

ABSTRACT
Dendritic cells (DCs) are crucial players in promoting immune responses. Logically, adoptive DC therapy is a promising approach in cancer immunotherapy. One of the major obstacles in cancer immunotherapy in general is the immunosuppressive tumor microenvironment, which hampers the maturation and activation of DCs. Therefore, human clinical outcomes with DC therapy alone have been disappointing. In this study, we use fully serotype 3 oncolytic adenovirus Ad3-hTERT-CMV-hCD40L, expressing human CD40L, to modulate the tumor microenvironment with subsequently improved function of DCs. We evaluated the synergistic effects of Ad3-hTERT-CMV-hCD40L and DCs in the presence of human peripheral blood mononuclear cells ex vivo and in vivo. Tumors treated with Ad3-hTERT-CMV-hCD40L and DCs featured greater antitumor effect compared with unarmed virus or either treatment alone. 100% of humanized mice survived to the end of the experiment, while mice in all other groups died by day 88. Moreover, adenovirally-delivered CD40L induced activation of DCs, leading to induction of Th1 immune responses. These results support clinical trials with Ad3-hTERT-CMV-hCD40L in patients receiving DC therapy.
Keywords: Dendritic cells, T-cells, oncolytic adenovirus, Ad3, CD40L
Introduction
The field of cancer immunotherapy has made tremendous progress recently and it has become a first or second line treatment option for many cancers. To establish a powerful anti-tumor immune response in patients, successful tumor antigen presentation through antigen-presenting cells (APCs), such as dendritic cells (DCs), to tumor-specific T cells is essential.1 DCs are APCs and key mediators of adaptive immune responses.2 Considering the key role of DCs in the initiation and regulation of immune responses, they are an attractive tool for immunotherapy.1 DC-based therapies have been investigated for various advanced-stage cancers such as prostate cancer, melanoma, renal cell carcinoma, and B-cell lymphoma.3 However, the typical tumor microenvironment (TME) is highly immunosuppressive and capable of impairing DC functions, thereby hampering the efficacy of DC therapies.4–6 Thus, despite promising preclinical results in DC therapy, clinical data has suggested that alone it may not be sufficient to reverse the immune-suppressive TME for meaningful responses in patients.7,8
For example, a randomized trial in colorectal cancer concluded that although anti-tumor immune responses could be induced with DC therapy, this did not result in anti-tumor efficacy or a survival advantage.9 Similarly, in melanoma, a survival advantage was not seen versus chemotherapy.10 Taken together with dozens of non-randomized trials, it appears that DC therapies are able to induce anti-tumor immunity but there is a limitation with efficacy, and tumor immunosuppression appears the likely culprit. This notion is supported by more promising trial results when DC therapy was given as an adjuvant therapy, in the context of minimal residual disease.11 If there is no macroscopic tumor, there is less immunosuppression caused by the TME.
Of note, it has repeatedly been suggested that patients responding immunologically to DC therapy have better outcomes.12–15 This finding could indicate that immune competent patients have better outcomes than highly immune suppressed patients,16–18 without DCs necessarily playing a role. An interesting outlier to lack of randomized efficacy is sipuleucel T, which is a mixed product containing T cells and DCs. It can be speculated that the survival advantage attributed to this cell product might relate to the presence of T cells in the product.19
Thus, with tumor immunosuppression identified as the likely reason for lack of efficacy of DC therapy, one option would be to sensitize the tumor milieu to DCs.20 Anti-tumor immune response depends on the amount and type of infiltrating immune cells, stromal cells, and MHC expression on tumor cells. During cancer progression, immunoediting and various escape tactics employed by tumors eventually prevent the host immune system from controlling tumors.21 Thus, for a successful cancer immunotherapy, it is important to revert the immunosuppressiveness of the TME.
Development of successful immune response requires multiple molecular signals. The primary signal is provided by binding of a tumor antigen to a T- or B-cell receptor, followed by secondary signals involving engagement of costimulatory proteins to their co-receptors on the surface of T or B lymphocytes. Additional signals, such as cytokine secretion, are necessary to further modify, enhance, and sustain the immune response against tumor cells. One of the key costimulatory molecules is the CD40 receptor22 . CD40 is a member of the tumor necrosis factor receptor family and expressed by antigen-presenting cells such as DCs and B cells, whereas its ligand CD40L is transiently expressed on T cells. CD40 engagement on the surface of DCs induces expression of costimulatory molecules and cytokine production. Thus, the activation licenses DCs to mature and to trigger immune responses.22
Oncolytic adenoviruses can be engineered to selectively replicate in and destroy tumor cells, providing an attractive platform for the treatment of cancer. In the larger context of cancer immunotherapy, oncolytic adenoviruses are especially promising for generating de novo immunity against tumors, and modifying the suppressive TME towards a proinflammatory status conducive to successful immunotherapy.23–26 Thus, viruses appear attractive companion therapies for approaches such as DC therapy, T-cell therapies, and checkpoint inhibitors, all of which are hindered by the immunosuppressive TME.
Arming the virus with immunostimulatory molecules such as CD40L enables efficient delivery of the therapeutic gene locally to the tumor, with local amplification and limited systemic exposure, which has proved to be an issue with recombinant CD40L. Then the recombinant molecule was given systemically, adverse events from non-target organs proved limiting to effective concentrations in tumors.27 High local levels of CD40L cause apoptosis of CD40+ tumor cells,28 but since many advanced tumors are apoptosis-resistant, the DC-activating effect of CD40L could be more relevant in the context of cancer.28–30
Previously, oncolytic adenovirotherapy has demonstrated safety and efficacy in preclinical studies and in patients.25,31–35 In one patient series, an oncolytic adenovirus coding for CD40L was used in advanced cancer patients refractory to available therapies,30 establishing safety of the approach. Possible signs of efficacy were reported in 83% of the treated patients. However, complete responses and long-term survival were rare, leaving room for improvement.
We have shown that Ad3-hTERT-CMV-hCD40L, a CD40L-coding oncolytic adenovirus fully based on serotype 3 (Ad3), can elicit potent antitumor efficacy by coupling the lytic function with production of high amounts of CD40L at the tumor.36 Importantly, the oncolytic platform restricts the expression of CD40L to cancer cells, reducing systemic exposure. Of note, Ad3 been shown to transduce tumors through the intravenous route both in patients and in animal models.25 Previously published in vitro, in vivo, and human data has additionally revealed that virally expressed CD40L is able to stimulate DCs.24–30 In this regard, we performed a pilot experiment where vectored delivery of mouse CD40L in a non-replicating virus was able to increase the efficacy of murine DC therapy.36 Delivery of human CD40L in an oncolytic virus has not been previously studied in the context of human DC therapy.
