

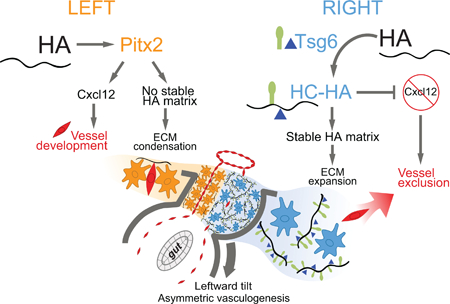
SUMMARY
For many years, biologists have focused on the role of Pitx2, expressed on the left side of developing embryos, in governing organ laterality. Here, we identify a different pathway during left-right asymmetry initiated by the right side of the embryo. Surprisingly, this conserved mechanism is orchestrated by the extracellular glycosaminoglycan, hyaluronan (HA), and is independent of Pitx2 on the left. Whereas HA is normally synthesized bilaterally as a simple polysaccharide, we show that covalent modification of HA by the enzyme Tsg6 on the right triggers distinct cell behavior necessary to drive the conserved midgut rotation and to pattern gut vasculature. HA disruption in chicken and Tsg6 −/− mice results in failure to initiate midgut rotation and perturbs vascular development predisposing to midgut volvulus. Our study leads us to revise the current symmetry-breaking paradigm in vertebrates and demonstrates how enzymatic modification of HA matrices can execute the blueprint of organ laterality.
Graphical Abstract
In-Brief/eTOC Blurb
Gut malrotation predisposes newborns to catastrophic strangulation of the intestine. Sivakumar et al. show that covalent modification of the extracellular matrix (ECM) component hyaluronan specifically on the right side of mouse and chick embryos regulates gut rotation key to intestinal morphogenesis. Disruption of modification of hyaluronan results in gut malrotation.
INTRODUCTION
Almost two centuries ago, the medical pupils of Dr. Matthew Baillie discovered the first reported case of situs inversus (Baillie, 1788). The reversed organ and vessel arrangement within the cadaver sparked an explosion of questions that still captivate biologists today. How do the organ primordia, which develop at different places and times during gestation, sense, interpret and execute the laterality instructions encoded by nature? In spite of enormous progress made to understand the molecular players controlling left-right (LR) asymmetry (Levin, 2005; Tabin, 2005), the mechanisms that lead to morphological symmetry breaking remain unresolved.
An important model to study organ laterality is the conserved process of midgut rotation (Figure 1A). Errors in gut chirality lead to gut malrotation and the catastrophic midgut volvulus in newborns, a self-strangulation of the gut tube with its mesenteric vasculature (Applegate, 2009). The direction of midgut rotation was long assumed intrinsic to the tube itself. However, we previously showed that in chickens and mice, rotation is instead driven by the dorsal mesentery (DM, Figure 1A), a mesodermal structure that suspends the gut tube along the dorsal body wall (Davis et al., 2008; Kurpios et al., 2008). A key property of the DM is its binary LR cellular organization, with distinct left and right compartments. Asymmetric changes in each cellular compartment cause the DM to deform and mechanically tilt the attached gut tube leftward (Davis et al., 2008; Hecksher-Sorensen et al., 2004; Kurpios et al., 2008). This critical leftward bias determines subsequent gut chirality, the disruption of which randomizes gut looping (Davis et al., 2008).
Figure 1: Expansion of the DM right side initiates gut rotation.
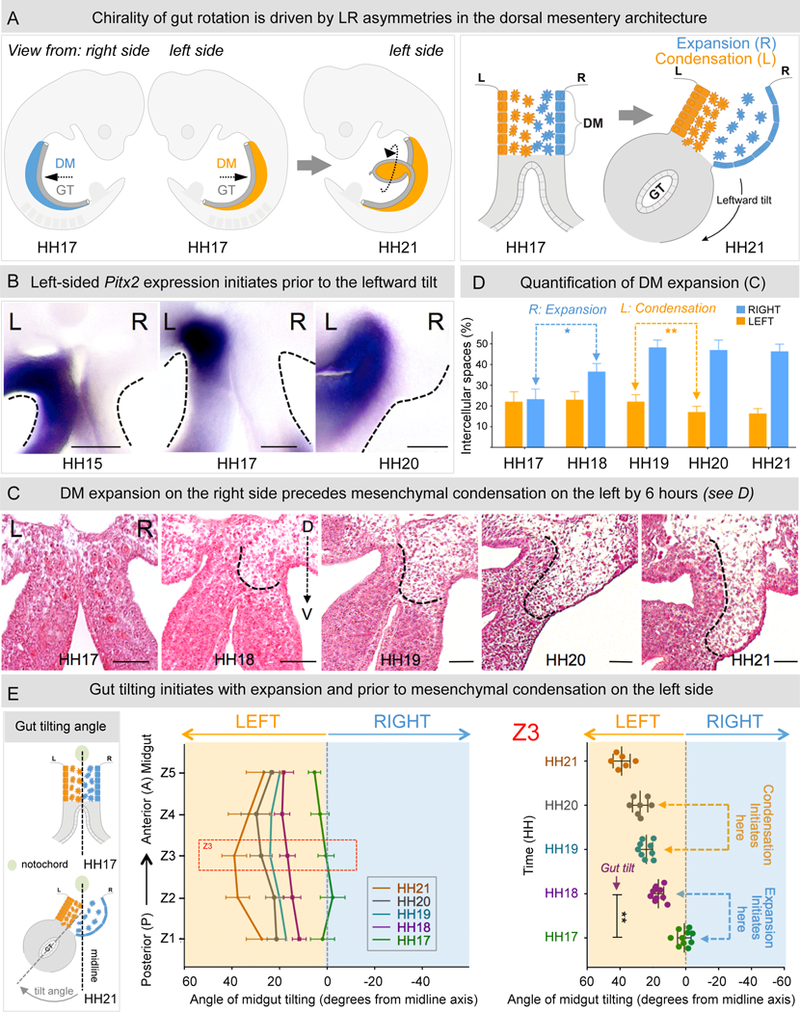
A The gut tube (GT, grey), in the chicken embryo at HH17 (mouse E9.5) suspended by the DM (blue-right; orange-left) undergoes a rotation to drive looping. This is driven by the leftward tilt (HH21, mouse E10.5) mediated by changes in cell architecture across the LR axis of the initially symmetric DM (HH17). B Pitx2 expression in the left DM appears prior to the initiation of the tilt (Pitx2 RNA ISH). C H&E staining of Z3 midgut sections from HH17–21 shows that right-sided expansion breaks the DM symmetry at HH18, prior to left-sided condensation at HH20. D Quantification of intercellular (cell-free) spaces from C; right expansion: p = 0.0001, left condensation: p = 0.0044, n = 10 embryos per stage. E Angle of midgut tilting from HH17–21, measured as displacement of GT from the midline (notochord) reveals significant increase in tilting at HH18 coincident with expansion (p = 0.0012, n = 10 embryos for HH17, HH18, n = 9 embryos for HH19, n = 7 embryos for HH20 and n = 6 embryos for HH21). Error bars represent mean ± SEM. See also Figure S1. Scale bars: B (100 µm); C (50 µm).
Initial LR symmetry-breaking decisions are made early during gastrulation via transient signaling by the conserved Nodal, a gene encoding a TGFβ-related secreted protein (Levin et al., 1995). This yields persistent expression of the bicoid-type homeobox transcription factor Pitx2 on the left side of the embryo. Pitx2 then specifies left side identity within individual organ primordia including the left side of the DM (Campione et al., 1999; Davis et al., 2008; Kurpios et al., 2008; Logan et al., 1998; Piedra et al., 1998; St Amand et al., 1998; Yoshioka et al., 1998). Based on the highly conserved asymmetric expression domain of Pitx2, the Nodal-Pitx2 axis has long been considered a common denominator of LR development. However, the role of Pitx2 during LR asymmetric organ morphogenesis is not well understood. For example, Pitx2 mutant mouse embryos exhibit laterality defects in many but not all visceral organs and Pitx2 is not required for asymmetric heart or gut looping in zebrafish (Ji et al., 2016; Kitamura et al., 1999; Lin et al., 1999; Liu et al., 2002; Lu et al., 1999; Shiratori et al., 2006). Moreover, Pitx2-dependent asymmetries in morphology appear long after the onset of Pitx2 expression, leaving unresolved how LR asymmetry is executed at the cellular level. In the DM, asymmetric Pitx2 expression does not cause morphological asymmetries in regions of the intestine that do not loop (Davis et al., 2008; Kurpios et al., 2008). Thus, Pitx2 expression alone is insufficient to break LR symmetry. Other local factors in the region of the looping gut must intersect with Pitx2 target genes to drive intestinal rotation.
After careful analyses in chicken and mouse embryos, we recently learned that morphological asymmetry in the DM is first broken on the right side of the DM, not the left. Unexpectedly, this LR symmetry-breaking event depends on the accumulation of hyaluronan (HA), a major component of the extracellular matrix (ECM), and is, surprisingly, independent of Pitx2 activity on the left. Whereas HA is synthesized bilaterally in the DM, we show that on the right side HA is covalently modified with heavy chain (HC) peptides by the enzyme Tsg6 (tumor necrosis factor α-stimulated gene 6) (Fülöp et al., 2003; Lauer et al., 2013c; Mukhopa dhyay et al., 2004), resulting in asymmetric accumulation of stable HC-HA matrices on the right side. HC-HA then triggers dramatic expansion of the right side of the DM necessary for midgut rotation and gut vascular development, linking vessel patterning with the morphogenesis of the host organ. Lastly, we show that on the left side, the role of nascent HA is distinct from its role on the right and is necessary to maintain proper Pitx2 expression and function within the left DM.
Collectively, our work uncovers a new role in vertebrates for HC-HA in the regulation of LR asymmetry and defines the biochemical principles underlying the assembly of mesenteric blood vessels. Our studies implicate the embryo’s right side as an active - rather than a passive “not left” - compartment and provide new mechanistic insights into the role of HA matrices during asymmetric organ morphogenesis.
RESULTS
Expansion of the right side of the DM initiates LR asymmetric midgut rotation
The LR DM asymmetries are transient and serve to initiate the critical midgut rotation by deforming the DM and tilting the gut tube leftward (Figure 1A) (Davis et al., 2008; Kurpios et al., 2008). This includes condensation of the left mesenchymal compartment and expansion of the right (Figure 1A). Pitx2 expression in the left lateral plate mesoderm initiates at E7.5 in mice and HH7–8 in chickens (Piedra et al., 1998), several days prior to the leftward tilt (HH20) (Figure 1B). This provides evidence that Pitx2 expression alone does not break the morphological symmetry within the DM to initiate the tilt (Davis et al., 2008).
To quantify the LR differences in the mesenchymal architecture of the DM prior to the formation of the leftward tilt, tissue sections from caudal (posterior, P) to rostral (anterior, A) midgut were stained with Hemotoxylin and Eosin (H&E) and the intercellular (cell-free) spaces in the left and right DM were quantified (Figures 1C, 1D, S1A, and S1B). To understand how these changes subsequently affect midgut tilt, the tilting angle was quantified with respect to the embryonic midline (Figure 1E). At HH17, the cellular architecture of the entire AP length of the midgut was symmetric with both the left and right mesenchymal compartments densely packed (Figures 1C, 1D and S1B) (Davis et al., 2008). Correspondingly, the axis of the midgut was closely aligned with the midline demonstrating that gut tilting had not yet initiated (Figure 1E). Within 4 hours, while the cellular architecture of the left mesenchyme remained unchanged, the mesenchymal compartment on the right became significantly expanded (Figures 1C and 1D, dotted line, percentage of intercellular spaces in HH18 right side DM is ~ 37% (36.57 ± 1.248) vs. HH17 right side DM, ~ 23% (23.27 ± 1.557), n = 10, p = 0.0007). Strikingly, the axis of the midgut also tilted significantly to the left (Figure 1E, angle of tilt of central midgut = 16.642 ± 3.256°). Thus, the morphological symmetry of the DM is broken by changes taking place on the right, which are associated with early displacement of the gut tube.
Surprisingly, it was not until HH20 (6 hours after expansion) that we observed mesenchymal condensation on the left (Figures 1C and 1D, percentage of intercellular spaces HH20 left side DM is ~ 17% (17.09 ± 2.769), n = 8 vs. HH17 left side DM, ~ 23% (22.11 ± 4.845), n = 10, p = 0.0205). A significant increase in the tilting angle was again observed (Figure 1E, HH21 (38.99 ± 2.123°), n = 6 vs. HH19 (23.95 ± 1.26°), n = 9, p = 0.001), and was attributed to the changes in the left DM. In conclusion, the leftward gut tilt is initiated by right DM expansion, independent of mesenchymal condensation within the left DM.
HA accumulation within the right DM coincides with DM expansion
We previously uncovered significant LR asymmetries in the accumulation of hyaluronan (HA) on the right side of the DM at HH21 (Kurpios, 2008). HA is a long glycosaminoglycan (GAG) polymer (Hascall and Esko, 2009) of thousands of glucuronic acid and N-acetyl-glucosamine disaccharide units (Fülöp et al., 1997). Unlike the other GAGs, HA is not sulfated (Hascall and Esko, 2009). HA is required for heart development and embryo viability, with functions in both tissue structure and signaling (Camenisch et al., 2000).
