

Abstract
Primary aldosteronism (PA) affects ~5–10% of hypertensive patients and has unilateral and bilateral forms. Most unilateral PA is caused by computed tomography (CT)-detectable aldosterone-producing adenomas (APA), which express CYP11B2 (aldosterone synthase) and frequently harbor somatic mutations in aldosterone-regulating genes. The etiology of the most common bilateral form of PA, idiopathic hyperaldosteronism (IHA), is believed to be diffuse hyperplasia of aldosterone-producing cells within the adrenal cortex. Herein, a multi-institution cohort of fifteen IHA adrenals were examined with CYP11B2 immunohistochemistry and next generation sequencing (NGS). CYP11B2 immunoreactivity in adrenal glomerulosa harboring non-nodular hyperplasia was only observed in 4/15 IHA adrenals suggesting that hyperplasia of CYP11B2 expressing cells may not be the major cause of IHA. However, the adrenal cortex of all IHA adrenals harbored at least one CYP11B2-positive aldosterone-producing cell cluster (APCC) or a micro-APA. The number of APCCs per case (and individual APCC area) in IHA adrenals was significantly larger than in normotensive controls. NGS of DNA from 99 IHA APCCs demonstrated somatic mutations in genes encoding the L-type calcium voltage-gated channel subunit alpha 1-D (CACNA1D, n=57; 58%) and potassium voltage-gated channel subfamily J-5 (KCNJ5, n=1; 1%). These data suggest that IHA may result from not only hyperplasia, but also the accumulation or enlargement of CT-undetectable APCC harboring somatic aldosterone-driver gene mutations. The high prevalence of mutations in the CACNA1D L-type calcium channel provides a potential actionable therapeutic target that could complement mineralocorticoid blockade and inhibit aldosterone overproduction in some IHA patients.
Keywords: Aldosterone, Aldosteronism, Hypertension, Adrenal Cortex, Calcium Channels
Introduction
Hypertension affects about 1 in 3 adults1 and is a major risk factor for stroke and heart disease, contributing to 1,000 deaths per day2, which is comparable to that of malignant diseases3. Primary aldosteronism (PA) affects ~5–10% of all hypertensive patients4, 5. Appropriate therapy of PA is important as PA patients are at greater risk of stroke, coronary artery disease, atrial fibrillation and heart failure6.
PA is classified into two subtypes based on laterality assessed by adrenal vein sampling4, 7 (Figure 1). Unilateral PA is predominantly caused by aldosterone-producing adenomas (APA) that are detectable by computed tomography (CT) and account for approximately 30% of PA5. A small percentage of unilateral PA results from CT-undetected aldosterone-producing areas, termed multiple unilateral adrenocortical micronodules (UMN) or microAPA8, 9. Similar to APA, such micronodules exhibit high CYP11B2 expression (aldosterone synthase, the enzyme responsible for the final step of aldosterone biosynthesis).
Figure 1. Classification of PA.
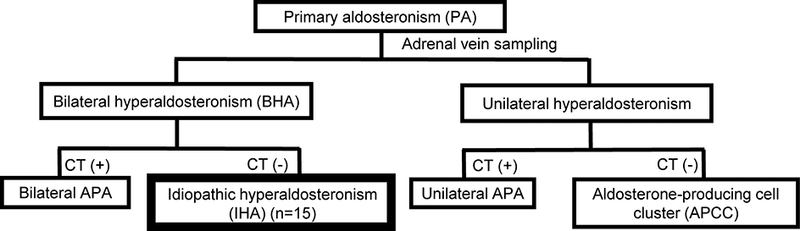
Primary aldosteronism (PA) is categorized into bilateral and unilateral forms based on the source of aldosterone production evaluated by adrenal vein sampling. Computed tomography (CT)-detectable PA is predominantly caused by aldosterone-producing adenoma (APA), whereas CT-negative unilateral PA is caused by CYP11B2 (aldosterone synthase) expressing aldosterone-producing cell clusters (APCCs). CT-negative bilateral PA, the most common subtype of PA (70%), is referred to as idiopathic hyperaldosteronism (IHA). Herein, examining a rare collection of IHA adrenals and combining histology, immunohistochemistry and next generation sequencing, we defined the role of somatic mutations in IHA.
The advent of next generation sequencing (NGS) has led to the identification of recurrent somatic mutations in APA. Approximately half of APA harbor somatic mutations in KCNJ5, which encodes potassium inwardly rectifying channel, subfamily J, member 5, while an additional 10–15% of APA harbor somatic mutations in CACNA1D (encoding calcium channel, voltage-dependent, L-type, α1D-subunit), ATP1A1 (encoding ATPase, Na/K transporting,1-polypeptide (A)), or ATP2B3 (encoding ATPase, Ca2-transporting, plasma membrane 3), respectively10–20. In vitro studies have shown that these mutations increase CYP11B2 expression and aldosterone production through increased intracellular calcium levels10–13. CACNA1D encodes Cav 1.3, a subunit of L-type calcium channels that normally mediates calcium ion entry into various cell types, including adrenocortical cells21. Cav 1.3 contains four repeated domains, each of which consists of six transmembrane segments (S1-S6). The Cav 1.3 voltage sensor (S4), the cytoplasmic S4-S5 linker coupling the voltage-sensing domain to the pore, and the channel activation gate (S6) have all been reported to harbor activating mutations in APA11.