In the present study, we explored the potential benefit of oncolytic Ad3-hTERT-CMV-hCD40L in a clinically relevant “humanized” model of DC therapy featuring human peripheral blood mononuclear cells (PBMCs) as a source of immune cells. Synergistic effects of this approach were shown to lead to enhanced DC maturation and antitumor immune response. Our findings highlight the potential therapeutic benefit of Ad3-hTERT-CMV-hCD40L as an enabling therapy in patients receiving DC therapy. These preclinical results set the stage for clinical translation.
Materials and methods
Cell lines
Human A549 lung adenocarcinoma cell line, LNCaP prostate cancer cell line and SKOV3 ovarian cancer were obtained from American Type Culture Collection (ATCC; LGS standards, USA). EJ human bladder cancer cell line was a kindly provided by A.G. Eliopoulos (University of Crete Medical School and Laboratory of Cancer Biology, Heraklion, Crete, Greece). All the cell lines except LNCaP were cultured in Dulbecco’s modified Eagles’s medium (DMEM) whereas LNCaP cells were cultured in Roswell Park Memorial Institute medium (RPMI). All the cell lines were maintained under a humidified 5% CO2 atmosphere at 37°C and media were supplemented with 1% Penicillin/Streptomycin (P/S), 1% L-Glutamine, 10% FBS.
Viruses
Two human oncolytic adenovirus based on serotype 3 were used: Ad3-hTERT-E1A34 and Ad3-hTERT-CMV-hCD40L.36 Both feature human telomerase reverse transcriptase promoter (hTERT), to restrict the virus replication in tumor cells.
Generation of human dcs
Generation of human DCs was done according to a protocol reported previously (Zafar et al., 2016). Briefly, human PBMCs were isolated from buffy coat of healthy donor obtained from Red Cross Blood Service (Helsinki, Finland). Isolation was done through density gradient centrifugation using lymphoprep (StemCell technologies). Isolated PBMCs were washed with PBS, and ACK lysis buffer (Sigma, St Louis, MO. A10492.01) was used to remove erythrocytes. CD14+ cells were isolated from PBMCs with CD14+ magnetic beads (Miltenyi Biotec, 130–050–201) according to the manufacturer’s instructions. 4.5 X106 CD14+ cells were cultured for 5–7 days in 10 ml of 10% RPMI supplemented with 1000U granulocyte-macrophage colony-stimulating factor (GMCSF, Peprotech) and 20ng interleukin 4 (IL4, Peprotech). Immature DCs were then incubated with 50 µg/ml tumor cell lysate for 24h, followed by incubation with lipopolysaccharide (LPS, 100ng) (Sigma, L4391-1MG) for 17-24h. Maturation markers (CD80, CD86, CD83) of DCs were analyzed with flow cytometry.
DC maturation and functionality assay
Freshly isolated monocytes from PBMCs were cultured in a medium containing recombinant human GMCSF and IL4 to obtain immature DCs. The immature DCs were used in two maturation assays: first in the presence of Ad3-hTERT-E1A and Ad3-hTERT-CMV-hCD40L infected cells, and second in the presence of cell culture media supernatants collected from virus-infected cells.
In the first assay, A549 cells were infected with Ad3-hTERT-E1A, Ad3-hTERT-CMV-hCD40L, or left uninfected. The cells were washed after 18h with PBS, and the infection media was replaced with fresh media containing monocyte-derived immature DCs. After 48h, maturation status of the DCs was assessed using flow cytometry. After this T cells isolated from fresh PBMCs through Pan T cell Isolation kit (Miltenyi Biotec, 130–096-535) were added to the mixture of DCs and virus-infected tumor cells. After 24h, T-cell activation was assessed with flow cytometry (see Supplementary Table 1 for the list of antibodies).
In the second assay, A549 cells were first infected with Ad3-hTERT-CMV-CD40L or Ad3-hTERT-E1A and supernatants were collected and filtered to remove the viruses 48 hours later. The supernatants were added to fresh A549 cells together with monocyte-derived DCs. Similarly to the first assay, DC maturation was assayed after 48h, followed by an addition of T cells into the wells containing DCs and cancer cells. T-cell activation was measured through flow cytometry 24h later. LPS (100 ng) (Sigma, L4391–1MG) and recombinant hCD40L (500 ng) (Abcam, ab51956) were used as positive controls in both of the assays. The assay was done in triplicates.
Cell viability assay
10,000 A549, EJ, SKOV3 or LNCaP cells were plated in growth medium containing 2% FBS on 96-well plates. After 24h, the cells were infected with Ad3-hTERT-CMV-hCD40L or Ad3-hTERT-E1A at concentrations of 1 viral particle (VP), 10 VP, 100 VP, or 1000 VP. Two days after the viral infection, DCs and human PBMCs were added in the wells. Tumor cells alone and DCs or PBMCs alone with virus were used as controls. Cell viability was normalized against the viability of controls. Cell viability was determined with MTS assay (CellTiter 96 AQueous One Solution, Promega, Madison, WI) starting from 24h to 96h after adding DCs and PBMCs.
Animal experiment
The experimental animal committee of the University of Helsinki and the Provincial Government of Southern Finland approved all animal protocols. Five weeks old immunodeficient SCID mice were implanted subcutaneously with 5 × 106 A549 cells. When the tumors become injectable 14 days after implantation,37 mice were divided into eight groups (n = 10/group). Mice received intravenous injection of 10 × 106 HLA-matched PBMCs on day 0. Intratumoral injections of viruses (108 VP) were administered on days 1, 3, and 5, followed by 1 × 106 DCs on days 2, 4, and 6. Tumor growth was measured with electronic caliper every other day until day 44 and the survival was followed until day 112. Mice were euthanized when tumor size reached the limit of 18 mm, and tumor ulceration was considered as an exclusion criteria (excluded mice are shown in the figure with reversed triangles). Tumors were collected, homogenized, filtered, and cultured overnight before analyzing with flow cytometry (See Supplementary Table 1 for the list of antibodies). Part of the tumor samples were snap frozen and homogenized, to analyze various cytokines with CBA Flex set cytokine beads using BD Accuri C6. Results were analyzed with FCAP array software.
Statistics
For statistical analyses, two tailed Student’s t-test, Two-way ANOVA (Tukey’s multiple comparisons test), and log-rank were performed using Graphpad Prism (Graphpad Software Inc. La Jolla, CA). Statistical significance was considered when p < 0.05.
Results
Tumor cells infected with Ad3-hTERT-CMV-hCD40L induce DC maturation, resulting in T-cell stimulation
After incubating immature DCs with cancer cells infected with hCD40L-armed or parental unarmed virus, we observed statistically significant upregulation of DC maturation markers CD83, CD80, and CD86 compared with the non-infected mock group (p < 0.0001; Figure 1A-C). Moreover, the DC maturation markers CD83 (p = 0.0005) and CD80 (p = 0.04) were significantly more upregulated if tumor cells were infected with Ad3-hTERT-CMV-hCD40L instead of the unarmed virus.