To test whether HA enrichment in the right DM could explain the observed DM expansion, we performed HA immunohistochemistry (de la Motte and Drazba, 2011) (Figures 2A and 2B). At HH17, the DM showed basal HA deposition only, with no detectable enrichment on either side and no observable DM expansion (Figures 2A and 2B). At HH18, we observed HA-rich ECM in the dorsal right DM overlapping with the domain of DM expansion, with lower levels of HA on the left side (Figures 2A and 2B). Intensity profiles across the LR axis revealed that the right DM showed ~ 5-fold (5.13±1.71, p = 0.007) enriched accumulation of HA (Figure 2B). By HH19, the HA-rich ECM covered the entire right side of the DM, resulting in ~ 12-fold (11.76 ± 3.43, p = 0.025) enrichment of HA levels in the right (Figures 2A and 2B). These data raised the possibility that HA accumulation may be the causative factor for DM expansion.
Figure 2: HA drives the right-sided mesenchymal expansion of the DM.
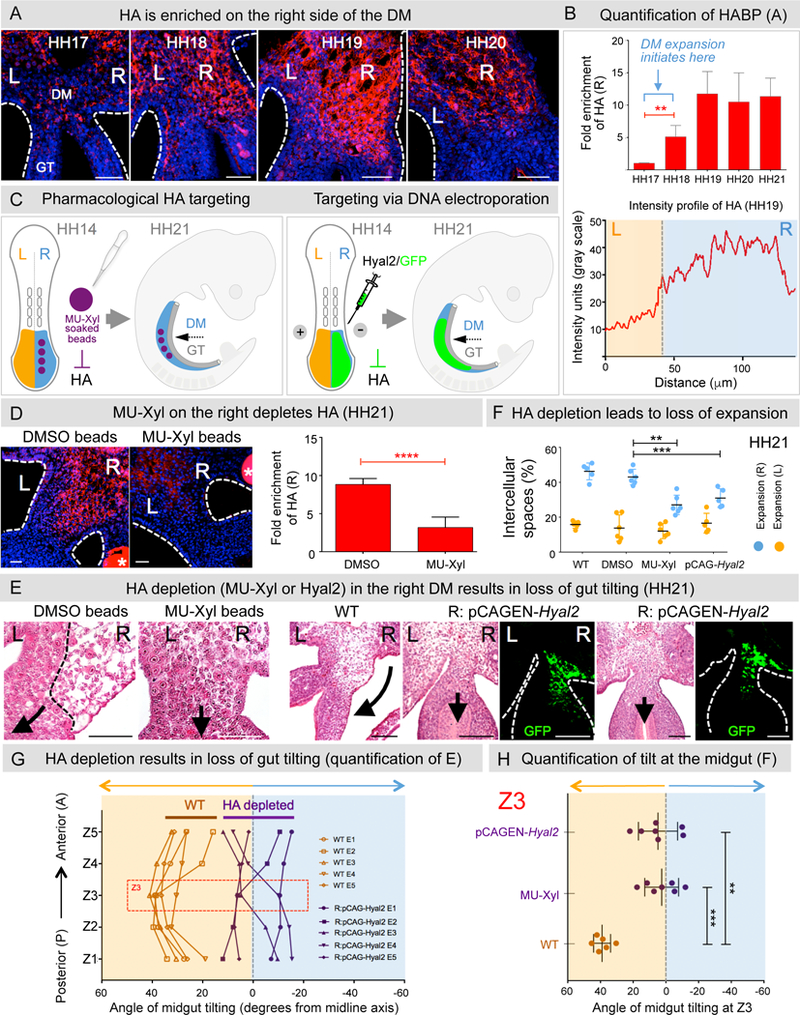
A HA staining using biotinylated HABP. B Quantification of HA accumulation: p = 0.007 between HH17–18, p = 0.025 between HH18–19, n = 6 embryos per stage. Average intensity profile of HA across the left (orange) and right (blue) axis at HH19. Fold enrichment calculated as area under intensity profile graph. C Left panel: MU-Xyl soaked beads (asterisks in D) are surgically inserted into the right coelomic cavity (HH14) prior to DM formation, Right panel: pCAGEN-Hyal2 DNA electroporation is utilized to degrade HA. D MU-Xyl treated DM shows significant reduction in HA accumulation (quantified in right panel, p = 0.0031, n = 7 embryos). E Depletion of HA in MU-Xyl treated embryos (panels 1–2) or pCAGEN-Hyal2 (panels 3–7) results in the loss of right-sided DM expansion. Quantification of E in F by measurements of intercellular spaces in the right panel: MU-Xyl vs DMSO: p = 0.0053, n = 6 embryos each for MU-Xyl, DMSO; WT vs pCAGEN-Hyal2, p = 0.0025, n = 5 embryos each for WT, pCAGEN-Hyal2). Tilting was quantified in G across the AP axis of the midgut (n = 5 embryos each for pCAGEN-Hyal2, WT) and in H in the Z3 region of the midgut: MU-Xyl vs WT, p = 0.00016, n =7 embryos each; pCAGEN-Hyal2 vs WT, p = 0.0025, n = 7 embryos each. Error bars represent mean ± SEM. See also Figure S3A. Scale bars: A (50 µm); D (30 µm); E (50 µm for panels 1–2 and 100 µm for panels 3–7).
Hyaluronan is required for ECM expansion and the leftward gut tilting
To test for a requirement of HA in the chicken DM, we surgically inserted resin beads soaked in 4-Methylumbelliferone-β-D-Xyloside (MU-Xyl), an inhibitor of HA synthesis (Hamati et al., 1989; Kakizaki et al., 2004), in the right coelomic cavity of the splanchnic mesoderm (DM precursor) prior to DM formation (HH14) (Figure 2C) (Mahadevan et al., 2014). Compared to vehicle DMSO controls, the MU-Xyl-treated embryos had significantly reduced levels of HA in the right DM (Figure 2D; DMSO beaded: ~ 9-fold R vs. L enrichment (8.83 ± 0.78) vs. MU beaded: ~ 3-fold enrichment (3.18 ± 1.36), n = 7, p = 0.0031), with a corresponding significant reduction of ECM expansion (Figure 2E and 2F; R: MU-Xyl vs. R: DMSO: n = 6 for each, p = 0.0053, percentage of intercellular spaces in MU-Xyl right side DM is ~ 28% [27.57 ± 5.09] vs. DMSO right side DM, ~ 42% [41.53 ± 4.48]).
MU-Xyl may also affect synthesis of other GAGs such as chondroitin sulfate, albeit to a much lesser extent (Hamati et al., 1989). To selectively deplete HA, we electroporated Hyaluronidase 2 (Hyal2), encoding a highly specific enzyme that degrades extracellular HA (Albeiroti et al., 2015; Harada and Takahashi, 2007) (Figure 2C, 2E and 2F). We marked targeted cells by co-electroporating GFP (Figure 2C and 2E, green). GFP-positive cells were found only on the right side at HH21, and electroporations with GFP alone had no effect on DM expansion (Figures 2E). However, similar to the MU-Xyl beading experiments, DM expansion was significantly reduced in Hyal2 electroporated embryos (Figures 2F, n = 5, p = 0.0025, percentage of intercellular spaces in right side DM of Hyal2 electroporated embryos is ~ 33% [33.19 ± 5.24] vs. right side DM of GFP electroporated embryos, ~ 44 % [43.75 ± 3.42]). We concluded that HA deposition is required for ECM expansion of the right DM.
Midgut tilting at HH21 was similarly affected, with loss of tilting angles in both the MU-Xyl-beaded and Hyal2 electroporated embryos (Figures 2G n = 5 embryos and 2H, n = 7, MU-Xyl vs. WT: p = 0.00016, Hyal2 vs. WT: p = 0.0025). Importantly, the expression of Pitx2 and its downstream target, Gpc3, remained restricted to the left DM in HA depleted embryos (Figure S3A, n = 8 for Pitx2, n = 3 Gpc3). Thus, loss of HA on the right disrupts midgut tilting despite the normal left-sided expression of Pitx2. These results firmly establish that HA directs ECM expansion of the right DM, and is required for the critical primary midgut tilt.
Hyaluronan is required for the normal process of vascular exclusion in the right DM
The gut vascular system derives from endothelial progenitors that form a bilaterally symmetric vascular endothelial dorso-ventral (DV) cords transiently residing in the left and right DM (Mahadevan et al., 2014; Pardanaud et al., 1989; Thomason et al., 2012). At the onset of gut tilting, the DV cords on the right regress, and at HH21 only the left-sided cords remain to become the major arterial branches supplying the intestine (Figure 3A) (Mahadevan et al., 2014). The first arterial branch, the primary longitudinal artery (1°LA, red arrowhead), forms at HH23 and becomes the ileocolic artery supplying the cecum, ileum, and appendix (human); the 2°LA forms at HH25 to become the middle colic artery (Figure 3D, green arrowhead).
Figure 3: Loss of HA from the right DM perturbs gut arterial vasculature.
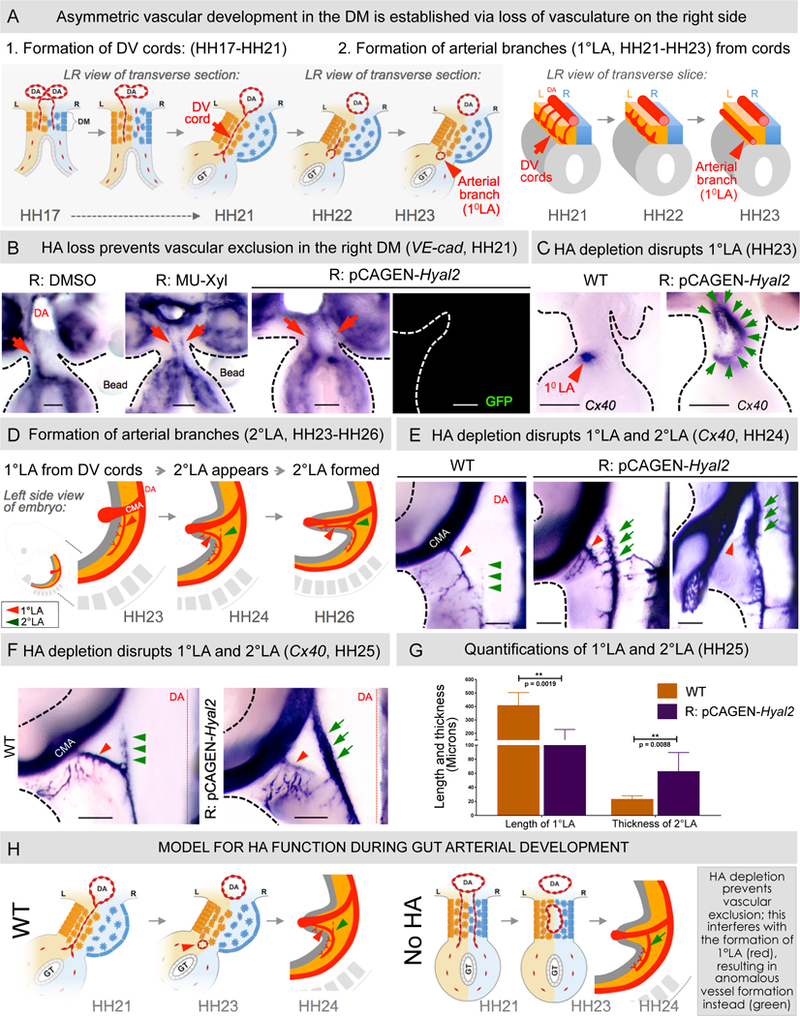
A WT asymmetric DM arteriogenesis. B VE-cadherin RNA ISH (HH21) shows ectopic maintenance of DV cords upon HA depletion (p = 0.0152 for DMSO vs MU-Xyl, n = 0/10 for DMSO, n = 6/12 for MU-Xyl, p = 0.0028 for WT vs pCAGEN- Hyal2, n = 0/12 for WT, n = 10/15 for pCAGEN-Hyal2 embryos). C Cx40 RNA ISH of HH23 embryo slices showing normal 1°LA in WT embryos (red arrowhead) and significantl y enlarged 2°LA-like vessel upon Hyal2 targeting (green arrows, p = 0.0325 for WT vs pCAGEN-Hyal2, n = 0/10 WT embryos, n = 5/10 pCAGEN-Hyal2 embryos). D Schematic of formation of arterial branches in WT embryos. (EF) Hyal2 targeted embryos (Cx40, HH24-HH25) lack (red arrowhead) or have significantly truncated 1°LA and show premature formation of anom alous 2°LA-like vessel, quantified in G: loss/truncation of 1°LA, p = 0.0019 for WT vs pCAGE N-Hyal2, n = 0/5 embryos WT, n = 7/7 pCAGEN-Hyal2; abnormal thickness, p = 0.0088 for WT vs pCAGEN-Hyal2, n = 0/5 embryos WT, n = 6/7 embryos pCAGEN-Hyal2). Error bars represent mean ± SEM. H Model for HA function during DM arterial patterning. CMV, cranial mesenteric artery; DA, dorsal aorta; DV cords, dorso-ventral cords. See also Figures S2 and S3A. Scale bars: B, C (100 µm); E, F (200 µm).