A recently developed monoclonal antibody for CYP11B222 enabled multiple research groups to identify small CYP11B2 positive cell clusters—termed aldosterone-producing cell clusters (APCC)—beneath the adrenal capsule23–25. Without CYP11B2 immunohistochemistry, these cells appear histologically identical to the adjacent zona glomerulosa (ZG) cells24. We have further demonstrated the presence and age-dependent accumulation of APCC in adrenals from American kidney donors25, 26 and normotensive Japanese autopsy adrenals27. Hypothesizing that APCC have similar mutations to that seen in APA, we performed NGS targeting the complete coding sequence of aldosterone-regulating genes that are commonly mutated in APA. We identified mutations in CACNA1D (preferentially affecting the same residues as in APA) in ~35% of APCC25, 27, suggesting autonomous aldosterone production in APCC similar to that proposed for APA. We further showed that the morphology and mutation spectrum of CYP11B2-expressing micronodules in CT-negative unilateral PA were essentially identical to APCC9. Of note, the number of APCC was significantly higher in CT-negative unilateral PA compared to that of normotensive adrenals27, supporting APCC as source of aldosterone excess in unilateral PA without CT- or grossly-identifiable tumors.
Bilateral hyperaldosteronism (BHA) most often presents without detectable masses by CT and is called idiopathic hyperaldosteronism (IHA), but can also be caused by bilateral APA28, 29. Here, we hypothesized that IHA aldosterone excess results from increased APCCs compared to normal adrenals9. To test this hypothesis, we performed CYP11B2 immunohistochemistry in a unique cohort of adrenals from patients with IHA who underwent adrenalectomy. CYP11B2 immunohistochemistry revealed that while diffuse CYP11B2 expressing hyperplasia was infrequent in our cohort, APCCs were more numerous (and larger) than observed in control adrenals. Furthermore, targeted NGS demonstrated that all IHA adrenals harbored at least one APCC or a micro-APA with a somatic mutation in a gene known to dysregulate aldosterone production (most frequently CACNA1D). Together these findings indicate that somatic mutation and APCC accumulation/enlargement may play a role in IHA.
Methods
Most of the data that support the findings of this study are available within the article and in the online-only Data Supplement. The remaining supporting data are available from the corresponding author on reasonable request.
Human IHA adrenal glands
We retrospectively reviewed cases with IHA from two institutes (Tohoku University, Sendai, Japan, and University of Queensland, Brisbane, Australia), and collected 10% formalin-fixed paraffin-embedded (FFPE) tissue blocks of resected adrenal glands that met all of the following inclusion criteria of IHA clinical diagnosis per guidelines30, 31: 1) patients who were diagnosed with sporadic (non-familial) PA and without cortisol co-secretion, 2) patients who were proven to have bilateral hypersecretion of aldosterone by adrenal vein sampling (AVS), 3) patients who did not have adrenal masses by computed tomography (CT), 4) patients who underwent unilateral adrenalectomy.
AVS methods are described in the Supplemental Methods. In all cases, the adrenal gland corresponding to the side with a highest aldosterone secretion was resected. Results from this IHA cohort were compared to previously described cohorts of 53 age-matched normotensive adrenals (29 males, 24 females)27 and 7 adrenals from CT-negative unilateral PA (unilateral APCC; 3 males, 4 females)9 evaluated similarly.
Patients with IHA are typically treated medically, most commonly with mineralocorticoid receptor antagonists. Surgery is only offered in exceptional situations, to provide individualized therapy in unique circumstances, such as difficult to control hypertension or hypokalemia, and in rare instances of intolerance to medical therapy. Our cohort consisted of fifteen such cases that were identified over a 30 year period in two very active PA referral centers; the circumstances for surgical consideration are provided in Table S1.
The research protocol was approved by the local ethics committee/institutional review board at Tohoku University, Sendai, Japan (Approval number: 2016–1-693), University of Queensland, Brisbane, Australia (Approval number: HREC/13/QPAH232), and University of Michigan, Ann Arbor, USA (Approval number: HUM00042749, HUM00106809). All participants provided written informed consent.
Adrenal sectioning, immunohistochemistry and next generation sequencing (NGS)
Adrenal specimens were fixed in 10% formalin and embedded in paraffin after surgery. FFPE blocks from one adrenal were selected for evaluation based on visual inspection of the block (largest amount of cortical tissue if more than one block was prepared) and availability. Selected blocks were serially sectioned and stained with hematoxylin and eosin (H&E) and CYP11B2 (Millipore, MABS1251) to detect and quantify APCC as previously described27. To enable comparison across the cohort (accounting for differences in section size), an APCC score was calculated as APCC number per section divided by the cortex area (cm2). Six micro-APA were observed and included with APCC for data analysis. While being CYP11B2-postive like APCC, these structures had histology that was consistent with an adenoma including nuclear atypia, a fibrous capsule, well circumscribed margins and disruption of adrenal zonation that was visible by H&E and CYP11B2 immunostaining, as previously described9, 32, 33. Targeted NGS (using a panel targeting the complete coding sequence of genes recurrently mutated in APA) was performed as previously described27. Detailed methods of sectioning, immunostaining and NGS are described in Supplemental Methods.
