

Abstract
Cyclooxygenase (COX)-derived prostaglandins regulate renal hemodynamics and salt and water homeostasis. Inhibition of COX activity causes blood pressure elevation. In addition, chronic analgesic abuse can induce renal injury, including papillary necrosis. COX-2 is highly expressed in the kidney papilla in renal medullary interstitial cell (RMICs). However, its role in blood pressure and papillary integrity in vivo has not been definitively studied. In mice with selective, inducible RMIC COX-2 deletion, a high salt diet led to an increase in blood pressure that peaked at 4–5 weeks and was associated with increased papillary expression of aquaporin 2 and epithelial sodium channel and decreased expression of cystic fibrosis transmembrane conductance regulator. With continued high salt feeding, the mice with RMIC COX-2 deletion had progressive decreases in blood pressure from its peak. After return to a normal salt diet for 3 weeks, blood pressure remained low and was associated with a persistent urinary concentrating defect. Within two weeks of institution of a high salt diet, increased apoptotic RMICs and collecting duct cells could be detected in papillae with RMIC deletion of COX-2, and by 9 weeks of high salt, there was a striking loss of the papillae. Therefore, renal medullary interstitial cell COX-2 expression plays a crucial role in renal handling water and sodium homeostasis, preventing salt-sensitive hypertension, and maintaining structural integrity of papilla.
Keywords: kidney, salt and water homeostasis, prostaglandin, chronic salt intake
Introduction
In the mammalian kidney, prostaglandins are important mediators of vascular tone and salt and water balance and are involved in the mediation and/or modulation of hormonal action, such as renin release and antagonism of vasopressin. Prostaglandins arise from enzymatic metabolism of free arachidonic acid, which is cleaved from membrane phospholipids by phospholipase A2 activity. Cyclooxygenase (prostaglandin synthase G2/H2, COX) is the rate-limiting enzyme in metabolizing arachidonic acid to prostaglandin G2 and subsequently to prostaglandin H2, which serves as the precursor for subsequent metabolism by prostaglandin and thromboxane synthases. Two isoforms of COX exist in mammals, “constitutive” COX-1 and inflammatory-mediated and glucocorticoid-sensitive COX-2. COX-1 is expressed in mammalian kidney in vasculature, glomerular mesangial cells and collecting duct. The highest expression of COX-2 in mammalian kidney is in the macula densa in the cortex and in lipid-rich interstitial cells in the medulla 1, although other cell types have also been shown to express detectable levels of the enzyme under some conditions and in some species, including afferent arterioles, podocytes, and intercalated collecting duct cells 2, 3.
Essential hypertension is a major source of morbidity and mortality in the general population, and a significant percentage of hypertensive patients manifest salt-sensitive hypertension. In addition, at least a quarter of normotensive individuals also show salt sensitivity 4. The etiology of salt-sensitive hypertension is undoubtedly multifactorial, but both experimental and epidemiologic evidence link abnormalities in the COX-2/prostaglandin system to its pathogenesis 5. COX-2 inhibitors, as well as non-selective non-steroidal anti-inflammatory drugs (NSAIDs), are among the most commonly prescribed and over-the-counter medications, and are known to elevate blood pressure and antagonize the blood pressure-lowering effect of antihypertensive medication to an extent that may potentially increase hypertension-related morbidity in many users 6–8.
Prostaglandins inhibit medullary sodium and water excretion by integrated actions to inhibit thick ascending limb NaCl reabsorption and to inhibit collecting duct sodium and water reabsorption. COX-2 expression increases in renal medullary interstitial cells (RMICs) in response to a high salt diet 9. However, it remains uncertain whether these cells are the major source of the prostaglandins that mediate the natriuretic and diuretic effects. The goal of the present studies was to determine whether selective deletion of RMIC COX-2 alters the kidney’s ability to regulate salt and water homeostasis using inducible Tenascin-C-CreER2:COX-2f/f mice (RMIC COX-2−/− mice).
Materials and Methods
The data will not be available to other researchers. We will make the mice available to other researchers.
Animal studies.
All animal experiments were performed in accordance with the guidelines and with the approval of the Institutional Animal Care and Use Committee of Vanderbilt University.
Generation of mice with COX-2 deletion in renal medullary interstitial cells.
COX-2f/f mice in which exons 6, 7, and 8 of Cox-2 gene are flanked by Lox P sites were originally generated in Dr. Fitzgerald’s laboratory 10. The COX-2f/f mice were crossed with inducible Tenascin-C-CreER2 mice, in which Cre recombinase activity is induced in adult mice by tamoxifen administration 11. Tenascin-C is an effective marker of medullary interstitial cells 12. Both mouse strains were on the C57BL/6 background, which is normally resistant to COX-2 deficiency-mediated hypertension 13. At 2 months of age, both COX-2f/f (wild type) and Tenascin-C-CreER2; COX-2f/f (RMIC COX-2−/−) mice were treated with tamoxifen (Sigma, T6648, dissolved in core oil) at a dose of 160 mg/kg by intraperitoneal injection for 5 consecutive days. Two weeks after the last injection, the mice were used for experiments.
Genotyping.
