

Abstract
G-protein-coupled receptors (GPCRs) have varying and diverse physiological roles, transmitting signals from a range of stimuli, including light, chemicals, peptides, and mechanical forces. More than 130 GPCRs are orphan receptors (i.e., their endogenous ligands are unknown), representing a large untapped reservoir of potential therapeutic targets for pharmaceutical intervention in a variety of diseases. Current deorphanization approaches are slow, laborious, and usually require some in-depth knowledge about the receptor pharmacology. In this study we describe a cell-based assay to identify small molecule probes of orphan receptors that requires no a priori knowledge of receptor pharmacology. Built upon the concept of pharmacochaperones, where cell-permeable small molecules facilitate the trafficking of mutant receptors to the plasma membrane, the simple and robust technology is readily accessible by most laboratories and is amenable to high-throughput screening. The assay consists of a target harboring a synthetic point mutation that causes retention of the target in the endoplasmic reticulum. Coupled with a beta-galactosidase enzyme-fragment complementation reporter system, the assay identifies compounds that act as pharmacochaperones causing forward trafficking of the mutant GPCR. The assay can identify compounds with varying mechanisms of action including agonists and antagonists. A universal positive control compound circumvents the need for a target-specific ligand. The veracity of the approach is demonstrated using the beta-2-adrenergic receptor. Together with other existing assay technologies to validate the signaling pathways and the specificity of ligands identified, this pharmacochaperone-based approach can accelerate the identification of ligands for these potentially therapeutically useful receptors.
Keywords: : pharmacochaperone, orphan GPCR, deorphanization, chemical biology, proteasome inhibitor
Introduction
In the whole of the human genome, the G-protein-coupled receptor (GPCR) super family is considered to be one of the most “druggable”; that is to say, the natural structure of these receptors is readily amenable to modulation with modern therapeutics (small molecules, peptides, and antibodies). Indeed, it is estimated that GPCRs comprise more than one-third of the targets of modern medications.1 Despite being common, even classic therapeutic targets, many GPCRs remain underexploited from both fundamental science and drug discovery perspectives. Indeed, approximately one-third of the 350 nonolfactory GPCRs in the human genome are orphan GPCRs (oGPCRs), whose endogenous ligands are unknown.2–4 Many have been inadequately characterized. Not surprisingly, many of these orphans lack a chemical probe or antibody suitable for interrogating their role in physiology and pathology, and thus remain underinvestigated.
Recent reports link the availability of high-quality chemical probes of a given receptor to the level of research activity.5,6 This is especially true of orphan receptors. For example, according to the National Institutes of Health (NIH) Pharos database7 the prototypical GPCR, beta-2-adrenergic receptor (ADRB2), is extensively represented in the literature. This is reflected in a very high PubMed score = 1614.13 and a similarly high PubTator score = 2620.80. In addition, ADRB2 is the subject of >600 NIH grants awarded in 2010 through 2014 totaling >$88 MM in research funding. Not surprisingly there is a plethora of commercially available reagents for its study including 43 Food and Drug Administration (FDA)-approved drugs, 313 chemical probes/ligands, and 779 antibodies. Pharos classifies this target as Tclin, meaning that it has known drugs with defined mechanisms of actions, known biological activities (gene ontology [GO] annotations) supported with experimental evidence, and sufficient tools for interrogating the role of this target in human biology and pathology. At the other end of the knowledge spectrum is the orphan GPR20. Compared with ADRB2, GPR20 is completely unknown and virtually unstudied. Applying the same analysis of the Pharos database said previously for ADRB2 to GPR20 illustrates the stark contrast between the information available for the two receptors. Despite being cloned more than 20 years ago,8 GPR20 is the subject of only five publications, yielding a PubMed score = 1.5 and a PubTator score = 2.2. GPR20 has no NIH funding (0 grants 2010–2014), and virtually no tools with which to study its function and pharmacology (0 FDA-approved drugs, 0 chemical probes/ligands, 174 antibodies). This simple bibliometric analysis demonstrates the importance of tools to the advancement of research on a given target and underscores the fact that a lack thereof permanently relegates orphan receptors to obscurity. Stuck in a fruitless circle, the lack of investigation is justified by the lack of tools and vice versa. As a result, much of the druggable genome remains an untapped resource of potentially fruitful therapeutic targets.
Unlike well-characterized receptors whose ligands are known, and whose function and pharmacology are defined, oGPCRs are rarely selected for fundamental research investigations or drug discovery efforts. This is because of several factors including, but not limited to (1) the lack of scientific impetus (i.e., unmet medical need or urgent scientific inquiry to be addressed) and (2) the lack of research tools that can be applied to testing a given hypothesis. Beyond these two intertwined limitations lie additional financial and technical impediments that render the application of advanced, high-throughput approaches impractical for the identification of probes for orphan receptors. The enormous cost to develop a single potent, selective chemical probe far exceeds the typical budget allowed for research grants. Funding agencies such as the NIH and Wellcome Trust are generally averse to the level of risk associated with such expensive efforts. Without a clear therapeutic indication and commercial market, pharmaceutical companies cannot justify the resources needed to screen for probes of an oGPCR. Even if financial concerns were eliminated and the generation of a particular probe desired, there remain significant technical limitations to overcome. Most significantly, at the technical level, standard high-throughput screening (HTS) assays typically used for the discovery of chemical probes require some knowledge of the receptor pharmacology, and a known ligand to act as a positive control. Furthermore, the downstream assays and technologies required to assess probe–receptor interactions and to determine receptor pharmacology in a biologically relevant context are virtually absent for oGPCRs. Therefore, much of the deorphanization efforts underway cannot leverage the power of automation and large chemical libraries to identify synthetic probe molecules, and thus remain stagnant. What is needed is a universal assay system that is amenable to HTS, does not require a priori knowledge of receptor pharmacology, is reasonably cost-effective, and can be integrated into a comprehensive strategy for identifying a chemical probe and validating its utility.