Figure 1.
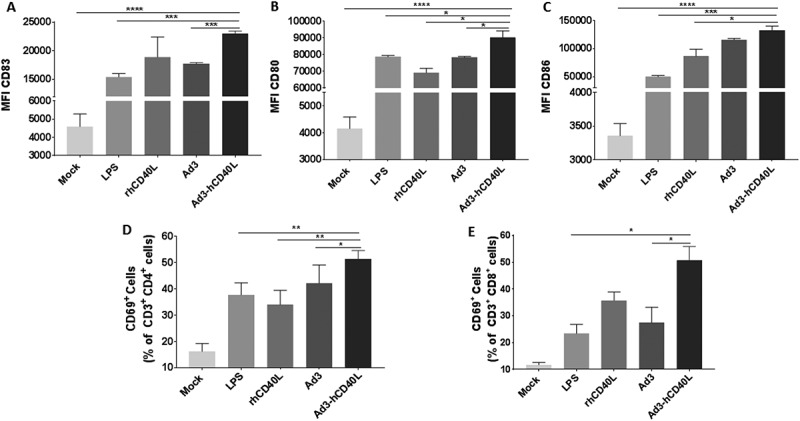
Ad3-hTERT-CMV-hCD40L infected tumor cells induce DC maturation and T-cell stimulation. A549 cells were infected with Ad3-hTERT-CMV-hCD40L, Ad3-hTERT-E1A, or left untreated. After 18 h, infection media were removed and cells were washed with PBS before adding monocyte-derived DCs added to co-cultures. LPS (100 ng) and recombinant hCD40L protein (500 ng) were used as positive controls. After 48 h, a portion of DCs was assayed for maturation by flow cytometry. Median fluores cence intensity (MFI) for CD83 (A), CD80 (B) and CD86 (C) of CD11c+ populations. T cells were added to the wells and the activation status of CD4 + T cells (D) or CD8 + T cells (E) was determined after 24 h by the expression of CD69. The assay was done in triplicates. MFI: Median fluorescence intensity, LPS: lipopolysaccharide, rhCD40L: recombinant human CD40L, Ad3-hCD40L and Ad3: cells infected with Ad3-hTERT-CMV-hCD40L and Ad3-hTERT-E1A viruses, respectively. Data presented as mean ± SEM *, P < 0.05. **, P < 0.01. ***, P < 0.001. ****, P < 0.0001 by two tailed Student’s t-test.
To evaluate the functional consequences of DC stimulation, T cells were added to co-cultures resulting in high-level T-cell activation as measured by CD69 expression (Figures 1D and 1E). Intriguingly, the group containing Ad3-hTERT-CMV-hCD40L infected tumor cells showed significantly higher levels of T-cell activation compared with the group containing Ad3-hTERT-E1A infected tumor cells (p < 0.05), indicating the importance of the arming device.
Virally expressed hCD40L induces DC maturation and T-cell activation ex vivo
To study the functionality of virally produced hCD40L, A549 cells were infected with hCD40L armed or unarmed virus and supernatants were collected and filtered for the assay. Immature DCs (CD14-, CD1a+) differentiated from CD14+ monocyte-enriched PBMCs were cultured with A549 tumor cells in the presence of filtered supernatants. After 48h, we evaluated co-cultured DCs for the expression of CD83, CD80, and CD86 (Figure 2A-C) with flow cytometry. We observed increased levels of maturation markers in groups incubated with filtered supernatants. Interestingly, co-culture of DCs in the presence of filtered supernatant containing hCD40L showed significant upregulation of DC maturation markers CD83 (p = 0.0134) and CD80 (p = 0.0052) compared to DCs co-cultured in the presence of filtered supernatant collected from cells infected with unarmed virus, again suggesting relevance of hCD40L arming.
Figure 2.
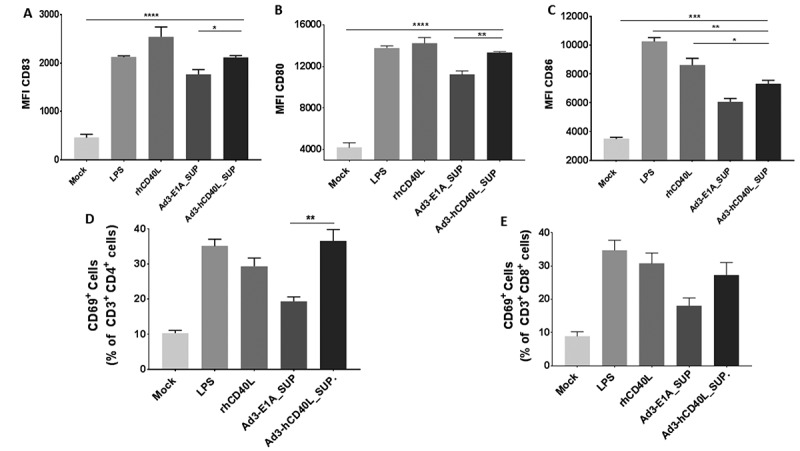
Virally expressed hCD40L induces DC maturation and T-cell activation ex vivo. A549 cells were infected with Ad3-hTERT-CMV-hCD40L or Ad3-hTERT-E1A and supernatants were collected and filtered. Immature DCs were cultured with filtered supernatants for 48hrs. LPS and recombinant hCD40L protein were used as positive controls. After 48h, a portion of DCs was evaluated for Median fluorescence intensity (MFI) for CD83 (A), CD80 (B) and CD86 (C) of CD11c± populationsor co-cultured with T cells. Activation status of CD4 + T cells (D) and CD8 + T (E) cells was assessed 24h later by the expression of CD69. Cells were stained and analyzed by flow cytometry. The assay was done in triplicates. Data presented as mean ± SEM. *, P < 0.05 **; P < 0.01. ***; P < 0.001****; P < 0.0001 by two tailed Student’s t-test.
We further assessed the activation capability of mature DCs to activate T cells in the presence of A549 tumor cells and filtered supernatants. Elevated levels of T-cell activation marker CD69 was observed on both CD3+ CD4+ T cells and CD3+ CD8 + T cells (2E and 2D). However, this increase in T cell activation between the positive control and treated groups has a trend towards significance. Especially CD3+ CD4+ T cells showed significantly (p<0.01) higher activation in a group containing filtered supernatant collected from Ad3-hTERT-CMV-hCD40L infected cells, compared with Ad3-hTERT-E1A infected supernatant.