DNA was isolated from tail snips. Cre and Cox-2 allele genotypes were determined as previously described 10, 11. All PCR reactions were carried out using an MJ Research thermal cycler.
TUNEL staining.
Apoptotic cells were detected by TACS®2 TdT-DAB in situ apoptosis detection kit (Trevigen, Catalog#: 4810–30-K) according to manufacturer’s instruction.
RNA isolation and quantitative RT-PCR.
Total RNAs from inner medullae/papillae were isolated using TRIzol reagents (Invitrogen). Quantitative RT-PCR was performed using TaqMan real time PCR (7900HT, Applied Biosystems). The Master Mix and all gene probes were also purchased from Applied Biosystems. The probes used in the experiments included mouse S18 (Mm02601778), COX-2 (Mm00478374), cystic fibrosis transmembrane conductance regulator (CFTR) (Mm00445197), aquaporin 2 (AQP2) (Mm00437575), Atp1a1 (Mm00523255), Atp1b1 (Mm00437612), epithelial sodium channel subunit α (ENaC-α) (Mm00803386), ENaC-β (Mm00441215), and ENaC-γ (Mm00441228).
Antibodies.
Rabbit anti-AQP2 antibody was purchased from Abcam (ab15116, Cambridge, MA), rabbit anti-COX-2 antibodies were from LifeSpan BioSciences (LS-C210609) as well as Thermo Fisher (MA5–14568), and monoclonal 4-Hydroxynonenal (4-HNE) antibody was from R&D systems (MAB3249).
Immunohistochemistry staining.
The animals were anesthetized with Nembutal (70 mg/kg, i.p.) and given heparin (1,000 units/kg, i.p.) to minimize coagulation. One kidney was removed for qRT-PCR, and the animal was perfused with FPAS (3.7% formaldehyde, 10 mM sodium m-periodate, 40 mM phosphate buffer, and 1% acetic acid) through the aortic trunk cannulated by means of the left ventricle. The fixed kidneys were dehydrated through a graded series of ethanols, embedded in paraffin, sectioned (4 μm), and mounted on glass slides. Immunostaining was carried out as in previous reports 14.
Blood pressure measurement using tail-cuff and carotid catheterization.
For tail-cuff blood pressure measurements, mice were trained for 3 consecutive days at room temperature before systolic blood pressure was recorded using a tail-cuff monitor (BP-2000 BP Analysis System, Visitech System). Systolic blood pressures recorded in 2 days were averaged and used as values from one mouse. For catheterization blood pressure measurements, blood pressure was recorded every minute for one hour, and the average of all recorded blood pressure was used as blood pressure from one mouse. Blood pressure measurement using carotid catheterization was performed through the Vanderbilt MMPC. Mice were anesthetized with 80 mg/kg of ketamine (Ft Dodge Laboratories) and 8 mg/kg of inactin (BYK) by intraperitoneal injection and were placed on a temperature-controlled pad. After tracheostomy, phycoerythrin 10 tubing was inserted into the right carotid artery. The catheter was tunneled under the skin, exteriorized, secured at the back of the neck, filled with heparinized saline, and sealed. The catheterized mouse was housed individually and 24 h later, data were collected with a Blood Pressure Analyzer (Micro-Med) 15. Systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial pressure (MAP), and heart rate were recorded every minute for 60 minutes. The average of the recorded data from each individual awake mouse was used.
Acute volume expansion
Mice (on a high salt diet for 2 week to increase RMIC COX-2) were intraperitoneally injected with isotonic saline (10% of their body weight), then immediately placed in a 96 well chamber as shown in supplemental Figure S1, and 4-hour urine was collected. Urinary sodium was determined using a flame photometer.
Urinary osmolality.
Mice were water deprived for 12 h (8:00 PM to 8:00 AM), and then urine was collected for measurement of urine osmolality using a microosmometer (μOsmette, Precision Systems,Natick, MA).
Statistics.
All values are presented as means, with error bars representing ± SEM. P values were calculated by Student’s t test.
Results
In order to delete COX-2 expression selectively in RMICs, we crossed COX-2f/f mice with inducible Tenascin-C-CreER2 transgenic mice (Tenascin-C-CreER2:COX-2f/f mice, RMIC COX-2−/−), in which Cre recombinase activity is induced in adult mice by tamoxifen administration 16. Tenascin C is an effective RMIC marker 12. Inducible deletion of COX-2 expression in RMIC in adult mice avoided any potential developmental abnormality of the inner medulla/papilla. As indicated in Figure 1A, COX-2 mRNA expression was markedly decreased in papillae after tamoxifen administration. To confirm further the effectiveness of COX-2 deletion in renal medullary interstitial cells, COX-2 immunostaining was performed in kidneys after high salt diet for 2 weeks, because high salt intake increases interstitial COX-2 expression 17, 18. As indicated in Figure 1B, strong COX-2 immunoreactivity was apparent in many interstitial cells (arrows) in wild type (COX-2f/f) mice, but only a few interstitial cells were COX-2 positive in kidneys of RMIC COX-2−/− mice. Therefore, COX-2 expression was effectively deleted in medullary interstitial cells in RMIC COX-2−/− mice.