A number of genetic diseases have been linked to mutations in cell surface receptors that cause improper folding and failure to traffic appropriately to the membrane.9–11 These misfolded receptors are retained in the endoplasmic reticulum (ER) and ultimately degraded. Many groups have demonstrated that cell-permeable small molecules can bind these mutant receptors and facilitate their trafficking to the plasma membrane (PM).9,12,13 These so called “pharmacochaperones” inspired us to apply the concept to the identification of ligands for oGPCRs. This approach addresses many of the limitations to the discovery of such important tool compounds. We have determined empirically that point mutations in conserved residues in the cholesterol consensus motif (CCM) found in many class A oGPCRs14 are tolerated by the cellular protein synthesis machinery, but remain trapped in the ER because of improper tertiary structure. These synthetic mutations are functionally analogous to those causing disease, but do not occur naturally. Combined with existing assay technologies that report on the trafficking of the receptor from the ER to the cell surface, a receptor mutated in this way, constitutes a cell-based assay suitable for interrogating large chemical libraries for the identification of novel target-specific chemical matter (e.g., agonists, antagonists, or allosteric modulators). Here we provide proof-of-concept of this approach using ADRB2.
Materials and Methods
Compounds
The library of pharmacologically active compounds (LOPAC) was purchased from Sigma-Aldrich (St Louis, MO) as 10 mM stock solutions in dimethylsulfoxide (DMSO) sixteen 96-well plates. The library was subsequently replated in a higher density plate format by transferring 30 μL of each compound into individual wells of four deep well 384-well plates using a Janus automated liquid handler (PerkinElmer, Waltham, MA). Next, 6 μL of the 10 mM stock was transferred into 384-well plates containing 24 μL of DMSO per well to create a 2 mM stock solution. To enable screening in 1,536-well plate format, 5 μL of this 2 mM stock was then transferred to a clear acoustic 1,536-well screening plate. Dry powder bortezomib, propranolol, formoterol, and procaterol were all purchased from Sigma-Aldrich and the identity and purity of each was verified by LC/MS before use. All dry powders were dissolved in 100% DMSO at 10 mM and plated at a range of concentrations in 1,536-well acoustic plates for use in hit confirmation.
Construct Generation
Human GPCR ADRB2 (NM_000024) was engineered into a retroviral vector as a full-length protein fused in-frame to the small ProLink (PK) beta-galactosidase (β-gal) fragment at the C-terminus. To enable retention of ADRB2 in the ER, a W158A mutation was engineered into the conserved tryptophan residue in the fourth transmembrane domain of ADRB2. Sequence analysis of the fusion construct confirmed the expected sequence of the mutant ADRB2(W158A) receptor.
Cell Generation and Validation
Osteosarcoma cell line, U-2 OS cells stably expressing a motif associated with early endosomes (from EEA1) fused to the large β-gal fragment, termed the enzyme acceptor (EA), were generated previously15 (Cat. No. 93-1102C3; Eurofins DiscoverX, Freemont, CA). These U-2 OS Endo-EA parental cells were subsequently transduced with retroviral constructs encoding either the wild type (ADRB2-PK) or mutant [ADRB2(W158A)-PK] each tagged with the balance of the β-gal enzyme, the PK fragment, and placed under antibiotic selection for 10 days to generate stable pools. Expression of the ADRB2-PK or ADRB2(W158A)-PK constructs in stable pools was evaluated with PathHunter® ProLink Detection Reagent (Cat. No. 93-0812E; Eurofins DiscoverX) in the presence or absence of exogenous EA. Stable clones, obtained by limiting dilution of both ADRB2-PK and ADRB2(W158A)-PK Endo-EA stable pools, were screened by in vitro enzyme-fragment complementation (EFC) assay, as previously described16 or by functional response to propranolol (described hereunder). This effort yielded multiple clones demonstrating an assay window >10. A single clone of each cell line was selected for expansion and further testing. The stability of ADRB2-PK and ADRB2(W158A)-PK expression over at least five passages was measured by in vitro EFC assay.
Cell Culture
U-2 OS Endo-EA (Cat. No. 93-1102C3; Eurofins DiscoverX) and derivative cell lines were maintained in Eagle's minimal essential medium (EMEM), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1 × penicillin/streptomycin/glutamine plus 250 μg/mL hygromycin (and 500 μg/mL neomycin for ADRB2-PK and ADRB2(W158A)-PK containing cell lines). For the screen, cells were placed in assay medium consisting of EMEM, 10% FBS, and 1% penicillin/streptomycin. Cells were incubated at 37°C (5% CO2, 95% relative humidity) and maintained only up to 80% confluence. Cells were passaged no more than five times before being discarded.
Immunofluorescence
U-2 OS Endo-EA cells expressing either ADRB2-PK or ADRB2(W158A)-PK were seeded onto an eight-well glass chamber slide (Cat. No. 155411; Thermo Scientific, Waltham, MA), allowed to adhere, and incubated at 37°C, 5% CO2 overnight or until cells were ∼80% confluent. Cells were washed with phosphate-buffered saline (PBS), fixed with 3.7% paraformaldehyde for 15 min at room temperature (RT), and permeabilized with PBS Tween (PBST); 0.2% Tween-20 and nonspecific binding was blocked with PBS diluted (1:1) Odyssey Blocking Buffer (P/N 927-50000; Li-Cor Biosciences) for 1 h at RT. Cells were then incubated with mouse anti-PK antibody (Cat. No. 92-0010, 1:200 diluted; Eurofins DiscoverX) and ER-ID Red Assay Detection Reagent (ENZ-51026-K500, 1:1,000 diluted; ENZO Life Sciences) for 1 h. Cells were next incubated with goat anti-mouse Alexa-488-conjugated secondary antibody (Cat. No. A11029, 1:500 diluted; Life Technologies, Carlsbad, CA) for 1 h in the dark. After washing again in PBS, coverslips were wet mounted with anti-fade Vectashield containing DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride; Cat. No. H-1200; Vector Laboratories, Burlingame, CA). Images were acquired on a Nikon A1R VAAS inverted confocal fluorescence microscope with either a 20 × or 63 × objective (60 × /1.40 Oil Nikon N), using a constant exposure time.