Statistics
The D’Agostino-Pearson method was used for normality test in each variable. Gaussian variables were reported as mean ±SD and non-Gaussian variables were described as median [IQR: interquartile range]. Two-sided Spearman rank test as used for correlation between the APCC mutation rate and plasma aldosterone levels. Two-sided Mann-Whitney test was used to compare APCC score and APCC area between IHA, CT-negative unilateral PA and age-matched normotensive adrenals. P < 0.05 was considered a significant difference. PRISM (version 7, GraphPad Software, Inc.) software was used for all statistical calculations.
Results
APCC Number and Area in IHA Adrenals
We collected 15 IHA cases from patients (10 men and 5 women) who underwent unilateral adrenalectomy with available surplus clinical FFPE tissue (8 from Tohoku University, 7 from University of Queensland) (Figure 1, Table S1). Clinical information for these cases is described in the Supplemental Results.
By histology, diffuse ZG hyperplasia and diffuse nodular hyperplasia of adrenocortical cells were observed in 13 (87%) and 9 (60%) of the 15 IHA cases, respectively. However, only 4/15 (27%) adrenals had diffuse positive CYP11B2 cells in either the ZG or ZF, indicating that the histologically observed hyperplastic adrenal cells were not aldosterone-producing cells (Table 1, Figure 2, C, D).
Table 1.
Somatic mutations detected in APCC from IHA adrenals.
| Sequenced IHA case (n=14) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | ||
| Mutated APML (n=58) | 6 | 6 | 4 | 7 | 4 | 3 | 4 | 2 | 9 | 2 | 4 | 3 | 3 | 1 | ||
| Sequenced APML (n=99) | 13 | 9 | 9 | 9 | 6 | 5 | 4 | 2 | 10 | 16 | 6 | 5 | 4 | 1 | ||
| Gene | Mutation | Frequency | ||||||||||||||
| CACNA1D | F747V* | 9 | 2 | 2 | 2 | |||||||||||
| G403R** | 8 | 2 | ||||||||||||||
| R990H** | 6 | 2 | 2 | |||||||||||||
| F747L** | 5 | 2 | ||||||||||||||
| F1248L | 3 | |||||||||||||||
| V1338M* | 3 | |||||||||||||||
| V401L* | 2 | |||||||||||||||
| S652L* | 2 | |||||||||||||||
| A998V* | 2 | |||||||||||||||
| E124K | 1 | |||||||||||||||
| L248F | 1 | |||||||||||||||
| V259G | 1 | |||||||||||||||
| L272R | 1 | |||||||||||||||
| G323R | 1 | |||||||||||||||
| S410L | 1 | |||||||||||||||
| L653P | 1 | |||||||||||||||
| S724L | 1 | |||||||||||||||
| L748S | 1 | |||||||||||||||
| 755_757del | 1 | |||||||||||||||
| S969L | 1 | |||||||||||||||
| A1011T | 1 | |||||||||||||||
| I1015V | 1 | |||||||||||||||
| F1147L | 1 | |||||||||||||||
| R1183H | 1 | |||||||||||||||
| D1273N | 1 | |||||||||||||||
| T1835I | 1 | |||||||||||||||
| KCNJ5 | G151R** | 1 | ||||||||||||||
Somatic mutations detected in IHA adrenals are shown and ordered by mutation frequency. The number in a box indicates the case had two APCC with that mutation. Frequency indicates the total number of APCC with that mutation in the whole cohort. Of 99 APCC sequenced in the cohort, 58% (57/99) harbored somatic mutations in CACNA1D, and 1% (1/99) in KCNJ5.
For each mutated residue observed herein, those previously reported as somatically mutated in sporadic APA are indicated by
and those reported as somatically mutated in APA and functionally characterized are indicated by
Figure 2. Representative images of APCC in IHA adrenals.
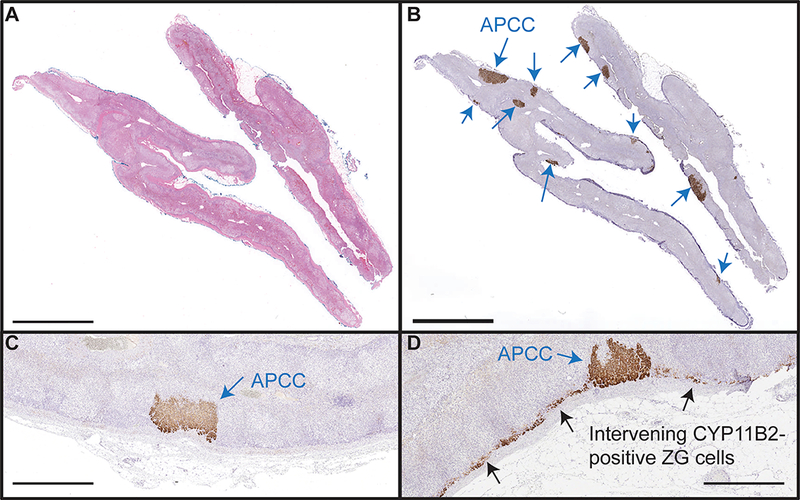
Representative images of hematoxylin and eosin (H&E, A) and CYP11B2 immunohistochemistry (B) are shown. All IHA cases harbored at least one APCC (blue arrows), supporting APCC as the common cause of IHA. Of the 15 adrenals studied, intervening zona glomerulosa (ZG) cells were negative (C) for CYP11B2 in 11/15 cases. Intervening ZG was diffusely positive (D, black arrow) for CYP11B2 only in 4/15 cases, arguing against diffuse aldosterone overproduction as the most common cause of IHA. Scale bars in A and B are 5mm, and in panels C and D are 1mm.
