downregulation of IDH2 exacerbates H2O2-mediated cell death and hypertrophy. Redox Rep 22: 35–41, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ku HJ, Kwon OS, Kang BS, Lee DS, Lee HS, and Park JW. IDH2 knockdown sensitizes tumor cells to emodin cytotoxicity in vitro and in vivo. Free Radic Res 50: 1089–1097, 2016 [DOI] [PubMed] [Google Scholar]
- 85.Kulkarni AP, Mittal SP, Devasagayam TP, and Pal JK. Oxidative stress perturbs cell proliferation in human K562 cells by modulating protein synthesis and cell cycle. Free Radic Res 43: 1090–1100, 2009 [DOI] [PubMed] [Google Scholar]
- 86.Kuznetsov AV, Margreiter R, Amberger A, Saks V, and Grimm M. Changes in mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin-induced cell death. Biochim Biophys Acta 1813: 1144–1152, 2011 [DOI] [PubMed] [Google Scholar]
- 87.Li G, Li X, Wu H, Yang X, Zhang Y, Chen L, Wu X, Cui L, Wu L, Luo J, and Liu XY. CD123 targeting oncolytic adenoviruses suppress acute myeloid leukemia cell proliferation in vitro and in vivo. Blood Cancer J 4: e194, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H, Wang Y, Pazhanisamy SK, Shao L, Batinic-Haberle I, Meng A, and Zhou D. Mn(III) meso-tetrakis-(N-ethylpyridinium-2-yl) porphyrin mitigates total body irradiation-induced long-term bone marrow suppression. Free Radic Biol Med 51: 30–37, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li S, Zou D, Li C, Meng H, Sui W, Feng S, Cheng T, Zhai Q, and Qiu L. Targeting stem cell niche can protect hematopoietic stem cells from chemotherapy and G-CSF treatment. Stem Cell Res Ther 6: 175, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liang Y, Van Zant G, and Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood 106: 1479–1487, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, Ahn BH, Kumar NG, Rovira II, Xu XL, van Lohuizen M, Motoyama N, Deng CX, and Finkel T. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459: 387–392, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mahbub AA, Le Maitre CL, Haywood-Small SL, Cross NA, and Jordan-Mahy N. Polyphenols act synergistically with doxorubicin and etoposide in leukaemia cell lines. Cell Death Discov 1: 15043, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mahbub AA, Le Maitre CL, Haywood-Small SL, Cross NA, and Jordan-Mahy N. Glutathione is key to the synergistic enhancement of doxorubicin and etoposide by polyphenols in leukaemia cell lines. Cell Death Dis 6: e2028, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maraldi T, Prata C, Vieceli Dalla Sega F, Caliceti C, Zambonin L, Fiorentini D, and Hakim G. NAD(P)H oxidase isoform Nox2 plays a prosurvival role in human leukaemia cells. Free Radic Res 43: 1111–1121, 2009 [DOI] [PubMed] [Google Scholar]
- 95.Maraldi T. Natural compounds as modulators of NADPH oxidases. Oxid Med Cell Longev 2013: 271602, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Matsuoka A, Tochigi A, Kishimoto M, Nakahara T, Kondo T, Tsujioka T, Tasaka T, Tohyama Y, and Tohyama K. Lenalidomide induces cell death in an MDS-derived cell line with deletion of chromosome 5q by inhibition of cytokinesis. Leukemia 24: 748–755, 2010 [DOI] [PubMed] [Google Scholar]
- 97.Mendelson A. and Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med 20: 833–846, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Merchant AA, Singh A, Matsui W, and Biswal S. The redox-sensitive transcription factor Nrf2 regulates murine hematopoietic stem cell survival independently of ROS levels. Blood 118: 6572–6579, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Miao W, Xufeng R, Park MR, Gu H, Hu L, Kang JW, Ma S, Liang PH, Li Y, Cheng H, Yu H, Epperly M, Greenberger J, and Cheng T. Hematopoietic stem cell regeneration enhanced by ectopic expression of ROS-detoxifying enzymes in transplant mice. Mol Ther 21: 423–432, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Minotti G, Menna P, Salvatorelli E, Cairo G, and Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56: 185–229, 2004 [DOI] [PubMed] [Google Scholar]
