

Abstract
The molecular circuitry directing tissue development and homeostasis is hardwired by genetic programs but may also be subject to fine-tuning or major modification by environmental conditions. It remains unclear whether such malleability is indeed at work—particularly in tissues directly in contact with the environment—and contributes to their optimal maintenance and resilience. The protein kinase p38α is activated by physiological cues that signal tissue damage and neoplastic transformation. Here, we found that p38α phosphorylated and thereby destabilized p63, a transcription factor essential for epidermal development. Through this regulatory mechanism, p38α limited the frequency of keratinocytes with stem cell properties and tumorigenic potential. Correspondingly, epidermal loss of p38α expression or activity promoted and correlated with carcinogenesis in mouse and human skin, respectively. These findings illustrate a novel epidermal tumor-suppressive mechanism in which stress-activated signaling induces the contraction of stem cell-like keratinocyte pools.
Introduction
Barrier tissues such as the skin have relatively high epithelial turnover rates and require stem and progenitor cells (SPCs) to constantly replenish cells lost due to exfoliation or injury (1, 2). The skin epithelium, or epidermis, comprises strata of stem/progenitor-like, proliferative, and differentiated keratinocytes that are positioned in a gradient of maturation. Epidermal SPCs are prone to acquisition and accumulation of oncogenic mutations: they are accessible to solar radiation and other environmental carcinogens and reside in the skin much longer than their transient progenies. Besides, mutations in SPCs are propagated more widely than those in the epidermal strata of later differentiation stages. Hence, cell-autonomous mechanisms suppressing skin carcinogenesis are particularly important and can become most effective in the SPC population. Few such mechanisms, however, have been demonstrated to date.
The protein kinase p38α was originally discovered due to its binding affinity for anti-inflammatory compounds (3) and its responsiveness to cytokines and microbial products (4–6) and has since been investigated as a driver and regulator of immune responses. Subsequent studies revealed that p38α was also activated by tissue-damaging and tumor-promoting stimuli and mediated cellular interpretation of them (7–9). Notably, it was found that conditional ablation of the p38α gene in mice resulted in hyperplasia and enhanced carcinogenesis in airway and intestinal epithelia as well as in hepatic parenchyma (10–13). It remains unclear how p38α signaling exerts control over tissue homeostasis and tumorigenesis and whether cell type-specific mechanisms are involved in this control.
In this study, we identified the role of p38α in SPC homeostasis in the stratified epithelium of the skin and its link to skin carcinogenesis. Furthermore, we discovered that p63, a master transcription factor governing the development and maintenance of the skin epithelium, is a key regulatory target of p38α signaling.
Results
Implications of intratumoral p38α activity for barrier tissue cancers
Tumors growing on cutaneous and mucosal surfaces are constantly exposed to environmental p38α-activating stimuli. We analyzed the Cancer Genome Atlas (TCGA) dataset to gauge the significance of p38α activation for the clinical outcomes of human patients with cancers of the skin and the ectodermal glandular, aerodigestive, respiratory, gastrointestinal, and lower genitourinary sites. The patients in each cancer type were divided into two groups based on their reverse-phase protein array (RPPA) signals (14) for intratumoral phosphorylated (p-) p38, a measure of p38α activation. The survival rates of the two groups, “p-p38 high” and “p-p38 low” (the 50th percentile as the cut-off), were different in some but not all cancer types (Fig. 1A). The p-p38 high groups exhibited prolonged survival compared to the p-p38 low counterparts, most significantly in lung squamous cell carcinoma and esophageal carcinoma (Fig. 1, B and C). We next examined p38 phosphorylation in tissues representing early stages of skin squamous cell carcinoma (SCC), a cancer type for which TCGA data did not exist. Most skin biopsies with a diagnosis of actinic keratosis (AK), a preneoplastic lesion that can progress to SCC, displayed epidermal p-p38 signals; in many SCC biopsies, however, p38 phosphorylation was drastically diminished in the tumor parenchyma (Fig. 1, D and E). The association between AK-to-SCC progression and the loss of p38α activity, along with the clinical benefit of intratumoral p38α activity observed in the TCGA dataset, prompted us to verify the tumor-suppressive role of p38α signaling in mice and pursue the underlying mechanisms.
Fig. 1. Loss of p38α signaling correlates with enhanced tumorigenesis in human barrier tissues.
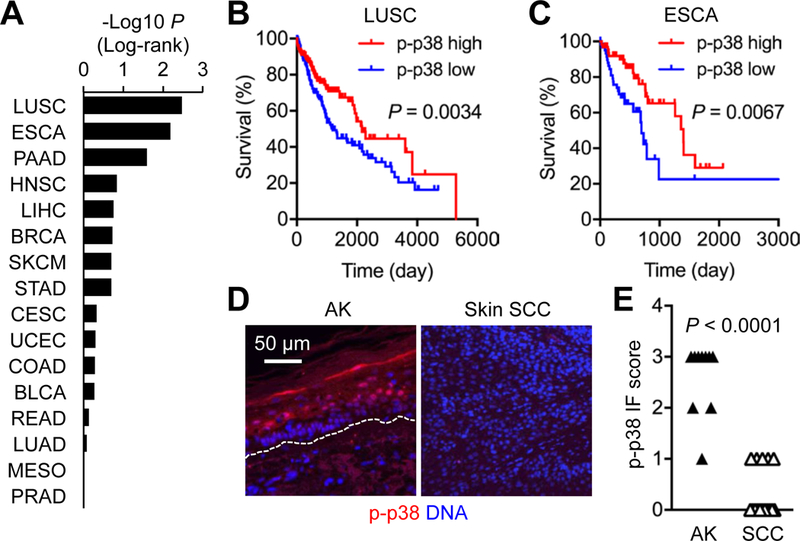
(A to C) Differential survival rates of the indicated TCGA patient groups with high and low phosphorylated (p-) p38 signals in their tumors are shown as P values by log-rank test. The cancer types are indicated by TCGA abbreviations, including LUSC (lung squamous cell carcinoma) and ESCA (esophageal carcinoma). The survival of LUSC and ESCA patients (n = 160 and 63 per group, respectively) is shown in Kaplan-Meier plots (B and C).
(D and E) Human actinic keratinosis (AK) skin and SCC tumor sections (n = 10 and 16, respectively) were analyzed by immunostaining (D). Dotted line, the epidermal-dermal boundary (D). p-p38 signals were graded from 0 to 3 based on immunofluorescence (IF) intensity per cell (E). P values by Mann-Whitney test.
Effects of p38α gene ablation on skin carcinogenesis
We generated mice in which deletion of the p38α gene was targeted to skin epithelial cells (p38αΔK) and subjected them to a skin carcinogenesis protocol that involved the chemical carcinogen 7,12-dimethyl-benz[a]anthracene (DMBA) and the chemical irritant 12-O-tetradecanoylphorbol-13 acetate (TPA). p38αΔK mice displayed a higher incidence of tumor development compared to wild-type (WT) mice (Fig. 2, A and B). The two groups of mice began to develop tumors at similar time points, but thereafter tumor incidence rose faster in the p38αΔK group. All tumors observed in WT and p38αΔK mice were papillomas; none of them underwent malignant progression during our chemical carcinogenesis experiment. Tumors from WT mice contained proliferating cells that were localized along the tumor-stroma boundary (Fig. 2C, left). By contrast, p38αΔK tumors exhibited broader areas of proliferation across tumor sections (Fig. 2C, right), suggesting an expansion of the SPC-like tumor-sustaining cell pool. WT tumors retained a stratified epidermal structure and contained large cells expressing the differentiation marker KRT1, whereas p38αΔK tumors did not manifest this phenotype (Fig. 2D). Furthermore, p38αΔK but not WT mice had extensive peritumoral vascularization and Ly6G+ myeloid cell infiltration (Fig. 2, E and F), which was notable in light of the previous findings that chemically induced skin tumors harbored tumor-sustaining cells that instigated the formation of tumor-associated vasculature (15), and that tumor-associated myeloid cell frequency correlates with tumor stemness (16). Intratumoral CD3+ T cell densities were similar in WT and p38αΔK mice (Fig. 2G).