In contrast, all IHA adrenals harbored at least one CYP11B2-postive APCC, with a total of 104 detected APCC (6.9 ± 4.1 APCC per IHA case) (Figure 2, A, B, Table S1). Of the 104 APCC, 6 fulfilled the criteria of micro-APA as described in the Methods and previously9 (Table S2). Spironolactone bodies were only detected in APCC but not in the intervening CYP11B2 positive/negative ZG cells.
The median APCC score (APCC number/adrenal cortex area, 6.1 vs 0 /cm2, p<0.0001) and APCC area (0.25 vs 0.16 mm2, p=0.0019) were significantly greater in IHA adrenals vs. a similarly evaluated cohort of normotensive adrenals (Figure 3). In contrast, the median APCC score (p=0.63) and APCC area (p=0.07) in IHA adrenals were not significantly different vs. adrenals from CT-negative unilateral PA (Figure 3).
Figure 3. APCC score and area in IHA adrenals.
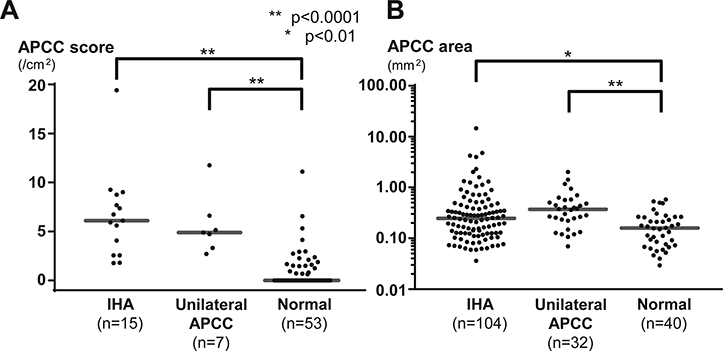
The median APCC (aldosterone-producing cell cluster) score (A) (6.1 vs 0 /cm2, p<0.0001, two-sided Mann-Whitney test) and the median APCC area (B) (0.25 vs 0.16 mm2, p<0.01, two-sided Mann-Whitney test) were significantly increased in IHA adrenals vs. previously described age-matched normotensive adrenals. IHA values were not significantly different vs. previously described CT-negative unilateral PA adrenals (unilateral APCC). The APCC score was defined as the number of APCC per section divided by adrenal cortex area to normalize section differences. APCC score (in log scale) is plotted for all samples according to the adrenal type (B).
NGS on APCC in IHA
To determine the somatic mutation spectrum of aldosterone-driving genes in IHA adrenals, we performed targeted NGS using a stringent two-panel approach on 99 APCC DNA samples (5/104 samples that did not pass DNA QC metrics were excluded). DNA from intervening CYP11B2 positive and negative ZG regions (two regions from each case) were also analyzed by NGS, with no somatic mutations detected in intervening ZG regions. In contrast, 14/14 (100%) sequenced cases had at least one APCC harboring a known somatic mutation in an aldosterone-driving gene. Specifically, 58% (57/99) and 1% (1/99) of APCC had somatic mutations in CACNA1D and KCNJ5, respectively (Table 1, Table S2). Importantly, the APCC mutation rate per case (number of APCC with mutations / total APCC number) was significantly positively correlated with preoperative plasma aldosterone concentration (PAC) (r=0.54, p=0.049). No significant differences in age and mutation frequency were observed between samples from the two institutes (Supplemental Results).
Of the 58 APCC with a somatic mutation, 38 (66%) harbored mutations previously reported in APA34 (Table 1). In total, 77% (44/57) of the CACNA1D mutations occurred at domains recurrently mutated in APA (9 [16%] in the voltage sensor S4, 9 [16%] in the cytoplasmic S4-S5 linker, and 26 [46%] in the channel S6 activation gate (Figure 4).
Figure 4. Somatic CACNA1D mutations found in IHA adrenals.
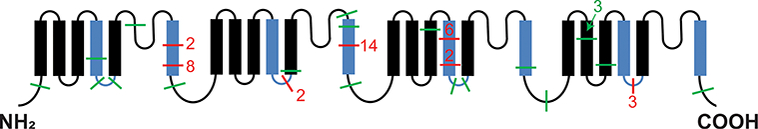
The α1 subunit of CACNA1D is shown with domains recurrently mutated in APA indicated in blue. Each line represents the location of a somatic mutation(s) detected in an APCC from an IHA case (the adjacent number indicates total number at that position in this cohort). Previously reported somatic mutations in APA are shown in red, and unreported mutations in green. In total, 77% (44/57) of the CACNA1D mutations were in the domains recurrently mutated in APA.
Discussion
While IHA was labeled as “idiopathic” in the 1960s35, the cause of hyperaldosteronism in these cases remains largely unknown, with diffuse hyperplasia of adrenocortical cells traditionally considered the driver of excess aldosterone. While diffuse hyperplasia of ZG cells in IHA adrenals has been reported often32, 33, 36–40, others reports described diffuse hyperplasia of multiple cortex zones concomitant with adrenocortical nodules35, 36, 39, 41–44. Such findings have been referred to as primary cortical hyperplasia (morphologically increased number of ZG and/or zona fasciculata cells) by some investigators. Importantly, these histologic evaluations were performed on limited series without the ability to assess for in situ aldosterone secretory capacity (enabled recently by CYP11B2 IHC)36, 38–40, 43, 45–47.