Because the timing of HA accumulation and DM expansion coincided with the progressive vascular exclusion observed on the right, we hypothesized that HA excludes vessel precursors in the right DM. Indeed, using VE-cadherin RNA in situ hybridization (ISH) in chicken embryos and eYFP immunohistochemistry in transgenic Tie1:H2B-eYFP quails we showed that vascular exclusion initiates at HH18, and by HH19–20 is lost completely (Figure S2 n = 5 per stage, chicken and quail embryos). Importantly, depletion of the HA-rich matrix with MU-Xyl-beaded or Hyal2 caused loss of right-sided avascularity and aberrant maintenance of DV cords (a “double-left” phenotype, p = 0.0152 for DMSO vs MU-Xyl, n = 0/10 for DMSO, n = 6/12 for MU-Xyl, p = 0.0028 for WT vs pCAGEN-Hyal2, n = 0/12 for WT, n = 10/15 for pCAGEN-Hyal2 embryos, Figure 3B, red arrow). We concluded that HA on the right side is anti-angiogenic and restricts vessel development to the left side.
While the “double-left” phenotype was previously ob served only when Pitx2 was misexpressed in the right DM (Mahadevan et al., 2014; Welsh et al., 2013), the expression of Pitx2 and a downstream target, Gpc3, remained restricted to the left DM in HA depleted embryos (Figure S3A n = 8 Pitx2, n = 3 Gpc3). Thus, loss of HA on the right disrupts the normal process of vascular exclusion without the loss of “ Pitx2-negative” right side identity.
Hyaluronan is required for the formation of gut arterial branches
To ask whether aberrant persistence of right-sided DV cords interferes with the proper formation of 1° and 2° LA we perturbed HA synthesis on the ri ght and followed arterial endothelium (ISH for Connexin 40, Cx40) on thick transverse midgut slices at HH23 and whole embryos at HH24-HH25. At HH23, all WT embryos formed a proper 1°LA located at the junction of the left DM and the gut tube (Figure 3C, red arrowhead). In contrast, in embryos electroporated with Hyal2 on the right, we observed a lack of 1°LA. Instead, a single and abnormally large arterial vessel formed, positioned along the DM midline (Figure 3C, green arrows, p = 0.0325 for WT vs pCAGEN-Hyal2, n = 0/10 WT embryos, n = 5/10 pCAGEN-Hyal2 embryos). Similarly, at HH24-HH25, the 1°LA was absent in three out of seven embryos electroporated with Hyal2 (Figures 3E and 3F, red arrowhead), and was reduced in length in the remaining four embryos (Figures 3E and 3F, red arrowhead). Again, we observed an abnormally large Cx40-positive arterial vessel anatomically resembling the 2°LA (Figures 3E-3G, green arrows, thickness of 1°LA in WT embryos = 23.4 ± 4.54 µm, p = 0.0019 for WT vs pCAGEN-Hyal2, n = 0/5 WT embryos showed abnormality, n = 7/7 embryos pCAGEN-Hyal2 showed abnormality; thickness of abnormal vessel resembling 2°LA in R: pCAGEN -Hyal2 embryos = 62.5 ± 26.58 µm, p = 0.0088 for WT vs pCAGEN-Hyal2, n = 0/5 for WT embryos, n = 6/7 for pCAGEN-Hyal2 embryos). It is important to note that in WT embryos, 2°LA formation does not initiate until HH24–25 and is only complete by HH26 (Mahadevan et al., 2014). In contrast, the anomalous vessel resembling 2°LA was fully developed at HH23– 24, a difference of ~ 12 hours (Figures 3C and 3E). These data indicate that HA-driven vascular exclusion is fundamental to the proper patterning of the major gut arteries. HA may also control the precise timing of blood vessel development and underscores the importance of coordinating organ development with that of its nascent vasculature (Figure 3H).
Tsg6 regulates HA-mediated expansion and vascular exclusion in the right DM
Whereas the expression of HA synthases is the first determinant of active HA synthesis, we were surprised to learn that expression of HA synthase 2 (Has2), the most active Has during embryogenesis (Camenisch et al., 2000) is not differentially expressed in the DM (Figure 4B graph). Consistent with bilateral Has2 expression in the DM, some HA staining can be observed on the left side of the DM (Figure 2A) (Kurpios et al., 2008). These observations suggest that HA synthesis alone may not account for the right-sided ECM expansion.
Figure 4: Tsg6 expression in the DM is right-sided and necessary for gut tilting.
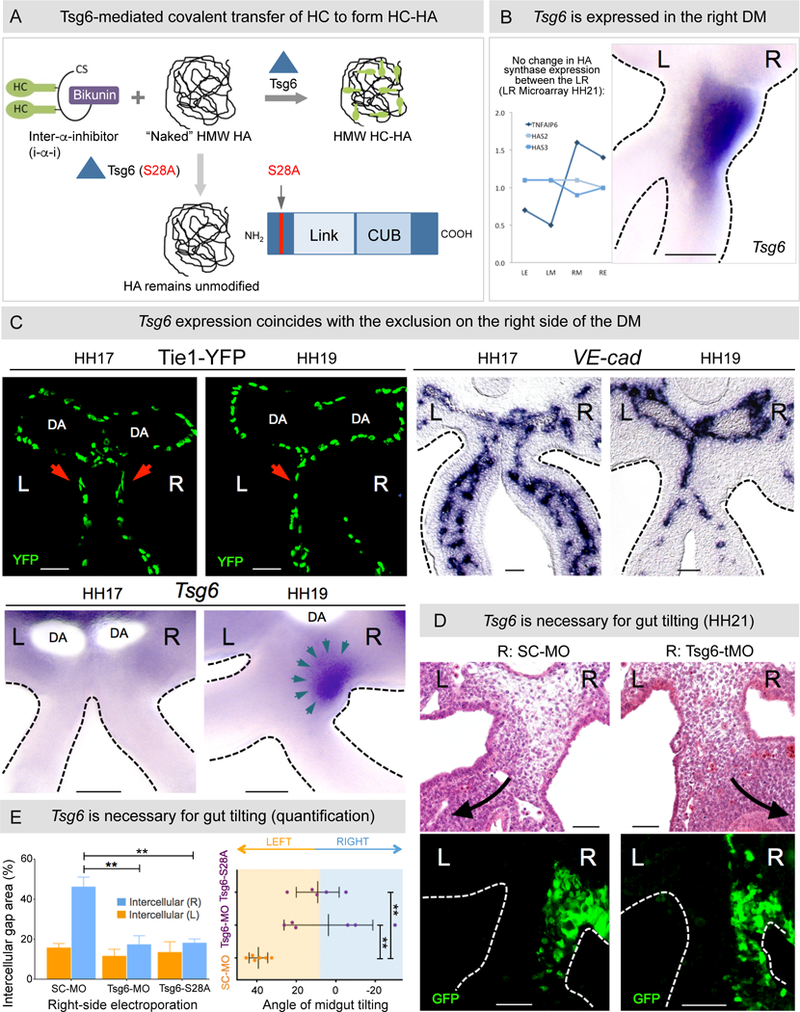
A Tsg6 modification of HA via transfer of HC complexes. A catalytically inactive form of Tsg6 (S28A) fails to covalently modify HA. B Laser capture microarray and RNA ISH for Tsg6 in the chicken DM reveal right-sided expression at HH21. C Onset of Tsg6 expression in the right DM at HH19 (bottom panel) coincides with vascular exclusion (top panel left: Tie1-H2B-YFP quail embryos; right: RNA ISH for VE-cadherin). D Tsg6-MO-knockdown, pCAGEN-Tsg6 S28A overexpression causes reduced expansion and subsequent loss of tilting, quantified in E: Left panel: p = 0.0021 for SC-MO vs Tsg6-tMO, p = 0.0015 for SC-MO vs pCAGEN-Tsg6 S28A; Right panel: p = 0.0036 for SC-MO vs Tsg6-tMO, p = 0.0032 for SC-MO vs pCAGEN-Tsg6 S28A, n = 6/6 embryos for SC-MO, n = 6/6 embryos for Tsg6-tMO, n = 5/5 embryos for pCAGEN-Tsg6 S28A. Error bars represent mean ± SEM. See also Figures S2, S4, S5C, and S6B. Scale bars: B, C (100 µm); D (50 µm).
The diversity of HA’s function in regulating cellular processes is thought to arise via the interaction of HA with HA-binding proteins (Milner and Day, 2003). Transcriptional profiling of the LR DM compartments (Welsh et al., 2013) has identified tumor necrosis factor α-stimulated gene 6 (Tsg6), which covalently modifies HA with heavy chain (HC) peptides to form a stable HA matrix known as HC-HA (Filston and Kirks, 1981) (Figures 4A and 4B). HC-HA has distinct biological activities capable of modulating signaling and cellular behavior (Fülöp et al., 2003; Milner and Day, 2003; Shen et al., 2006b). Whereas HC-HA plays critical roles in inflammation, the only known developmental function of HC-HA is to drive ECM expansion of the cumulus oocyte complex required for ovulation (Fülöp et al., 2003; Salustri et al., 1989).
Spatiotemporal analyses confirmed that Tsg6 mRNA is restricted to the DM right side at HH21 (Figure 4B). Tsg6 expression was not detected in the right DM at HH17, when the DM is bilaterally symmetric (Figures 4C). Importantly, it was faintly expressed in the dorsal right DM at HH18 (Figure S4), a region where significant ECM expansion, HA accumulation, and vascular exclusion are first detected. By HH19, when the right side of the DM is avascular, Tsg6 expression became prominent throughout the right DM (Figures 4C, teal arrows).
To demonstrate a requirement for Tsg6 in modulating the effects of HA, two distinct non-overlapping Tsg6 translation-blocking morpholinos (Tsg6-tMO) were electroporated on the right at HH14 (Simoes-Costa and Bronner, 2016). Histology revealed significantly reduced DM expansion and loss of gut tilting in embryos electroporated with Tsg6-tMO compared to scrambled (SC-MO) control embryos (Figures 4D and 4E, n = 6 embryos for both, percentage of intercellular spaces in right Tsg6-tMO DM is ~ 17% [17.42±4.32] p = 0.0021 vs. control right SC-tMO DM is, ~ 46% [46.26 ±4.81]; tilting angle in Tsg6-tMO is ~ 4° [3.71±22.49°] vs. control SC-MO is ~ 39° [39.16+4.71°], p = 0.0036). Importantly, as shown above with HA depletion, the expression of Pitx2 was not perturbed in embryos electroporated with Tsg6-specific morpholinos, arguing that molecular events on the right can disrupt midgut tilting despite the normal left-sided expression of Pitx2 (Figure S5C, p = 0.99 for WT vs Tsg6-tMO, n = 8/8 for WT, n = 5/5 for Tsg6-tMO).
Next, studying VE-cadherin expression at HH21 after Tsg6-tMO electroporation, we found that control embryos only harbored left sided DV cords (Figure 5A, red arrow). In contrast, abnormal bilateral DV cords were observed in Tsg6-tMO embryos (Figure 5A, red arrows, n = 6/8 embryos). Analyses of arterial branches in both Tsg6-tMO as well as Tsg6 splice blocking morpholinos (Tsg6-sMO) electroporated embryos at a later time point (HH25) confirmed loss of the 1°LA (Figures 5B, S5A and S5B, red arrowhead, n = 11/15, p = 0.0001) and appearance of an abnormal 2°LA-like vessel (Figures 5B, S5A, and S5B, green arrows, n = 9/11, p = 0.0078), a phenotype matching that of HA depletion via Hyal2 electroporation. Collectively, the functions of Tsg6 and HA in the DM are linked.
Figure 5: HC-HA is necessary and sufficient for gut vascular development.
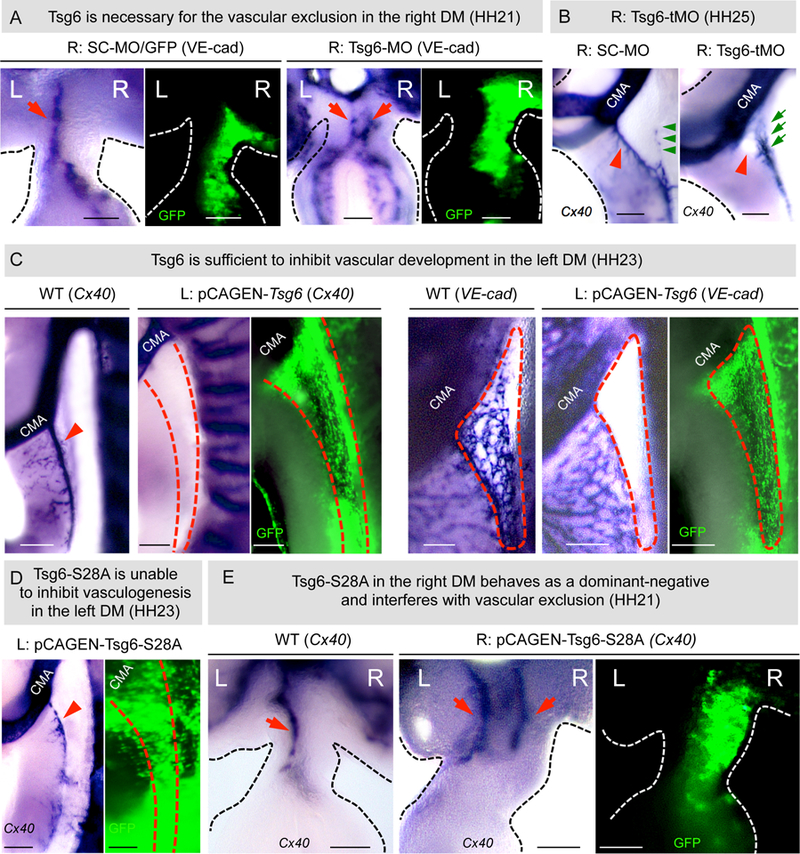
A Tsg6-MO-knockdown, but not scrambled control, prevents vascular exclusion in the right DM at HH21 (red arrowhead, right DM, p = 0.0068 for SC-MO vs Tsg6-MO, n = 0/8 for SC-MO, n = 6/8 embryos for Tsg6-MO), leading to the loss of 1° LA (B, red arrowheads, right panel, p = 0.0001 for SC-MO vs Tsg6-MO, n = 0/11 for SC- MO, n = 11/11 for Tsg6-MO embryos) and formation of the anomalous 2°LA-like vessel (green arrows) at HH25 (p = 0.0078 for SC- MO vs Tsg6-MO, n = 0/8 for SC-MO, n = 9/11for Tsg6-MO). RNA ISH for Cx40; GFP marks electroporated cells. C Ectopic left Tsg6 expression is sufficient to inhibit DM vascular arteriogenesis (Cx40 and VE-cadherin, p = 0.0005 for WT vs pCAGEN-Tsg6, n = 0/15 for WT, n = 11/19 for pCAGEN-Tsg6 embryos). D Catalytically dead Tsg6 mutant (S28A) fails to inhibit arteriogenesis (red arrow) on the left DM (Cx40) (p = 0.99 WT vs pCAGEN-S28A Tsg6, n = 15/ 15 WT, n = 7/7 pCAGEN-S28A). E Exogenous right-sided expression of S28A competes with endogenous Tsg6, preventing right-sided vascular exclusion (red arrow) at HH21 (Cx40, n = 5 embryos). See also Figure S6A. Scale bars: A, C, D, E (100 µm); B (200 µm).