Flow Cytometry
U-2 OS Endo-EA coexpressing wild-type ADRB2-PK or ADRB2(W158A)-PK cells (∼50,000/sample) were incubated with a 1:50 dilution of rabbit anti-ADRB2 antibody (Cat. No. ab36956; Abcam, Cambridge, United Kingdom) for 0.5 h on ice, washed twice with PBS/1% FBS, then incubated with a phycoerythrin (PE)-conjugated goat anti-rabbit secondary antibody (Cat. No. 111-116-144; Jackson ImmunoResearch Laboratories) for 20 min on ice. Cells were washed with cold PBS/1% FBS and diluted 1:5 in cold PBS/1% FBS before analyzing samples on a Guava flow cytometer (Millipore, Burlington, MA).
Primary Pharmacochaperone Assay Development
ADRB2(W158A)-PK cells were seeded into a white opaque 384-well microtiter plate (Cat. No. 781080; Greiner Bio-One, Kremsmünster, Austria) at a density of 5,000 cells/well in 20 μL cell culture medium without antibiotics and returned to the incubator where they remained overnight at 37°C, 5% CO2. The next day, cells were stimulated with the addition of propranolol and returned to the incubator for up to 16 h. To determine the time at which the luminescence peaked, the incubation period was stopped at 1.5, 3.0, 8.0, and 16.0 h after stimulation with propranolol. Cell lysis and EFC were achieved by the addition of 20 μL PathHunter Detection Reagent (Cat. No. 93-0001; Eurofins DiscoverX) that had been prepared following the manufacturer's protocol. After 1.5 h the luminescence intensity was measured using an Envision (PerkinElmer) multimode plate reader instrument with a 1 s/well integration time.
Cell-Seeding Density
ADRB2(W158A)-PK cells were dispensed into a white opaque 1,536-well plate (Cat. No. 4571; Corning) at a range of densities (500, 1,000, 1,500, and 2,000 cells/well) in 4 μL of assay medium using a Combi multidrop liquid dispenser (Thermo Fisher). Forward trafficking of ADRB2(W158A)-PK to the cell surface was induced by the proteasome inhibitor, bortezomib. Bortezomib (100 nM final concentration) or vehicle (DMSO, 0.0%–2.0% v/v final concentration) was added to the cells using a direct dispense protocol on an Echo555 acoustic dispenser. After a 16-h incubation period, the EFC assay was performed as described previously.
DMSO Tolerance
To assess the effect of DMSO on overall assay performance and determine the assay performance statistics, ADRB2(W158A)-PK cells were dispensed into a white opaque 1,536-well plate at a density of 1,500 cells/well as described previously. The control compound bortezomib (100 nM) and increasing concentrations of DMSO (0%–2.0%, v/v) were added to columns 1–8 of the plate, and repeated in columns 9–16, before being returned to the incubator for 16 h. Detection reagent was then added in the same manner previously described, and luminescence signal was measured.
Serum Interference
The effects of FBS (0%–20%) were tested in a similar manner using 1,500 cells/well and an incubation time of 16 h in the presence or absence of bortezomib (100 nM).
Concentration–Response Curve and Plate Statistics
A concentration–response curve was obtained by repeating this experiment using a range of concentrations (0.0–10.0 μM) of bortezomib and that of the known ADRB2 ligands, propranolol and procaterol. As described previously, the cell-seeding density was 1,500 cells/well; the final DMSO concentration was 1% v/v. FBS (10% v/v) was included in the cell medium, and the incubation time was 16 h. Positive control compound (bortezomib, 100 nM) was added to columns 1–16, whereas DMSO (1% v/v final) was added to columns 32–48. These high and low controls were used to calculated plate statistics as described hereunder. Bortezomib, propranolol, and procaterol were all individually dispensed at a range of concentrations in three columns each. All compound and vehicle dispenses were performed using the Echo555. The plate was returned to the incubator for 16 h before being removed and the EFC assay performed.
High-Throughput Pilot Screen
ADRB2(W158A)-PK cells were seeded in 4 μL of assay media at 1,500 cells/well in columns 1–48 of a white opaque 1,536-well plate (Cat. No. 4571; Corning). Cells were dispensed using the Combi multidrop dispenser and allowed to attach to the bottom of the plate for a period of 4 h at 37°C before the addition of compound. Positive control (high signal) wells (columns 1–4) included the addition of 100 nM bortezomib. Negative control (low signal) wells (columns 45–48) had DMSO only added to them (1%, v/v final concentration). All DMSO-based dispenses (vehicle, control, and compounds) were performed with an Echo555, using an acoustic plate (Cat. No. LP-400; Labcyte, San Jose, CA). Test compounds were dispensed in columns 5–44 at a final concentration of 10 μM. The plate was then covered using a stainless-steel lid (Kalypsys, San Diego, CA) and returned to the incubator at 37°C. After 16 h detection reagent was reconstituted as per the manufacturer's protocol and then 3 μL was added to each well using a BioRAPTR dispenser (Beckman Coulter Life Sciences, Brea, CA). After 1.5 h luminescence output was measured on an EnVision Plate reader using a 1 s integration time. Hits were then retested in the primary assay at a range of concentrations (0.0–100.0 μM final). The activity of the compounds was reconfirmed using the fully optimized, and HTS compatible assay protocol described previously. The final assay protocol is presented in Table 1.
Table 1.
High-Throughput Screening Assay Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Plate cells | 4 μL | 1,500 cells/well in CP5 medium |
| 2 | Spin plates | 1 min | Centrifuge at room temperature |
| 3 | Incubation time | 4 h | At 37°C, 50% CO2 |
| 4 | Library compounds | 20 nL | Single-dose singlets. 10 μM final concentration |
| 5 | Controls | 40 nL | DMSO as negative (low) control. Bortezomib (2 μM initial; 20 nM final) |
| 6 | Spin plates | 1 min | Centrifuge at room temperature |
| 7 | Incubation time | 16 h | At 37°C, 50% CO2 |
| 8 | Reporter reagent | 3 μL | DiscoverX detection reagent |
| 9 | Spin plates | 1 min | Centrifuge at room temperature |
| 10 | Incubation | 1 h | Room temperature |
| 11 | Assay readout | Luminescence | Read luminescence |
Step Notes
1. Solid white, 1,536-well plate (Cat. No. 4571; Corning). Plate using Multidrop Combi Dispenser into all wells.
2. Spin at 96 rcf.
3. Cover plates with stainless steel, vented plate lid to avoid edge effect from evaporation.
4. Use LabCyte Echo555, transfer 20 nL of each compound in the 1,536 compound library to its correspondent well in the assay plate. Columns 5–44.