Through a unique cohort of rarely resected adrenals from IHA patients, we demonstrated that most cases did not exhibit hyperplastic adrenocortical cells expressing CYP11B2, inconsistent with the concept of diffuse hyperplasia of aldosterone-producing cells causing IHA. Instead, by CYP11B2 IHC paired with targeted NGS, we observed that all IHA adrenals had small CT-undetectable CYP11B2 positive cell areas (APCCs), which were significantly increased in number and size compared to normal adrenals. In addition, APCC in the IHA adrenals frequently harbored aldosterone-driving somatic mutations, nearly exclusively in CACNA1D, consistent with the somatic mutation spectrum of APCC observed in normal adrenals27 and CT-negative unilateral PA adrenals9. This mutation spectrum is markedly different in APA, where mutations are much more frequent in KCNJ5 vs. CACNA1D13–20. Importantly, APCC score, area and mutation spectrum in IHA were essentially identical to those we previously described in CT-negative unilateral PA9, supporting APCC as a common cause of both unilateral and bilateral forms of CT-negative PA. Taken together, our findings support recurrent somatic CACNA1D mutations in APCC as the main cause of autonomous aldosterone production in IHA.
A crucial and necessary pharmacotherapy for IHA is mineralocorticoid receptor (MR) blockade. Although MR blockade is the only currently available specific treatment for IHA, in some cases these drugs provide insufficient blood pressure control and side effects48–51. In addition, alternative antihypertensive drugs, including calcium channel blockers (CCB), have not been extensively evaluated in IHA30. Thus, understanding the pathobiology of IHA to develop additional targeted therapeutic approaches, when MR blockade is not sufficient, could be beneficial. Intriguingly, CACNA1D, which was the most frequently mutated gene in IHA, encodes a L-type calcium channel that is inhibited by FDA approved CCB52. In addition, a recent in vitro study showed that aldosterone levels were significantly decreased by nifedipine in human adrenocortical cells transfected with mutated CACNA1D53. These results support CACNA1D mutation-stimulated aldosterone production in APCC as a potential therapeutic target that may directly inhibit the mechanism of increased aldosterone production in CT-negative PA. The positive correlation between APCC CACNA1D mutation rate and preoperative aldosterone levels suggests that CCB targeted therapy may be more effective for IHA patients with higher aldosterone secretion, who are most likely to need antihypertensive drugs beyond MR blockers.
Limitations of the present study includes the small numbers of the IHA adrenals available and the unique nature of the patients requiring surgery for bilateral PA. Therefore the results shown in the present study need to be confirmed in a larger cohort, potentially in a multicenter study using adrenals from well characterized CT-negative unilateral patients that continue to have PA following surgery. The specific patients used in this study may also impact the findings, as many were severe IHA cases that required additional pharmacotherapy to complement the MR-blockade approach and were often refractory to all therapies. The current results, however, remain foundational by demonstrating that IHA adrenals have gene mutations and a buildup of APCC. In addition, the study provides the methods to localize aldosterone-producing cells, isolate DNA and define mutations using FFPE tissues, providing a platform for others to use archival surgical IHA adrenals and expand such studies.
In conclusion, our results support increased autonomous aldosterone production driven by somatic mutations in aldosterone-regulating genes (nearly exclusively CACNA1D) and APCC accumulation/enlargement as an underlying cellular and molecular cause of IHA. Coupled with previous studies in APA and CT-negative unilateral PA, our findings in IHA suggest that the majority of PA results from somatic mutations in aldosterone-regulating genes that influence intracellular calcium homeostasis. Importantly, as opposed to APA, CT-negative PA is primarily associated with mutations in CACNA1D. The availability of widely used anti-hypertensive CCBs supports investigations into the efficacy of these drugs to inhibit aldosterone production in IHA.
Perspective
Worldwide, more than 100 million patients suffer from idiopathic hyperaldosteronism (IHA), yet its underlying cause remain unknown. In this manuscript, we show that IHA adrenals have expansion/enlargement of aldosterone-producing cells (aldosterone-producing cell clusters [APCCs]) with somatic gene mutations in the L-type calcium channel, CACNA1D. Importantly, these genetic findings provide a potential cause for the most common subtype of primary aldosteronism as well as the potential for rapid translation to clinical applications. Combined with previous studies of aldosterone-producing adenomas, this study supports somatic mutation-harboring adrenal cell lesions as the underlying cause of the vast majority of primary aldosteronism.
Supplementary Material
Novelty and Significance
What Is New?
This study demonstrates that aldosterone-producing cell clusters (APCCs) are increased/enlarged in idiopathic hyperaldosteronism (IHA) adrenals.
This study is also the first to demonstrate aldosterone-driver somatic gene mutations (nearly exclusively in CACNA1D) in IHA adrenals.
What Is Relevant?
The presence of CACNA1D aldosterone-driving somatic mutations in IHA adrenals provides a novel therapeutic target for inhibiting aldosterone overproduction that could complement current MR-based treatment approaches.
Summary:
The etiology of idiopathic hyperaldosteronism (IHA), the most common cause of endocrine hypertension, has long been believed to result from diffuse hyperplasia of aldosterone-producing cells within the adrenal cortex. Here, we demonstrate that IHA results, not only from diffuse hyperplasia, but also from somatic gene mutations that increase aldosterone production in discrete foci of aldosterone-synthase expressing adrenocortical cells.