Tsg6 catalysis on HA is necessary for ECM expansion and vascular exclusion
To understand whether the anti-vascular effects of Tsg6 on the right side depend on its covalent modification of HA, we obtained catalytically inactive human Tsg6 (Tsg6-S28A) mutated at a residue required for the transfer of HC to HA (Figure 4A). In vitro, this one residue change ablates HC transfer activity without affecting HA binding capacity of Tsg6 (Sanggaard et al., 2008). First, we electroporated the WT Tsg6 on the left side of the (vascularized) DM, to learn whether Tsg6 is sufficient to cause avascularity. Tsg6 on the left resulted in the loss of the 1°LA at HH23 (ISH to Cx40 and VE-cadherin, Figure 5C, p = 0.0005, n = 11/19 embryos showing complete loss of 1°LA, n = 8/19 with partial loss o f 1°LA). Tsg6-mediated loss of vasculature depended on the dosage of electroporated Tsg6 in the DM. At low Tsg6 doses in the left DM, vascular development was only partially inhibited (Figure S6A, n = 8/19).
To understand whether Tsg6 ablation of vessels on the left depends on its enzymatic activity on HA, we repeated this experiment with the Tsg6-S28A mutant. This had no effect on DM vasculature suggesting that the covalent modification of HA by Tsg6 is responsible for its anti-vascular function in the DM (ISH to Cx40, Figure 5D). This result also argues that endogenous HA in the left DM, however weak in HA staining, may have a Tsg6-independent role, consistent with bilateral Has2 expression in the DM (Figure 4B, graph). Interestingly, electroporating either the pCAGEN-Tsg6 or pCAGEN-Tsg6-S28A on the left was insufficient to induce ECM expansion on the left (Figure S6B, n = 15), suggesting that there are additional molecules needed to regulate HA enrichment and ECM expansion on the right.
Next, to prevent the formation of HC-HA on the right, we electroporated the right side with the inactive pCAGEN-Tsg6-S28A construct to compete with the WT Tsg6 protein for HA binding (to act as a dominant-negative) (Sanggaard et al., 2008). H&E sections revealed a significant reduction of DM expansion and loss of gut tilting in embryos electroporated with pCAGEN-Tsg6-S28A (Figures 4E and S6C, n = 5, p = 0.0015, percentage of intercellular spaces in right Tsg6-S28A DM is ~ 18% [18.2 ± 1.93] vs. right control GFP DM ~ 44 % [43.75 ± 3.42], n = 6 embryos each, p = 0.0001; gut tilting angles for Tsg6-S28A DM is ~ 9° [9.12 ± 10.82°] vs. control GFP DM ~39° [38.992 ± 5.200°], p = 0.0032, n = 6 embryos each).
ISH staining for VE-cadherin revealed abnormal double-left cords in pCAGEN-Tsg6-S28A embryos and normal left-sided DV cords in WT controls (Figure 5E, red arrows). Collectively, these experiments suggest that Tsg6 catalysis on HA is necessary for ECM expansion and vascular exclusion.
HA negatively regulates Cxcl12 mRNA expression
Vascular exclusion in the right DM is dependent on endothelial migration, driven by the chemokine Cxcl12, a ligand for the G protein-coupled receptor Cxcr4 and a direct target of Pitx2 (Mahadevan et al., 2014). Mice lacking Pitx2 or the Cxcr4/Cxcl12 axis have severely perturbed arterial patterning (Mahadevan et al., 2014; Tachibana et al., 1998).
When DV cords are initially bilateral, Cxcl12 mRNA expression in the DM is also bilateral (Figure 6A, HH17) but becomes lost on the right side commensurate with HA accumulation and vascular exclusion (Figure 6A, HH19) (Mahadevan et al., 2014). Moreover, Cxcl12 expression on the right was abnormally maintained in HA-depleted chicken embryos suggesting that HA negatively regulates Cxcl12 at the mRNA level (Figure 6B, p = 0.0108 for WT vs pCAGEN-Hyal2, n = 0/10 WT, n = 6/10 pCAGEN-Hyal2 embryos).
Figure 6: HA negatively regulates Cxcl12 mRNA expression.
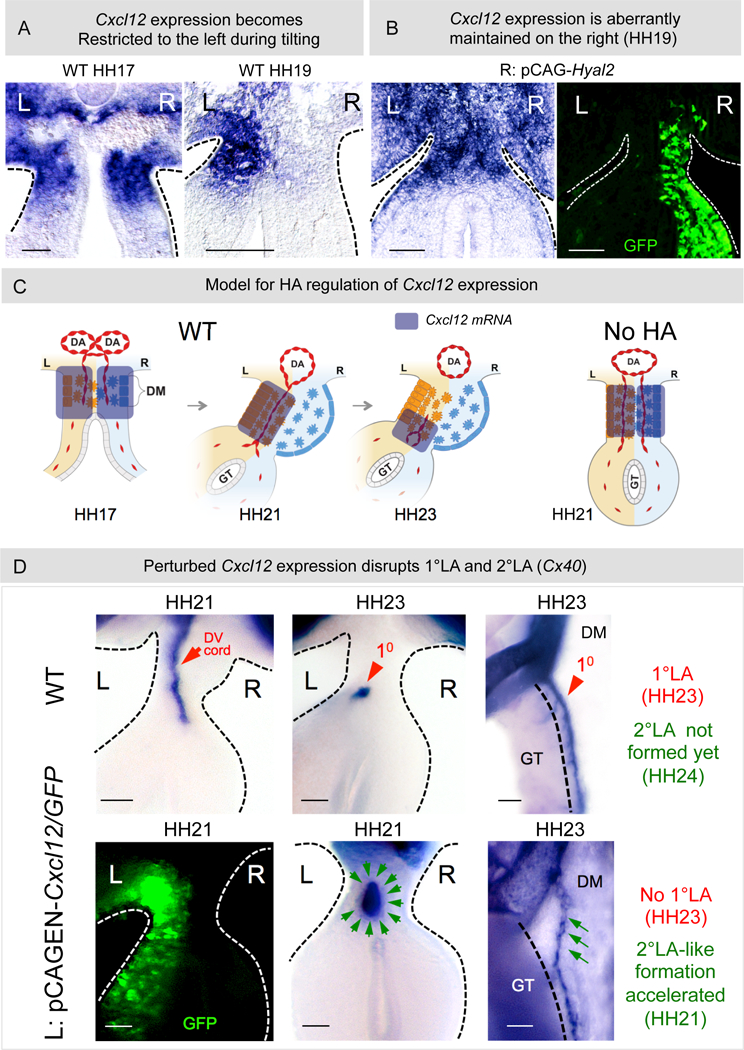
A Bilateral Cxcl12 at HH17 in WT embryos becomes restricted to the left concurrent with expansion and vascular exclusion at HH19. B Depletion of HA from the right causes abnormal maintenance of Cxcl12 on the right at HH19 (p = 0.0108 for WT vs pCAGEN-Hyal2, n = 0/10 for WT, n = 6/10 for pCAGEN-Hyal2). C Model showing dynamics of Cxcl12 expression in WT embryos. Loss of HA aberrantly maintains Cxcl12 on the right and perturbs the DV gradient of Cxcl12 on the left. D pCAGEN-Cxcl12 in the left DM perturbs the normal 1°LA morphogenesis (red arrowhead: WT 1°LA formed from DV cords [red arrow]. Green arrows: L: pCAGEN-Cxcl12, GFP marks electroporated cells (left panel); middle panel: transverse slices: p = 0.0278 WT vs pCAGEN-Cxcl12, n = 0/5 slices for WT, n = 5/7 slices for pCAGEN-Cxcl12; right panel: whole embryo, p = 0.022 WT vs pCAGEN-Cxcl12, n = 0/10 embryos for WT, n = 3/5 embryos for pCAGEN-Cxcl12, RNA ISH for Cx40. Scale bars: A, B (100 µm); D (50 µm).
In WT embryos, accompanying the formation of the 1°LA from DV cords on the left, Cxcl12 expression subsequently develops DV gradient asymmetry in the left DM, with its highest concentration ventrally where the 1°LA forms (Figure 6C, HH23) (Mahadevan et al., 2014). To understand whether perturbed gradient of Cxcl2 expression could be the cause of perturbed arterial patterning observed in HA-depleted embryos, we overexpressed Cxcl12 in the left DM. This resulted in the loss of the 1°LA and accelerated formation of the 2°LA-like vessel, phenotypes similar to those obtained when HA or Tsg6 was depleted from the right (Figure 6D, green arrows, left, middle panel: transverse slices: p = 0.0278, n = 0/5 WT embryos, n = 5/7 pCAGEN-Cxcl12 embryos, right panel: whole mount p = 0.022, n = 0/10 WT embryos, n = 3/5 pCAGEN-Cxcl12 embryos). Thus, loss of the Cxcl12/Cxcr4 angiogenic signal may be one mechanism by which HA accumulation on the right mediates vascular exclusion and hence restricts vascular development to the left DM.
Tsg6-HA pathway is conserved between chicken and mice
To learn whether Tsg6-HA function is conserved in the mouse intestine we first characterized the molecular distribution of HA, Tsg6, and also the inter-α-trypsin inhibitor (I-α-I), the critical substrate for Tsg6 to form HC-HA (Figure 4A) (Milner and Day, 2003), in the mouse DM. As in the chicken, the mesenchymal cells within the mouse DM at E10.5 are less densely packed on the right than the left side (Davis et al., 2008). However, these cellular differences are more pronounced towards the cranial end of the mouse DM (at the level of the future duodenum) and are even more transient than in the chicken (Davis et al., 2008). Importantly, both HA staining and I-α-I expression were significantly enriched on the right side of the mouse DM when compared to the left (Figure S7, n = 5 embryos). Because there are currently no reliable Tsg6 antibodies that work on mouse tissue, we characterized Tsg6 protein expression in the DM of embryonic rats (Coulson-Thomas et al., 2016). Consistent with the above data, Tsg6 expression was also restricted the right side of the rat DM (Figure S7, n = 8 embryos). Thus, the molecular machinery involved in the formation of the HC-HA matrix are conserved between the chicken, mouse, and rat.
Tsg6 −/− and bikunin −/− mouse embryos fail to initiate midgut rotation
Tsg6-null mice are viable but females are infertile because the cumulus cell-oocyte ECM fails to expand in the absence of HC-HA (Fülöp et al., 2003; Salustri et al., 1989). To study the role of Tsg6 in the mouse intestine, we first characterized Tsg6 +/+, Tsg6 +/− and Tsg6 −/− mouse embryos at E18.5, when the final topology of the digestive tract is achieved. Our preliminary genetic characterization of Tsg6-null mice suggested a significant departure from expected Mendelian inheritance ratios, highlighting potentially lethal or sub-lethal phenotypes associated with Tsg6 loss, in agreement with findings from other labs (Table S1). In Tsg6 +/+ and Tsg6 +/−mouse embryos at E18.5, we observed stereotypical looping topology with jejunal and ileal loops tightly packed without entanglement (Figure 7A). By contrast, in Tsg6 −/− embryos we observed a spectrum of striking perturbations in gut looping topology (Figure 7A, p = 0.0172 for WT vs Tsg6 −/− mouse embryos, n = 0/7 for WT, n = 4/5 for Tsg6 −/−) with the ileum traversing a loop of distal jejunum, causing entanglement of the loops (Figure 7A, n = 2/4).
Figure 7: Tsg6 −/− and bikunin −/− mice fail to initiate gut rotation.