Figure 1. Renal medullary interstitial cell COX-2 was effectively deleted in Tenascin-C-CreER2: COX-2f/f (RMIC COX-2f/f) mice.
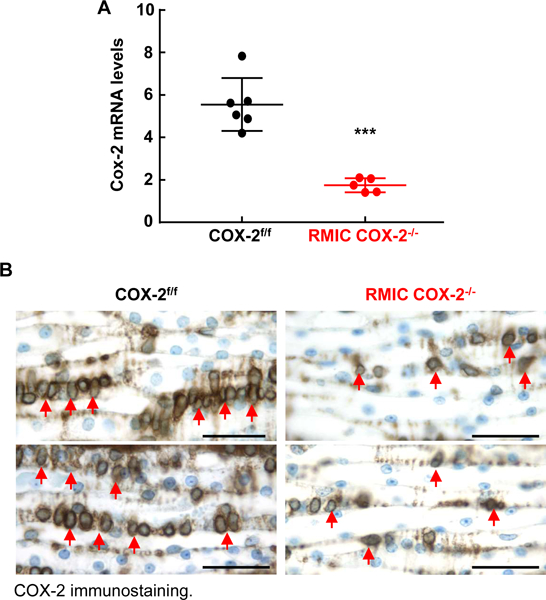
A: After tamoxifen administration for 2 weeks, renal inner medullae/papillae were dissected and COX-2 mRNA was quantitated by qPCR. COX-2 mRNA levels in renal medulla/papilla were significantly reduced in RMIC COX-2−/− mice. ***P < 0.001, n = 6 in COX-2f/f mice and n = 5 in RMIC COX-2−/− mice. All values are means ± SEM. P value was calculated by Student’s t test.
B: After administration of a high salt diet for 2 weeks, COX-2 positive cells were found in many inner medullary interstitial cells in COX-2f/f mice (arrows), but only a few inner medullary interstitial cells were COX-2 positive in RMIC COX-2−/− mice. Scale bar: 50 μM.
To investigate the role of RMIC COX-2 in regulation of salt and water excretion (natriuresis and diuresis) in response to acute volume expansion, mice with or without a high salt diet for 2 weeks in order to increase RMIC COX-2 expression were intraperitoneally administrated 10% body weight of warm isotonic saline. At baseline or after high salt diet for 2 weeks, RMIC COX-2−/− mice had impaired ability to rapidly excrete salt and water, compared to littermate COX-2f/f mice (Figure 2). Therefore, RMIC COX-2 plays an important role to facilitate sodium and water excretion efficiently in response to acute volume expansion.
Figure 2. RMIC COX-2 deficiency led to impaired diuresis and natriuresis in response to acute volume expansion.
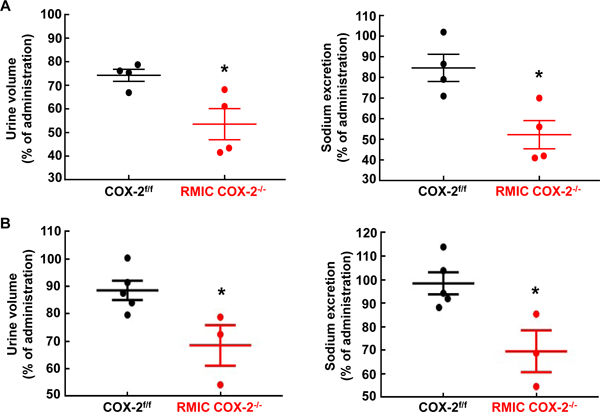
Acute volume expansion was carried out in mice with normal chow or mice on a high salt diet for 2 weeks. A: RMIC COX-2 deletion led to decreases in urine volume and sodium excretion at baseline. *P < 0.05, n = 4 in both groups.
B: RMIC COX-2 deficiency decreased urine volume and sodium excretion 2 weeks after high salt diet. *P < 0.05, n = 5 in COX-2f/f mice and n = 3 in RMIC COX-2−/− mice. All values are means ± SEM. P values were calculated by Student’s t test.
To investigate the potential mechanisms underlying renal medullary interstitial cell COX-2 mediated-natriuresis and diuresis, we determined the expression of transporters expressed in the inner medulla. Expression and activation of the water channel, aquaporin 2 (AQP2) is known to be down-regulated by medullary prostaglandins 19–22. As indicated in Figure 3A, AQP2 mRNA levels were markedly higher in high salt-treated RMIC COX-2−/− mice than high salt-treated COX-2f/f mice. In addition, immunostaining indicated that the density of AQP2 expression in renal medullary collecting duct epithelial cells (particularly, inner medulla) was more intense in RMIC COX-2−/− mice than in COX-2f/f mice. Furthermore, more AQP2 immunoreactivity was observed in the apical membrane of collecting duct epithelial cells from RMIC COX-2−/− mice, consistent with higher water channel activity.
Figure 3. RMIC COX-2 deficiency led to increases in expression of sodium transporters and aquaporin 2 but decreases in CFTR.