Fig. 2. Loss of p38α signaling results in enhanced skin tumorigenesis in mice.
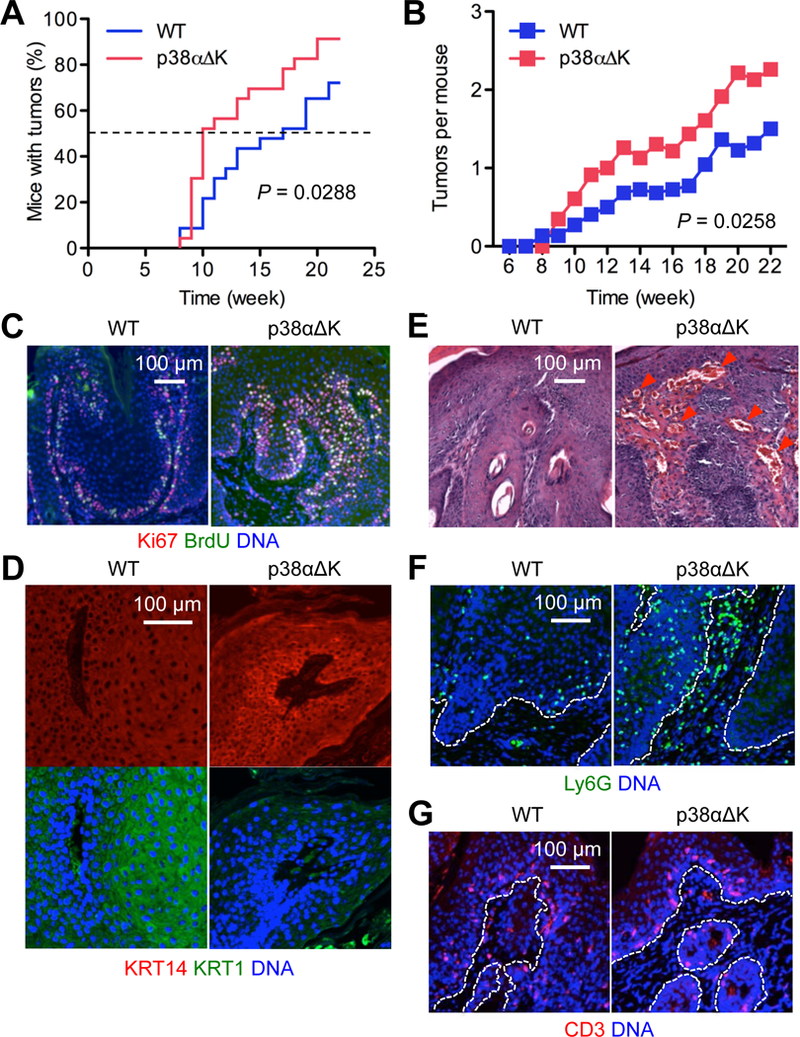
(A and B) Mice (n = 23) were subjected to DMBA-TPA skin tumorigenesis. Tumor incidence (A) and multiplicity (B) were determined over the indicated period. P values by log-rank test (A) and two-tailed unpaired Student’s t test (B).
(C to G) DMBA-TPA-induced tumor sections from mice were analyzed by immunostaining/counterstaining for the indicated molecules (C, D, F and G) and by H&E staining (E). Red arrowhead, peritumoral vasculature (E). Dotted line, the tumor-stroma boundary (F and G). Images are representative of three to five tissue sections.
Inflammation promotes tumorigenesis, including in mouse models of skin carcinogenesis (17, 18). We examined whether keratinocyte-specific p38α deficiency enhanced inflammatory responses to TPA. Similar extents of acanthosis (epidermal thickening), edema formation (dermal thickening), and leukocyte infiltration were observed in TPA-treated WT and p38αΔK skin (fig. S1, A-C). The expression of molecular markers of inflammation in p38αΔK skin was comparable to or lower than that in WT skin (fig. S1D). Therefore, the higher cancer susceptibility of p38αΔK skin was not likely due to an increase in TPA-induced tumor-promoting inflammation.
Effects of p38α gene ablation on epidermal regeneration
Tumorigenesis and wound healing entail similar tissue remodeling processes and are driven by overlapping molecular mechanisms (19). This resemblance is in part attributable to their shared reliance on SPC activity. To determine the effect of epidermal p38α deficiency on skin wound repair, we created punch-biopsy wounds in WT and p38αΔK mice. Macroscopically, wound closure was moderately more efficient in p38αΔK than WT mice until five days after wounding (Fig. 3, A and B), a time point around which dermal granulation tissue begins to contract, pulling together the wound margins and obfuscating the impact, if any, of concomitant epithelial events. Histological analysis, however, revealed a striking difference in re-epithelialization rate between the two animal groups: 24 hours after wounding, the leading edge of regenerating epidermis was rapidly penetrating the open wound area underneath the eschar in all p38αΔK mice examined, whereas re-epithelialization had barely started in WT mice (Fig. 3, C-E). The linear speed of re-epithelialization in WT skin was ~3-fold less than that in p38αΔK skin (50 and 178 μm/hour on average, respectively).
Fig. 3. Loss of p38α signaling results in enhanced epidermal regeneration after punch biopsy wounding.
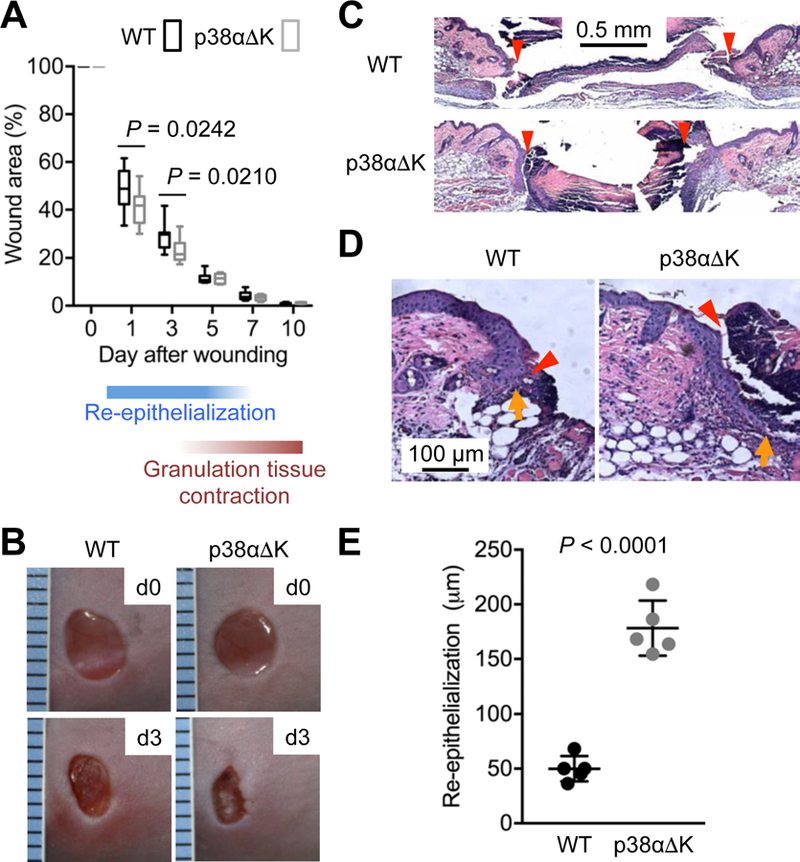
(A to E) Mice (n = 12) were subjected to punch-biopsy wounding in the dorsal skin. Wound closure rates were determined over the indicated period and are shown in box and whisker plots (A). Wounds were photographed immediately (d0) and three days (d3) after wounding (B). Wound skin sections prepared one day after wounding were analyzed by H&E staining (C and D). Red arrowhead, wound margin; orange arrow, leading edge of regenerating epidermis (C and D). The extent of re-epithelialization one day after wounding was quantified based on the distance between wound margin and epidermal leading edge and is shown as means ± SD (E). P values by two-tailed unpaired Student’s t test (A and E). Images are representative of five tissue sections.