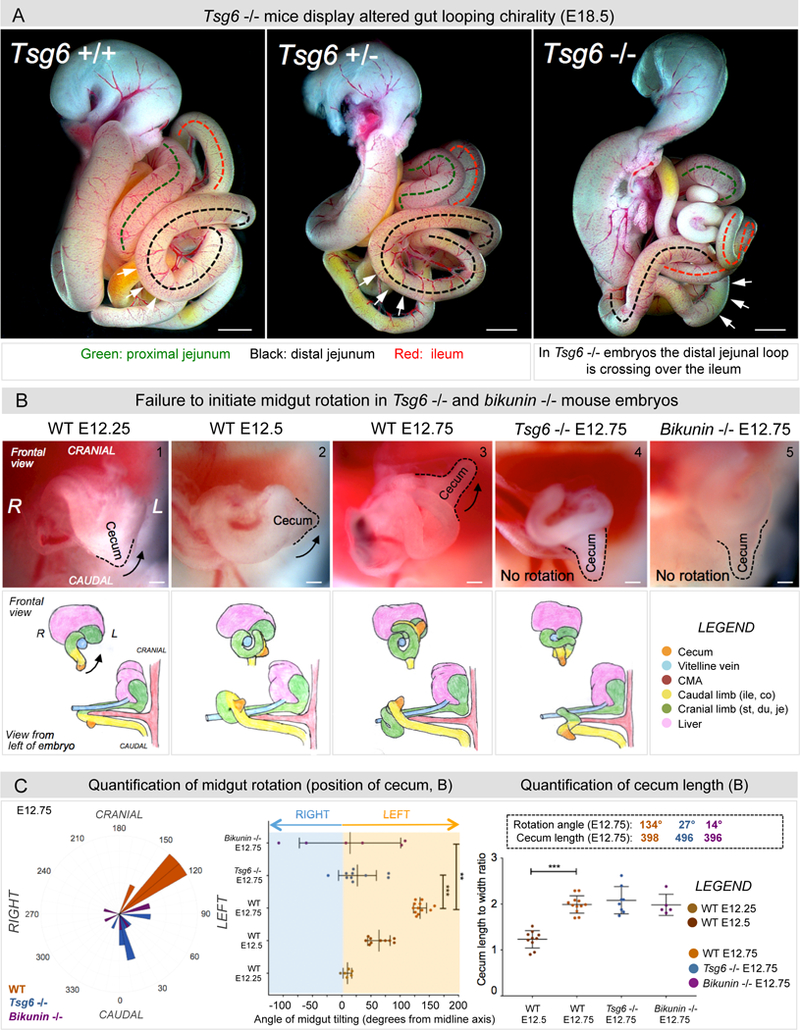
A E18.5 embryos: In Tsg6 −/− embryos the distal jejunal loop crosses over the ileum, indicating malrotation (p = 0.0172 for WT vs Tsg6 −/− embryos, n = 0/7 WT, n = 4/5 Tsg6 −/−). B Midgut rotation in mice initiates at E12 and is restricted to the midgut apex. Unlike WT embryos (panel 1–3), Tsg6 −/− and bikunin −/− embryos fail to initiate rotation as seen by the cecal position (compare panel 3 with 4–5). C Left panel: Polar histogram of cecal angle with respect to the cranial-caudal axis also shown as a dot plot (Middle panel). Right panel: Cecum length-to-width ratios indicating no delay in gut development. Numbers within boxes summarize mean angles of cecum rotation and length at E12.25–12.75 (p = 0.00019 for WT E12.75 vs Tsg6 −/− E12.75, p = 0.0021 for WT E12.75 vs bikunin −/− E12.75, n = 6 embryos for WT E12.25, n = 10 embryos for WT E12.5, n = 12 embryos for WT E12.75, n = 10 embryos for Tsg6 −/− E12.75, n = 6 embryos for bikunin −/− E12.75). Error bars represent mean ± SEM. See also Figure S7, S8, and Table S1. Scale bars: A (1000 µm); B (100 µm).
Whereas most intestinal malrotations do not lead to volvulus, some possess characteristics that are strongly associated with strangulation (Langer, 2017; Pelayo and Lo, 2016). For example, mispositioning of the duodenojejunal junction (DJJ) and the cecum shortens the separation of the DJJ from the ileocecal junction (ICJ) to less than half the width of the abdomen, thus narrowing the mesenteric stalk (Langer, 2017). Remarkably, out of five Tsg6 −/− embryos obtained from multiple litters, four of the mutant embryos satisfied these criteria, indicating malrotation due to a narrow mesenteric stalk (Figure S8A).
Consistent with the above analyses, younger Tsg6 −/− embryos at E13.5 (p = 0.035 for WT vs Tsg6 −/− at E13.5, n = 0/8 for WT) showed large deviations from the highly reproducible looping geometry observed in age-matched Tsg6 +/− embryos (Figure S8B). These included altered positioning and orientation of the distal jejunal and ileal loops and mispositioning of the cecum, linking early topology errors with subsequent malrotation phenotypes (Langer, 2017; Pelayo and Lo, 2016).
To learn whether looping defects in Tsg6-null mouse embryos derive from the catalytic activity of Tsg6 during covalent HA modification, we quantified midgut rotation of WT, Tsg6 −/−, and bikunin −/− mouse embryos (Figure 7B and 7C). Bikunin is the molecular precursor to the HC donor and Tsg6 substrate, I-α-I. Like Tsg6 mutants, bikunin-null animals fail to form HC-HA with nearly identical phenotypes (Lauer et al., 2013b; Salier et al., 1996; Zhuo et al., 2001). We hypothesized that if the role of Tsg6 in the mouse intestine is dependent on HA catalysis, altered gut looping in bikunin −/− mouse embryos would be observed.
Our analyses of mouse embryos starting at the onset of midgut rotation (E12 - E12.75) revealed that 100% of WT embryos underwent proper rotation, with a highly reproducible average rotation of 134° at E12.75 (WT angle: 134.2 ± 3.213°, n = 12; Figures 7B and 7C). In contrast, 70% of Tsg6 −/− embryos at E12.75 were unable to initiate counterclockwise rotation (Tsg6 angle: 26.89 ± 10.22°, n = 7/10, p = 0.00019; Figures 7B and 7C). Similarly, 66% of bikunin −/− mouse embryos failed to initiate rotation (bikunin angle: 14.3 ± 35.4, n = 4/6, p = 0.0021; Figures 7B and 7C). This defect was not due to a developmental delay, as the cecum length - to - width ratio was not affected in the mutants (Figure 7C). Thus, both Tsg6 and bikunin are necessary for midgut rotation in mice, a result consistent with the right-sided localization of the heavy chain I-α-I in the mouse DM (Figure S7).
Collectively, the Tsg6-HA pathway plays a critical and conserved role in initiating midgut rotation. These findings also connect multiple members of the Tsg6-HA pathway to the right side of the DM.
Tsg6-HA pathway is independent of Pitx2 on the left
Although most situs-specific organogenesis depends on Pitx2, the mechanism of Pitx2 action remains unknown. We hypothesized that in the DM, Pitx2 functions to repress Tsg6-catalyzed HA enrichment on the left side. To test this, we performed morpholino-based knockdown of Pitx2 on the left side of the chicken DM (Figure 8). Intriguingly, this had no effect on the levels of HA on the left side, showing that HA accumulation is independent of Pitx2 status (Figures 8A and 8C, p = 0.1558 for WT vs Pitx2-MO, n = 5/5 for WT, n = 11/11 for Pitx2-MO). In contrast, knockdown of Pitx2 dramatically reduced the expression of the canonical Pitx2 target gene, Islet1 (Davis et al., 2008), validating the knockdown efficacy of Pitx2-specific morpholinos (Figures 8B and 8D, p = 0.0029 for WT vs Pitx2- MO, n = 0/6 WT, n = 6/6 Pitx2-MO). Thus, perturbed Pitx2 expression on the left does not de-repress the Tsg6-catalyzed HA enrichment. This conclusion is consistent with data presented above showing that electroporating WT Tsg6 on the left side does not drive DM expansion (Figure S6B).
Figure 8: HA-Tsg6 pathway is independent of Pitx2 activity on the left.
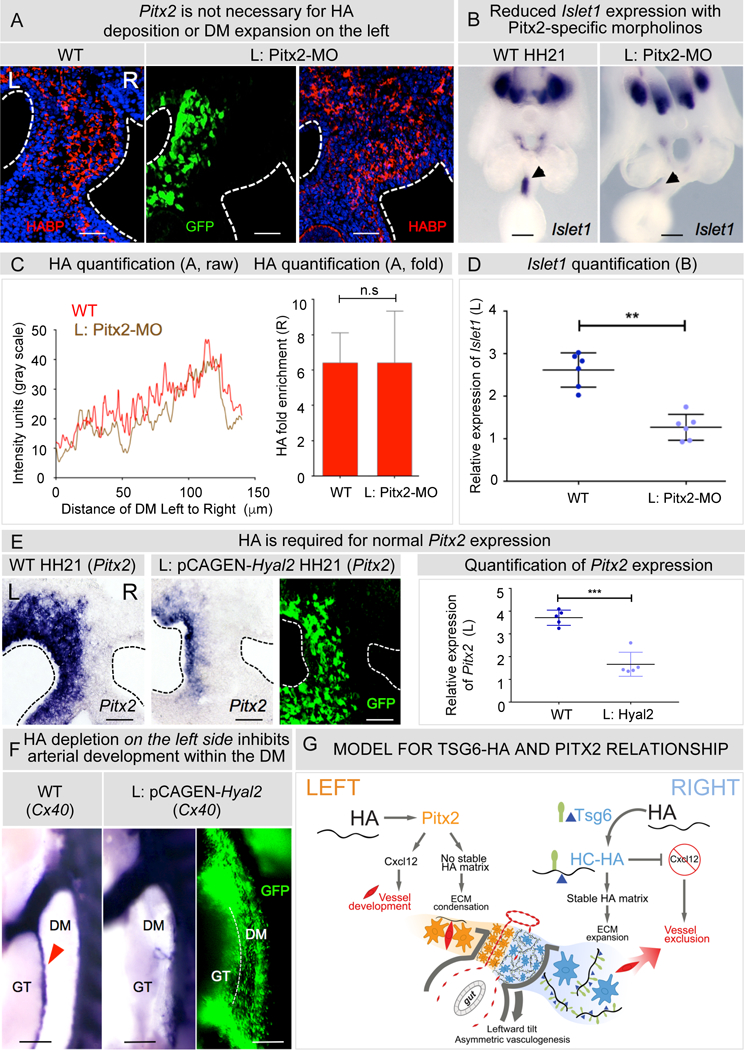
A Knockdown of Pitx2 in left DM with Pitx-MO does not affect HA deposition (quantified in C using average intensity profiles of HA distribution and fold enrichment of HA, p = 0.1558 for WT vs Pitx2-MO, n = 5/5 WT, n = 11/11 Pitx2-MO). B Validation of Pitx2-MO showing reduction of Islet1 expression (quantified in D using imageJ and integrated density ratios of Islet1 expression p = 0.0029 for WT vs Pitx2-MO, n = 0/6 WT, n = 6/6 Pitx2-MO,). E Degradation of HA on the left by pCAGEN-Hyal2 electroporation reduces normal Pitx2 expression, quantified as integrated density ratios (p = 0.0084 for WT vs pCAGEN-Hyal2, n = 0/5 WT, n = 5/5 pCAGEN-Hyal2). F pCAGEN-Hyal2 inhibits arteriogenesis in left DM (p = 0.0001 for WT vs pCAGEN-Hyal2, n = 0/16 WT, n = 8/10 pCAGEN-Hyal2) G Model for the Tsg6-HA (right) and Pitx2 (left) pathway in the DM: HA production and Tsg6 catalysis on the right makes stable HC-HA and initiates rotation. The HC-HA matrix negatively regulates Cxcl12 in right DM leading to vascular exclusion and left-side restricted arteriogenesis. On the left, Pitx2 regulates Cxcl12-dependent vascular remodeling. Nascent HA deposition on the left is required to maintain Pitx2. Error bars represent mean ± SEM. Scale bars A, E (50 µm); B (200µm); F (100 µm). See also Figure S5, S6.
In summary, loss of Pitx2 activity is not sufficient to induce HA enrichment on the left. There are distinct molecules and mechanisms contributing to the enrichment of HA and ECM expansion on the right side that are not activated on the left side when the function of Pitx2 is perturbed.
Following the observation that nascent HA is deposited on both sides of the DM while HC-HA accumulates only on the right in the absence of Pitx2 (Figure 2A), we asked whether HA can regulate Pitx2 expression in the DM. We expressed Hyal2 on the left side by electroporation of chicken embryos to ablate HA in the Pitx2-positive microenvironment. Left-sided hyaluronidase expression in these embryos caused a striking reduction in the number of Pitx2-positive cells (Figure 8E, p = 0.0084 for WT vs pCAGEN-Hyal2, n = 0/5 for WT, n = 5/5 for pCAGEN-Hyal2), while also completely disrupting Pitx2-driven arterial vasculogenesis in the left DM (Figure 8F, p = 0.0001 for WT vs pCAGEN-Hyal2, n = 0/16 for WT, n = 8/10 embryos for pCAGEN-Hyal2, HH25). This effect was specific to HA lysis, because misexpression of Tsg6 (Figure S3B, p = 0.8592 for WT vs pCAGEN-Tsg6, n = 5/5 embryos for both) or overexpression of the HA synthase Has2 (n = 6, p = 0.5468, data not shown) in the left DM had no effect on Pitx2 expression. Thus, nascent left-sided HA deposition is required to maintain the normal spatial domain and function of Pitx2 in the DM.
DISCUSSION
In the present study, we show that the conserved counterclockwise rotation of the chicken and mammalian midgut results from a break in midline morphological symmetry initiated by changes on the right side, independent of Pitx2 activity on the left. Our major findings are that the Tsg6-HA pathway triggers DM expansion on the right to initiate midgut rotation and patterns the major blood vessels that supply the intestine, linking vessel patterning with the morphogenesis of the parent organ itself. Our studies implicate the embryo’s right side as an active compartment and provide new mechanistic insights into the role of HA matrices in organ laterality.