- 101.Minotti G, Recalcati S, Menna P, Salvatorelli E, Corna G, and Cairo G. Doxorubicin cardiotoxicity and the control of iron metabolism: quinone-dependent and independent mechanisms. Methods Enzymol 378: 340–361, 2004 [DOI] [PubMed] [Google Scholar]
- 102.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M, Chen C, Hosokawa K, Nakauchi H, Nakayama K, Nakayama KI, Harada M, Motoyama N, Suda T, and Hirao A. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1: 101–112, 2007 [DOI] [PubMed] [Google Scholar]
- 103.Morotti A, Parvis G, Cilloni D, Familiari U, Pautasso M, Bosa M, Messa F, Arruga F, Defilippi I, Catalano R, Rosso V, Carturan S, Bracco E, Guerrasio A, and Saglio G. CD7/CD56-positive acute myeloid leukemias are characterized by constitutive phosphorylation of the NF-kB subunit p65 at Ser536. Leukemia 21: 1305–1306, 2007 [DOI] [PubMed] [Google Scholar]
- 104.Morrison SJ, Wright DE, and Weissman IL. Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc Natl Acad Sci U S A 94: 1908–1913, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, Evans WE, Shurtleff SA, and Downing JR. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446: 758–764, 2007 [DOI] [PubMed] [Google Scholar]
- 106.Muranaka S, Fujita H, Fujiwara T, Ogino T, Sato EF, Akiyama J, Imada I, Inoue M, and Utsumi K. Mechanism and characteristics of stimuli-dependent ROS generation in undifferentiated HL-60 cells. Antioxid Redox Signal 7: 1367–1376, 2005 [DOI] [PubMed] [Google Scholar]
- 107.Naka K, Muraguchi T, Hoshii T, and Hirao A. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid Redox Signal 10: 1883–1894, 2008 [DOI] [PubMed] [Google Scholar]
- 108.Nakanishi K, Tajima F, Nakamura A, Yagura S, Ookawara T, Yamashita H, Suzuki K, Taniguchi N, and Ohno H. Effects of hypobaric hypoxia on antioxidant enzymes in rats. J Physiol 489 (Pt 3): 869–876, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nogueira-Pedro A, Cesario TA, Dias CC, Origassa CS, Eca LP, Paredes-Gamero EJ, and Ferreira AT. Hydrogen peroxide (H2O2) induces leukemic but not normal hematopoietic cell death in a dose-dependent manner. Cancer Cell Int 13: 123, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oberley LW. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed Pharmacother 59: 143–148, 2005 [DOI] [PubMed] [Google Scholar]
- 111.Oishi M, Iizumi Y, Taniguchi T, Goi W, Miki T, and Sakai T. Apigenin sensitizes prostate cancer cells to Apo2L/TRAIL by targeting adenine nucleotide translocase-2. PLoS One 8: e55922, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Orsolic N, Golemovic M, Quintas-Cardama A, Scappini B, Manshouri T, Chandra J, Basic I, Giles F, Kantarjian H, and Verstovsek S. Adaphostin has significant and selective activity against chronic and acute myeloid leukemia cells. Cancer Sci 97: 952–960, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Patwardhan RS, Sharma D, Checker R, and Sandur SK. Mitigation of radiation-induced hematopoietic injury via regulation of cellular MAPK/phosphatase levels and increasing hematopoietic stem cells. Free Radic Biol Med 68: 52–64, 2014 [DOI] [PubMed] [Google Scholar]
- 114.Pazhanisamy SK, Li H, Wang Y, Batinic-Haberle I, and Zhou D. NADPH oxidase inhibition attenuates total body irradiation-induced haematopoietic genomic instability. Mutagenesis 26: 431–435, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, O'Dwyer KM, Li Z, Shi L, Greninger P, Settleman J, Benes C, Hagen FK, Munger J, Crooks PA, Becker MW, and Jordan CT. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem 288: 33542–33558, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pennathur-Das R. and Levitt L. Augmentation of in vitro human marrow erythropoiesis under physiological oxygen tensions is mediated by monocytes and T lymphocytes. Blood 69: 899–907, 1987 [PubMed] [Google Scholar]