Effects of p38α gene ablation on epidermal SPC frequency
Seeking explanations for enhanced carcinogenesis and epidermal regeneration in p38αΔK skin, we examined its epidermis for signs of increased SPC frequency. In contrast to the wealth of knowledge about hair follicle stem cells, little definitive information is available that would enable the detection of interfollicular epidermal SPCs based on their molecular properties or microanatomical locations. Our analysis was therefore directed toward keratinocytes with functional properties of SPCs. The skin of adult p38αΔK mice harbored greater numbers of cells that, when plated and grown in culture, gave rise to holocolonies, large clonal aggregates derived from cells with sustained proliferative potential (Fig. 4, A and B). Stem cells and other long-lived cells in adult tissue exhibit high expression of ABC family transporters, and thus can pump out the DNA-binding dye Hoechst33342 (20), likely serving their need for an enhanced capacity to defend against environmental genotoxins. Mouse keratinocytes with SPC properties have been shown to exclude and resist labeling by Hoechst33342 in what is known as the “side population” discrimination assay (21, 22). Greater percentages of side population cells were detected in p38αΔK relative to WT epidermis (Fig. 4, C and D; fig. S2A). Hoechst33342 exclusion by epidermal side population cells in this experiment was sensitive to verapamil, an ABC family transporter inhibitor (fig. S2B). Finally, epidermal cells that possessed slow-cycling (label-retaining) property, another characteristic of SPCs, were more abundant in p38αΔK than WT skin (Fig. 4, E and F). Of note, interfollicular epidermal label retention lasted for at least fifteen days in our experimental condition but was not observed when we used a protocol with a longer chase period (seventy days) optimized for detecting hair follicle stem cells in the mouse tail skin (23), suggesting a relatively high proliferative state of interfollicular epidermal SPCs. Together, these observations indicated that p38α functioned to limit the size of the epidermal SPC pool, and that elimination of this regulatory mechanism resulted in their expansion.
Fig. 4. p38α restricts the frequency of keratinocytes with stem cell properties.
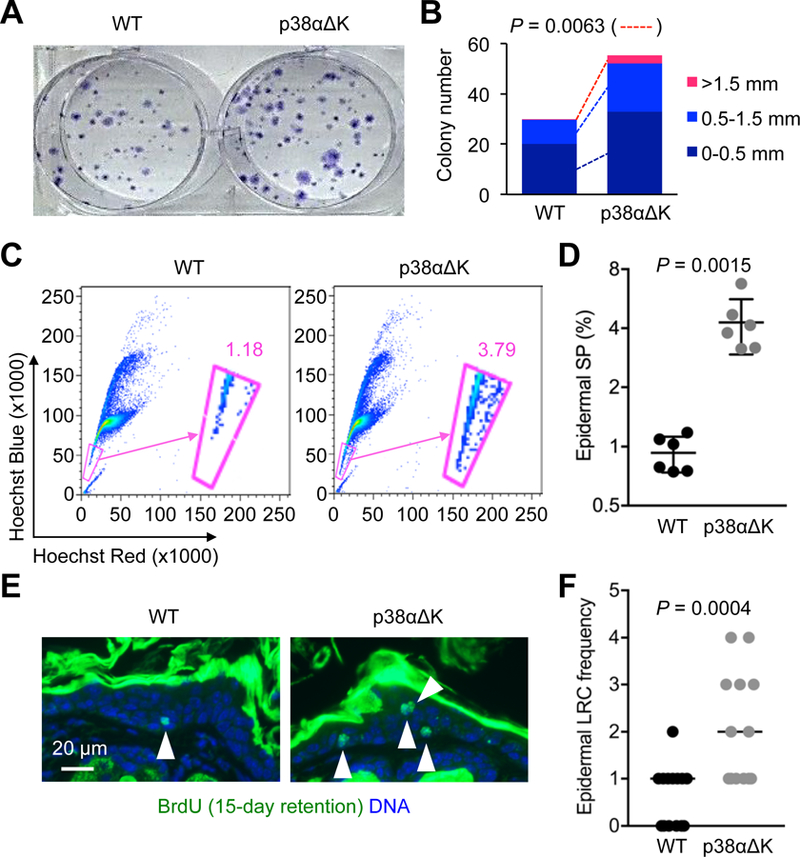
(A and B) Epidermal cells from adult mice were plated and grown for colony formation. Colonies stained with crystal violet were photographed (A) and quantified from three experiments (B). P values by two-tailed unpaired Student’s t test.
(C and D) Steady-state epidermal cells from adult mice (n = 6 per group) were incubated with Hoechst33342. Label-refractory side populations (encircled in pink line) were detected by flow cytometry (C). The percentage of the epidermal side population (SP) was quantified and is shown as individual values and means ± SD (D). P values by two-tailed unpaired Student’s t test.
(E and F) Steady-state skin sections (n = 13–14 per group) from adult mice were analyzed by immunostaining/counterstaining for the indicated molecules 15 days after BrdU injection (E). White arrowhead, BrdU label-retaining cells (E). Epidermal label-retaining cell (LRC) frequency was determined based on label-retaining cell numbers per image field and is shown as individual values and medians (F). P values by Mann-Whitney test.
Effects of p38α gene ablation on p63 protein abundance
To obtain clues about the molecular mechanism linking p38α signaling to epidermal SPC maintenance, we examined the effect of p38α deficiency on a panel of transcription factors dictating the identity and fate of skin epithelium. Intriguingly, the amounts of ΔNp63α protein, the most abundant p63 isoform in the epidermis, were higher in p38αΔK than WT keratinocytes under a steady-state culture condition as well as after stimulation with TPA (Fig. 5A). Calcium-induced differentiation, ultraviolet-B radiation (UVB) exposure, and anisomycin treatment, all of which induced p38α activation, resulted in decreased ΔNp63α amounts in mouse keratinocytes, yet these responses were greatly attenuated in the absence of p38α (Fig. 5, B and C). ΔNp63α amount in human keratinocytes was also reduced upon anisomycin treatment (Fig. 5D). This response was blocked by pharmacological inhibition of TAK1 (upstream of and necessary for p38α activation) or p38α but not JNK (downstream of TAK1 and activated in parallel with p38α). The effect of inhibiting these protein kinases on p38α signaling in human keratinocytes was mirrored by the extent of phosphorylation of MK2, a substrate of p38α (Fig. 5D).
Fig. 5. p38α activity reduces p63 protein amounts and p63+ cell numbers in the epidermis and epidermal-derived tumors.