New role for the Tsg6-HA pathway during LR asymmetry
HA is essential during many processes including heart development, angiogenesis, cancer, diabetes, and inflammation, but the mechanisms by which HA mediates these diverse processes are challenging to study in vivo, and remain largely unknown. The structural variability of HA adapts it to a complex spectrum of interactions with multiple HA receptors and binding proteins (Cowman and Matsuoka, 2005; Jaime et al., 2015), while variably-sized HA polymers in vivo regulate distinct signaling pathways that can induce opposing biological responses (Jaime et al., 2015; Pardue et al., 2008). In this study, we present evidence that, in the right side of the DM, a unique form of HA accumulates bearing covalent HC peptide modifications. The addition of HC to HA is catalyzed by the HA-binding protein Tsg6, forming stable HC-HA matrices that trigger dramatic expansion of the right side of the DM, driving midgut rotation and gut vascular development. While the Tsg6-catalyzed production of HC-HA is a well-described inflammatory event during rheumatoid and osteoarthritis, ovarian cancer, cervical ripening, and chronic liver disease (Fujimoto et al., 2002; Lauer et al., 2013a; Shen et al., 2006a; Wu et al., 2002), the only known developmental function of HC-HA is during cumulus-oocyte ECM expansion necessary for ovulation (Fülöp et al., 2003; Salustri et al., 1989).
An essential player in the formation of HC-HA is bikunin. Bikunin is a small chondroitin sulfate proteoglycan and the precursor of the critical HC donor, i-α-i. Bikunin-null mice lack i-α-i and, like Tsg6 mutants, fail to form HC-HA with nearly identical phenotypes (Lauer et al., 2013b; Salier et al., 1996; Zhuo et al., 2001). Our experiments demonstrate that HA, Tsg6, and i-α-i accumulate on the right side in the mouse DM and both Tsg6- and bikunin-null mouse embryos fail to initiate midgut rotation. Thus, our studies reveal a critical and conserved role for the Tsg6-HA pathway in initiating midgut rotation in vertebrates.
Interestingly, functional HA is also synthesized on the left side of the DM, in the presence of endogenous Pitx2 expression. Importantly, the roles of HA on the left side are distinct from those on the right. Unexpectedly, degradation of HA on the left not only reduced the spatial domain of Pitx2 expression but it also affected Pitx2-driven arterial vasculogenesis within the left DM. Therefore, whereas HA on the right is anti-angiogenic (drives vascular exclusion), HA on the left is required for DM arteriogenesis. This observation is consistent with published literature that the angiogenic responses to HA are highly dependent on its binding partners (like Tsg6) (Asano et al., 2017; Deokbum et al., 2012; Köwitsch et al., 2 018). These data support our proposed model that HA is differentially modified by Tsg6 only on the right side, and produces opposing results across the LR axis. Thus, it is not only the presence of HA but rather the type of HA that is important to execute LR asymmetry.
Tsg6-HA pathway patterns the midgut vasculature
Vasculogenesis is among the most important processes in human biology and vascular anomalies are the leading cause of gestational failure but by their nature are very difficult to study. Here we have characterized LR vascular asymmetry as the defining feature of DM vessel patterning, which is achieved primarily via tightly controlled timing of endothelial cell migration (Bentley and Chakravartula, 2017). Whereas this process also involves spatial distribution of specific molecular cues (chemoattractant/repellant cues) within the local microenvironment, it is the temporal regulation of these cues that is responsible for establishing the architecture of organ-specific vascular patterns (Bentley and Chakravartula, 2017; Kim et al., 2011; Potente et al., 2011; Zambrano et al., 2016). In the DM, the emigration of endothelial cells from the right side and the subsequent remodeling of left-sided vascular cords pattern the DM arterial vasculature dependent on Cxcl12 expression (Mahadevan et al., 2014) in tight temporal coordination with the rapid morphogenesis of the developing midgut. Our current study shows that vascular exclusion on the right requires the anti-vascular activity of HC-HA, while depletion of HA alters Cxcl12 expression and causes persistence of the right-sided vasculature and subsequent defects in arterial patterning. This confirms that midgut vessel morphogenesis requires molecular asymmetry in the DM with the HA-rich ECM as a key spatiotemporal modulator, and highlights how carefully the tissue environment within the DM must be tuned for vasculogenesis that is both accurate and properly coordinated with the developing gut.
The dynamic nature of developmental vasculogenesis makes this paradigm difficult to approach in vivo. Moreover, the study of HA during this process adds additional challenge, given the nature of HA as a non-encoded carbohydrate with diverse isoforms and modifications in vivo. Thus, while the roles of HA in vessel development and disease have long been appreciated, the complex mechanisms of HA biology are mainly unexplored, principally for lack of a robust system to manipulate HA in vivo. Here we present the DM as a simple binary, planar structure that is readily amenable to the dissection of discrete HA mechanisms in vivo, across multiple species, and relevant to several HA-associated human pathologies.
Mechanisms downstream of HA during vascular gut patterning
HA has been known for decades to modulate the vascularity of developing tissues (Feinberg and Beebe, 1983). The specific mechanisms involved have not been clearly defined, owing to the wide diversity of contexts within which HA operates and their challenging access in vivo. Our finding that HA regulates Cxcl12 expression on the right side is intriguing, and may suggest HA serves as a molecular sink to modulate endothelial morphogens. Indeed, canonical (Tamura et al., 2011) and noncanonical (Tamura et al., 2011; Witze et al., 2008) Wnts, hypoxia-inducible factor-1 and −2 (Ceradini et al., 2004; Martin et al., 2010), and VegfA (Hong et al., 2006) are all known to regulate Cxcl12 expression, while our previous studies identified active VegfA and Wnt signaling both in the arterial endothelium of left DV cords, and in the surrounding mesenchyme (HH21) (Mahadevan et al., 2014; Welsh et al., 2013). Moreover, we have recently shown that targeted ectopic expression of Cxcl12 in the right DM induces VegfA expression (unpublished data), following existing literature that suggests VegfA expression exists in a positive feedback loop with Cxcl12 (Hong et al., 2006; Ping et al., 2011; van Weel et al., 2007; Zagzag et al., 2006). Thus, HA on the right may inhibit VEGF signaling either transcriptionally in a circuit with Cxcl12 modulation, or may act as an extracellular sink to sequester VEGF from its receptor. Future work is needed to determine the nuances of HA-dependent vascular behavior in the right DM and the specific effectors involved, questions the DM model is ideally suited to address.
Of note, our studies suggest that HA likely employs distinct downstream signaling mechanisms to drive ECM expansion and vascular exclusion during gut looping. Whereas Tsg6 misexpression on the left was sufficient to inhibit vasculature, it failed to induce expansion on the left. This argues additional factors are differentially expressed across the LR axis that may affect HA biological function, driving asymmetry of HA molecular structures in the DM. Tsg6 is a soluble chemokine-binding protein known for its ability to inhibit chemokine presentation on endothelial cells (Dyer et al., 2016; Dyer et al., 2014). This is mediated not only through a direct binding of Tsg6 with chemokines but also by the ability of Tsg6 to bind GAGs and compete for chemokine adsorption in these matrices (Dyer et al., 2016; Dyer et al., 2014). Thus, in addition to its catalysis on HA, Tsg6 may mediate loss of vasculature by interfering with the Cxcl12/Cxcr4 interaction, a possibility that should be pursued in future work.
Tsg6 and bikunin during intestinal malrotation and volvulus
Despite a large body of clinical and more recently molecular studies on correct and aberrant gut looping, the field lacks genetic mouse models to study the process of malrotation and its postnatal effects in living animals. The current mouse models that exhibit defects in gut looping chirality include Pitx2ΔASE/ΔASE (lack the asymmetric Pitx2c isoform expression) (Shiratori et al., 2006) and Nodalneo/neo (hypomorphic for Nodal) (Saijoh et al., 2003). However, the phenotypes of these mutant embryos are restricted to the aberrant looping of the duodenum (a foregut derivative) and most suffer from early embryonic lethality while others die immediately after birth (Saijoh et al., 2003; Shiratori et al., 2006). Hence, whether these malrotations predispose to volvulus during later development cannot be studied. In contrast, Tsg6 −/− and bikunin −/− mice survive to adulthood, albeit at non-Mendelian inheritance ratios, suggesting lethal or sub-lethal phenotypes associated with loss of Tsg6. Whether this is a consequence of HA-dependent or -independent Tsg6 function is not known, but it is fascinating to speculate that gut and/or vascular anomalies are the underlying cause of these phenotypes. Collectively, Tsg6 −/− and bikunin −/− mice may provide an important genetic model where the clinical consequences of early gut malrotation and volvulus can be followed throughout adult life.
Taken together, our studies unravel a key LR symmetry-breaking event in chickens and mice that triggers evolutionarily conserved midgut rotation and vasculogenesis. We identify a right side restricted and ECM-derived pathway during LR organogenesis initiated by HA and its enzymatic modification by Tsg6. Our work offers new insights into the mechanisms regulating LR asymmetry and HA function and addresses a critical area of vascular developmental biology and medicine, with potential relevance for other developmental defects and organogenesis paradigms.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Natasza. A. Kurpios (natasza.kurpios@cornell.edu)
Experimental Model and Subject Details
Animals
Chicken embryos and transgenic quail embryos:
Fertile chicken eggs (White Leghorn/ISA brown) obtained from the Sunrise Farms/Charles River/ Westwind farms, Interlaken, NY as well as the transgenic Tie1-H2b-YFP quail eggs obtained from Ozark egg company were incubated at 38°C, and staged accordingly (Hamburger and Hamilton, 1992). The embryos were harvested between HH15-HH27 (2.5–5.5 d.p.f). No discrimination was done between the sex of the embryos for the experiments conducted.
Mice and rats:
The Tsg6 mice were kindly provided by Dr. Hascall (BALB/cJ inbred background) (Fülöp et al., 2003) and were maintained in the pathogen free Cornell animal facility. The mutant embryos were obtained from a cross of Tsg6 (+/−) females to either Tsg6 (−/−) males or Tsg6 (+/−) males. WT BALB/cJ mice were purchased from Jackson Labs (Stock number 000651) and were maintained under similar conditions to the Tsg6 mice colony. Bikunin-null mouse embryos and corresponding WT embryos of the same background were harvested at the laboratory of Dr. Stavros Garantziotis (NIEHS) with appropriate permissions from Dr. Koji Kimata ‘s group where the mice were generated (Zhuo et al., 2001). Both male and female mice embryos were collected and used for experiments at the following stages: E10.5, E12.25, E12.5, E12.75, E13.5, and E18.5. No gender associations with the obtained phenotypes were observed. Timed pregnant Sprague Dawley rats were obtained from Charles River, and E11.5 embryos were harvested according to standard protocols provided by the Cornell rodent housing facility. All animal experiments adhered to guidelines of the Institutional Animal Care and Use Committee of Cornell University and were conducted within the scope of an approved animal protocol. Cornell University operates its animal care and use program under the Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare.
Methods Details
In ovo electroporation
Candidate genes were delivered to the developing DM using in ovo DNA electroporation as previously described (Welsh et al., 2013). Briefly, full-length genes or mutant constructs were delivered using the pCAGEN vector system (Matsuda and Cepko, 2004). A solution of plasmid DNA (encoding Hyal2, Tsg6, S28ATsg6, Cxcl12) was microinjected into the left or right coelomic cavity of HH14 chicken embryos (prior to the formation of the DM) and 3 pulses of 50 V, 10 mA were applied using a BTX electroporator. Eggs were then sealed and incubated until the desired stage. The pCAGEN vector was co-electroporated with pCAGEN-GFP for identification of target cells.
Morpholino design and electroporation
Tsg6 knockdown experiments were carried out by electroporation of targeted morpholinos (Gene Tools) targeting the right side of the DM. To target Tsg6, the following 5’ UTR targeting FITC-labeled translation-blocking morpholinos were designed: 5’-AGGGCAATCATCTCTTCCGAACTGT-3′ and 5’-CCTGTTAAAACACTCAGGGTAACCT-3’. In addition, the following splice block morpholino for targeting Tsg6 was designed: 5’-TTTGGGATTAAATTATGTCTTACCT-3’. To target Pitx2c isoform the following morpholinos were designed: 5’-GTCCTTCATGCAACTCATCGGCGAG-3’ As an experimental control, FITC-conjugated standard scrambled RNA-MO (Sc-MO) (Gene Tools) was used. The morpholinos were injected into the right coelomic cavity at HH14 and were electroporated at a concentration of 1.0 mM along with carrier DNA (pSlax) at a concentration of 1 µg/µl and 10 mM Tris (pH 8). The pulsing conditions were three 10 ms pulses of 50 V spaced out by a 100 ms time interval.
In ovo bead implantation
For inhibition of HA synthesis, vehicle (DMSO) or 40 mM solution of 4 Methyl umbelliferone-B-D-Xyloside (Sigma, #M7008) was adsorbed onto AG resin beads (Bio-Rad, #143–1255) overnight (ON) at 4°C, washed and inserted into the coelom with fine forceps (Dumont 55).
Histology
WT embryos were dissected in cold 1X PBS and fixed ON using Bouin’s fixative. The embryos were washed in 70% ethanol and embedded in paraffin. 6 µm tissue sections were collected on Superfrost Plus slides and stained with H&E. Color development was moderated by controlling the exposure in 1% HCl/70% EtOH and ammonium hydroxide solution. Images of the sections taken using a 40X plan/achromat objective and the Zeiss Observer Z1 microscope.