- 117.Pervaiz S, Taneja R, and Ghaffari S. Oxidative stress regulation of stem and progenitor cells. Antioxid Redox Signal 11: 2777–2789, 2009 [DOI] [PubMed] [Google Scholar]
- 118.Piccoli C, D'Aprile A, Ripoli M, Scrima R, Lecce L, Boffoli D, Tabilio A, and Capitanio N. Bone-marrow derived hematopoietic stem/progenitor cells express multiple isoforms of NADPH oxidase and produce constitutively reactive oxygen species. Biochem Biophys Res Commun 353: 965–972, 2007 [DOI] [PubMed] [Google Scholar]
- 119.Pollyea DA. and Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood, 2017 [DOI] [PubMed] [Google Scholar]
- 120.Reiling C. MRP1: A Target for Hematopoeitic Stem Cell Diseases. Thesis and Dissertations-Toxicology and Cancer Biology University of Kentucky; Paper 8, 2014 [Google Scholar]
- 121.Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P, and Preudhomme C. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia 22: 915–931, 2008 [DOI] [PubMed] [Google Scholar]
- 122.Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, Small D, and Rassool F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood 111: 3173–3182, 2008 [DOI] [PubMed] [Google Scholar]
- 123.Schafer FQ. and Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30: 1191–1212, 2001 [DOI] [PubMed] [Google Scholar]
- 124.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, Casero RA, Jr., Marton L, Woster P, Minden MD, Dugas M, Wang JC, Dick JE, Muller-Tidow C, Petrie K, and Zelent A. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med 18: 605–611, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Scott MT, Korfi K, Saffrey P, Hopcroft LE, Kinstrie R, Pellicano F, Guenther C, Gallipoli P, Cruz M, Dunn K, Jorgensen HG, Cassels JE, Hamilton A, Crossan A, Sinclair A, Holyoake TL, and Vetrie D. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov 6: 1248–1257, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sekeres MA. Lenalidomide in MDS: 4th time's a charm. Blood 118: 3757–3758, 2011 [DOI] [PubMed] [Google Scholar]
- 127.Shao L, Feng W, Li H, Gardner D, Luo Y, Wang Y, Liu L, Meng A, Sharpless NE, and Zhou D. Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arf-independent manner. Blood 123: 3105–3115, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shastri A, Will B, Steidl U, and Verma A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood 129: 1586–1594, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shi X, Zhang Y, Zheng J, and Pan J. Reactive oxygen species in cancer stem cells. Antioxid Redox Signal 16: 1215–1228, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, McLeod JL, Doedens M, Medeiros JJ, Marke R, Kim HJ, Lee K, McPherson JD, Hudson TJ, Consortium HP-LGP, Brown AM, Yousif F, Trinh QM, Stein LD, Minden MD, Wang JC, and Dick JE. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506: 328–333, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Skouby AR. Haematologic adaptation in patients with chronic bronchitis and pulmonary insufficiency. Acta Med Scand 199: 185–190, 1976 [DOI] [PubMed] [Google Scholar]
- 132.Spasojevic I, Chen Y, Noel TJ, Yu Y, Cole MP, Zhang L, Zhao Y, St Clair DK, and Batinic-Haberle I. Mn porphyrin-based superoxide dismutase (SOD) mimic, MnIIITE-2-PyP5+, targets mouse heart mitochondria. Free Radic Biol Med 42: 1193–1200, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R, Yusuf R, Cote D, Vinogradov SA, Scadden DT, and Lin CP. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 508: 269–273, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Squellerio I, Caruso D, Porro B, Veglia F, Tremoli E, and Cavalca V. Direct glutathione quantification in human blood by LC-MS/MS: comparison with HPLC with electrochemical detection. J Pharm Biomed Anal 71: 111–118, 2012 [DOI] [PubMed] [Google Scholar]