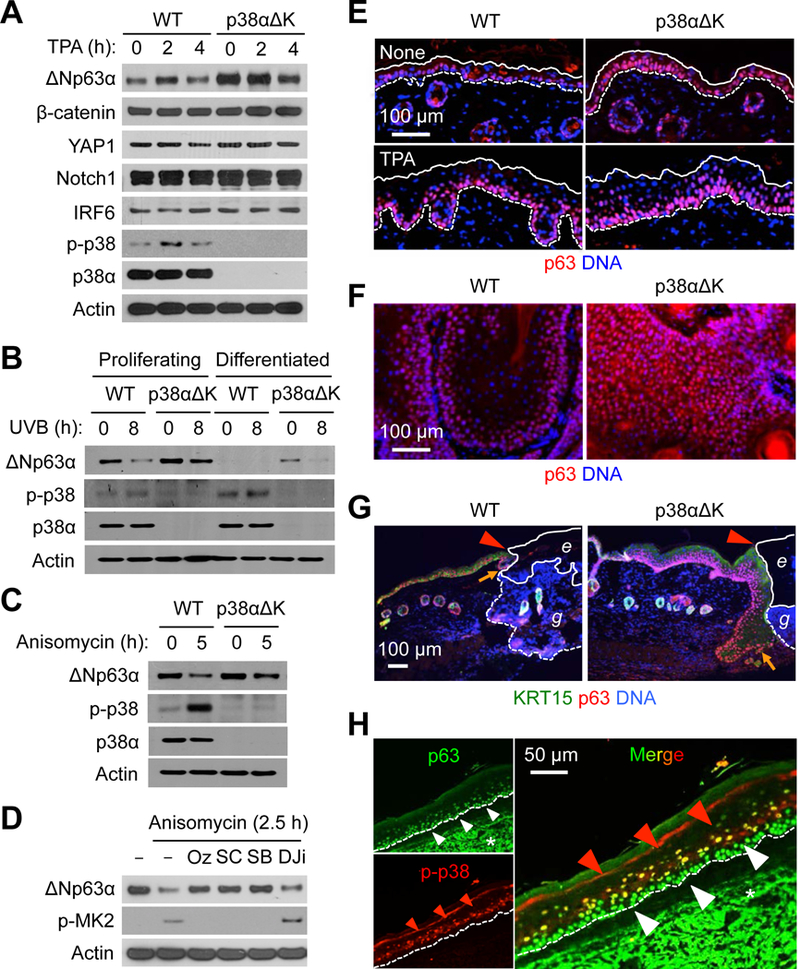
(A to D) Mouse (A-C) and human (D) keratinocytes were left unstimulated or stimulated with TPA (100 nM), UVB (75 mJ/cm2), and anisomycin (10 μg/ml). Whole cell lysates were prepared after the indicated durations of stimulation and analyzed by immunoblotting. p-, phosphorylated. Cells were preincubated with 5Z-7-oxozeaenol (Oz), SC409 (SC), SB202190 (SB), and D-JNKi (DJi) before stimulation (D). Blots are representative of three experiments.
(E to G) Steady-state (“None”) and TPA-treated skin sections (E), DMBA-TPA-induced tumor sections (F), and wound skin sections (G) from mice were analyzed by immunostaining/counterstaining for the indicated molecules. Solid and dotted line, epidermal margins and the epidermal-dermal boundary, respectively (E). Red arrowhead, wound margin; orange arrow, leading edge of regenerating epidermis; solid and dotted line, margins of eschar (e) and granulation tissue (g), respectively (G). Images are representative of five to seven tissue sections.
(H) Human AK skin sections were analyzed by immunostaining for the indicated molecules. Red and white arrowheads, epidermal cell layers with differential immunofluorescence signals; dotted line, the epidermal-dermal boundary; *, non-specific staining. Images are representative of three tissue sections.
p63 is a master transcription factor for epidermal SPC gene expression and plays a pivotal role in the development of stratified epithelia and ectodermal appendages (24–26), oncogene-induced keratinocyte transformation (27), and SCC cell survival (28, 29). The p63-expressing basal epidermal layer was markedly expanded in both unprovoked and TPA-treated p38αΔK skin (Fig. 5E). Skin tumors from p38αΔK mice exhibited the distribution of p63+ cells across entire intratumoral areas, which contrasted with their restricted occurrence in WT tumors (Fig. 5F). KRT15+ regenerating epidermis in the wound skin of p38αΔK mice was hyperproliferative and contained expanded p63+ cell populations (Fig. 5G). The differences in p63 expression in WT and p38αΔK cells were not due to changes in mRNA amount (fig. S3), suggesting control at the protein level.
Analysis of the TCGA RNA-Seq dataset revealed cancer type-specific p63 expression, with the highest expression in SCC of the head and neck, lung, and cervix, and esophageal carcinoma (fig. S4A). We also observed intense p63-specific immunofluorescence signals from the parenchyma of human SCC (fig. S4B). Intriguingly, these human cancer types with high p63 expression partially overlapped with those in which intratumoral p38α activity was associated with clinical outcomes (Fig. 1, A and E). In human AK epidermis, p63 expression was spatially excluded from areas with strong p38 phosphorylation (Fig. 5H). Similarly, p63 protein amounts examined in a large set of human cancer cell lines (30) were inversely correlated with phosphorylated p38 signals (fig. S4C). Together, our findings indicated that p38α signaling exerted control on p63 protein abundance in mouse and human cells.
Phosphorylation and destabilization of p63 by p38α
We tested whether p38α limited p63 protein abundance by direct phosphorylation. p38α was able to phosphorylate ΔNp63α in a reaction involving purified recombinant proteins and ATP (Fig. 6A). To examine the effect of p38α hyperactivity on ΔNp63α in cells normally devoid of its expression, we ectopically expressed in 293T cells ΔNp63α together with p38αCA, a p38α mutant locked in an active state due to amino acid substitutions [Asp176 to Ala (D176A) and Phe327 to Ser (F327S)]. In this setting, p38α hyperactivity led to a precipitous decline in ΔNp63α amounts, which was prevented by the proteasome inhibitor MG132 (Fig. 6B). The ΔNp63α protein spared from destruction manifested a slightly decreased electrophoretic mobility, possibly indicative of phosphorylation. Taken together, our findings suggested that p38α, through its basal and stimulus-induced kinase activity, modulated ΔNp63α protein turnover by signaling proteasome-mediated degradation of ΔNp63α.
Fig. 6. p38α phosphorylates and destabilizes p63.
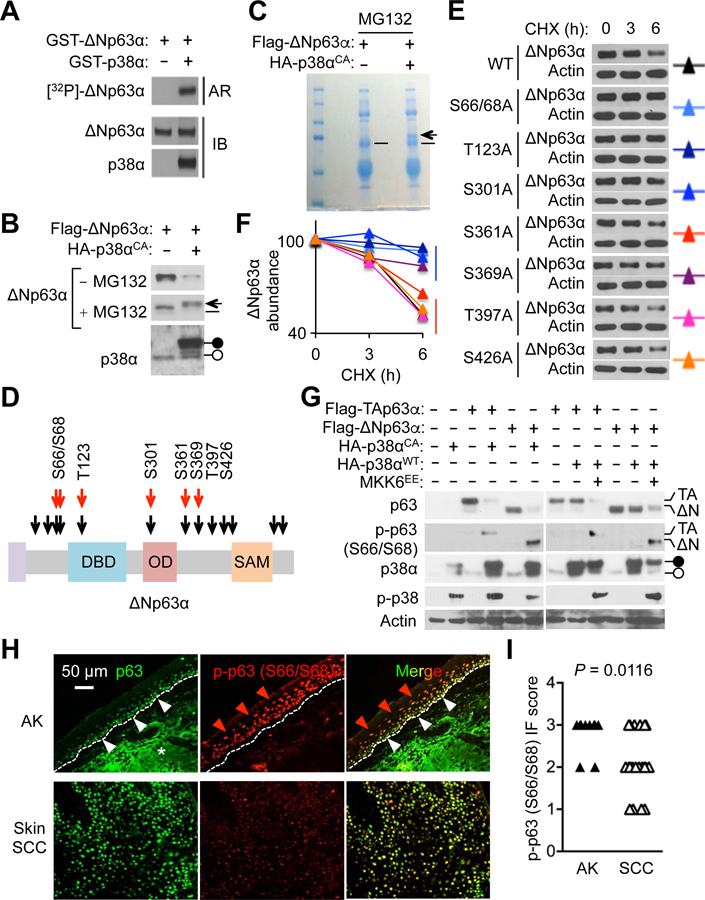
(A) 32P-radiolabel p63 from in vitro kinase reactions was analyzed by autoradiography and immunoblotting. Autoradiographic images and blots are representative of two experiments.
(B and C) 293T cells were transfected with plasmids expressing the indicated proteins. Whole cell lysates prepared 24 hours after transfection were analyzed by immunoblotting; where indicated, cells were incubated with MG132 (5 μM) for 12 hours before lysate preparation (B). p63 was immunoprecipitated from transfected/MG132-treated cell lysates and analyzed by SDS-PAGE and Coomassie Blue staining (C). Solid and open circles, plasmid-expressed and endogenous p38α, respectively (B); arrow and dash, putative phosphorylated and unphosphorylated p63, respectively (B and C). Blots and gel images are representative of two experiments.