Ad3-hTERT-CMV-hCD40L improves DC- and PBMC-mediated cancer cell killing ex vivo
The cytotoxic potency of Ad3-hTERT-CMV-hCD40L or Ad3-hTERT-E1A virus with DCs and PBMCs was assessed in two CD40 positive cell lines (LNCaP and EJ) and two CD40 negative cell lines (SKOV3 and A549). Ad3-hTERT-CMV-hCD40L together with DCs and PBMCs induced complete cell killing at 1000 VP/cell in LNCaP (Figure 3A) and EJ cells (Figure 3B) 24h after adding DCs and PBMCs. In A549 cells (Figure 3D) and SKOV3 cells (Figure 3C) killing was observed 72h after adding DCs and PBMCs.
Figure 3.
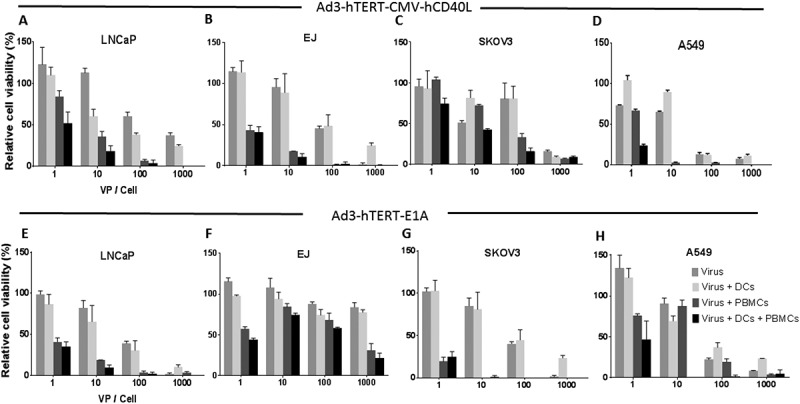
Ad3-hTERT-E1A, DCs and PBMCs efficiently kill tumor cells ex vivo. Tumor-killing potency of Ad3-hTERT-CMVhCD40L, DCs and PBMCs was assessed after 1 day (in LNCaP and EJ cells) and 3 days (in SKOV3, and A549 cells), after adding DCs and PBMCs in co-culture. The assay was done in triplicates. Oncolytic potency of Ad3-hTER-E1A with DCs and PBMCs was evaluated after 3 days (in LNCaP cells), 2 days (in EJ cells) and 4 days (in SKOV3, and A549 cells), after adding DCs and PBMCs in co-culture. Data presented as mean ± SEM. Cell viability was normalized against the viability of controls (not shown).
The cytotoxic capacity of Ad3-hTERT-E1A, DCs, and PBMCs was less pronounced than the corresponding Ad3-hTERT-E1A-hCD40L triple therapy in all the cell lines except Skov3 (Figure 3 E-H). Moreover, triple therapy with either armed or unarmed virus showed more prominent cell killing than double therapy (virus and DCs or virus and T cells) or virus alone groups. Thus, the CD40L-armed virus was able to enhance PBMCs-mediated cell killing even ex vivo when DCs were present.
As expected, CD40L armed virus was more potent in CD40+ EJ and LNCaP cells compared with the unarmed virus. This was probably due to the proapoptotic effect of CD40L on CD40+ cancer cells.28 There was no difference in the oncolytic potency of armed and unarmed virus alone in CD40- cells, suggesting that addition of transgene does not hamper the cell killing capacity of virus, which is in accordance with our previous findings (14).
Ad3-hTERT-E1A-hCD40L and human DCs therapy results in antitumor effects and 100% survival of humanized mice
To mimic the situation in humans, the ability of the virus to enhance DC therapy was studied in mice humanized by injection of human PBMCs intravenously.38,39 Intratumoral injections of Ad3-hTERT-CMV-hCD40L, Ad3-hTERT-E1A, or PBS, and maturated DCs was performed on alternate days. As, the goal of DC vaccines in the clinical use is to use ex vivo “trained” DCs, appropriately activated and loaded with tumor antigen, and thus capable of inducing strong antitumor T-cell responses, we chose to use mature DCs in the in vivo experiment to mimick the clinical setting. Tumor growth was followed until day 44 when the tumor growth in control groups reached the criteria determined by animal regulations. DCs or PBMCs alone were not able to inhibit tumor growth compared with the mock control group (Figure 4A). The group treated with the combination of PBMCs and DCs (Figure 4 and Supplementary Figure 1A) showed some tumor control but only the addition of oncolytic adenovirus (either hCD40L-armed or unarmed) inhibited tumor growth significantly (Figure 4 and Supplementary Figure 1A).
Figure 4.
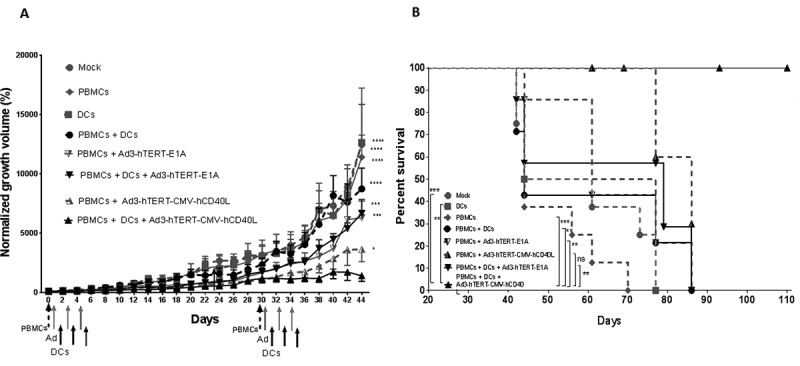
Ad3-hTERT-E1A-hCD40L, human PBMCs, and human DCs therapy enhanced antitumor effects and survival in mice. Antitumor efficacy (A) and cancer specific survival (B) of humanized mice receiving DC therapy and injections of Ad3-hTERT-CMV-hCD40L or the unarmed control virus Ad3-hTERT-E1A. A549 tumors were implanted subcutaneously in immunodeficient SCID mice lacking B and T-cells. To humanize the white blood cell compartment of the mice, 10 × 106 PBMCs were injected intravenously on day 0 (dashed arrow). Viruses (gray arrows) were injected at 1 × 108 VP and DCs (black arrows), 1X106, were injected intratumorally three times alternatively. Tumor growth was monitored every other day. Ad3-hTERT-CMV-hCD40L and DCs therapy significantly reduced tumor growth as compared with other groups. Tumor growth is expressed as normalized tumor volume based on the values from the first day of virus injection. Data is presented as mean ± SEM. ***, P < 0.001; ****, P < 0.0001. 1A by Two-way ANOVA (Tukey’s post-hoc test) and 1B Kaplan-Meier survival was analyzed bylog-rank test.
The double therapy or the triple therapy showed significant anti-tumor effect as compared with mock group (p < 0.0001). However, tumor control was best in the group treated with hCD40L-armed virus, PBMCs, and DCs (Ad3-hTERT-E1A + PBMCs + DCs Vs Ad3-hTERT-CMV-hCd40L + PBMCs + DCs p < 0.001).