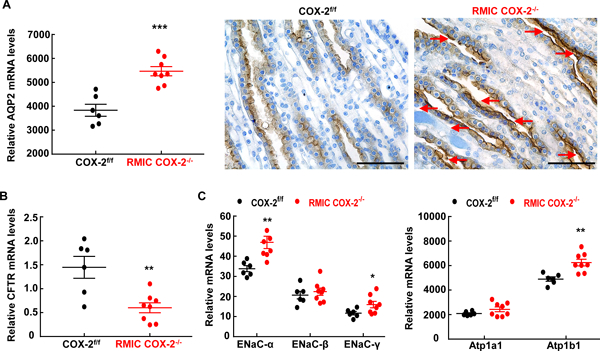
Mice were fed with high salt diet for 2 weeks. A: RMIC COX-2 deficiency led to increases in aquaporin 2 (AQP2) mRNA and protein expression in the inner medullae/papillae. In addition, the immunostaining density in the apical plasma membrane was higher in RMIC COX-2−/− mice than in COX-2f/f mice (arrows), an indication of increased AQP2 activity. Original magnification: x 400. ***P < 0.001, n = 6 in the COX-2f/f group and n = 8 in the RMIC COX-2−/− group. Scale bar: 50 μM.
B: The mRNA levels of CFTR decreased in RMIC COX-2−/− mice. **P < 0.01, n = 6 in the COX-2f/f group and n = 8 in the RMIC COX-2−/− group.
C: The mRNA levels of ENaCα, EnaCγ, and Atp1b1 were increased in RMIC COX-2−/− mice. *P < 0.05, **P < 0.01, n = 6 in the COX-2f/f group and n = 8 in the RMIC COX-2−/− group. All values are means ± SEM. P values were calculated by Student’s t test.
The cystic fibrosis transmembrane conductance regulator (CFTR) is expressed along all the nephron segments and facilitates chloride secretion in the medullary collecting duct 23, 24. We found that CFTR mRNA levels were markedly lower in RMIC COX-2−/− mice than in COX-2f/f mice (Figrue 3B), suggesting that COX-2-mediated prostanoids from renal medullary interstitial cells may increase chloride secretion through upregulation of CFTR in the medulla. In addition, deletion of RMIC COX-2 led to increases in renal medullary mRNA levels of α and γ subunits of ENaC and β subunit of Na+/K+ ATPase (Figure 3C).
Global COX-2 deletion did not affect blood pressure in C57/BL6 mice on a normal salt diet 13, and high salt alone did not markedly increase blood pressure in wild type mice or rats while high salt plus selective COX-2 inhibition increased blood pressure in both 25, 26. In preliminary studies, we also found that RMIC COX-2−/− did not affect blood pressure when fed a normal salt diet. However, when mice were administered a high salt diet and blood pressure was continuously measured by tail cuff plethysmography, COX-2f/f mice had minimal increases in systolic blood pressure, but RMIC COX-2−/− mice developed progressively increased systolic blood pressure for the first 3–5 weeks of treatment (Figure 4A). Similarly, increased SBP, DBP, and MAP were seen when blood pressure was measured with indwelling carotid catheterization in awake mice on a high salt diet for 4 weeks (DBP: 101.1 ± 0.9 vs. 88.8 ± 2.4 mmHg, P < 0.005; SBP: 131.6 ± 1.5 vs. 114.3 ± 3.0 mmHg, P < 0.001; MAP: 117.2 ± 4.0 vs. 101.8 ± 1.8 mmHg, P < 0.005. N = 8 in COX-2f/f group and n = 6 in RMIC COX-2−/− group) (Figure 4B). Heart rate difference was minimal between two groups (524 ± 13 vs. 473 ± 17 of COX-2f/f, P > 0.05).
Figure 4. RMIC COX-2 deficiency caused salt-sensitive hypertension and papillary damage in response to chronic salt loading Both COX-2f/f and RMIC COX-2−/− mice were fed with a high salt diet (8% NaCl) after tamoxifen administration for 2 weeks.
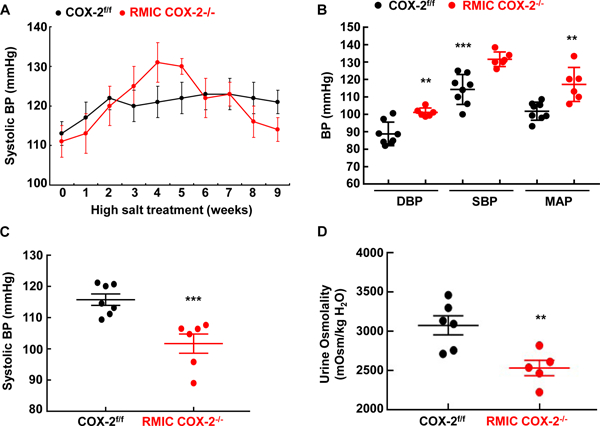
A: Systolic blood pressure (SBP) was monitored with tail cuff microphonic manometer following initiation of high salt treatment (8% NaCl). High salt administration had no apparent effect on blood pressure in COX-2f/f mice throughout experiment period, but increased blood pressure in RMIC COX-2 deficient mice for the first 5 weeks, and then led to progressively decreased blood pressure. N= 7 in COX-2f/f mice and n = 6 RMIC COX-2−/− mice. All values are means ± SEM.