(D) Phosphorylation sites on ΔNp63α are depicted. Black and red arrows, putative and experimentally determined p38α phosphorylation sites, respectively.
(E and F) KERT cells were infected with lentiviruses expressing the indicated p63 mutant derivatives. Whole cell lysates prepared 48 hours after lentiviral infection were analyzed by immunoblotting; cells were incubated with cycloheximide (CHX; 20 μg/ml) for the indicated durations before lysate preparation (E). p63 amounts were quantified by densitometry (F). Blots are representative of three experiments.
(G) 293T cells were transfected with plasmids expressing the indicated proteins. Whole cell lysates prepared 24 hours after transfection were analyzed by immunoblotting. Solid and open circles, plasmid-expressed and endogenous p38α, respectively; p-, phosphorylated. Blots are representative of three experiments.
(H and I) Human AK skin and skin SCC tumor sections (n = 10 and 20, respectively) were analyzed by immunostaining for the indicated molecules (H). Red and white arrowheads, epidermal cell layers with differential immunofluorescence (IF) signals; dotted line, the epidermal-dermal boundary; *, non-specific staining (H). p-p63 signals were graded from 0 to 3 based on IF intensity per cell (I). P values by Mann-Whitney test.
We mapped the p38α-phosphorylated residues in ΔNp63α by endoproteinase digestion and mass spectrometry of the samples recovered from an in vitro kinase reaction and immunoprecipitated from an extract of MG132-treated 293T cells (Fig. 6, A and C; table S1). In addition, we performed immunoblot analysis using phosphorylation-specific p63 antibodies to examine some consensus p38 target sites whose phosphorylation would likely escape detection by mass spectrometry due to the lack of nearby endoproteinase cleavage sites (fig. S5). These analyses identified six ΔNp63α residues (Ser66, Ser68, Thr123, Ser301, Ser361, and Ser369) targeted for phosphorylation by p38α (Fig. 6D).
We generated a series of lentiviruses expressing ΔNp63α variants in which individual phosphoacceptor residues were removed by site-directed mutagenesis. The turnover rates of these proteins in transduced human KERT keratinocytes were determined by tracking their decay after blocking de novo protein synthesis with cycloheximide. Substitution of Ser66/68, Thr123, and Ser301 to non-phosphorylatable Ala prolonged the half-life of ΔNp63α (Fig. 6, E and F). Phosphorylation of ΔNp63α at Ser66/68 and of TAp63α (a p63 isoform with an alternative, long amino-terminal domain) at the corresponding site occurred in 293T cells expressing p38αCA or a constitutively active form of MKK6, a kinase immediately upstream of p38α (Fig. 6G). Furthermore, AK epidermis exhibited immunostaining signals for Ser66/68-phosphorylated p63, but their intensity diminished in SCC specimens (Fig. 6, H and I), an observation paralleling the patterns of p38 phosphorylation (Fig. 1D).
Regulation of p63 target gene expression by p38α
Given the capacity of p38α to modulate p63 availability, we examined its impact on the gene expression program in keratinocytes. Global gene expression profiling indeed identified genes whose expression, either constitutive or UVB-inducible, was attenuated by p38α deficiency (Fig. 7A). Many of these p38α-dependent genes were functionally related to epidermal homeostasis, inflammation, and cancer, and included Mmp13, S100a8, S100a9, Il1a, and Ptgs2. Their induction in response to TPA and interleukin-1 was also dependent on p38α (fig. S6).
Fig. 7. p38α reverses p63-mediated repression of tumor suppressor gene expression.
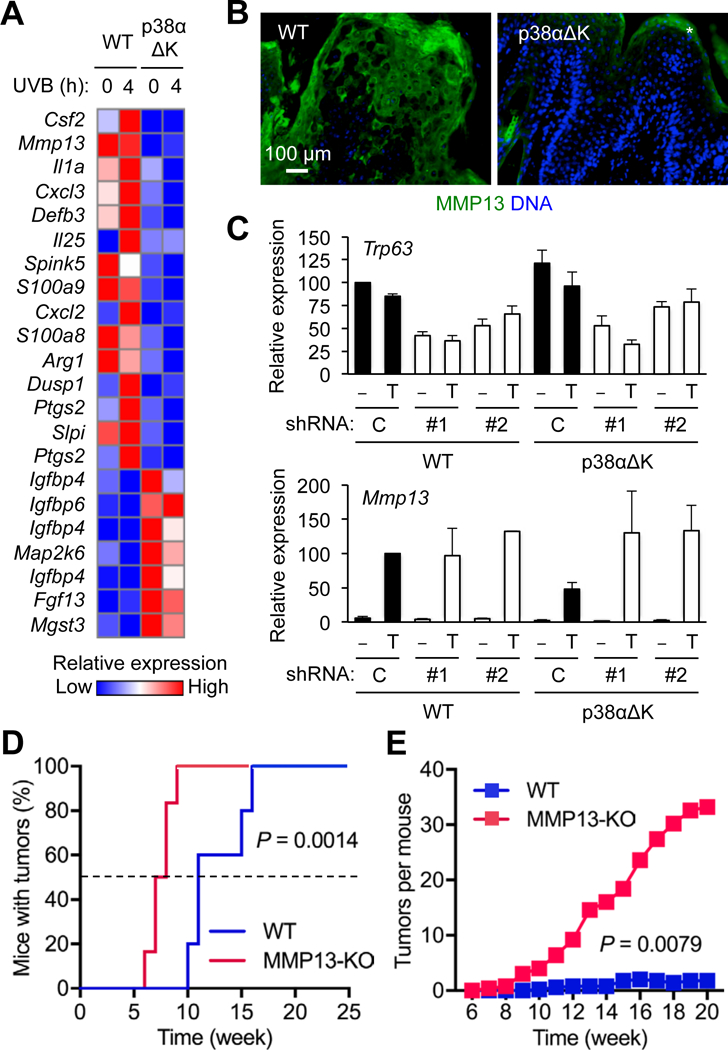
(A) Mouse keratinocytes were left unstimulated or stimulated with UVB (75 mJ/cm2). RNA prepared 4 hours after stimulation was analyzed using DNA microarrays.
(B) DMBA-TPA-induced tumor sections from mice were analyzed by immunostaining/counterstaining for the indicated molecules. *, non-specific staining. Images are representative of five tissue sections.
(C) Mouse keratinocytes were infected with control (Con) or p63 shRNA-expressing (#1 and #2) lentiviruses, and left unstimulated or stimulated with TPA (100 nM). RNA prepared 4 hours after stimulation was analyzed by qPCR. Data are means ± SD of three biological replicates.
(D and E) Mice (n = 6) were subjected to DMBA-TPA skin tumorigenesis. Tumor incidence (D) and multiplicity (E) were determined over the indicated period. P values by log-rank test (A) and two-tailed unpaired Student’s t test (B).