5. Use LabCyte Echo555, transfer 40 nL to columns 1–4 bortezomib, and 45–48 DMSO.
6. Spin at 96 rcf.
7. Cover plates with stainless steel, vented plate lid to avoid edge effect from evaporation.
8. Use Beckman BioRAPTR dispenser to add 3 μL/well of the reporter reagent to all wells.
9. Spin at 790 rpm.
10. Keep plate covered and stored away from light.
11. Read plate on Envision plate reader using a luminescence protocol with 1 s/well integration time.
DMSO, dimethylsulfoxide; rcf, relative centrifugal field.
Data Analysis
Results from the pilot screen were calculated as percent activity (from the positive control) and Z-score (deviations from the plate mean). The activity of the compound in the library was visualized by plotting a histogram of the Z-score for each compound using Spotfire v7.6.1 (Tibco, Palo Alto, CA). Active compounds or “hits” were defined as those with a Z-score ≥2.9. Raw plate data were captured and maintained in our Chemical and Biological Information System (CBIS) biological database (ChemInnovation, San Diego, CA). Assay performance statistics including Z′-factor, assay window coefficient, and signal-to-background ratio were calculated using bortezomib (max) and DMSO columns (min). The following formulas were used to calculate these parameters:
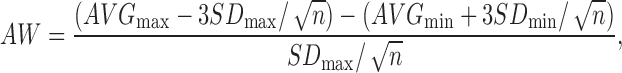 |
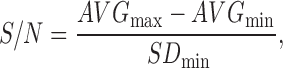 |
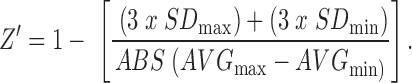 |
Concentration–response curves were obtained by nonlinear regression analysis using a four-parameter logistic with either CBIS or Prism 7 (GraphPad, La Jolla, CA). Results from the assay development and miniaturization experiments and also hit confirmation data were plotted and analyzed using Prism 7.
Results
Assay Concept and Reagent Construction
We set out to build an assay platform suitable for HTS that would enable the identification of chemical probes for oGPCRs using the concept of pharmacochaperone-induced trafficking of mutant receptors. To demonstrate the feasibility of this approach, we selected the ADRB2 as our test receptor. ADRB2 was chosen for the following reasons: ADRB2 is a widely studied class A GPCR whose pharmacology is known and ADRB2 harbors a conserved tryptophan (W158) in the CCM (Fig. 1A) that when substituted with an alanine creates a mutant receptor that is retained in the ER. There are numerous commercially available cell lines and chemical probe compounds necessary to validate the assay function and to confirm the activities of hits emerging from the HTS campaign. Many of the ADRB2 probe compounds are included in the LOPAC, a small compound collection typically used to demonstrate the suitability of an assay for chemical library screening (www.sigmaaldrich.com/life-science/cell-biology/bioactive-small-molecules/lopac1280-navigator.html).
Fig. 1.

Snake plot of ADRB2. Full-length snake plot showing the amino acid sequence of the human ADRB2 receptor. The tryptophan residue residing in the fourth transmembrane domain (W158) that is mutated to facilitate ER retention is highlighted in red. Membrane spanning domains are numbered sequentially (N-term to C-term) in blue (A). Diagram of the cell-based ADRB2 pharmacochaperone assay. Mutated ADRB2 harboring the single amino acid substitution (W158A) and the pro-link tag is retained in the ER by the cell's quality control systems. Upon binding to the target, a cell-permeable ligand (such as propranolol, given here) induces forward trafficking through the ER (red) and Golgi (gray), and ultimately to the plasma membrane. There it is internalized through the endosome (yellow) where the PK-tagged receptor can physically interact with the EA reconstituting a functional β-gal enzyme. The addition of lysis buffer and substrate produces light (B). ADRB2, beta-2-adrenergic receptor; β-gal, beta-galactosidase; EA, enzyme acceptor; ER, endoplasmic reticulum; PK, ProLink.
Figure 1B illustrates the EFC-based pharmacochaperone assay concept. U-2 OS cells engineered to express the large β-gal fragment (EA) anchored in the early endosome by fusion to an EEA1 domain, and expressing a mutant GPCR (the intended target, in this example ADRB2) fused to the small β-gal fragment (PK) at the C-terminus. In the absence of a cell-permeable ligand or probe, the mutant receptor is sequestered in the ER, preventing the complementation between the EA and PK fragments of β-gal in the endosome. When the cells are incubated with a cell-permeable small molecule that binds to the receptor, the compound induces the mutant receptor to adopt a confirmation that allows trafficking of the target through the ER and ultimately to the PM. Once there, passive internalization through natural turnover of the membrane or agonist-mediated internalization promotes receptor internalization through the early endosome. It is in the early endosome that the PK-tagged receptor can then associate with the EA-tagged endosomal protein and the full complementation of β-gal occurs. The formation of the reconstituted enzyme is then detected by the addition of substrate and chemiluminescent detection reagent. The resulting luminescence is read on a standard microtiter plate reader and reflects the extent of mutant receptors that were successfully trafficked to the endosome in the presence of compound.
We chose the split β-gal EFC reporter system17–19 for this pharmacochaperone approach because it has been successfully utilized for HTS in a wide range of applications.16,20–22 For the assay to distinguish between compounds that are functional agonists from those that merely bind to the target (e.g., an antagonist and allosteric modulators), we chose to anchor the EA tag in the early endosome. An agonist will promote active internalization yielding a highly efficacious response in the assay. In contrast, an antagonist or allosteric modulator will bind the target and facilitate trafficking to the PM, but will yield a lower level of response because it will be internalized through passive mechanisms.