Acknowledgments
We thank Yasuko Tsukada (Tohoku University) for her management of clinical database, and Kazue Ise and Kumi Kikuchi (Tohoku University) for their work on slide preparation.
Sources of Funding
This work was supported by a grant from American Heart Association (17POST33410759) to K.O. and grants from the NIDDK (R01 DK106618 and R01 DK043140) to S. A. T. and W. E. R.
Footnotes
Conflicts of Interest/Disclosures Statement
S. A. T. is supported as the A. Alfred Taubman Emerging Scholar by the A. Alfred Taubman Medical Research Institute. S. A. T. has received travel support from Thermo Fisher Scientific and had a separate sponsored research agreement with Thermo Fisher Scientific. None of the study described herein was supported by Thermo Fisher Scientific and they had no role in the data collection, interpretation, or analysis, and did not participate in the study design or the decision to submit for publication. The remaining authors have declared that no conflict of interest exists.
References
- 1.Merai R, Siegel C, Rakotz M, Basch P, Wright J, Wong B, Dhsc, Thorpe P. Cdc grand rounds: A public health approach to detect and control hypertension. MMWR Morb. Mortal. Wkly. Rep 2016;65:1261–1264 [DOI] [PubMed] [Google Scholar]
- 2.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C, Stroke Statistics S. Heart disease and stroke statistics−−2015 update: A report from the american heart association. Circulation. 2015;131:e29–322 [DOI] [PubMed] [Google Scholar]
- 3.Kochanek KD, Xu J, Murphy SL, Minino AM, Kung HC. Deaths: Final data for 2009. Natl. Vital Stat. Rep 2011;60:1–116 [PubMed] [Google Scholar]
- 4.Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, Ganzaroli C, Giacchetti G, Letizia C, Maccario M, Mallamaci F, Mannelli M, Mattarello MJ, Moretti A, Palumbo G, Parenti G, Porteri E, Semplicini A, Rizzoni D, Rossi E, Boscaro M, Pessina AC, Mantero F, Investigators PS. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J. Am. Coll. Cardiol 2006;48:2293–2300 [DOI] [PubMed] [Google Scholar]
- 5.Mulatero P, Stowasser M, Loh KC, Fardella CE, Gordon RD, Mosso L, Gomez-Sanchez CE, Veglio F, Young WF Jr. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J. Clin. Endocrinol. Metab. 2004;89:1045–1050 [DOI] [PubMed] [Google Scholar]
- 6.Monticone S, D’Ascenzo F, Moretti C, Williams TA, Veglio F, Gaita F, Mulatero P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018;6:41–50 [DOI] [PubMed] [Google Scholar]
- 7.Nanba K, Tsuiki M, Sawai K, Mukai K, Nishimoto K, Usui T, Tagami T, Okuno H, Yamamoto T, Shimatsu A, Katabami T, Okumura A, Kawa G, Tanabe A, Naruse M. Histopathological diagnosis of primary aldosteronism using cyp11b2 immunohistochemistry. J. Clin. Endocrinol. Metab 2013;98:1567–1574 [DOI] [PubMed] [Google Scholar]
- 8.Omura M, Sasano H, Fujiwara T, Yamaguchi K, Nishikawa T. Unique cases of unilateral hyperaldosteronemia due to multiple adrenocortical micronodules, which can only be detected by selective adrenal venous sampling. Metabolism. 2002;51:350–355 [DOI] [PubMed] [Google Scholar]
- 9.Yamazaki Y, Nakamura Y, Omata K, Ise K, Tezuka Y, Ono Y, Morimoto R, Nozawa Y, Gomez-Sanchez CE, Tomlins SA, Rainey WE, Ito S, Satoh F, Sasano H. Histopathological classification of cross-sectional image-negative hyperaldosteronism. J. Clin. Endocrinol. Metab 2017;102:1182–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Akerstrom G, Wang W, Carling T, Lifton RP. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W, Shaikh LH, Brighton CA, Teo AE, Davenport AP, Dekkers T, Tops B, Kusters B, Ceral J, Yeo GS, Neogi SG, McFarlane I, Rosenfeld N, Marass F, Hadfield J, Margas W, Chaggar K, Solar M, Deinum J, Dolphin AC, Farooqi IS, Striessnig J, Nissen P, Brown MJ. Somatic mutations in atp1a1 and cacna1d underlie a common subtype of adrenal hypertension. Nat. Genet 2013;45:1055–1060 [DOI] [PubMed] [Google Scholar]
- 12.Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, Walther A, Tauber P, Schwarzmayr T, Diener S, Graf E, Allolio B, Samson-Couterie B, Benecke A, Quinkler M, Fallo F, Plouin PF, Mantero F, Meitinger T, Mulatero P, Jeunemaitre X, Warth R, Vilsen B, Zennaro MC, Strom TM, Reincke M. Somatic mutations in atp1a1 and atp2b3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet 2013;45:440–444, 444e441–442 [DOI] [PubMed] [Google Scholar]