Cancer specific survival data (Figure 4B and Supplementary Figure 1B) mirrored tumor control data. Mice treated with hCD40L-armed virus, PBMCs, and DCs showed a significant improvement in survival. Impressively, all mice remained alive until the end of the experiment. Thus, these results indicate that CD40L-armed virus is a potent enhancer of DC therapy when human T cells are present.
DC therapy and Ad3-hTERT-CMV-hCD40L induce anti-tumor immune responses in the tumor microenvironment
To investigate mechanism-of-action, four mice from each group were euthanized one week after the last administration of DCs. Analysis of the microenvironment revealed robust upregulation of DC maturation markers CD83, CD80, and CD86 in tumors treated with triple therapy (Figures 5A-C). Moreover, infiltration of significantly high levels of B and T lymphocytes in the same groups were also observed (Figures 5D and 5E). The immune modulation of the tumor microenvironment towards Th1 phenotype was further confirmed through the presences of high levels of TNF alpha, IFN gamma, IL2, IL12, granzyme B and IL6 in the same groups (Supplementary Figure 3); In summary, our findings suggest that expression of CD40L in the tumor induces maturation of DCs, leading to activation of adaptive immune response against the tumor.
Figure 5.
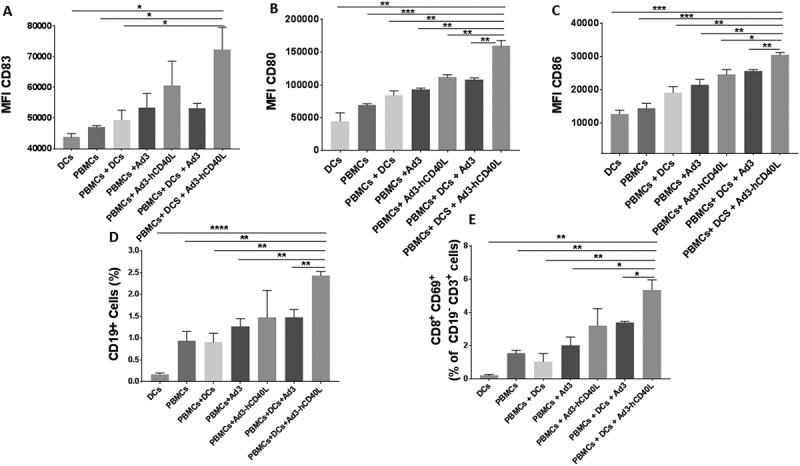
Immune response in the tumor microenvironment. Median fluorescence intensity (MFI) for CD83 (A), CD80 (B) and CD86 (C) of CD11c± populations. Percentage of the CD19 + B cell population (D) and CD8+ CD69+ lymphocytes of the CD19-CD3+ parent population (E). Data is presented as mean ± SEM. *, P < 0.05 **, P < 0.01. ***, P < 0.001, ****, P < 0.0001.
Discussion
The highly immunosuppressive tumor microenvironment is a major obstacle to successful cancer immunotherapy in general and for DC therapy in particular.40–42 Suppression results from complex interplay between soluble factors such as TGF-β, IL10, and VEGF,43–47 cell-bound molecules such as PD-L1, and cellular factors including regulatory T cells, myeloid-derived suppressor cells, and tumor-associated neutrophils.48 Immunosuppression is associated with poor prognosis.16–18 With regard to DC therapy, which is a promising approach with a solid theoretical basis, immunosuppressive factors hamper the ability of DCs to present antigens, thwarting the stimulation of tumor-specific T cells.49 Therefore, DC immunotherapy has not yet been successful enough to become a routine therapy in humans.42
CD40, as a target for cancer immunotherapy, has gained interest due to its capacity for activation of Th1 type immunity through DC maturation.28 Interaction of CD40 with its natural ligand CD40L leads to activation of DCs, which is needed for T-cell activation.50 Without this crucial signal for T-cell priming and proliferation, tumor-infiltrating T cells would undergo apoptosis.36,51,52 Furthermore, CD40-CD40L interaction induces high levels of IL12 which in turn is responsible for the initiation of Th1 responses.53 In addition, the interaction enhances DC capacity to promote IFN-gamma production by T cells.50–53
In preclinical studies, it has been reported that murine CD40L upregulates DC co-stimulatory receptors and induces antitumor immune responses.54,55 In clinical use, CD40L has been used in different forms with encouraging results.27–30,56–58 However, it has also been recognized that systemic administration is suboptimal as normal tissue damage seen, for example, as liver enzyme elevation, limits the concentration that can be achieved in tumors. Nevertheless, this creates the rationale for local production of CD40L, which has been explored in a few human pilot cohorts with promising results.30–59 Although this approach seems to have anti-tumor activity, patients were not cured, providing the rationale for further improvements.30 Of note, the oncolytic platform may provide many advantages over non-replicating vector approaches.28–30
Oncolytic adenoviruses are an attractive platform for cancer immunotherapy due to their tumor-specific replication, ability to infect different tumors, good stability in vivo, and favorable safety profile in humans.60,61 In this study, we studied CD40L-armed adenovirus serotype 3 Ad3-hTERT-CMV-hCD40L. It features the following important aspects: fully serotype 3 to enhance tumor transduction through the intravenous route, tumor selectivity due to the presence of hTERT promoter, and induction of apoptosis in CD40+ tumors.36 As discussed before, the serotype 3 platform may be advantageous to the ubiquitous Ad5 in several ways.25–36 The primary receptor for Ad3, desmoglein-2, is highly expressed in advanced tumors,25–36 allowing enhanced tumor transduction. Moreover, it has been reported that fully Ad3 capsid allows effective intravenous delivery in animals and humans.25–36
Virally expressed CD40L has previously shown to induce apoptosis of CD40+ tumors and also activates antigen-presenting cells.28,36,62 We have shown previously that Ad3-hTERT-CMV-hCD40L virus as well as virally coded hCD40L induces maturation of DCs ex vivo.36 In the present study, we demonstrated the ability of Ad3-hTERT-CMV-hCD40L to facilitate DC therapy in a clinically relevant setting using human DCs, human PBMCs and human tumor cells or xenografts ex vivo and in vivo. The purpose of the ex vivo study was to evaluate the capability of virally produced CD40L to mediate tumor cell killing by enhancing the activation of DCs. Ad3-hTERT-CMV-hCD40L demonstrated significantly higher DC activation seen as high expression of CD80, CD86, and CD83 in comparison to other groups. Furthermore, in co-cultures Ad3-hTERT-CMV-hCD40L and DCs activated CD4+ T cells and CD8+ T cells.