B: Diastolic blood perssure (SBP), systolic blood pressure (SBP) and mean arterial pressure (MAP) measured by carotid catheterization after a high salt diet for 4–5 weeks was markedly higher in RMIC COX-2−/− mice than in COX-2f/f mice. **P < 0.01; n = 8 in COX-2f/f mice and n = 6 in RMIC COX-2−/− mice.
C and D: Both COX-2f/f and RMIC COX-2−/− mice were fed with a high salt diet (8% NaCl) for 9 weeks, and then fed with normal chow for 3 weeks. C: Blood pressure was markedly lower in RMIC COX-2−/− mice than in COX-2f/f mice. ***P < 0.001; n= 7 in COX-2f/f mice and n = 6 RMIC COX-2−/− mice.
D: Urine osmolality was markedly lower in RMIC COX-2−/− mice than in COX-2f/f mice after 16 hours of water deprivation. **P < 0.01; n= 6 in COX-2f/f mice and n = 5 RMIC COX-2−/− mice.
All values are shown as mean ± SEM. P values were calculated by Student’s t test.
When the mice were treated with high salt for an extended period of time (up to 9 weeks), blood pressure in the RMIC COX-2−/− mice progressively decreased from its peak, and eventually fell to levels lower than in the COX-2f/f mice (Figure 4A). Even after return to a normal salt diet for 3 weeks after 9-weeks of high salt diet, RMIC COX-2−/− mice still had persistently lower blood pressures (Figure 4C), and a persistent urinary concentrating defect compared to COX-2f/f mice (Figure 4D). However, baseline urinary concentrating ability was comparable between COX-2f/f mice and RMIC COX-2−/− mice (mOsm/kg H2O: 3300 ± 95 vs. 3129 ± 113 of COX-2f/f, P > 0.05. N = 8 in COX-2f/f group and n = 9 in COX-2−/− group).
Morphologic examination either by MRI or direct examination indicated striking loss of papillae in the RMIC COX-2−/− mice in response to chronic high salt exposure (Figure 5), which was not seen in RMIC COX-2−/− mice maintained on the normal salt diet (not shown). Of note, COX-2f/f mice fed the chronic high salt diet exhibited normal papillae.
Figure 5. Chronic high salt intake caused renal papillary loss in RMIC COX-2 deficient mice.
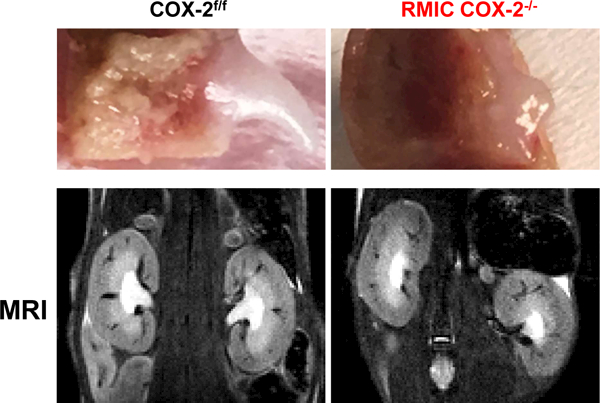
Direct morphologic examination (upper panels) or magnetic resonance imaging (MRI, lower panels) showed striking papillary loss after long-term high salt exposure (9–10 weeks) in RMIC COX-2−/− mice, but not in COX-2f/f mice.
To investigate whether RMIC COX-2-derived prostanoids exerted cytoprotective effects on structures in the inner medulla, we determined that there was increased TUNEL staining in papillae of RMIC COX-2−/− mice as early as two weeks after institution of a high salt diet (Figure 6A) and increased oxidative stress, as indicated by increased papillary 4-HNE immunostaining (Figure 6B).
Figure 6. RMIC COX-2 deficiency led to apoptosis in the inner medullae/papillae in response to high salt diet.
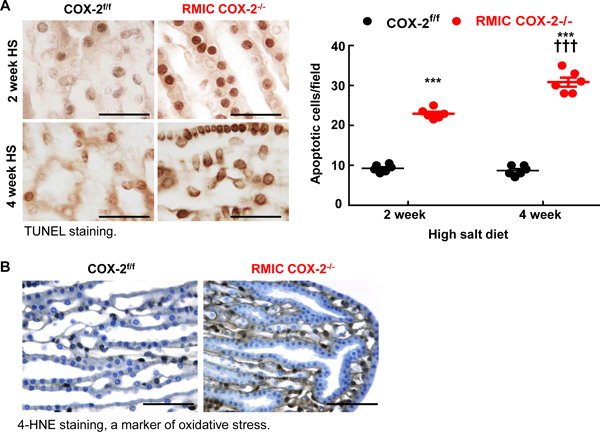
A: RMIC COX-2−/− mice had increased papillary apoptotic cells as early as 2 weeks after a high salt diet, and more apoptotic cells were evident in both interstitial and epithelial cells 4 weeks after a high salt diet. ***P < 0.001 vs. corresponding COX-2f/f mice, †††P < 0.001 vs. 2 weeks of RMIC COX-2−/− mice; n = 6 in each group. Scale bar: 25 μM.