- 135.Srdic-Rajic T, Tisma-Miletic N, Cavic M, Kanjer K, Savikin K, Galun D, Konic-Ristic A, and Zoranovic T. Sensitization of K562 Leukemia Cells to Doxorubicin by the Viscum album Extract. Phytother Res 30: 485–495, 2016 [DOI] [PubMed] [Google Scholar]
- 136.Stanicka J, Russell EG, Woolley JF, and Cotter TG. NADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cells. J Biol Chem 290: 9348–9361, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Suda T, Arai F, and Hirao A. Hematopoietic stem cells and their niche. Trends Immunol 26: 426–433, 2005 [DOI] [PubMed] [Google Scholar]
- 138.Suda T, Takubo K, and Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9: 298–310, 2011 [DOI] [PubMed] [Google Scholar]
- 139.Sun JY, Xu L, Tseng H, Ciccarelli B, Fulciniti M, Hunter ZR, Maghsoudi K, Hatjiharissi E, Zhou Y, Yang G, Zhu B, Liu X, Gong P, Ioakimidis L, Sheehy P, Patterson CJ, Munshi NC, O'Connor OA, and Treon SP. Histone deacetylase inhibitors demonstrate significant preclinical activity as single agents, and in combination with bortezomib in Waldenstrom's macroglobulinemia. Clin Lymphoma Myeloma Leuk 11: 152–156, 2011 [DOI] [PubMed] [Google Scholar]
- 140.Suzuki F, Hashimoto K, Kikuchi H, Nishikawa H, Matsumoto H, Shimada J, Kawase M, Sunaga K, Tsuda T, Satoh K, and Sakagami H. Induction of tumor-specific cytotoxicity and apoptosis by doxorubicin. Anticancer Res 25: 887–893, 2005 [PubMed] [Google Scholar]
- 141.Tabassum H, Parvez S, Pasha ST, Banerjee BD, and Raisuddin S. Protective effect of lipoic acid against methotrexate-induced oxidative stress in liver mitochondria. Food Chem Toxicol 48: 1973–1979, 2010 [DOI] [PubMed] [Google Scholar]
- 142.Tagde A, Singh H, Kang MH, and Reynolds CP. The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J 4: e229, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thomas D. and Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 129: 1577–1585, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Thompson JE, Conlon JP, Yang X, Sanchez PV, and Carroll M. Enhanced growth of myelodysplastic colonies in hypoxic conditions. Exp Hematol 35: 21–31, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tiu RV. and Sekeres MA. Lenalidomide in del 5q MDS: responses and side effects revisited. Leuk Res 35: 1440–1441, 2011 [DOI] [PubMed] [Google Scholar]
- 146.Toledano MB. and Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A 88: 4328–4332, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tovmasyan A, Carballal S, Ghazaryan R, Melikyan L, Weitner T, Maia CG, Reboucas JS, Radi R, Spasojevic I, Benov L, and Batinic-Haberle I. Rational design of superoxide dismutase (SOD) mimics: the evaluation of the therapeutic potential of new cationic Mn porphyrins with linear and cyclic substituents. Inorg Chem 53: 11467–11483, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tovmasyan A, Sampaio RS, Boss MK, Bueno-Janice JC, Bader BH, Thomas M, Reboucas JS, Orr M, Chandler JD, Go YM, Jones DP, Venkatraman TN, Haberle S, Kyui N, Lascola CD, Dewhirst MW, Spasojevic I, Benov L, and Batinic-Haberle I. Anticancer therapeutic potential of Mn porphyrin/ascorbate system. Free Radic Biol Med 89: 1231–1247, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tovmasyan A, Sheng H, Weitner T, Arulpragasam A, Lu M, Warner DS, Vujaskovic Z, Spasojevic I, and Batinic-Haberle I. Design, mechanism of action, bioavailability and therapeutic effects of mn porphyrin-based redox modulators. Med Princ Pract 22: 103–130, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Trachootham D, Alexandre J, and Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8: 579–591, 2009 [DOI] [PubMed] [Google Scholar]