CD40L stimulates and recruits DCs, leading to direct cytotoxic T-cell activation and skewing the immune response towards Th1 phenotype.28 Accordingly, in our study stimulated DCs were able to activate T cells in co-cultures. Cell killing with armed or unarmed virus together with DCs and PBMCs was more prominent compared with single agent treatments. As expected, CD40+ tumor cells treated with Ad3-hTERT-CMV-hCD40L, DCs, and PBMCs were more susceptible to the treatment compared to the CD40- tumor cells, although cell killing was achieved also in this group. This is in accordance with our previous findings, indicating that potential application of this virus is not restricted to CD40+ tumors.36
Next, we tested the ability of Ad3-hTERT-CMV-hCD40L to sensitize the tumor microenvironment to DC therapy in vivo. The specificity of Ad3-hTERT-CMV-hCD40L virus and its human transgene hCD40L restricted the choice of animal model to immunodeficient SCID mice bearing human xenografts, as human CD40L would not activate mouse CD40.28 Key components of the human immune system were introduced by intravenous injections of human PBMCs (SCID mice lack murine B and T cells). We were also able to demonstrate the in vivo ability of Ad3-hTERT-CMV-hCD40L to polarize an immunosuppressive microenvironment towards a more immunogenic phenotype as upregulation of Th1 immune-stimulatory cytokines was observed. Even the unarmed Ad3-hTERT-E1A virus alone was able to stimulate DCs as seen by high expression of CD80, CD86, and CD83 and to activate T-cell and B-cell responses. The engagement of CD40 expressed on B cells and CD40L is also important for the initiation of humoral immune response. Moreover, it has been shown that this interaction leads to germinal center formation, antibody isotype switching and affinity maturation.63 Thus, CD40 pathway is essential for the survival of many cell types and is crucial in the generation of humoral immune response.22–64 These responses, however, were more pronounced with Ad3-hTERT-CMV-hCD40L administered with DCs leading to the best tumor control and prolonged survival. We think that it is a promising starting point for human translation that death due to cancer could be prevented in 100% of mice in the key experimental group.
In summary, we provide preclinical proof of principle for using Ad3-hTERT-CMV-hCD40L in cancer patients receiving DC therapy. Thus, Ad3-hTERT-CMV-hCD40L is a promising candidate for human clinical trials.
Acknowledgments
We thank Minna Oksanen and Susanna Grönberg-Vähä-Koskela for expert assistance. This study was supported by University of Helsinki Doctoral Programme in Clinical Research (KLTO), Jane and Aatos Erkko Foundation, HUCH Research Funds (EVO), Sigrid Juselius Foundation, Finnish Cancer Organizations, University of Helsinki, TILT Biotherapeutics Ltd, European Commission Marie Curie Innovative Training Network (ITN) grant VIRION (H2020-MSCA-ITN-2014 project number 643130).
Conflict of interest
A.H. and O.H. are shareholders in Targovax ASA and TILT Biotherapeutics Ltd. A.H., S.S., M.S., R.H., V.C.C., and J.M.S. are employees of TILT Biotherapeutics Ltd.
Abbreviations
| ACK | Ammonium-Chloride-Potassium lysis buffer |
| Ad5 | Serotype 5 adenoviruses |
| Ad3 | Serotype 3 adenoviruses |
| APCs | Antigen presenting cells |
| BD | Becton Dickinson |
| CBA | Cytometric bead array |
| DCs | Dendritic cells |
| DMEM | Dulbecco’s modified Eagle’s medium |
| GMCSF | Granulocyte macrophage colony stimulation factor |
| hCD40L | Human CD40 Ligand |
| hTERT | Human telomerase reverse transcriptase |
| IFN-gamma | Interferon gamma |
| IL4 | Interleukin 4 |
| IL6 | Interleukin 6 |
| IL2 | Interleukin 2 |
| IL10 | Interleukin 10 |
| IL12 | Interleukin 12 |
| LPS | Lipopolysaccharide |
| NK | Natural killer cells |
| rhCD40L | Recombinant human CD40 Ligand |
| Th1 | T helper type 1 cells |
| Th2 | T helper type 2 cells |
| TGF-β | Transforming growth factor - beta |
| TME | Tumor microenvironment |
| VEGF | Vascular endothelial growth factor |
Supplemental data for this article can be here.
References
- 1.Kim Y, Clements DR, Sterea AM, Jang HW, Gujar SA.. PWK L: dendritic cells in oncolytic virus-based anti-cancer therapy In: Chiocca EA, Lamfers MLM, Eds. Viruses. 2015. Vol. 7 p. 6506–6525. Ocolytic Viruses: Viruses / MDPI AG, Basel, Switzerland. doi: 10.3390/v7122953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman RM. Hemmi H: dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. [DOI] [PubMed] [Google Scholar]
- 3.Fong L. Engleman EG: dendritic cells in cancer immunotherapy. Annu Rev Immunol. 2000;18:245–273. doi: 10.1146/annurev.immunol.18.1.245. [DOI] [PubMed] [Google Scholar]
- 4.Rabinovich GA, Gabrilovich D. Sotomayor EM: immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gervais A, Leveque J, Bouet-Toussaint F, Burtin F, Lesimple T, Sulpice L, Patard JJ, Genetet N. Catros-Quemener V: dendritic cells are defective in breast cancer patients: a potential role for polyamine in this immunodeficiency. Breast Cancer Res. 2005;7:R326–335. doi: 10.1186/bcr1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerundolo V, Hermans IF. Salio M: dendritic cells: a journey from laboratory to clinic. Nat Immunol. 2004;5:7–10. doi: 10.1038/ni0104-7. [DOI] [PubMed] [Google Scholar]
- 7.Wong KK, Li WA, Mooney DJ. Dranoff G: advances in Therapeutic Cancer Vaccines. Adv Immunol. 2016;130:191–249. doi: 10.1016/bs.ai.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Lesterhuis WJ, Aarntzen EH, De Vries IJ, Schuurhuis DH, Figdor CG, Adema GJ. Punt CJ: dendritic cell vaccines in melanoma: from promise to proof? Crit Rev Oncol Hematol. 2008;66:118–134. doi: 10.1016/j.critrevonc.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Caballero-Banos M, Benitez-Ribas D, Tabera J, Varea S, Vilana R, Bianchi L, Ayuso JR, Pages M, Carrera G, Cuatrecasas M, et al. Phase II randomised trial of autologous tumour lysate dendritic cell plus best supportive care compared with best supportive care in pre-treated advanced colorectal cancer patients. Eur J Cancer. 2016;64:167–174. doi: 10.1016/j.ejca.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Brocker EB, Grabbe S, Rittgen W, Edler L, Sucker A, et al. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol. 2006;17:563–570. doi: 10.1093/annonc/mdj138. [DOI] [PubMed] [Google Scholar]