B: Four weeks after high salt intake, there was increased papillary oxidative stress in RMIC COX-2−/− mice, as indicated by 4-HNE-staining. Original magnification: x 160. All values are shown as mean ± SEM. P values were calculated by Student’s t test. Scale bar: 160 μM.
Discussion
With salt loading, PGE2 production in the medulla increases and inhibits water reabsorption by antagonizing vasopressin action and decreasing AQP2 expression 22 and stimulates salt excretion by inhibiting sodium reabsorption by decreasing ENaC expression and stimulating chloride excretion through activation of cystic fibrosis transmembrane conductance regulator (CFTR) 24. The current studies utilized inducible COX-2 deletion in renal medullary interstitial cells to investigate the potential role of RMIC COX-2 expression in renal handling of sodium and water homeostasis and maintenance of the structural integrity of the papilla. RMIC COX-2−/− mice exhibited impaired renal sodium and water excretion in response to acute volume expansion; inner medullary AQP2 mRNA and protein levels were higher in RMIC COX-2−/− mice than in COX-2f/f mice in response to a high salt intake, and mRNA expression of other relevant sodium transporters, including α and γ subunits of eNaC and β subunit of Na+/K+-ATPase were also increased in RMIC COX-2−/− mice while expression of CFTR was decreased. The other striking finding was that RMIC COX-2−/− mice initially developed salt-sensitive hypertension, but long-term high salt intake led to papillary loss in RMIC COX-2−/− mice, in association with lower blood pressure and impaired urinary concentrating ability, at least in part due to increased apoptosis and oxidative stress in both RMIC and surrounding collecting duct cells. Of note, the persistent low blood pressure and the impaired urinary concentrating ability seen in RMIC COX-2−/− mice after long-term high salt diet is due to papillary damage or loss, rather than a physiological effect.
The adult mammalian kidney is a particularly rich source of prostaglandin as well as an important biological target of these prostaglandins 27. In the medulla, COX-2 is primarily expressed in medullary interstitial cells, particularly in the papilla tip, and a high salt diet has been shown to up-regulate COX-2 expression in renal medullary interstitial cells 1, 28. COX-2 inhibitors reduce urinary sodium excretion during an initial period of salt loading 25, 29–32. In rats with uninephrectomy plus high salt diet, medullary infusion of a selective COX-2 inhibitor resulted in salt-sensitive hypertension 33. However, these studies did not determine the source of COX-2 derived prostanoids. It has been suggested that NSAID/COX-2 inhibitor-induced increases in blood pressure might be due in part to inhibition of endothelial COX-2-derived prostacyclin/PGI2 34, 35 or myeloid COX-2-derived PGE2 26. Our current study provides clear proof of the importance of COX-2 expression in RMIC in mediating renal salt and water regulation and demonstrate that inhibition of renal medullary interstitial cell COX-2 contributes to salt-sensitive hypertension.
In addition to their role in regulating salt and water homeostasis, there is evidence that inner medullary prostanoids provide an important survival function for inner medullary structures since papillary necrosis or apoptosis has been reported with both non-selective and COX-2-selective inhibitors 36, 37. In mouse renal inner medullary collecting duct cells, inhibition of PGE2 production did not affect cell proliferation or apoptosis 38, indicating that collecting duct was not the source of cytoprotective prostanoids. Although cultured medullary fibroblasts have been shown to undergo apoptosis in response to hypertonic stress following COX-2 inhibition 39, the role of COX-2 derived prostaglandins from RMICs in maintenance of papillary integrity in vivo was previously undetermined. The present studies clearly demonstrate that COX-2 in RMICs plays an important role in maintaining structural integrity of all papillary structures subjected to the stress of a high salt diet.
An intriguing recent study examining the role of COX-2 vs. COX-1 found that although COX-1 generates orders of magnitude more prostanoids than COX-2, it cannot compensate for loss of COX-2 in renal development, suggesting that COX-2 may serve other functions in addition to, or instead of prostanoid function 40. Further studies will be necessary to determine the mechanisms by which RMIC COX-2 provides papillary cytoprotection.
In summary, renal medullary interstitial cell COX-2 expression plays an important role in renal handling water and sodium homeostasis, preventing salt-sensitive hypertension, and maintaining structural integrity of papilla.
Perspectives
Our data clearly demostarte that COX-2 is essential to promote sodium and water excretion in response to acute salt loading as well as chronic salt loading. Specific inhibition of cyclooxygenase 2 in interstitial cells in the kidney medulla is an important mediator of NSAID-induced hypertension and analgesic nephropathy.
Supplementary Material
Novelty and Significance
What is New?
Using an inducible COX-2 knockout mdoel, we investigated the potential role of renal medullary interstitial cell COX-2 in renal handling of sodium and water homeostasis and maintenance of the structural integrity of the papilla. We definitively determined that renal medullary interstitial cell COX-2 is essential to excrete excessive sodium in response to acute salt loading and to prevent salt-sensitive hypertension and to protect against papillary damage in response to chronic high salt intake.