HA staining and immunohistochemistry
The distribution of HA was determined by incubating frozen or paraffin sections with biotinylated HA-binding protein (bHABP; 5 µg/ml; EM D Millipore, Cat No. 385911) in 3% Heat Inactivated Goat Serum in 1X PBS at 4°C ON, fo llowed by washing and incubation with AlexaFluor 568 conjugated streptavidin (Life Technologies #S11226) for 1 hour at room temperature (RT).
For immunohistochemistry, embryos were dissected in cold 1X PBS, fixed ON in 4% PFA/PBS and were washed through a graded series of 30% sucrose/PBS solutions prior to freezing in a 1:1 mixture of 30% sucrose/OCT. Sections of 8–30 µm were collected, dried ON, and stored at −80°C. Rehydrated cryosections of DM were then subjected to blocking with 3% Heat Inactivated Goat Serum for 1 hour at RT. Sections were then incubated with the following primary antibodies: rabbit polyclonal anti-GFP (Abcam, AB290, 1:1000), polyclonal rabbit anti human Tsg6 (RAH1 1:250, gift from Dr. Anthony Day), anti I-α-I antibody (Dako, A0301, 1:100) for ON at 4°C. Sections were incubated with appropriate secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 568 (Life Technologies) and DAPI (1:2000) for 1 hour at RT. Sections were imaged using Zeiss Observer Z1 with Apotome and/or Leica SP5 confocal microscope and images were subsequently processed using Imaris (Bitplane).
RNA in situ hybridization, cloning, plasmids and oligonucleotides
For RNA in situ hybridization (ISH) of chicken embryos, 400 µm embryo slices were collected with a McIlwain tissue chopper (Campden Instruments), fixed in 4% PFA/PBS ON, dehydrated, and stored in 100% methanol prior to processing. ISH on embryo slices, whole embryos or frozen cryosections was performed by following standard protocols as previously described (Welsh et al., 2013). Probes for RNA ISH were cloned using a TA cloning kit (Qiagen, #231124) or oligo-dT primed cDNA reverse transcription (Superscript III, Life Technologies, #4368814) using RNA pooled from HH19 and 21 whole chicken embryos. The following ESTs or cDNA clones were used to generate antisense riboprobes: chicken Tsg6 (NM_001037837.2 Fw: 5’ to 3’ GACTATGGGGATTCGCCTCA Rv: 5’ to 3’ CGTATGTCCCTGCCTGA TCT); chicken Cx40 (NM_205504.1, Fw: 5’ to 3’ TGTCGGTGTGATCTGTCTCC Rv: 5’ to 3’ CGTCTGGGAGGTTACAGTGG); chicken VE-cadherin (NM_204227.1 Fw: 5’ to 3’ CAGATAGGTATCAAGAGCCTGCC Rv: 5’ to 3’ CTCCTGCCACGAAA GTACTGTG). The RNA ISH in mouse embryos was carried out on 6µm paraffin sections using the RNAscope multiplex fluorescent kit along with the RNAscope mM–Pitx2 probe (Cat No. 412841). Human HYAL2 cDNA was amplified from pCMV6-HYAL2 (sc117754 OriGene) using the following primers: Fw: 5’ to 3’ AAAACTCGAGATGCGGGCAGGCCCA and Rv: 5’ to 3’ AAAAGCGGCCGCCTACAAGGTCCAGGTAAAGGCCAG (restriction enzyme sites are underlined). It was then subcloned into pCAGEN plasmid (Matsuda and Cepko, 2004) using XhoI and NotI sites. Chicken Tsg6 cDNA was synthesized and cloned into pCAGEN using the following primers: Fw: 5’ to 3’ AAAAGAATTCGGGAGTATT TACAGCCTAAC and Rv: 5’ to 3’ TTTTGCGGCCGCTATTCTGCTCACATAAT and cloned using EcoRI and NotI sites. The catalytically inactive Tsg6-S28A mutant construct (pcDNA5/FRT S28A mutant Tsg6) was a gift from Jan J. Enghild’s lab (Department of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark). The insert was a full length human Tsg6 whose serine 28 was mutated to an alanine. The original construct was cloned between KpnI (5’-end) and Xho1 (3’-end) in the pCDNA5/FRT vector system and was subsequently subcloned into the pCAGEN system via EcoRI digest followed by sticky end ligation. All DNA clones were verified by sequencing (Cornell University Life Sciences Core Laboratories Center).
Quantification and statistical analysis
Histology quantifications
To quantify cell-free spaces within the DM (acellular compartment) sections were processed using the ImageJ program for calculating the fraction of the acellular compartment. To do this, random regions within the left and right DM were selected and duplicated for processing (Figure S1A). The cell-free spaces were identified by negative thresholding using the ImageJ Thresholding Tool with the dark background option, segmented into smaller parts using the ImageJ Watershed Algorithm and the sum total of their areas were estimated using an Image J Particle Analyzer (Figure S1A). The percent ratio of the cell-free area to the total area was identified as the parameter that quantifies extent of DM expansion in the left and right DM. Student’s t-test was used to test the significance of the differences.
HA-fold enrichment quantifications
1µm interval z-stacks of 15µm thick HA stained sections were acquired using Zeiss Observer Z1 with Apotome and/or Leica SP5 confocal microscope. 3-D reconstruction of the z-stacks were obtained using the maximum intensity projections in Imaris and were subsequently used for analysis. To quantify the differences in the spatial distribution of HA fluorescence, rectangular sections of the whole DM across the left right axis were created and the intensity profiler of ImageJ was used to generate the average intensity profile across the left right axis. The area under the curve for the left and the right sides were used to calculate the relative fold enrichment of HA in the DM.
RNA in situ hybridization quantifications
Quantifications for the RNA in situ hybridization (ISH, Figure 8) were performed using ImageJ. The colorimetric images were inverted, and the integrated densities (IntDen) of the stained side of the DM as well as the unstained side of the DM was calculated using the analyze toolbar of ImageJ. The ratio of the integrated density of the DM stained side to the unstained side was then taken as a normalized parameter for comparison between the WT and the perturbed samples.
Midgut rotation quantifications
To quantify midgut rotation in mouse embryos we used the orientation of the cecum with respect to the cranial-caudal axis of the embryo as a marker of gut rotation. Quantification of cecum orientation (Figures 7B and 7C) was scored from frontal images of whole embryos of WT, Tsg6 −/− and bikunin −/− at E12.25, E12.5, and E12.75. Line vectors were drawn from the center of the cecum to intersect with the cranial-caudal axis drawn through the midline of the embryo (ImageJ). The vector coordinates were used to calculate the angle orientation of the cecum with respect to the cranial-caudal axis, and were plotted on a polar angle histogram using Matlab, (Mathworks). Vectors were grouped in 10° “bins” bas ed on angle of orientation across the cranial-caudal axis (caudal, 0°; cranial: 180° coun terclockwise). Cecum measurements to determine developmental stages as well as DJJ-ICJ lengths (Figure S8) were measured using the line tool and analyze toolbar of ImageJ.
Statistical analysis
Statistical analysis was performed using the unpaired two-tailed Student’s t-test (Microsoft Excel and Graphpad Prism software). A p value of 0.05 or lower was considered as significant. Data were presented as averages ± SEM (Figures 1D, 1E, 2B, 2D, 2F, 2G, 2H, 3G, 4E, 7C, 8C, 8D, 8E, S1B, S3B and S8A). For all assays, the number of samples (n) is indicated in figure legends.
Supplementary Material
Highlights.
Left-right asymmetry is initiated by the right side of the embryo
Extracellular hyaluronan determines gut and vascular laterality
Tsg6-catalyzed hyaluronan modification on the right is independent of Pitx2 on left
Tsg6 null mice fail to initiate gut rotation predisposing to volvulus
ACKNOWLEDGMENTS
We thank Drs. Aaron Petrey, Drew Noden, and David Gludish for reading the manuscript and excellent suggestions. We thank Drs. Marcos Simoes-Costa for expertise and training with in ovo morpholinos, Jan J. Enghild for providing the S28A construct, Anthony J. Day for RAH-1 antibodies, and Amitabha Bandyopadhyay for plasmids. We are grateful to Melissa Werner, Athina Angel, Brittany Laslow, Beth Mahler, Laura Miller, Brittany Scott, Ligon Perrow, Cindy Westmiller and the Cornell Imaging Core for technical assistance. This work was supported by March of Dimes 1-FY11-520 (N.A.K.), NIDDK R01 DK092776 (N.A.K.), NHLBI Program of Excellence in Glycosciences P01 HL107147 (V.C.H.) and the Division of Intramural Research, NIEHS, 1ZIAES102605 (S.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albeiroti S, Ayasoufi K, Hill DR, Shen B, and de la Motte CA (2015). Platelet hyaluronidase-2: an enzyme that translocates to the surface upon activation to function in extracellular matrix degradation. Blood 125(9), 1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegate KE (2009). Evidence-based diagnosis of malrotation and volvulus. Pediatr Radiol Suppl 2, S161–163. [DOI] [PubMed] [Google Scholar]
- Asano K, Nelson CM, Nandadasa S, Aramaki-Hattori N, Lindner DJ, Alban T, Inagaki J, Ohtsuki T, Oohashi T, Apte SS, et al. (2017). Stromal Versican Regulates Tumor Growth by Promoting Angiogenesis. Scientific Reports 7, 17225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie M (1788). An Account of a Remarkable Transposition of the Viscera. By Matthew Baillie, M. D. In a Letter to John Hunter, Esq. F. R. S. Philosophical Transactions of the Royal Society of London 78, 350–363. [Google Scholar]
- Bentley K, and Chakravartula S (2017). The temporal basis of angiogenesis. Philos Trans R Soc Lond B Biol Sci 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Kubalak S, Klewer SE, and McDonald JA (2000). Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. Journal of Clinical Investigation 106, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campione M, Steinbeisser H, Schweickert A, Deissler K, van Bebber F, Lowe LA, Nowotschin S, Viebahn C, Haffter P, Kuehn MR, et al. (1999). The homeobox gene Pitx2: mediator of asymmetric left-right signaling in vertebrate heart and gut looping. Development (Cambridge, England) 126, 1225–1234. [DOI] [PubMed] [Google Scholar]
- Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, and Gurtner GC (2004). Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10, 858–864. [DOI] [PubMed] [Google Scholar]
- Coulson-Thomas VJ, Lauer ME, Soleman S, Zhao C, Hascall VC, Day AJ, and Fawcett JW (2016). Tumor Necrosis Factor-stimulated Gene-6 (TSG-6) Is Constitutively Expressed in Adult Central Nervous System (CNS) and Associated with Astrocyte-mediated Glial Scar Formation following Spinal Cord Injury. The Journal of biological chemistry 291, 19939–19952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman MK, and Matsuoka S (2005). Experimental approaches to hyaluronan structure. Carbohydrate Research 340, 791–809. [DOI] [PubMed] [Google Scholar]
- Davis NM, Kurpios NA, Sun X, Gros J, Martin JF, and Tabin CJ (2008). The chirality of gut rotation derives from left-right asymmetric changes in the architecture of the dorsal mesentery. Developmental cell 15, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Motte CA, and Drazba JA (2011). Viewing Hyaluronan: Imaging Contributes to Imagining New Roles for This Amazing Matrix Polymer. Journal of Histochemistry and Cytochemistry 59, 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deokbum P, Youngmi K, Hyunah K, kyungjong K, Yun-Sil L, Jongseon C, Jang-Hee H, Hansoo L, Jongwook J, Chulhee C, et al. (2012). Hyaluronic Acid Promotes Angiogenesis by Inducing RHAMM-TGFβ Receptor Interaction via CD44-PKCδ. Mol Cells 33, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg RN, and Beebe DC (1983). Hyaluronate in vasculogenesis. Science (New York, NY 220, 1177–1179. [DOI] [PubMed] [Google Scholar]
- Filston HC, and Kirks DR (1981). Malrotation - the ubiquitous anomaly. J Pediatr Surg 16, 614–620. [DOI] [PubMed] [Google Scholar]
- Fujimoto T, Savani RC, Watari M, Day AJ, and Strauss JF 3rd (2002). Induction of the hyaluronic acid-binding protein, tumor necrosis factor-stimulated gene-6, in cervical smooth muscle cells by tumor necrosis factor-alpha and prostaglandin E(2). The American journal of pathology 160, 1495–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fülöp C, Salustri A, and Hascall VC (1997). Coding sequence of a hyaluronan synthase homologue expressed during expansion of the mouse cumulus-oocyte complex. Archives of Biochemistry and Biophysics 337, 261–266. [DOI] [PubMed] [Google Scholar]
- Fülöp C, Szántó S, Mukhopadhyay D, Bárdos T, Kamath RV, Rugg MS, Day AJ, Salustri A, Hascall VC, Glant TT, et al. (2003). Impaired cumulus mucification and female sterility in tumor necrosis factor-induced protein-6 deficient mice. Development 130, 2253–2261. [DOI] [PubMed] [Google Scholar]
- Hamati HF, Britton EL, and Carey DJ (1989). Inhibition of proteoglycan synthesis alters extracellular matrix deposition,proliferation, and cytoskeletal organization of rat aortic smooth muscle cells in culture. J Cell Biol 108(6), 2495–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, and Hamilton HL (1992). A series of normal stages in the development of the chick embryo. 1951. Dev Dyn 195, 231–272. [DOI] [PubMed] [Google Scholar]