With the design considerations described previously, one would expect that in the absence of compound, there is little or no EFC signal of the mutant receptor that is completely sequestered in the ER. In practice, we have observed a modest but consistent level of basal activity in this assay. This presumably reflects incomplete retention of the mutated receptor in the ER, or some level of intrinsic inefficiency in the ER quality control machinery. This “leakage” of ADRB2(W158A) from the ER to the endosome indicates that the EFC system can indeed report translocation of ADRB2(W158A) to the PM. Moreover, it provides an opportunity to use chemical stressors of ER quality control to force the translocation of the target receptor to the PM. This serendipitous characteristic of the assay is critically useful because it allows for a target independent positive control for the assay. As described hereunder, we exploited this “leakage” using the proteasome inhibitor bortezomib as a successful, universal positive control. It is important to note that bortezomib does not act directly at ADBR2 or the oGPCR of interest. However, we hypothesize that a proteasome inhibitor, when applied to cells expressing a mutant oGPCR retained in the ER would enhance the basal “leakage” of the mutant receptor to the PM, thus providing the necessary and important high-signal positive control required for a robust HTS.
After producing mutant expression clones, we confirmed that ADRB2(W158A)-PK was retained in the ER by evaluating subcellular localization of both wild-type and mutant ADRB2 receptors in the engineered cells using the anti-PK antibody. PK immunoreactivity was observed in cells expressing both wild-type and mutant ADRB2. In wild-type cells, PK immunoreactivity was observed throughout the cell and was enriched in regions of cell-to-cell contacts (Fig. 2A–E). In contrast, staining of ADRB2(W158A)-PK-expressing cells is concentrated around the nucleus (Fig. 2F–J). This is consistent with retention of the mutant receptor in the ER.
Fig. 2.

Localization of wild-type and mutant ADRB2 in U-2 OS cells. Confocal images in the Z and Y planes of wild-type U-2 OS ADRB2 (A–E) and ADRB2(W158A) (F–J) in U-2 OS cells harboring the β-gal EFC components. The distribution of ADRB2(W158A) and co-localization with the ER-specific dye verify retention of mutant receptors within the ER. Boxes indicate regions of interest selected for Y-axis projections inset. ADRB2 receptor (green) was visualized using an anti-PK-tag primary and Alexa-488-conjugated secondary antibody. The ER is stained red. Nuclei stained with DAPI are given in blue. Scale bar size is 10 μm. DAPI, 4′,6-diamidino-2-phenylindole, dihydrochloride; EFC, enzyme-fragment complementation.
Next, we demonstrated that propranolol, a known ADRB2 blocker, facilitates ADRB2(W158A) trafficking to the cell surface. Localization of wild-type ADRB2 and ADRB2(W158A) receptors in propranolol-treated cells was assayed by flow cytometry. ADRB2(W158A) is not found at the cell surface when cells are exposed to DMSO (vehicle), but robust surface localization is detected when the cells are treated with propranolol (Fig. 3A). The ability of the EFC system to report ligand-dependent trafficking of ADRB2(W158A) to the early endosome was determined in a similar manner. Initially, the same concentration of propranolol and incubation time used for the flow cytometry assay was applied to the EFC assay. These conditions failed to yield a response that was significantly higher than vehicle-treated cells. We hypothesized that adherent cells in the microtiter plate may require a longer exposure period to facilitate forward trafficking of ADRB2(W158A). A time-course experiment revealed that incubation times of 8 and 16 h were required to elicit a statistically significant response to ligand (Fig. 3B). Using a 16-h incubation time, propranolol induced forward trafficking of ADRB2(W158A) to the early endosome in a concentration-dependent manner (0–10 μM). As given in Figure 3C, incubation of U-2 OS Endo-EA ADRB2(W158A) cells to propranolol yielded a robust luminescence signal with ∼10 × assay window, and an EC50 = 9.0 nM.
Fig. 3.

Cell surface distribution of wild-type and mutant ADRB2. (A) U-2 OS Endo-EA cells expressing wild-type ADRB2-PK (top row) or mutant ADRB2(W158A) (bottom row) were treated with vehicle or 10 μM propranolol for 1.5 h. Cells were stained for surface expression of ADRB2 with an anti-ADRB2 antibody (Cat. No. ab36956; Abcam) and evaluated by flow cytometry using a Guava flow cytometer. Purple bar represents the range of cell surface fluorescence. Time-course response of ADRB2(W158) to propranolol (B) and propranolol concentration response curve (C) in the EFC trafficking assay. U-2 OS Endo-EA cells expressing ADRB2(W158A)-PK were treated with an 11-point dose response of propranolol, each dose run in quadruplicate, for 16 h at 37°C. The lowest dose in the curve corresponds to vehicle only. Each data point represents the mean RLU ± SEM of triplicate data points from at least two independent experiments. Curves represent the best fit of a four-parameter logistic generated using GraphPad Prism 7. RLU, raw luminescence units; SEM, standard error of the mean.
Assay Optimization for HTS
Having demonstrated that the assay functions in principle, we next began to adapt it for automation and HTS. Using a white, opaque 1,536-well plate, we determined the optimal cell-seeding density. Given the U-2 OS cell size and the assay signal intensity observed in previous experiments, we selected 2,000, 1,500, 1,000, and 500 cells/well for testing. The maximum luminescence obtained in response to bortezomib (100 nM) at each cell-seeding density is given in Figure 4A. Despite a cell-seeding density of 2,000 cells/well providing the largest assay window (signal-to-noise ratio [S/N] = 38.3), 1,500 cells/well yielded an acceptable window (S/N = 31.3) and was selected as the cell density for HTS because of the larger demands on cell production for HTS at scale. The effect of DMSO on assay performance was determined using the same protocol as described previously. Final DMSO concentration was also determined using the same assay format as previously described. Figure 4B shows that DMSO has an effect on the assay signal when used at >1% final. Owing to volume transfer accuracy and fixed compound library concentration constraints, 1% DMSO final was selected as the maximal DMSO concentration for the screen (Fig. 4B).
Fig. 4.