What is Relevant?
These studies demonstrate that specific inhibition of cyclooxygenase 2 in interstitial cells in the kidney medulla is an important mediator of NSAID-induced hypertension and analgesic nephropathy.
Summary
Renal medullary interstitial cell COX-2-mediated prostaglandin plays an important role in handing water and sodium homeostasis, preventing salt-sensitive hypertension, and maitaining structural integrity of papilla.
Acknowledgments
Sources of Funding
This work was supported by NIH grants DK-95785, DK-51265, and DK-114809 (to MZZ and RCH); DK-62794 and DK-103067 (to RCH); and funds from the Department of Veterans Affairs (to RCH). Carotid catheterization was performed through the Vanderbilt Mouse Metabolic Phenotyping Center (MMPC, DK059637).
Footnotes
Disclosures
None.
References
- 1.Harris RC. Cyclooxygenase-2 in rat nephron development. Am J Physiol. 1997;273:F994–1002. [DOI] [PubMed] [Google Scholar]
- 2.Harris RC. Cox-2 and the kidney. J Cardiovasc Pharmacol. 2006;47 Suppl 1:S37–42. [DOI] [PubMed] [Google Scholar]
- 3.Stegbauer J, Chen D, Herrera M, Sparks MA, Yang T, Konigshausen E, Gurley SB, Coffman TM. Resistance to hypertension mediated by intercalated cells of the collecting duct. JCI Insight. 2017;2:e92720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanbay M, Chen Y, Solak Y, Sanders PW. Mechanisms and consequences of salt sensitivity and dietary salt intake. Curr Opin Nephrol Hypertens. 2011;20:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kohsaka S, Volcik KA, Folsom AR, Wu KK, Ballantyne CM, Willerson JT, Boerwinkle E. Increased risk of incident stroke associated with the cyclooxygenase 2 (cox-2) g-765c polymorphism in african-americans: The atherosclerosis risk in communities study. Atherosclerosis. 2008;196:926–930. [DOI] [PubMed] [Google Scholar]
- 6.Johnson AG, Nguyen TV, Day RO. Do nonsteroidal anti-inflammatory drugs affect blood pressure? A meta-analysis. Ann Intern Med. 1994;121:289–300. [DOI] [PubMed] [Google Scholar]
- 7.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin e synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elliott WJ. Do the blood pressure effects of nonsteroidal antiinflammatory drugs influence cardiovascular morbidity and mortality? Curr Hypertens Rep. 2010;12:258–266. [DOI] [PubMed] [Google Scholar]
- 9.Abou-Issa HM, Alshafie GA, Seibert K, Koki AT, Masferrer JL, Harris RE. Dose-response effects of the cox-2 inhibitor, celecoxib, on the chemoprevention of mammary carcinogenesis. Anticancer Res. 2001;21:3425–3432. [PubMed] [Google Scholar]
- 10.Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J, Zhang H, Hui Y, Cheng Y, Petrenko N, Yu Y, FitzGerald GA. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci U S A. 2009;106:7548–7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He W, Xie Q, Wang Y, Chen J, Zhao M, Davis LS, Breyer MD, Gu G, Hao CM. Generation of a tenascin-c-creer2 knockin mouse line for conditional DNA recombination in renal medullary interstitial cells. PLoS One. 2013;8:e79839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neelisetty S, Alford C, Reynolds K, Woodbury L, Nlandu-Khodo S, Yang H, Fogo AB, Hao CM, Harris RC, Zent R, Gewin L. Renal fibrosis is not reduced by blocking transforming growth factor-beta signaling in matrix-producing interstitial cells. Kidney Int. 2015;88:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang T, Huang YG, Ye W, Hansen P, Schnermann JB, Briggs JP. Influence of genetic background and gender on hypertension and renal failure in cox-2-deficient mice. Am J Physiol Renal Physiol. 2005;288:F1125–1132. [DOI] [PubMed] [Google Scholar]
- 14.Cheng H, Wang S, Jo YI, Hao CM, Zhang M, Fan X, Kennedy C, Breyer MD, Moeckel GW, Harris RC. Overexpression of cyclooxygenase-2 predisposes to podocyte injury. J Am Soc Nephrol. 2007;18:551–559. [DOI] [PubMed] [Google Scholar]
- 15.Yao B, Harris RC, Zhang MZ. Intrarenal dopamine attenuates deoxycorticosterone acetate/high salt-induced blood pressure elevation in part through activation of a medullary cyclooxygenase 2 pathway. Hypertension. 2009;54:1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abu El-Asrar AM, Mohammad G, Nawaz MI, Siddiquei MM, Kangave D, Opdenakker G. Expression of lysophosphatidic acid, autotaxin and acylglycerol kinase as biomarkers in diabetic retinopathy. Acta Diabetol. 2013;50:363–371. [DOI] [PubMed] [Google Scholar]