- Harada H, and Takahashi M (2007). CD44-dependent Intracellular and Extracellular Catabolism of Hyaluronic Acid by Hyaluronidase-1 and −2. Journal of Biological Chemistry 282, 5597–5607. [DOI] [PubMed] [Google Scholar]
- Hascall V, and Esko JD (2009). Hyaluronan. In In Essentials of Glycobiology, C.R.D Varki A, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, ed. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; ), pp. 219–228. [PubMed] [Google Scholar]
- Hecksher-Sorensen J, Watson RP, Lettice LA, Serup P, Eley L, De Angelis C, Ahlgren U, and Hill RE (2004). The splanchnic mesodermal plate directs spleen and pancreatic laterality, and is regulated by Bapx1/Nkx3.2. Development (Cambridge, England) 131, 4665–4675. [DOI] [PubMed] [Google Scholar]
- Hong X, Jiang F, Kalkanis SN, Zhang ZG, Zhang XP, DeCarvalho AC, Katakowski M, Bobbitt K, Mikkelsen T, and Chopp M (2006). SDF-1 and CXCR4 are up-regulated by VEGF and contribute to glioma cell invasion. Cancer Lett 236, 39–45. [DOI] [PubMed] [Google Scholar]
- Jaime MC, Carol ST, and Garantziotis S (2015). Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int J Cell Biol 563818. [DOI] [PMC free article] [PubMed]
- Ji Y, Buel SM, and Amack JD (2016). Mutations in zebrafish pitx2 model congenital malformations in Axenfeld-Rieger syndrome but do not disrupt left-right placement of visceral organs. Developmental biology 416, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizaki I, Kojima K, Takagaki K, Endo M, Kannagi R, Ito M, Maruo Y, Sato H, Yasuda T, Mita S, et al. (2004). A Novel Mechanism for the Inhibition of Hyaluronan Biosynthesis by 4-Methylumbelliferone. The Journal of Biological Chemistry 279, 33281–33289. [DOI] [PubMed] [Google Scholar]
- Kim J, Oh WJ, Gaiano N, Yoshida Y, and Gu C (2011). Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev 25, 1399–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Miura H, Miyagawa-Tomita S, Yanazawa M, Katoh-Fukui Y, Suzuki R, Ohuchi H, Suehiro A, Motegi Y, Nakahara Y, et al. (1999). Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development (Cambridge, England) 126, 5749–5758. [DOI] [PubMed] [Google Scholar]
- Köwitsch A, Zhou G, and Groth T (2018). Medical application of glycosaminoglycans: a review. Journal of Tissue Engineering and Regenerative Medicine 12, e23–e41. [DOI] [PubMed] [Google Scholar]
- Kurpios NA, Ibañes M, Davis NM, Lui W, Katz T, Martin JF, Izpisúa Belmonte JC, and Tabin CJ (2008). The direction of gut looping is established by changes in the extracellular matrix and in cell:cell adhesion. PNAS 105, 8499–8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer JC (2017). intestinal Rotation Abnormalities and Midgut Volvulus. Surgical clinics of North America 97, 147–159. [DOI] [PubMed] [Google Scholar]
- Lauer ME, Cheng G, Swaidani S, Aronica MA, Weigel PH, and Hascall VC (2013a). Tumor necrosis factor-stimulated gene-6 (TSG-6) amplifies hyaluronan synthesis by airway smooth muscle cells. The Journal of biological chemistry 288, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer ME, Glant TT, Mikecz K, DeAngelis PL, Haller FM, Husni ME, Hascall VC, and Calabro A (2013b). Irreversible heavy chain transfer to hyaluronan oligosaccharides by tumor necrosis factor-stimulated gene-6. The Journal of biological chemistry 288, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer ME, Glant TT, Mikecz K, DeAngelis PL, Haller FM, Husni ME, Hascall VC, and Calabro A (2013c). Irreversible Heavy Chain Transfer to Hyaluronan Oligosaccharides by Tumor Necrosis Factor-stimulated Gene-6. J Biol Chem 288, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M (2005). Left-right asymmetry in embryonic development: a comprehensive review. Mech Dev 122, 3–25. [DOI] [PubMed] [Google Scholar]
- Levin M, Johnson RL, Stern CD, Kuehn M, and Tabin C (1995). A molecular pathway determining left-right asymmetry in chick embryogenesis. Cell 82, 803–814. [DOI] [PubMed] [Google Scholar]
- Lin CR, Kioussi C, O’Connell S, Briata P, Szeto D, Liu F, Izpisua-Belmonte JC, and Rosenfeld MG (1999). Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature 401, 279–282. [DOI] [PubMed] [Google Scholar]
- Liu C, Liu W, Palie J, Lu MF, Brown NA, and Martin JF (2002). Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development (Cambridge, England) 129, 5081–5091. [DOI] [PubMed] [Google Scholar]
- Logan M, Pagan-Westphal SM, Smith DM, Paganessi L, and Tabin CJ (1998). The transcription factor Pitx2 mediates situs-specific morphogenesis in response to left-right asymmetric signals. Cell 94, 307–317. [DOI] [PubMed] [Google Scholar]
- Lu MF, Pressman C, Dyer R, Johnson RL, and Martin JF (1999). Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature 401, 276–278. [DOI] [PubMed] [Google Scholar]
- Mahadevan A, Welsh IC, Sivakumar A, Gludish DW, Shilvock AR, Noden DM, Huss D, Lansford R, and Kurpios NA (2014). The left-right Pitx2 pathway drives organ-specific arterial and lymphatic development in the intestine. Developmental Cell 31, 690–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SK, Diamond P, Williams SA, To LB, Peet DJ, Fujii N, Gronthos S, Harris AL, and Zannettino AC (2010). Hypoxia-inducible factor-2 is a novel regulator of aberrant CXCL12 expression in multiple myeloma plasma cells. Haematologica 95, 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, and Cepko CL (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Prot Natl Acad Sci U S A 101, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner CM, and Day AJ (2003). TSG-6: a multifunctional protein associated with inflammation. J Cell Sci 116, 1863–1873. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Asari A, Rugg MS, Day AJ, and Fülöp C (2004). Specificity of the Tumor Necrosis Factor-induced Protein 6-mediated Heavy Chain Transfer from Inter alpha trypsin Inhibitor to Hyaluronan: Implications for the assembly of the cumulus extracellular matrix. J Biol Chem 279, 11119–11128. [DOI] [PubMed] [Google Scholar]
- Pansky B (1982). Review of Medical Embryology (McGraw-Hill; ). [Google Scholar]
- Pardanaud L, Yassine F, and Dieterlen-Lievre F (1989). Relationship between vasculogenesis, angiogenesis and haemopoiesis during avian ontogeny. Development 105, 473–485. [DOI] [PubMed] [Google Scholar]
- Pardue EL, Ibrahim S, and Ramamurthi A (2008). Role of hyaluronan in angiogenesis and its utility to angiogenic tissue engineering. Organogenesis 4, 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelayo JC, and Lo A (2016). Intestinal Rotation Anomalies. Pediatr Ann 45, e247–250. [DOI] [PubMed] [Google Scholar]
- Piedra ME, Icardo JM, Albajar M, Rodriguez-Rey JC, and Ros MA (1998). Pitx2 participates in the late phase of the pathway controlling left-right asymmetry. Cell 94, 319–324. [DOI] [PubMed] [Google Scholar]
- Ping YF, Yao XH, Jiang JY, Zhao LT, Yu SC, Jiang T, Lin MC, Chen JH, Wang B, Zhang R, et al. (2011). The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J Pathol 224, 344–354. [DOI] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, and Carmeliet P (2011). Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887. [DOI] [PubMed] [Google Scholar]
- Saijoh Y, Oki S, Ohishi S, and Hamada H (2003). Left-right patterning of the mouse lateral plate requires nodal produced in the node. Developmental biology 256, 160–172. [DOI] [PubMed] [Google Scholar]
- Salier JP, Rouet P, Raguenez G, and Daveau M (1996). The inter-alpha-inhibitor family: from structure to regulation. The Biochemical journal 315 (Pt 1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salustri A, Yanagishita M, and Hascall VC (1989). Synthesis and accumulation of hyaluronic acid and proteoglycans in the mouse cumulus cell-oocyte complex during follicle-stimulating hormone-induced mucification. The Journal of biological chemistry 264, 13840–13847. [PubMed] [Google Scholar]
- Sanggaard KW, Sonne-Schmidt CS, Krogager TP, Kristensen T, Wisniewski HG, Thøgersen IB, and Enghild JJ (2008). TSG-6 Tr ansfers Proteins between Glycosaminoglycans via a Ser28-mediated Covalent Catalytic Mechanism. Journal of Biological Chemistry 283, 33919–33926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Zhuo L, Okumura A, Ishikawa T, Miyachi M, Owa Y, Ishizawa T, Sugiura N, Nagata Y, Nonami T, et al. (2006a). The SHAP-hyaluronan complex in serum from patients with chronic liver diseases caused by hepatitis virus infection. Hepatology research : the official journal of the Japan Society of Hepatology 34, 178–186. [DOI] [PubMed] [Google Scholar]
- Shen L, Zhuo L, Okumura A, Ishikawa T, Miyachi M, Owa Y, Ishizawa T, Sugiura N, Nagata Y, Nonami T, et al. (2006b). The SHAP-hyaluronan complex in serum from patients with chronic liver diseases caused by hepatitis virus infection. Hepatol Res 34, 178–186. [DOI] [PubMed] [Google Scholar]
- Shiratori H, Yashiro K, Shen MM, and Hamada H (2006). Conserved regulation and role of Pitx2 in situs-specific morphogenesis of visceral organs. Development (Cambridge, England) 133, 3015–3025. [DOI] [PubMed] [Google Scholar]
- Simoes-Costa M, and Bronner ME (2016). Reprogramming of avian neural crest axial identity and cell fate. Science 352, 1570–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soffers JH, J.P. H, Mekonen HK, Koehler SE, and Lamers WH (2015). The growth pattern of the human intestine and its mesentery. BMC Dev Biol 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Amand TR, Ra J, Zhang Y, Hu Y, Baber SI, Qiu M, and Chen Y (1998). Cloning and expression pattern of chicken Pitx2: a new component in the SHH signaling pathway controlling embryonic heart looping. Biochemical and biophysical research communications 247, 100–105. [DOI] [PubMed] [Google Scholar]
- Tabin C (2005). Do we know anything about how left-right asymmetry is first established in the vertebrate embryo? J Mol Histol 36, 317–323. [DOI] [PubMed] [Google Scholar]
- Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, et al. (1998). The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393, 591–594. [DOI] [PubMed] [Google Scholar]
- Tamura M, Sato MM, and Nashimoto M (2011). Regulation of CXCL12 expression by canonical Wnt signaling in bone marrow stromal cells. Int J Biochem Cell Biol 43, 760–767. [DOI] [PubMed] [Google Scholar]
- Thomason RT, Bader DM, and Winters NI (2012). Comprehensive timeline of mesodermal development in the quail small intestine. Developmental Dynamics 241, 1678–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weel V, Seghers L, de Vries MR, Kuiper EJ, Schlingemann RO, Bajema IM, Lindeman JH, Delis-van Diemen PM, van Hinsbergh VW, van Bockel JH, et al. (2007). Expression of vascular endothelial growth factor, stromal cell-derived factor-1, and CXCR4 in human limb muscle with acute and chronic ischemia. Arterioscler Thromb Vasc Biol 27, 1426–1432. [DOI] [PubMed] [Google Scholar]
- Welsh IC, Thomsen M, Gludish DW, Alfonso-Parra C, Bai Y, Martin JF, and Kurpios NA (2013). Integration of L-R Pitx2 transcription and Wnt signaling drives asymmetric gut morphogenesis via Daam2. Developmental Cell 26, 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witze ES, Litman ES, Argast GM, Moon RT, and Ahn NG (2008). Wnt5a control of cell polarity and directional movement by polarized redistribution of adhesion receptors. Science 320, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Kirschmeier P, Hockenberry T, Yang TY, Brassard DL, Wang L, McClanahan T, Black S, Rizzi G, Musco ML, et al. (2002). Transcriptional regulation during p21WAF1/CIP1-induced apoptosis in human ovarian cancer cells. The Journal of biological chemistry 277, 36329–36337. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Meno C, Koshiba K, Sugihara M, Itoh H, Ishimaru Y, Inoue T, Ohuchi H, Semina EV, Murray JC, et al. (1998). Pitx2, a bicoid-type homeobox gene, is involved in a lefty-signaling pathway in determination of left-right asymmetry. Cell 94, 299–305. [DOI] [PubMed] [Google Scholar]
- Zagzag D, Lukyanov Y, Lan L, Ali MA, Esencay M, Mendez O, Yee H, Voura EB, and Newcomb EW (2006). Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: implications for angiogenesis and glioma cell invasion. Lab Invest 86, 1221–1232. [DOI] [PubMed] [Google Scholar]
- Zambrano S, De Toma I, Piffer A, Bianchi ME, and Agresti A (2016). NF-kappaB oscillations translate into functionally related patterns of gene expression. Elife 5, e09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Yoneda M, Zhao M, Yingsung W, Yoshida N, Kitagawa Y, Kawamura K, Suzuki T, and Kimata K (2001). Defect in SHAP-hyaluronan complex causes severe female infertility. A study by inactivation of the bikunin gene in mice. The Journal of biological chemistry 276, 7693–7696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.