Optimization of pharmacochaperone assay in 1,536 well plates. (A) Cell-seeding density: U-2 OS-ADRB2(W158A) (black bars) were seeded at the indicated densities and exposed to control compounds for 16 h. (B) DMSO tolerance: U-2 OS-ADRB2(W158A) cells were seeded at 1,500 cells/well and allowed to attach for 4 h. Cells were incubated in the presence of bortezomib (100 nM) and the corresponding amounts of DMSO indicated as final percent v/v for 16 h. (C) Bortezomib concentration–response curve, each dose run in triplicate. (D) Procaterol and propranolol in the EFC trafficking assay: U-2 OS-ADRB2(W158A)-PK were incubated in the presence of various concentrations (11-point curve) of propranolol or procaterol, each dose run in quadruplicate, for 16 h at 37°C. Note, the Y-axis here is plotted as raw luminescence units rather than normalized percent activity (as in C). This is to better illustrate the higher maximal response elicited by procaterol versus propranolol. The lowest dose in the curve corresponds to vehicle only. Data plotted are mean ± SEM of triplicate data points from at least two independent experiments. Curves represent the best fit of a four-parameter logistic generated using GraphPad Prism 7. DMSO, dimethylsulfoxide.
During our previous experience with the EFC system, we observed that serum interferes with β-gal enzyme activity. This was also observed with the ADRB2 pharmacochaperone assay (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/adt). Normally, FBS would be eliminated from the assay media, but the cells cannot tolerate complete serum depletion for the 16-h incubation period. Therefore, we elected to keep the FBS at 10%, despite some expected interference in the assay.
As a final validation of the miniaturized assay protocol, we next tested three positive control compounds propranolol, procaterol, and bortezomib using these conditions. The known ADRB2 ligands propranolol and procaterol each elicited a concentration-dependent EFC response, confirming the successful adaptation of the assay to 1,536-well plates. The EFC response to the agonist procaterol is dramatically stronger than that of propranolol, supporting the notion that compounds that promote active internalization could be readily discriminated from antagonists in this assay model. Of importance, the proteasome inhibitor bortezomib similarly stimulated forward trafficking of ADRB2(W158A) in a concentration-dependent manner (Fig. 4C). This confirms our hypothesis that a nonspecific positive control compound that enhances the basal “leakage” of ADRB2(W158A) could be used to validate assay performance and serve as a positive control when this technology is applied to oGPCRs for which no positive control ligand is available. The final assay protocol is given in Table 1. Using this final, validated protocol, we next measured the ability of the assay to respond to two known ADRB2 ligands (Fig. 4D). As observed during assay development, when U-2 OS ADRB2(W158A) cells were exposed to propranolol EFC-dependent luminescence increased in a concentration-dependent manner. The ADRB2 agonist procaterol also elicited a robust, concentration-dependent response from these cells. Of importance, the efficacy of procaterol was significantly greater than that of propranolol (procaterol RLUmax = 155,802 ± 790 vs. propranolol RLUmax = 24,353 ± 296; p < 0.001).
Pilot Screen
After optimization and miniaturization, we next executed a pilot screen to demonstrate the robustness of the assay and suitability for HTS. Despite its small size, the accessibility, low cost, and the abundance of ADRB2 ligands in LOPAC made this library appropriate for our pilot screen. The 1,280 compounds compromising the LOPAC were tested at a final concentration of 10 μM. The screen performed well with S/B (signal/background) = 9.2 and Z′-factor = 0.585. The screen performance statistics are given in Table 2. The results of the pilot screen are displayed in histogram and a three-dimensional scatter plot using SpotFire (Fig. 5). To account for the lack of specificity of our positive control (bortezomib) at ADRB2, the hit selection was based on Z′-score and not on percent activity of control. Compounds with Z-score ≥2.9 standard deviation of the mean plate signal were considered to be “hits.” Using this criterion, a hit rate of 1.09% was obtained, yielding a total of 16 compounds. The hits included several compounds with known agonist or antagonist activity of ADRB2 including propranolol, pindolol, formoterol, salmeterol, and carvedilol among others. Of the 16 hits only 2, propafinone hydrochloride, a K+ channel blocker and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) a nitric oxide synthase inhibitor, had no reported activity at ADRB2. All of the remaining compounds were known ligands of ADRB2. A complete list of hits is provided in Supplementary Table S1. These results demonstrate that this pharmacochaperone approach can identify a variety of on-target ligands that can exhibit a range of pharmacological activities and mechanisms of action.
Table 2.
Library of Pharmacologically Active Compounds Pilot Screen Performance Statistics
| Parameter | |
|---|---|
| Positive (bortezomib) control | 118,875 ± 13,027 |
| Negative (DMSO) control | 12,920 ± 1,619 |
| Signal/background (S/B) | 9.2 |
| Signal-to-noise ratio (S/N) | 65.4 |
| Assay window | 4.76 |
| Z′-factor | 0.585 |
| Hits (hit rate) at 10 μM | 16 (0.0125%) |
Screening statistics were calculated automatically by CBIS.
Fig. 5.

HTS results. Histogram showing the distribution of hits from the proof-of-concept pilot screen of the LOPAC. Positive control (bortezomib 20 nM) is given in red. Negative control DMSO (1%) is given in yellow. Compounds are given in blue. A three-dimensional scatter plot of the same data (inset) is provided as an alternative view of the data. HTS, high-throughput screening; LOPAC, library of pharmacologically active compounds.
Hit Confirmation
From the hit set, we selected two compounds for further confirmation in the primary assay formoterol and propranolol. Formoterol is a highly potent and selective ADRB2 agonist that is typically used clinically to mitigate bronchospasm in asthma or chronic obstructive pulmonary disease.23 Propranolol is a potent, nonselective ADRB2 antagonist that is widely used for the therapy of hypertension, cardiac arrhythmias, angina pectoris, and hyperthyroidism.24 These compounds were purchased from Sigma-Aldrich, and their identity and purity were verified by liquid chromatography–mass spectrometry (LC/MS) and proton nuclear magnetic resonance (data not given) before use. In the primary pharmacochaperone assay, both formoterol and propranolol displayed full concentration–response curves with EC50 = 50.4 ± 0.1 and = 512.8 ± 0.1 nM, respectively (Fig. 6A, B). As expected, because of passive versus active internalization of the two types of ligands, formoterol showed a higher maximal signal (RLUmax = 200,540 ± 3,579) than that of propranolol (RLUmax = 45,645 ± 1,571) (Fig. 6C).
Fig. 6.