- 151.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, and Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 10: 241–252, 2006 [DOI] [PubMed] [Google Scholar]
- 152.Tsai CC, Yew TL, Yang DC, Huang WH, and Hung SC. Benefits of hypoxic culture on bone marrow multipotent stromal cells. Am J Blood Res 2: 148–159, 2012 [PMC free article] [PubMed] [Google Scholar]
- 153.Tsai JJ, Dudakov JA, Takahashi K, Shieh JH, Velardi E, Holland AM, Singer NV, West ML, Smith OM, Young LF, Shono Y, Ghosh A, Hanash AM, Tran HT, Moore MA, and van den Brink MR. Nrf2 regulates haematopoietic stem cell function. Nat Cell Biol 15: 309–316, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Uetaki M, Tabata S, Nakasuka F, Soga T, and Tomita M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci Rep 5: 13896, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Urao N. and Ushio-Fukai M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic Biol Med 54: 26–39, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, Chomienne C, Ishikawa F, Schuringa JJ, Stassi G, Huntly B, Herrmann H, Soulier J, Roesch A, Schuurhuis GJ, Wohrer S, Arock M, Zuber J, Cerny-Reiterer S, Johnsen HE, Andreeff M, and Eaves C. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer 12: 767–775, 2012 [DOI] [PubMed] [Google Scholar]
- 157.Valenzuela M, Glorieux C, Stockis J, Sid B, Sandoval JM, Felipe KB, Kviecinski MR, Verrax J, and Buc Calderon P. Retinoic acid synergizes ATO-mediated cytotoxicity by precluding Nrf2 activity in AML cells. Br J Cancer 111: 874–882, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Valez V, Cassina A, Batinic-Haberle I, Kalyanaraman B, Ferrer-Sueta G, and Radi R. Peroxynitrite formation in nitric oxide-exposed submitochondrial particles: detection, oxidative damage and catalytic removal by Mn-porphyrins. Arch Biochem Biophys 529: 45–54, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 10: 671–684, 2011 [DOI] [PubMed] [Google Scholar]
- 160.Wakabayashi N, Slocum SL, Skoko JJ, Shin S, and Kensler TW. When NRF2 talks, who's listening? Antioxid Redox Signal 13: 1649–1663, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang AM, Ku HH, Liang YC, Chen YC, Hwu YM, and Yeh TS. The autonomous notch signal pathway is activated by baicalin and baicalein but is suppressed by niclosamide in K562 cells. J Cell Biochem 106: 682–692, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang XJ, Hayes JD, Henderson CJ, and Wolf CR. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc Natl Acad Sci U S A 104: 19589–19594, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang Y, Liu L, Pazhanisamy SK, Li H, Meng A, and Zhou D. Total body irradiation causes residual bone marrow injury by induction of persistent oxidative stress in murine hematopoietic stem cells. Free Radic Biol Med 48: 348–356, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang Y, Miao H, Li W, Yao J, Sun Y, Li Z, Zhao L, and Guo Q. CXCL12/CXCR4 axis confers adriamycin resistance to human chronic myelogenous leukemia and oroxylin A improves the sensitivity of K562/ADM cells. Biochem Pharmacol 90: 212–225, 2014 [DOI] [PubMed] [Google Scholar]
- 165.Waterstrat A. and Van Zant G. Effects of aging on hematopoietic stem and progenitor cells. Curr Opin Immunol 21: 408–413, 2009 [DOI] [PubMed] [Google Scholar]