- 11.Cho DY, Yang WK, Lee HC, Hsu DM, Lin HL, Lin SZ, Chen CC, Harn HJ, Liu CL, Lee WY, et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: a phase II clinical trial. World Neurosurg. 2012;77:736–744. doi: 10.1016/j.wneu.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 12.Sakai K, Shimodaira S, Maejima S, Sano K, Higuchi Y, Koya T, Sugiyama H. Kazuhiro Hongo: clinical effect and immunological response in patients with advanced malignant glioma treated with WT1-pulsed dendritic cell-based immunotherapy: A report of two cases. Interdisciplinary Neurosurgery. 2017;9:24–29. doi: 10.1016/j.inat.2017.02.004. [DOI] [Google Scholar]
- 13.Cui Y, Yang X, Zhu W, Li J, Wu X. Pang Y: immune response, clinical outcome and safety of dendritic cell vaccine in combination with cytokine-induced killer cell therapy in cancer patients. Oncol Lett. 2013;6:537–541. doi: 10.3892/ol.2013.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, Wang Q, Li WF, Wang HY, Zhang HJ. Zhu JJ: different antitumor immunity roles of cytokine activated T lymphocytes from naive murine splenocytes and from dendritic cells-based vaccine primed splenocytes: implications for adoptive immunotherapy. Eksp Onkol. 2004;26:55–62. [PubMed] [Google Scholar]
- 15.Chang XH, Cheng HY, Cheng YX, Ye X, Guo HF, Fu TY, Zhang L, Zhang G. Cui H: [specific immune cell therapy against ovarian cancer in vivo and in vitro]. Ai Zheng. 2008;27:1244–1250. [PubMed] [Google Scholar]
- 16.Saito H, Tsujitani S, Oka S, Kondo A, Ikeguchi M, Maeta M. Kaibara N: an elevated serum level of transforming growth factor-beta 1 (TGF-beta 1) significantly correlated with lymph node metastasis and poor prognosis in patients with gastric carcinoma. Anticancer Res. 2000;20:4489–4493. [PubMed] [Google Scholar]
- 17.Hasegawa Y, Takanashi S, Kanehira Y, Tsushima T, Imai T. Okumura K: transforming growth factor-beta1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer. 2001;91:964–971. [PubMed] [Google Scholar]
- 18.Ghellal A, Li C, Hayes M, Byrne G, Bundred N. Kumar S: prognostic significance of TGF beta 1 and TGF beta 3 in human breast carcinoma. Anticancer Res. 2000;20:4413–4418. [PubMed] [Google Scholar]
- 19.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 20.Tahtinen S, Gronberg-Vaha-Koskela S, Lumen D, Merisalo-Soikkeli M, Siurala M, Airaksinen AJ, Vaha-Koskela M. Hemminki A: adenovirus improves the efficacy of adoptive T-cell therapy by recruiting immune cells to and promoting their activity at the tumor. Cancer Immunol Res. 2015;3:915–925. doi: 10.1158/2326-6066.CIR-14-0220-T. [DOI] [PubMed] [Google Scholar]
- 21.Lawler SE, Speranza MC, Cho CF. Chiocca EA: oncolytic viruses in cancer treatment: A review. JAMA Oncol. 2017;3:841–849. doi: 10.1001/jamaoncol.2016.2064. [DOI] [PubMed] [Google Scholar]
- 22.Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y. Noelle RJ: molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taipale K, Liikanen I, Juhila J, Turkki R, Tahtinen S, Kankainen M, Vassilev L, Ristimaki A, Koski A, Kanerva A, et al. Chronic activation of innate immunity correlates with poor prognosis in cancer patients treated with oncolytic adenovirus. Mol Ther. 2016;24:175–183. doi: 10.1038/mt.2015.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parviainen S, Ahonen M, Diaconu I, Hirvinen M, Karttunen A, Vaha-Koskela M, Hemminki A. Cerullo V: CD40 ligand and tdTomato-armed vaccinia virus for induction of antitumor immune response and tumor imaging. Gene Ther. 2014;21:195–204. doi: 10.1038/gt.2013.73. [DOI] [PubMed] [Google Scholar]
- 25.Hemminki O, Diaconu I, Cerullo V, Pesonen SK, Kanerva A, Joensuu T, Kairemo K, Laasonen L, Partanen K, Kangasniemi L, et al. Ad3-hTERT-E1A, a fully serotype 3 oncolytic adenovirus, in patients with chemotherapy refractory cancer. Mol Ther. 2012;20:1821–1830. doi: 10.1038/mt.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cervera-Carrascon V, Siurala M, Santos JM, Havunen R, Tähtinen S, Karell P, Sorsa S, Kanerva A. Hemminki A: tNFa and IL-2 armed adenoviruses enable complete responses by anti-PD-1 checkpoint blockade. Oncoimmunology. 2018;7. doi: 10.1080/2162402X.2017.1412902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vonderheide RH, Dutcher JP, Anderson JE, Eckhardt SG, Stephans KF, Razvillas B, Garl S, Butine MD, Perry VP, Armitage RJ, et al. Phase I study of recombinant human CD40 ligand in cancer patients. J Clin Oncol. 2001;19:3280–3287. doi: 10.1200/JCO.2001.19.13.3280. [DOI] [PubMed] [Google Scholar]
- 28.Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, Bramante S, Parviainen S, Kanerva A, Loskog AS, et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012;72:2327–2338. doi: 10.1158/0008-5472.CAN-11-2975. [DOI] [PubMed] [Google Scholar]
- 29.Sun Y, Peng D, Lecanda J, Schmitz V, Barajas M, Qian C. Prieto J: in vivo gene transfer of CD40 ligand into colon cancer cells induces local production of cytokines and chemokines, tumor eradication and protective antitumor immunity. Gene Ther. 2000;7:1467–1476. doi: 10.1038/sj.gt.3301264. [DOI] [PubMed] [Google Scholar]
- 30.Pesonen S, Diaconu I, Kangasniemi L, Ranki T, Kanerva A, Pesonen SK, Gerdemann U, Leen AM, Kairemo K, Oksanen M, et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res. 2012;72:1621–1631. doi: 10.1158/0008-5472.CAN-11-3001. [DOI] [PubMed] [Google Scholar]
- 31.Alemany R, Balague C. Curiel DT: replicative adenoviruses for cancer therapy. Nat Biotechnol. 2000;18:723–727. doi: 10.1038/77283. [DOI] [PubMed] [Google Scholar]
- 32.Dummer R, Hassel JC, Fellenberg F, Eichmuller S, Maier T, Slos P, Acres B, Bleuzen P, Bataille V, Squiban P, et al. Adenovirus-mediated intralesional interferon-gamma gene transfer induces tumor regressions in cutaneous lymphomas. Blood. 2004;104:1631–1638. doi: 10.1182/blood-2004-01-0360. [DOI] [PubMed] [Google Scholar]
- 33.Sangro B, Mazzolini G, Ruiz J, Herraiz M, Quiroga J, Herrero I, Benito A, Larrache J, Pueyo J, Subtil JC, et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol. 2004;22:1389–1397. doi: 10.1200/JCO.2004.04.059. [DOI] [PubMed] [Google Scholar]