- 17.Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest. 1994;94:2504–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CL, Yu X, James IO, Zhang HY, Yang J, Radulescu A, Zhou Y, Besner GE. Heparin-binding egf-like growth factor protects intestinal stem cells from injury in a rat model of necrotizing enterocolitis. Lab Invest. 2012;92:331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia Z, Wang H, Yang T. Mice lacking mpges-1 are resistant to lithium-induced polyuria. Am J Physiol Renal Physiol. 2009;297:F1689–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soodvilai S, Jia Z, Wang MH, Dong Z, Yang T. Mpges-1 deletion impairs diuretic response to acute water loading. Am J Physiol Renal Physiol. 2009;296:F1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim GH, Choi NW, Jung JY, Song JH, Lee CH, Kang CM, Knepper MA. Treating lithium-induced nephrogenic diabetes insipidus with a cox-2 inhibitor improves polyuria via upregulation of aqp2 and nkcc2. Am J Physiol Renal Physiol. 2008;294:F702–709. [DOI] [PubMed] [Google Scholar]
- 22.Cheng X, Zhang H, Lee HL, Park JM. Cyclooxygenase-2 inhibitor preserves medullary aquaporin-2 expression and prevents polyuria after ureteral obstruction. J Urol. 2004;172:2387–2390. [DOI] [PubMed] [Google Scholar]
- 23.Souza-Menezes J, da Silva Feltran G, Morales MM. Cftr and tnr-cftr expression and function in the kidney. Biophys Rev. 2014;6:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajagopal M, Thomas SV, Kathpalia PP, Chen Y, Pao AC. Prostaglandin e2 induces chloride secretion through crosstalk between camp and calcium signaling in mouse inner medullary collecting duct cells. Am J Physiol Cell Physiol. 2014;306:C263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao B, Harris RC, Zhang MZ. Interactions between 11beta-hydroxysteroid dehydrogenase and cox-2 in kidney. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1767–1773. [DOI] [PubMed] [Google Scholar]
- 26.Zhang MZ, Yao B, Wang Y, Yang S, Wang S, Fan X, Harris RC. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J Clin Invest. 2015;125:4281–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang MZ, Wang JL, Cheng HF, Harris RC, McKanna JA. Cyclooxygenase-2 in rat nephron development. Am J Physiol. 1997;273:F994–1002. [DOI] [PubMed] [Google Scholar]
- 28.Schnermann J, Traynor T, Yang T, Arend L, Huang YG, Smart A, Briggs JP. Tubuloglomerular feedback: New concepts and developments. Kidney Int Suppl. 1998;67:S40–45. [DOI] [PubMed] [Google Scholar]
- 29.Catella-Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, Lasseter KC, Quan H, Gertz BJ, FitzGerald GA. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther. 1999;289:735–741. [PubMed] [Google Scholar]
- 30.Rodriguez F, Llinas MT, Gonzalez JD, Rivera J, Salazar FJ. Renal changes induced by a cyclooxygenase-2 inhibitor during normal and low sodium intake. Hypertension. 2000;36:276–281. [DOI] [PubMed] [Google Scholar]
- 31.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and −2 activity on the pressor response to angiotensin ii. J Clin Invest. 2002;110:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zewde T, Mattson DL. Inhibition of cyclooxygenase-2 in the rat renal medulla leads to sodium-sensitive hypertension. Hypertension. 2004;44:424–428. [DOI] [PubMed] [Google Scholar]
- 33.Zewde T, Mattson DL. Inhibition of cyclooxygenase-2 in the rat renal medulla leads to sodium-sensitive hypertension. Hypertension. 2004;44:424–428. [DOI] [PubMed] [Google Scholar]
- 34.Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim H-S, Rockman HA, FitzGerald GA, Koller BH, Coffman TM. Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metabolism. 2005;2:201–207. [DOI] [PubMed] [Google Scholar]
- 35.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Pure E, Funk CD, FitzGerald GA. Vascular cox-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4:132ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segasothy M, Samad SA, Zulfigar A, Bennett WM. Chronic renal disease and papillary necrosis associated with the long-term use of nonsteroidal anti-inflammatory drugs as the sole or predominant analgesic. Am J Kidney Dis. 1994;24:17–24. [DOI] [PubMed] [Google Scholar]
- 37.Akhund L, Quinet RJ, Ishaq S. Celecoxib-related renal papillary necrosis. Arch Intern Med. 2003;163:114–115. [DOI] [PubMed] [Google Scholar]
- 38.Rocha GM, Michea LF, Peters EM, Kirby M, Xu Y, Ferguson DR, Burg MB. Direct toxicity of nonsteroidal antiinflammatory drugs for renal medullary cells. Proc Natl Acad Sci U S A. 2001;98:5317–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez-Rodriguez C, Antos CL, Shelton JM, Richardson JA, Lin F, Novobrantseva TI, Bronson RT, Igarashi P, Rao A, Olson EN. Loss of nfat5 results in renal atrophy and lack of tonicity-responsive gene expression. Proc Natl Acad Sci U S A. 2004;101:2392–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Mazaleuskaya LL, Ballantyne LL, Meng H, FitzGerald GA, Funk CD. Differential compensation of two cyclooxygenases in renal homeostasis is independent of prostaglandin-synthetic capacity under basal conditions. FASEB J. 2018:fj201800252R. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.