Based on the diverse functions of matrix metalloproteinase (MMP) family proteins in tissue remodeling and tumorigenesis (31, 32), we suspected the involvement of MMP13 in mediating the tumor-suppressive function of p38α, and subjected the mechanism regulating its expression to scrutiny. Similar to p38α-deficient keratinocytes, p38αΔK tumors showed decreased MMP13 expression (Fig. 7B). To demonstrate that p38α signaling drove MMP13 expression through reducing p63 availability, we determined the effect of loss of p63 on MMP13 expression. Given the essentiality of p63 in keratinocyte identity, proliferation, and survival, complete ablation of p63 expression might not permit the investigation of viable keratinocytes preserving their original phenotype. To bypass this potential problem, we used a small hairpin RNA-mediated gene knockdown (KD) approach, which could reduce but not completely abolish p63 gene (Trp63) expression. p63 KD resulted in an enhancement in TPA-induced MMP13 expression in p38αΔK keratinocytes, restoring its magnitude to that of WT keratinocytes (Fig. 7C). The analysis of chromatin immunoprecipitation-sequencing data illustrating genome-wide p63 occupancy in human keratinocytes (Gene Expression Omnibus GSE59827; 33) revealed p63 binding near the MMP13 gene as well as in the genomic loci of other p38α target gene homologs (fig. S7A). Further, KD of the p63 gene led to increased MMP13 expression in human keratinocytes (fig. S7, B and C), indicating a conserved role for p63 in repressing epidermal MMP13 expression in mice and humans. Lastly, we examined skin cancer development in Mmp13-knockout (KO) mice. In a chemical carcinogenesis experiment, tumors developed much faster and with a higher incidence in MMP13-KO relative to WT mice (Fig. 7, D and E).
Discussion
We have shown that p38α signaling suppresses skin carcinogenesis and wound skin re-epithelialization and restrains the size of the epidermal SPC pool. Mechanistically, p38α serves as a molecular switch for p63 protein turnover and the reprogramming of p63-governed gene expression in the epidermis. Our findings represent a novel mechanism that enables modification of epidermal-intrinsic genetic circuitry by external stimuli and a stress-activated intracellular signaling cascade. Similar mechanisms are likely to operate in other tissues and exert a key control at the nexus of tissue development, homeostasis, and tumorigenesis.
Earlier studies sought to identify the role of p38α in skin carcinogenesis by using transgenic mice expressing a dominant-negative p38α mutant (Thr180 to Ala and Tyr180 to Ala; 34, 35) and the small-molecule p38 inhibitor SB203580 (36), yet yielded incongruous findings— the dominant-negative mutant diminishing and the inhibitor enhancing tumorigenesis. Decreased activation of the transcription factor AP-1 and increased reactive oxygen species production by the NADPH oxidase NOX2 were proposed as possible explanations for diminished and enhanced tumorigenesis in the respective studies. Our study used mice in which the p38α gene was specifically ablated in keratinocytes. These different experimental approaches as well as other dissimilarities between our and the earlier studies, such as the carcinogenesis protocols used (chemically induced versus UV-induced) and the genetic backgrounds of mice (C57BL/6 versus SKH-1), may have led to contrasting conclusions about the in vivo effects and molecular changes resulting from loss of p38α function.
It has been shown that DMBA-TPA-induced papillomas in C57BL/6 mice rarely undergo malignant conversion (37). None of the papillomas formed in WT and p38αΔK mice, which were in a C57BL/6 background, progressed to squamous cell carcinoma or other malignant forms of skin cancer during our chemical carcinogenesis experiment. Therefore, although p38α deficiency enhanced tumor initiation, it did not appear to overcome the constraints imposed by the C57BL/6 genotype that limited malignant conversion. To address whether p38α plays a role in tumor progression independently of its role in tumor initiation, it will be necessary to generate and investigate an inducible p38α-KO mouse line in a genetic background prone to malignant conversion.
The transcriptional function of p63 is essential for driving epidermal SPC cycling and preventing their senescence (26, 38, 39), particularly under conditions of oncogene activation (27). A genome-wide association study (GWAS) of skin SCC discovered that genetic variation at the TP63 locus (rs6791479) was associated with the risk of this cancer type (40), a finding replicated in another independent GWAS study (41). Similar genetic evidence linking p63 to cancer susceptibility in human populations exists for bladder, lung, and pancreatic cancer (42–44). Unlike somatic gene mutations, deletions, and amplifications that drive clonal neoplastic transformation, GWAS-identified genetic variations may increase cancer risk by creating conditions that predispose normal tissues to cancer development. It remains to be determined whether p63 genetic variants associated with cancer susceptibility serve to produce such predispositions by elevating the frequency of SPCs that could become tumor-initiating cells after acquiring oncogenic genetic alterations.
Our study has demonstrated that phosphorylation by p38α enhances steady-state turnover and stimulus-induced degradation of p63. The three p38α-phosphorylated sites, Ser66/68, Thr123, and Ser301, were found to play a role in destabilizing ΔNp63α protein. This phosphorylation-dependent regulatory mechanism appears to be crucial for reprogramming p63-governed gene expression in the epidermis. In particular, p38α permits MMP13 gene induction by relieving p63-mediated transcriptional repression. p38α is an actionable signal transducer, amenable to inhibition and activation by small molecules. Pharmacological modulation of p38α signaling may serve as a novel approach to directed modification of epidermal and cancer stem cell phenotype for translation in regenerative medicine and clinical oncology.
Most of the known functions of MMPs in cancer are related to creating a tissue microenvironment conducive to tumor growth and progression by processing extracellular matrix proteins or cell-surface molecules. There is some evidence, however, that MMPs also serve antitumor functions (45). Our study presents a novel tumor-suppressive function of MMP13 and beckons further investigation about its action mechanisms. In our DMBA-TPA skin tumorigenesis experiments, MMP13-KO mice exhibited far greater tumor incidence and multiplicity than p38αΔK mice. This discrepancy may be due to a difference in the extent of ablation of epidermal MMP13 expression. MMP13 produced in p38αΔK keratinocytes, albeit in diminished amounts, may still have functional significance. Moreover, MMP13 expression in other cell types, such as dermal fibroblasts and myeloid cells, is likely intact in p38αΔK skin; MMP13 produced from these alternative cellular sources could compensate the loss of keratinocyte-derived MMP13.
Materials and Methods
Mice
p38αΔK mice (Mapk14fl/fl-K14Cre; Mapk14tm1.2Otsu and Tg(KRT14-cre)8Brn) were generated by crossing mice harboring loxP-flanked p38α alleles (46) with mice carrying a Cre recombinase transgene expressed under the control of the human KRT14 promoter (47). MMP13-KO (Mmp13−/−; Mmp13tm1Smk) mice had a systemic deficiency of MMP13 (48). These mice were in a C57BL/6J background and maintained in specific pathogen-free conditions. All animal experiments were conducted under an Institutional Animal Care and Use Committee-approved protocol.
Primary cells and cell lines
Primary mouse keratinocytes isolated from the skin of C57BL/6 newborn mice and primary human keratinocytes (a gift from G. Paolo Dotto) were cultured as described (49, 50). KERT cells (a gift from Anna Mandinova; originally from the American Type Culture Collection) and 293T cells were cultured in Keratinocyte-SFM and DMEM (both from Thermo Fisher Scientific), respectively.
Keratinocyte colony formation
Pinnae of ten- to twelve-week-old mice were separated into dorsal and ventral halves, and incubated with trypsin (0.5%; Thermo Fisher Scientific) for 1 hour and then with DNase I (10 U/ml; Sigma) for 20 min at 37°C. Keratinocytes thus isolated were plated at a density of 105 cell/ml in a 6-well plate and grown in CnT-07 medium (CELLnTECH) for ten days with medium changed every other day. Keratinocyte colonies were visualized by crystal violet staining.
Clinical tissue samples
Cases with a diagnosis of AK and SCC were identified from the Massachusetts General Hospital pathology archives. De-identified AK/SCC sections were prepared and provided by the Massachusetts General Hospital Dermatopathology Unit.