- 166.Weitzel DH, Tovmasyan A, Ashcraft KA, Rajic Z, Weitner T, Liu C, Li W, Buckley AF, Prasad MR, Young KH, Rodriguiz RM, Wetsel WC, Peters KB, Spasojevic I, Herndon JE, 2nd, Batinic-Haberle I, and Dewhirst MW. Radioprotection of the brain white matter by Mn(III) n-Butoxyethylpyridylporphyrin-based superoxide dismutase mimic MnTnBuOE-2-PyP5+. Mol Cancer Ther 14: 70–79, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Woolley JF, Naughton R, Stanicka J, Gough DR, Bhatt L, Dickinson BC, Chang CJ, and Cotter TG. H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling. PLoS One 7: e34050, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wu RP, Hayashi T, Cottam HB, Jin G, Yao S, Wu CC, Rosenbach MD, Corr M, Schwab RB, and Carson DA. Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 107: 7479–7484, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Xiao y, Chen Y, Chen B, Huang M, Yu P, and Lin J. Nispex inhibits myeloid leukemia cell HL-60 growth in vitro and in mice. China Oncol; 6, 2007 [Google Scholar]
- 170.Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, Fujiwara Y, Qi J, Cheloni G, Das PP, Nguyen M, Li S, Bradner JE, and Orkin SH. Chronic myelogenous leukemia- initiating cells require polycomb group protein EZH2. Cancer Discov 6: 1237–1247, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yahata T, Takanashi T, Muguruma Y, Ibrahim AA, Matsuzawa H, Uno T, Sheng Y, Onizuka M, Ito M, Kato S, and Ando K. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 118: 2941–2950, 2011 [DOI] [PubMed] [Google Scholar]
- 172.Yang F, Teves SS, Kemp CJ, and Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta 1845: 84–89, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yao JC. and Link DC. Concise review: The malignant hematopoietic stem cell niche. Stem Cells 35: 3–8, 2017 [DOI] [PubMed] [Google Scholar]
- 174.Yulyana Y, Tovmasyan A, Ho IA, Sia KC, Newman JP, Ng WH, Guo CM, Hui KM, Batinic-Haberle I, and Lam PY. Redox-Active Mn Porphyrin-based Potent SOD Mimic, MnTnBuOE-2-PyP(5+), Enhances Carbenoxolone-Mediated TRAIL-Induced Apoptosis in Glioblastoma Multiforme. Stem Cell Rev 12: 140–155, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zeng Z, Zheng L, Lin J, and Chen J. Successful bortezomib treatment in combination with dexamethasone and thalidomide for previously untreated epidural plasmacytoma. Oncol Lett 3: 557–559, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang BP, Zhao J, Li SS, Yang LJ, Zeng LL, Chen Y, and Fang J. Mangiferin activates Nrf2-antioxidant response element signaling without reducing the sensitivity to etoposide of human myeloid leukemia cells in vitro. Acta Pharmacol Sin 35: 257–266, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang H, Trachootham D, Lu W, Carew J, Giles FJ, Keating MJ, Arlinghaus RB, and Huang P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia 22: 1191–1199, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, and Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 18: 1639–1642, 2012 [DOI] [PubMed] [Google Scholar]
- 179.Zhao Y, Carroll DW, You Y, Chaiswing L, Wen R, Batinic-Haberle I, Bondada S, Liang Y, and St Clair DK. A novel redox regulator, MnTnBuOE-2-PyP5+, enhances normal hematopoietic stem/progenitor cell function. Redox Biol 12: 129–138, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhao Y, Chaiswing L, Oberley TD, Batinic-Haberle I, St Clair W, Epstein CJ, and St Clair D. A mechanism-based antioxidant approach for the reduction of skin carcinogenesis. Cancer Res 65: 1401–1405, 2005 [DOI] [PubMed] [Google Scholar]
- 181.Zhong W, Yan T, Webber MM, and Oberley TD. Alteration of cellular phenotype and responses to oxidative stress by manganese superoxide dismutase and a superoxide dismutase mimic in RWPE-2 human prostate adenocarcinoma cells. Antioxid Redox Signal 6: 513–522, 2004 [DOI] [PubMed] [Google Scholar]
- 182.Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, Koh TL, Yu Q, and Chng WJ. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 118: 2830–2839, 2011 [DOI] [PubMed] [Google Scholar]