- 34.Hemminki O, Bauerschmitz G, Hemmi S, Lavilla-Alonso S, Diaconu I, Guse K, Koski A, Desmond RA, Lappalainen M, Kanerva A, et al. Oncolytic adenovirus based on serotype 3. Cancer Gene Ther. 2011;18:288–296. doi: 10.1038/cgt.2010.79. [DOI] [PubMed] [Google Scholar]
- 35.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 36.Zafar S, Parviainen S, Siurala M, Hemminki O, Havunen R, Tahtinen S, Bramante S, Vassilev L, Wang H, Lieber A, et al. Intravenously usable fully serotype 3 oncolytic adenovirus coding for CD40L as an enabler of dendritic cell therapy. Oncoimmunology. 2017;6:e1265717. doi: 10.1080/2162402X.2016.1265717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wen FT, Thisted RA, Rowley DA. Schreiber H: A systematic analysis of experimental immunotherapies on tumors differing in size and duration of growth. Oncoimmunology. 2012;1:172–178. doi: 10.4161/onci.1.2.18311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mosier DE, Gulizia RJ, Baird SM. Wilson DB: transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. 1988;335:256–259. doi: 10.1038/335256a0. [DOI] [PubMed] [Google Scholar]
- 39.Brehm MA, Shultz LD. Greiner DL: humanized mouse models to study human diseases. Curr Opin Endocrinol Diabetes Obes. 2010;17:120–125. doi: 10.1097/MED.0b013e328337282f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 41.Banchereau J. Palucka AK: dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 42.Lim DS, Kim JH, Lee DS, Yoon CH. Bae YS: DC immunotherapy is highly effective for the inhibition of tumor metastasis or recurrence, although it is not efficient for the eradication of established solid tumors. Cancer Immunol Immunother. 2007;56:1817–1829. doi: 10.1007/s00262-007-0325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amoils KD. Bezwoda WR: TGF-beta 1 mRNA expression in clinical breast cancer and its relationship to ER mRNA expression. Breast Cancer Res Treat. 1997;42:95–101. [DOI] [PubMed] [Google Scholar]
- 44.Asselin-Paturel C, Echchakir H, Carayol G, Gay F, Opolon P, Grunenwald D, Chouaib S. Mami-Chouaib F: quantitative analysis of Th1, Th2 and TGF-beta1 cytokine expression in tumor, TIL and PBL of non-small cell lung cancer patients. Int J Cancer. 1998;77:7–12. [DOI] [PubMed] [Google Scholar]
- 45.Conrad CT, Ernst NR, Dummer W, Brocker EB. Becker JC: differential expression of transforming growth factor beta 1 and interleukin 10 in progressing and regressing areas of primary melanoma. J Exp Clin Cancer Res. 1999;18:225–232. [PubMed] [Google Scholar]
- 46.Yang L. Carbone DP: tumor-host immune interactions and dendritic cell dysfunction. Adv Cancer Res. 2004;92:13–27. doi: 10.1016/S0065-230X(04)92002-7. [DOI] [PubMed] [Google Scholar]
- 47.Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D. Carbone DP: production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–1103. [DOI] [PubMed] [Google Scholar]
- 48.Lindau D, Gielen P, Kroesen M, Wesseling P. Adema GJ: the immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138:105–115. doi: 10.1111/imm.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobie JJ, Wu RS, Kurt RA, Lou S, Adelman MK, Whitesell LJ, Ramanathapuram LV, Arteaga CL. Akporiaye ET: transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003;63:1860–1864. [PubMed] [Google Scholar]
- 50.Korniluk A, Kemona H. Dymicka-Piekarska V: multifunctional CD40L: pro- and anti-neoplastic activity. Tumour Biol. 2014;35:9447–9457. doi: 10.1007/s13277-014-2407-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I. Banchereau J: activation of human dendritic cells through CD40 cross-linking. J Exp Med. 1994;180:1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peguet-Navarro J, Dalbiez-Gauthier C, Rattis FM, Van Kooten C, Banchereau J. Schmitt D: functional expression of CD40 antigen on human epidermal langerhans cells. J Immunol. 1995;155:4241–4247. [PubMed] [Google Scholar]
- 53.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A. Alber G: ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grewal IS. Flavell RA: CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 55.Grewal IS, Xu J. Flavell RA: impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature. 1995;378:617–620. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- 56.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruter J, Antonia SJ, Burris HA, Huhn RD. Vonderheide RH: immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10:983–993. doi: 10.4161/cbt.10.10.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takahashi S, Rousseau RF, Yotnda P, Mei Z, Dotti G, Rill D, Hurwitz R, Marini F, Andreeff M. Brenner MK: autologous antileukemic immune response induced by chronic lymphocytic leukemia B cells expressing the CD40 ligand and interleukin 2 transgenes. Hum Gene Ther. 2001;12:659–670. doi: 10.1089/104303401300057360. [DOI] [PubMed] [Google Scholar]
- 59.Loskog A, Maleka A, Mangsbo S, Svensson E, Lundberg C, Nilsson A, Krause J, Agnarsdottir M, Sundin A, Ahlstrom H, et al. Immunostimulatory AdCD40L gene therapy combined with low-dose cyclophosphamide in metastatic melanoma patients. Br J Cancer. 2016;114:872–880. doi: 10.1038/bjc.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cerullo V, Vaha-Koskela M. Hemminki A: oncolytic adenoviruses: A potent form of tumor immunovirotherapy. Oncoimmunology. 2012;1:979–981. doi: 10.4161/onci.20172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, Nokisalmi P, Raki M, Rajecki M, Guse K, et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther. 2010;18:1874–1884. doi: 10.1038/mt.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noguchi M, Imaizumi K, Kawabe T, Wakayama H, Horio Y, Sekido Y, Hara T, Hashimoto N, Takahashi M, Shimokata K, et al. Induction of antitumor immunity by transduction of CD40 ligand gene and interferon-gamma gene into lung cancer. Cancer Gene Ther. 2001;8:421–429. doi: 10.1038/sj.cgt.7700320. [DOI] [PubMed] [Google Scholar]
- 63.Danese S, Sans M. Fiocchi C: the CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut. 2004;53:1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bishop GA, Moore CR, Xie P, Stunz LL. Kraus ZJ: TRAF proteins in CD40 signaling. Adv Exp Med Biol. 2007;597:131–151. doi: 10.1007/978-0-387-70630-6_11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.