Reagents
DMBA, TPA, 5-bromo-2’-deoxyuridine (BrdU), cycloheximide, 5Z-7-Oxozeaenol, and verapamil were from Sigma-Aldrich; MG132, anisomycin, SB202190, and SC409 from EMD Millipore; D-JNKi from BML; and murine IL-1α from R&D systems. Antibodies against the following proteins were used in immunoblotting after 1:1000 dilution: p38α (sc-535) and p63 4A4 (sc-8431; both from Santa Cruz Biotechnology); p-p38 (9211), p-MK2 (3007), and p-Ser66/68-p63 (4981; all from Cell Signaling Technology); p-Ser301-p63 (PA5–39827) and p-Ser361-p63 (PA5–38380; both from Thermo Fisher Scientific); and actin (A4700; Sigma-Aldrich). Antibodies specific to the following proteins were used in immunofluorescence staining after 1:50 to 1:1000 dilution: p38α (sc-535), p63 H-137 (sc-8343), and p63 4A4 (sc-8431; all from Santa Cruz Biotechnology); p-p38 (9211) and p-Ser66/68-p63 (4981; both from Cell Signaling Technology); Ki67 (ab16667) and MMP13 (ab39012; both from Abcam); KRT14 (PRB-155P) and KRT15 (PCK-153P; both from Covance); CD3e (SP7) and KRT1 (ab9286; both from Abcam); and Ly6G/Gr-1 (553125; BD Biosciences). Hoechst 33324 and antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 595 were from Molecular Probes.
Chemical carcinogenesis and TPA-induced inflammation
Shaved dorsal back skin of eight- to ten-week-old mice was subjected to a single treatment of topical DMBA (100 μg) for tumor initiation and semiweekly treatment of topical TPA (10 μg) for twenty weeks for tumor promotion (51). The first treatment with TPA was one week after DMBA exposure. To induce acute skin inflammation, shaved dorsal back skin was treated with TPA (10 μg) daily for two consecutive days; inflamed skin was analyzed two days later.
Punch-biopsy wound healing
Shaved and depilated dorsal skin of seven- to eight-week-old mice was subjected to full-thickness wounding (epidermis to panniculus carnosus) using a 4-mm biopsy punch. Each wound circle was digitally photographed, and the wound area was calculated using Adobe Photoshop.
BrdU label retention
Ten-day-old mice were injected with BrdU (50 mg/kg) every twelve hours for a total of four injections; skin was collected after a chase period of fifteen days to analyze epidermal BrdU+ cells.
Side population discrimination assay
Keratinocytes from the pinna of ten- to twelve-week-old mice were incubated with Hoechst 33324 (5 μg/ml) for 1 hour, washed with ice-cold phosphate-buffered saline, stained with propidium iodide (1 μg/ml), and analyzed using Fortessa X-20 (BD Biosciences) and FlowJo (Tree Star).
Plasmid transfection and lentiviral transduction
Plasmid vectors expressing HA-tagged p38α and p38αCA (gifts from David Engelberg), MKK6EE (a gift from Jiahuai Han), and FLAG-tagged ΔNp63α (Addgene) were used in 293T cell transfection using Lipofectamine 2000 (Thermo Fisher Scientific). Lentiviral vectors expressing ΔNp63α and its variants were constructed using the backbone vector pCDH-CMV-MCS-EF1-copGFP (System Biosciences). To generate ΔNp63α variants harboring amino acid substitutions, mutations were introduced into the coding sequence using GeneArt Site-Directed Mutagenesis System (Thermo Fisher Scientific). Mouse p63-specific shRNA constructs (TRCN0000012749 and TRCN0000423330; Thermo Fisher Scientific) were among The RNAi Consortium clones. The human p63-specific shRNA construct was specific to 5’-acacacatggtatccagatgac-3’ in TP63. Lentiviral vectors expressing mouse and human p63-specific shRNA were based on pLKO.1. Lentiviral particles were generated by 293T cell transfection and quantified using the QuickTiter Lentivirus Titer Kit (Cell Biolabs). Keratinocytes were infected with lentiviruses in the presence of polybrene (8 μg/ml) for four hours, and used for subsequent analyses two days later.
Histology and immunofluorescence
Mouse skin/tumor tissues and human AK/SCC biopsies were fixed with formalin and embedded in paraffin. Tissue sections were analyzed by hematoxylin and eosin staining or by immunofluorescence using marker-specific primary antibodies, Alexa Fluor 488/595-conjugated secondary antibodies, and Hoechst 33324. BrdU in tissue sections were analyzed using the BrdU In Situ Detection Kit (BD Biosciences).
Protein and RNA analysis
Whole cell lysates were prepared and analyzed by immunoblotting as described (52). For in vitro kinase reactions, recombinant p38α and ΔNp63α proteins fused with glutathione-S-transferase (SignalChem) were incubated in a reaction buffer containing [γ−32P]ATP at 30°C for 30 min, and analyzed by SDS-PAGE and autoradiography. To map phosphorylated ΔNp63α residues, SDS-PAGE-separated and Coomassie Blue-stained proteins were subjected to trypsin or chymotrypsin digestion and mass spectrometry (LC MS/MS; the BIDMC Mass Spectrometry Facility). Total RNA was isolated using the RNeasy Mini Kit (Qiagen) and analyzed by real-time quantitative PCR using gene-specific primers (table S2). DNA microarray analysis was performed using GeneChip Mouse Genome 430 2.0 Array (Affymetrix) and the GENE-E matrix visualization and analysis platform (Broad Institute).
TCGA data analysis
TCGA data (clinical, mRNASeq, and RPPA) were downloaded from the GDAC Firehose, the cBioPortal for Cancer Genomics, and the Cancer Proteome Atlas Portal. Survival analysis of patient groups with differential RPPA signals was performed using GraphPad Prism.
Statistical Analysis
Data values are expressed as mean ± S.D. unless indicated otherwise. P values were obtained with the unpaired two-tailed Student’s t-test, the Mann-Whitney test, and the Log-rank (Mantel-Cox) test.
Supplementary Material
Acknowledgments:
We thank Yasuyo Sano, Dario Antonini, Ravi Mylvaganam, Xunwei Wu, Anna Mandinova, and Yang Brooks for technical support and materials; Paolo Dotto, Kristin White, Bruce Morgan, and Chun Kim for discussion and criticism. The results published here are in part based on data generated by the TCGA Research Network.
Funding: This study was supported by a National Institutes of Health grant to J.M.P (AI127768), by a Harvard Department of Dermatology Institutional training grant from the National Institutes of Health to M.-K.C. (T32AR7098), and by the Italian Association for Cancer Research (AIRC; IG2011-N.11369) to C.M.
Footnotes
Competing interests: None.