Hit confirmation. Freshly prepared dry powders of both formoterol (A) and propranolol (B) were subjected to reconfirmation in the primary assay. Both compounds exhibited concentration-dependent responses in the U-2 OS-ADRB-2(W158) cells. (C) The agonist formoterol exhibited a significantly higher maximal signal relative to the antagonist propranolol. Data plotted are mean ± SEM of triplicate data points from at least two independent experiments. Curves represent the best fit of a four-parameter logistic generated using GraphPad Prism 7.
Discussion
GPCR signaling receptors constitute one of the most frequently targeted families for therapeutic drugs.1 Still, ∼120 of them remain orphans.25 The dearth of knowledge regarding function and pharmacology, as well as the lack of an endogenous ligand to act as a positive control have stymied the application of high-throughput approaches to the deorphanization effort. The efforts are at an impasse; standard HTS assays require a robust positive control and some a priori knowledge of receptor function to facilitate a quantifiable outcome of receptor activation. The lack of functional downstream assays to validate hits also limits the confirmation of the probe compound specificity, and ultimately the elucidation of receptor pharmacology. In this study, we preset a novel approach that addresses two of the critical limitations of current deorphanization efforts, lack of a positive control and limited understanding of receptor signaling.
We demonstrate the technical methods and strategic approach that exploits pharmacochaperone concepts to elucidate new ligands of oGPCRs, both agonist and antagonist, applicable at HTS scale. Taking advantage of the established EFC technology and the pharmacochaperone concept, we designed an in vitro system capable of providing a quantitative response to receptor-ligand binding without a priori knowledge of receptor function or the availability of a positive control, thus circumventing the primary limitation of the deorphanization process. The implementation of an EFC system ensures specificity by limiting signal production to only that obtained by the PK-tagged receptor and the EA-tagged early endosome. It is also suitable for automated screening platforms, cost-effective, and accessible to most research groups because of its compatibility with common instrumentation. The use of a proteasome inhibitor bortezomib as a positive control allowed us to bypass the need for a surrogate ligand; it should be noted, however, that such control acts on the quality control machinery and not on the target receptor, thus it is nonspecific and cannot be used for hit assessment. We applied this approach to ADRB2, where a proof-of-concept screen successfully identified known chemical ligands of the receptor. Of importance, the pharmacochaperone assay was capable of identifying both agonist and antagonist compounds and distinguishing between the two. Collectively, these data presented herein support the conclusion that our pharmacochaperone assay platform constitutes an HTS compatible assay suitable for discovering probes of orphan receptors, the assay format and readout does not require any a priori knowledge of the receptor signaling pathway, and a proteasome inhibitor such as bortezomib can serve as a universal positive control for the assay acting on the cellular machinery and not on the target itself. Furthermore, as it has been well understood that understanding that pharmacochaperones can rescue trafficking of enzymes, transporters, and ion channels, our approach could theoretically be valid to such targets as well.9
Despite the technical challenges to find chemical probes of oGPCRs, efforts have gained momentum in the last decade. In silico approaches have emerged as one solution. The possibility of screening several millions of structures in a matter of days, including different orientations and conformations, for fitting in the receptor pocket is an attractive solution that can be cost-effective and rapid. Indeed, recent efforts using computer-aided structure-based docking screens have significantly added to the library of known GPCR ligands. This technique, however, lacks the capacity to distinguish agonist from antagonist and is often biased for highly refined ligand binding sites such as those found in hormone receptors.26 In addition, a high-resolution crystal structure is required that is something not often available for oGPCRs. The use of bacterial and yeast systems for physical testing of molecules identified through docking has been limited by the ability of such systems to express functional receptors. Similarly, phenotypic screens have been of limited utility to deorphanization efforts because of their innate inability to report receptor-specific events.
In this context the need for a system that is amenable to automation for large-scale screening (>100,000 compounds) and does not require knowledge about receptor signaling becomes evident. Such individual platform technologies also must be complemented with a strategy that uses traditional assay technologies to further characterize probe–receptor complexes in relevant systems.
Although readily accessible and versatile, some receptors will not be amenable to this approach. As the altered receptor sequence, in our case a CCM in the ADRB2 receptor, is conserved in many class 2 oGPCRs, we expect it to be widely applicable to this family. However, it is likely that some GPCRs and members of other target classes will not be readily amenable to such manipulation. Multimeric receptor proteins in which the introduction of a point mutation does not effectively retain the receptor in the ER, or results in a significant loss of tertiary structure, will not be successfully deorphanized using this approach. In addition, the determination of probe function (agonist, antagonist, allosteric modulator, etc.), relies on the comparative analysis of the Emax of hits. Such evaluation is dependent on the hits obtained, limiting the utility of this technology as a stand-alone approach. Indeed, any HTS campaign must have adequate orthogonal and secondary counter and confirmation assays to verify the specificity and function of the hits. The pharmacochaperone approach is no exception. Finally, hit identification is but the first step in the deorphanization effort and therefore cannot be a stand-alone assay for full receptor characterization. Like all probe discovery campaigns, a series of secondary assays must complement the screen to confirm activity, determine potency, verify selectivity, and to further elucidate the probe mechanism of action. Cyclic adenosine monophosphate (cAMP) and β-arrestin, Ca2+-flux, receptor internalization, GTPγS are a few of the technologies available. Label-free technologies such as cellular impedance,27 surface plasma resonance,28 and back-scattering interferometry have also been used to characterize probe–receptor pharmacology.29 Taking the advantages and limitations of this pharmacochaperone approach into consideration, we propose one possible workflow that utilizes this novel screening platform as a primary assay, combined with downstream assays required to validate probe pharmacology and ultimately its potential utility to the scientific community (Fig. 7).
Fig. 7.

Proposed workflow for a pharmacochaperone HTS to identify surrogate ligands of oGPCRs.
Supplementary Material
Abbreviations Used
- β-gal
beta-galactosidase
- ADRB2
beta-2-adrenergic receptor
- AVG
average
- AW
assay window
- CBIS
Chemical and Biological Information System
- CCM
cholesterol consensus motif
- DAPI
4′,6-diamidino-2-phenylindole, dihydrochloride
- DMSO
dimethylsulfoxide
- EA
enzyme acceptor
- EFC
enzyme-fragment complementation
- EMEM
Eagle's minimal essential medium
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- FDA
Food and Drug Administration
- GPCR
G-protein-coupled receptor
- GTP
guanosine triphosphate
- HTS
high-throughput screening
- LC/MS
liquid chromatography mass spectroscopy
- LOPAC
library of pharmacologically active compounds
- NIH
National Institutes of Health
- oGPCR
orphan GPCR
- PBS
phosphate-buffered saline
- PK
ProLink
- PM
plasma membrane
- RLU
raw luminescence units
- RT
room temperature
- S/N
signal-to-noise ratio
- U-2 OS
osteosarcoma cell line
Acknowledgments
The authors thank Kyle C. Ziegler for editorial assistance. This work was funded in part by NIH HG005033 and the Florida Translational Research Program (COHK8).