- 13.Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson-Williams C, Libutti SK, Mane S, Hellman P, Westin G, Akerstrom G, Bjorklund P, Carling T, Fahlke C, Hidalgo P, Lifton RP. Somatic and germline cacna1d calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet 2013;45:1050–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, Strom TM, Monticone S, Amar L, Meatchi T, Mantero F, Cicala MV, Quinkler M, Fallo F, Allolio B, Bernini G, Maccario M, Giacchetti G, Jeunemaitre X, Mulatero P, Reincke M, Zennaro MC. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension. 2014;64:354–361 [DOI] [PubMed] [Google Scholar]
- 15.Zheng FF, Zhu LM, Nie AF, Li XY, Lin JR, Zhang K, Chen J, Zhou WL, Shen ZJ, Zhu YC, Wang JG, Zhu DL, Gao PJ. Clinical characteristics of somatic mutations in chinese patients with aldosterone-producing adenoma. Hypertension. 2015;65:622–628 [DOI] [PubMed] [Google Scholar]
- 16.Wu VC, Huang KH, Peng KY, Tsai YC, Wu CH, Wang SM, Yang SY, Lin LY, Chang CC, Lin YH, Lin SL, Chu TS, Wu KD. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-taiwanese population. Sci. Rep 2015;5:11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitamoto T, Suematsu S, Matsuzawa Y, Saito J, Omura M, Nishikawa T. Comparison of cardiovascular complications in patients with and without kcnj5 gene mutations harboring aldosterone-producing adenomas. J Atheroscler Thromb. 2015;22:191–200 [DOI] [PubMed] [Google Scholar]
- 18.Akerstrom T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, Stalberg P, Robinson B, Alexander Iwen K, Dralle H, Walz MK, Lehnert H, Sidhu S, Gomez-Sanchez C, Hellman P, Bjorklund P. Activating mutations in ctnnb1 in aldosterone producing adenomas. Sci. Rep 2016;6:19546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong AR, Kim JH, Song YS, Lee KE, Seo SH, Seong MW, Shin CS, Kim SW, Kim SY. Genetics of aldosterone-producing adenoma in korean patients. PLoS One. 2016;11:e0147590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scholl UI, Healy JM, Thiel A, Fonseca AL, Brown TC, Kunstman JW, Horne MJ, Dietrich D, Riemer J, Kucukkoylu S, Reimer EN, Reis AC, Goh G, Kristiansen G, Mahajan A, Korah R, Lifton RP, Prasad ML, Carling T. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin. Endocrinol. (Oxf.). 2015;83:779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International union of pharmacology. Xlviii. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev 2005;57:411–425 [DOI] [PubMed] [Google Scholar]
- 22.Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, Satoh F, Maekawa T, Nakamura Y, Sasano H, Gomez-Sanchez EP. Development of monoclonal antibodies against human cyp11b1 and cyp11b2. Mol. Cell. Endocrinol 2014;383:111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, Shibata H, Itoh H, Mitani F, Yamazaki T, Ogishima T, Suematsu M, Mukai K. Adrenocortical zonation in humans under normal and pathological conditions. J. Clin. Endocrinol. Metab 2010;95:2296–2305 [DOI] [PubMed] [Google Scholar]
- 24.Nakamura Y, Maekawa T, Felizola SJ, Satoh F, Qi X, Velarde-Miranda C, Plonczynski MW, Ise K, Kikuchi K, Rainey WE, Gomez-Sanchez EP, Gomez-Sanchez CE, Sasano H. Adrenal cyp11b1/2 expression in primary aldosteronism: Immunohistochemical analysis using novel monoclonal antibodies. Mol. Cell. Endocrinol 2014;392:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, Liu CJ, Sanjanwala AR, Edwards MA, Gomez-Sanchez CE, Nanba K, Rainey WE. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc. Natl. Acad. Sci. U. S. A 2015;112:E4591–4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nanba K, Vaidya A, Williams GH, Zheng I, Else T, Rainey WE. Age-related autonomous aldosteronism. Circulation. 2017;136:347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omata K, Anand SK, Hovelson DH, Liu C-J, Yamazaki Y, Nakamura Y, Ito S, Satoh F, Sasano H, Rainey WE, Tomlins SA. Aldosterone-producing cell clusters frequently harbor somatic mutations and accumulate with age in normal adrenals. Journal of the Endocrine Society. 2017;1:787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satoh F, Morimoto R, Seiji K, Satani N, Ota H, Iwakura Y, Ono Y, Kudo M, Nezu M, Omata K, Tezuka Y, Kawasaki Y, Ishidoya S, Arai Y, Takase K, Nakamura Y, McNamara K, Sasano H, Ito S. Is there a role for segmental adrenal venous sampling and adrenal sparing surgery in patients with primary aldosteronism? Eur. J. Endocrinol 2015;173:465–477 [DOI] [PubMed] [Google Scholar]
- 29.Wu VC, Chueh SC, Chang HW, Lin WC, Liu KL, Li HY, Lin YH, Wu KD, Hsieh BS. Bilateral aldosterone-producing adenomas: Differentiation from bilateral adrenal hyperplasia. QJM. 2008;101:13–22 [DOI] [PubMed] [Google Scholar]
- 30.Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, Stowasser M, Young WF Jr. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab 2016;101:1889–1916 [DOI] [PubMed] [Google Scholar]