Data and materials availability: The microarray data are available in the NCBI GEO database (GSE51206). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes:
- 1.Blanpain C, Fuchs E, Stem cell plasticity: plasticity of epithelial stem cells in tissue regeneration. Science 344, 1242281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donati G, Watt FM, Stem cell heterogeneity and plasticity in epithelia. Cell Stem Cell 16, 465–476 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Land vatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR, A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Han J, Lee JD, Bibbs L, Ulevitch RJ, A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265, 808–811 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR, A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78, 1027–1037 (1994). [DOI] [PubMed] [Google Scholar]
- 6.Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J, Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 78, 1039–1049 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ Jr., Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR, p38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 11, 191–205 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Freund A, Patil CK, Campisi J, p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 30, 1536–1548 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, Komnenovic V, Scheuch H, Beug H, Wagner EF, p38α suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet 39, 741–749 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Ventura JJ, Tenbaum S, Perdiguero E, Huth M, Guerra C, Barbacid M, Pasparakis M, Nebreda AR, p38α MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet 39, 750–758 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M, Hepatocyte necrosis induced by oxidative stress and IL-1α release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 14, 156–165 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta J, del Barco Barrantes I, Igea A, Sakellariou S, Pateras IS, Gorgoulis VG, Nebreda AR, Dual function of p38α MAPK in colon cancer: suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell 25, 484–500 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, Yang JY, Broom BM, Verhaak RG, Kane DW, Wakefield C, Weinstein JN, Mills GB, Liang H, TCPA: a resource for cancer functional proteomics data. Nat. Methods 10, 1046–1047 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A, Sotiropoulou PA, Loges S, Lapouge G, Candi A, Mascre G, Drogat B, Dekoninck S, Haigh JJ, Carmeliet P, Blanpain C, A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 478, 399–403 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, Wan S, Wei S, Wang Y, Liu Y, Staroslawska E, Szubstarski F, Rolinski J, Grywalska E, Stanisławek A, Polkowski W, Kurylcio A, Kleer C, Chang AE, Wicha M, Sabel M, Zou W, Kryczek I, Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res 76, 3156–3165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D, Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev 13, 1382–1397 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, Holdsworth H, Turner L, Rollins B, Pasparakis M, Kollias G, Balkwill F, Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med 5, 828–831 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Arwert EN, Hoste E, Watt FM, Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 12, 170–180 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Golebiewska A, Brons NH, Bjerkvig R, Niclou SP, Critical appraisal of the side population assay in stem cell and cancer stem cell research. Cell Stem Cell 8, 136–147 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Yano S, Ito Y, Fujimoto M, Hamazaki TS, Tamaki K, Okochi H, Characterization and localization of side population cells in mouse skin. Stem Cells 23, 834–841 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Redvers RP, Li A, Kaur P, Side population in adult murine epidermis exhibits phenotypic and functional characteristics of keratinocyte stem cells. Proc. Natl. Acad. Sci. U S A 103, 13168–13173 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braun KM, Watt FM, Epidermal label-retaining cells: background and recent applications. J. Investig. Dermatol. Symp. Proc 9, 196–201 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F, p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A, p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Senoo M, Pinto F, Crum CP, McKeon F, p63 is essential for the proliferative potential of stem cells in stratified epithelia. Cell 129, 523–536 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, Guo X, Garcia EL, Michurina TV, Enikolopov G, Muthuswamy SK, Mills AA, ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 8, 164–176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW, p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9, 45–56 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Ramsey MR, Wilson C, Ory B, Rothenberg SM, Faquin W, Mills AA, Ellisen LW, FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J. Clin. Invest 123, 3525–3538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S, Vellano CP, Roebuck P, Yu Q, Eterovic AK, Byers LA, Davies MA, Deng W, Gopal YN, Chen G, von Euw EM, Slamon D, Conklin D, Heymach JV, Gazdar AF, Minna JD, Myers JN, Lu Y, Mills GB, Liang H, Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell 31, 225–239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Page-McCaw A, Ewald AJ, Werb Z, Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol 8, 221–233 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kessenbrock K, Plaks V, Werb Z, Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kouwenhoven EN, Oti M, Niehues H, van Heeringen SJ, Schalkwijk J, Stunnenberg HG, van Bokhoven H, Zhou H, Transcription factor p63 bookmarks and regulates dynamic enhancers during epidermal differentiation. EMBO Rep 16, 863–878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dickinson SE, Olson ER, Zhang J , Cooper SJ, Melton T, Criswell PJ, Casanova A, Dong Z, Hu C, Saboda K, Jacobs ET, Alberts DS, Bowden GT, p38 MAP kinase plays a functional role in UVB-induced mouse skin carcinogenesis. Mol Carcinog 50, 469–478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu K, Yu D, Cho YY, Bode AM, Ma W, Yao K, Li S, Li J, Bowden GT, Dong Z, Dong Z, Sunlight UV-induced skin cancer relies upon activation of the p38α signaling pathway. Cancer Res 73, 2181–2188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Rezvani HR, Back JH, Hosseini M, Tang X, Zhu Y, Mahfouf W, Raad H, Ragi G, Athar M, Kim AL, Bickers DR, Inhibition of p38 MAPK signaling augments skin tumorigenesis via NOX2 driven ROS generation. PLoS One 9, e97245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woodworth CD, Michael E, Smith L, Vijayachandra K, Glick A, Hennings H, Yuspa SH, Strain-dependent differences in malignant conversion of mouse skin tumors is an inherent property of the epidermal keratinocyte. Carcinogenesis 25, 1771–1778 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, Biernaskie JA, Sinha S, Prives C, Pevny LH, Miller FD, Flores ER, TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 5, 64–75 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, Sinha S, ΔNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 139, 772–782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asgari MM, Wang W, Ioannidis NM, Itnyre J, Hoffmann T, Jorgenson E, Whittemore AS, Identification of Susceptibility Loci for Cutaneous Squamous Cell Carcinoma. J. Invest. Dermatol 136, 930–937 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chahal HS, Lin Y, Ransohoff KJ, Hinds DA, Wu W, Dai HJ, Qureshi AA, Li WQ, Kraft P, Tang JY, Han J, Sarin KY, Genome-wide association study identifies novel susceptibility loci for cutaneous squamous cell carcinoma. Nat. Commun 7, 12048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN, Gudmundsson J, Jakobsdottir M, Bergthorsson JT, Sigurdsson A, Blondal T, Witjes JA, Vermeulen SH, Hulsbergen-van de Kaa CA, Swinkels DW, Ploeg M, Cornel EB, Vergunst H, Thorgeirsson TE, Gudbjartsson D, Gudjonsson SA, Thorleifsson G, Kristinsson KT, Mouy M, Snorradottir S, Placidi D, Campagna M, Arici C, Koppova K, Gurzau E, Rudnai P, Kellen E, Polidoro S, Guarrera S, Sacerdote C, Sanchez M, Saez B, Valdivia G, Ryk C, de Verdier P, Lindblom A, Golka K, Bishop DT, Knowles MA, Nikulasson S, Petursdottir V, Jonsson E, Geirsson G, Kristjansson B, Mayordomo JI, Steineck G, Porru S, Buntinx F, Zeegers MP, Fletcher T, Kumar R, Matullo G, Vineis P, Kiltie AE, Gulcher JR, Thorsteinsdottir U, Kong A, Rafnar T, Stefansson K, Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat. Genet 40, 1307–1312 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miki D, Kubo M, Takahashi A, Yoon KA, Kim J, Lee GK, Zo JI, Lee JS, Hosono N, Morizono T, Tsunoda T, Kamatani N, Chayama K, Takahashi T, Inazawa J, Nakamura Y, Daigo Y, Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat. Genet 42, 893–896 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, Li D, Neale RE, Olson SH, Scelo G, Amundadottir LT, Bamlet WR, Bijlsma MF, Blackford A, Borges M, Brennan P, Brenner H, Bueno-de-Mesquita HB, Canzian F, Capurso G, Cavestro GM, Chaffee KG, Chanock SJ, Cleary SP, Cotterchio M, Foretova L, Fuchs C, Funel N, Gazouli M, Hassan M, Herman JM, Holcatova I, Holly EA, Hoover RN, Hung RJ, Janout V, Key TJ, Kupcinskas J, Kurtz RC, Landi S, Lu L, Malecka-Panas E, Mambrini A, Mohelnikova-Duchonova B, Neoptolemos JP, Oberg AL, Orlow I, Pasquali C, Pezzilli R, Rizzato C, Saldia A, Scarpa A, Stolzenberg-Solomon RZ, Strobel O, Tavano F, Vashist YK, Vodicka P, Wolpin BM, Yu H, Petersen GM, Risch HA, Klein AP, Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat. Genet 47, 911–916 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.López-Otín C, Matrisian LM, Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 7, 800–808 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, Takeda T, Osuka S, Morita T, Kondoh G, Uno Y, Kashiwase K, Taniike M, Nakai A, Matsumura Y, Miyazaki J, Sudo T, Hongo K, Kusakari Y, Kurihara S, Chien KR, Takeda J, Hori M, Otsu K, p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol 24, 10611–10620 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A, Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet 29, 418–425 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, López-Otín C, Krane SM, Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc. Natl. Acad. Sci. U S A 101, 17192–17197 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, Lawrence T, Otsu K, Brissette JL, Arthur JS, Park JM, The kinase p38α serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat. Immunol 9, 1019–1027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu X, Nguyen BC, Dziunycz P, Chang S, Brooks Y, Lefort K, Hofbauer GF, Dotto GP, Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature 465, 368–372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, Holdsworth H, Turner L, Rollins B, Pasparakis M, Kollias G, Balkwill F, Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med 5, 828–831 (1999). [DOI] [PubMed] [Google Scholar]
- 52.Park JM, Ng VH, Maeda S, Rest RF, Karin M, Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med 200, 1647–1655 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.