Disclosure Statement
J.L. and D.B. are employees of Eurofins DiscoverX. At the time this work was initiated, T.W. was employed by Eurofins DiscoverX. He is currently employed by PrimityBio.
References
- 1.Hopkins AL, Groom CR: The druggable genome. Nat Rev Drug Discov 2002;1:727–730 [DOI] [PubMed] [Google Scholar]
- 2.Bjarnadottir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schioth HB: Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 2006;88:263–273 [DOI] [PubMed] [Google Scholar]
- 3.Fredriksson R, Schioth HB: The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol Pharmacol 2005;67:1414–1425 [DOI] [PubMed] [Google Scholar]
- 4.Vassilatis DK, Hohmann JG, Zeng H, et al. : The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci U S A 2003;100:4903–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH: Too many roads not taken. Nature 2011;470:163–165 [DOI] [PubMed] [Google Scholar]
- 6.Roth BL, Kroeze WK: Integrated approaches for genome-wide interrogation of the druggable non-olfactory G protein-coupled receptor superfamily. J Biol Chem 2015;290:19471–19477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen DT, Mathias S, Bologa C, et al. : Pharos: collating protein information to shed light on the druggable genome. Nucleic Acids Res 2017;45:D995–D1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Dowd BF, Nguyen T, Jung BP, et al. : Cloning and chromosomal mapping of four putative novel human G-protein-coupled receptor genes. Gene 1997;187:75–81 [DOI] [PubMed] [Google Scholar]
- 9.Leidenheimer NJ, Ryder KG: Pharmacological chaperoning: a primer on mechanism and pharmacology. Pharmacol Res 2014;83:10–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ulloa-Aguirre A, Zarinan T, Dias JA, Conn PM: Mutations in G protein-coupled receptors that impact receptor trafficking and reproductive function. Mol Cell Endocrinol 2014;382:411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab 2004;15:222–228 [DOI] [PubMed] [Google Scholar]
- 12.Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM: Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic 2004;5:821–837 [DOI] [PubMed] [Google Scholar]
- 13.Bernier V, Lagace M, Lonergan M, Arthus MF, Bichet DG, Bouvier M: Functional rescue of the constitutively internalized V2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059. Mol Endocrinol 2004;18:2074–2084 [DOI] [PubMed] [Google Scholar]
- 14.Hanson MA, Cherezov V, Griffith MT, et al. : A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008;16:897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammer MM, Wehrman TS, Blau HM: A novel enzyme complementation-based assay for monitoring G-protein-coupled receptor internalization. FASEB J 2007;21:3827–3834 [DOI] [PubMed] [Google Scholar]
- 16.Bassoni DL, Raab WJ, Achacoso PL, Loh CY, Wehrman TS: Measurements of beta-arrestin recruitment to activated seven transmembrane receptors using enzyme complementation. Methods Mol Biol 2012;897:181–203 [DOI] [PubMed] [Google Scholar]
- 17.Olson KR, Eglen RM: Beta galactosidase complementation: a cell-based luminescent assay platform for drug discovery. Assay Drug Dev Technol 2007;5:137–144 [DOI] [PubMed] [Google Scholar]
- 18.Wehrman T, He X, Raab B, Dukipatti A, Blau H, Garcia KC: Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 2007;53:25–38 [DOI] [PubMed] [Google Scholar]
- 19.Wehrman TS, Casipit CL, Gewertz NM, Blau HM: Enzymatic detection of protein translocation. Nat Methods 2005;2:521–527 [DOI] [PubMed] [Google Scholar]
- 20.Bassoni DL, Jafri Q, Sastry S, Mathrubutham M, Wehrman TS: Characterization of G-protein coupled receptor modulators using homogeneous cAMP assays. Methods Mol Biol 2012;897:171–180 [DOI] [PubMed] [Google Scholar]
- 21.Patel A, Murray J, McElwee-Whitmer S, Bai C, Kunapuli P, Johnson EN: A combination of ultrahigh throughput PathHunter and cytokine secretion assays to identify glucocorticoid receptor agonists. Anal Biochem 2009;385:286–292 [DOI] [PubMed] [Google Scholar]
- 22.Zhao X, Vainshtein I, Gellibolian R, et al. : Homogeneous assays for cellular protein degradation using beta-galactosidase complementation: NF-kappaB/IkappaB pathway signaling. Assay Drug Dev Technol 2003;1:823–833 [DOI] [PubMed] [Google Scholar]
- 23.National Center for Biotechnology Information: PubChem compound database. https://pubchem.ncbi.nlm.nih.gov/compound/3410 (Last accessed on July30, 2018)
- 24.National Center for Biotechnology Information: PubChem compound database. https://pubchem.ncbi.nlm.nih.gov/compound/4946 (Last accessed July30, 2018)
- 25.Sriram K, Insel PA: G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol Pharmacol 2018;93:251–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schapira M, Abagyan R, Totrov M: Nuclear hormone receptor targeted virtual screening. J Med Chem 2003;46:3045–3059 [DOI] [PubMed] [Google Scholar]
- 27.Hennen S, Wang H, Peters L, et al. : Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Sci Signal 2013;6:ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen K, Obinata H, Izumi T: Detection of G protein-coupled receptor-mediated cellular response involved in cytoskeletal rearrangement using surface plasmon resonance. Biosens Bioelectron 2010;25:1675–1680 [DOI] [PubMed] [Google Scholar]
- 29.Latham JC, Stein RA, Bornhop DJ, McHaourab HS: Free-solution label-free detection of alpha-crystallin chaperone interactions by back-scattering interferometry. Anal Chem 2009;81:1865–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.