- 31.Rossi GP, Auchus RJ, Brown M, Lenders JW, Naruse M, Plouin PF, Satoh F, Young WF Jr. An expert consensus statement on use of adrenal vein sampling for the subtyping of primary aldosteronism. Hypertension. 2014;63:151–160 [DOI] [PubMed] [Google Scholar]
- 32.Neville AM, O’Hare MJ. 15 hyperaldosteronism and related syndromes of mineralocorticoid excess The human adrenal cortex. Berlin; New York: Springer-Verlag; 1982:202–241. [Google Scholar]
- 33.Lack EE. Functional endocrine pathology. Malden, MA: Blackwell Science; 1998:596–636. [Google Scholar]
- 34.Azizan EA, Brown MJ. Novel genetic determinants of adrenal aldosterone regulation. Curr. Opin. Endocrinol. Diabetes Obes 2016;23:209–217 [DOI] [PubMed] [Google Scholar]
- 35.Ledingham JG, Laragh JH, Sommers SC. Secondary aldosteronism and reduced plasma renin in hypertensive disease. Trans. Assoc. Am. Physicians. 1967;80:168–182 [PubMed] [Google Scholar]
- 36.Biglieri EG, Schambelan M, Slaton PE, Stockigt JR. The intercurrent hypertension of primary aldosteronism. Circ. Res 1970;27:195–202 [PubMed] [Google Scholar]
- 37.Ross EJ. Conn’s syndrome due to adrenal hyperplasia with hypertrophy of zona glomerulosa, relieved by unilateral adrenalectomy. Am. J. Med 1965;39:994–1002 [DOI] [PubMed] [Google Scholar]
- 38.Nakamura Y, Felizola SJ, Satoh F, Konosu-Fukaya S, Sasano H. Dissecting the molecular pathways of primary aldosteronism. Pathol. Int 2014;64:482–489 [DOI] [PubMed] [Google Scholar]
- 39.Baer L, Sommers SC, Krakoff LR, Newton MA, Laragh JH. Pseudo-primary aldosteronism. An entity distinct from true primary aldosteronism. Circ. Res 1970;27:203–220 [PubMed] [Google Scholar]
- 40.Enberg U, Volpe C, Hoog A, Wedell A, Farnebo LO, Thoren M, Hamberger B. Postoperative differentiation between unilateral adrenal adenoma and bilateral adrenal hyperplasia in primary aldosteronism by mrna expression of the gene cyp11b2. Eur. J. Endocrinol 2004;151:73–85 [DOI] [PubMed] [Google Scholar]
- 41.Davis WW, Newsome HH Jr., Wright LD Jr., Hammond WG, Easton J, Bartter FC. Bilateral adrenal hyperplasia as a cause of primary aldosteronism with hypertension, hypokalemia and suppressed renin activity. Am. J. Med 1967;42:642–647 [DOI] [PubMed] [Google Scholar]
- 42.Katz FH. Primary aldosteronism with suppressed plasma renin activity due to bilateral nodular adrenocortical hyperplasia. Ann. Intern. Med 1967;67:1035–1042 [DOI] [PubMed] [Google Scholar]
- 43.Ganguly A Primary aldosteronism. N. Engl. J. Med 1998;339:1828–1834 [DOI] [PubMed] [Google Scholar]
- 44.Doorenbos H, Elings HS, Van Buchem FS. Primary aldosteronism due to adrenocortical hyperplasia. Lancet. 1956;271:335–337 [DOI] [PubMed] [Google Scholar]
- 45.Weinberger MH, Grim CE, Hollifield JW, Kem DC, Ganguly A, Kramer NJ, Yune HY, Wellman H, Donohue JP. Primary aldosteronism: Diagnosis, localization, and treatment. Ann. Intern. Med 1979;90:386–395 [DOI] [PubMed] [Google Scholar]
- 46.Conn JW, Conn ES. Primary aldosteronism versus hypertensive disease with secondary aldosteronism. Recent Prog. Horm. Res 1961;17:389–414 [PubMed] [Google Scholar]
- 47.Young WF. Primary aldosteronism: Renaissance of a syndrome. Clin. Endocrinol. (Oxf.). 2007;66:607–618 [DOI] [PubMed] [Google Scholar]
- 48.Wehling M, Neylon CB, Fullerton M, Bobik A, Funder JW. Nongenomic effects of aldosterone on intracellular ca2+ in vascular smooth muscle cells. Circ. Res 1995;76:973–979 [DOI] [PubMed] [Google Scholar]
- 49.Juurlink DN, Mamdani MM, Lee DS, Kopp A, Austin PC, Laupacis A, Redelmeier DA. Rates of hyperkalemia after publication of the randomized aldactone evaluation study. N. Engl. J. Med 2004;351:543–551 [DOI] [PubMed] [Google Scholar]
- 50.Douma S, Petidis K, Doumas M, Papaefthimiou P, Triantafyllou A, Kartali N, Papadopoulos N, Vogiatzis K, Zamboulis C. Prevalence of primary hyperaldosteronism in resistant hypertension: A retrospective observational study. Lancet. 2008;371:1921–1926 [DOI] [PubMed] [Google Scholar]
- 51.Vinson GP, Coghlan JP. Expanding view of aldosterone action, with an emphasis on rapid action. Clin. Exp. Pharmacol. Physiol 2010;37:410–416 [DOI] [PubMed] [Google Scholar]
- 52.Kang S, Cooper G, Dunne SF, Dusel B, Luan CH, Surmeier DJ, Silverman RB. Cav1.3-selective l-type calcium channel antagonists as potential new therapeutics for parkinson’s disease. Nat Commun. 2012;3:1146. [DOI] [PubMed] [Google Scholar]
- 53.Xie CB, Haris Shaikh L, Garg S, Tanriver G, Teo AE, Zhou J, Maniero C, Zhao W, Kang S, Silverman RB, Azizan EA, Brown MJ. Regulation of aldosterone secretion by cav1.3. Sci Rep. 2016;6:24697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.