

Abstract
Over the past six decades the inflation-adjusted cost to bring a new drug to market has been increasing constantly and doubles every 9 years - now reaching in excess of $2.5 billion. Overall, the likelihood of FDA approval for a drug (any disease indication) that has entered phase I clinical trials is a mere 9.6%, with the approval rate for oncology far below average at only 5.1%. Lack of efficacy or toxicity is often not revealed until the later stages of clinical trials, despite promising preclinical data. This indicates that the current in vitro systems for drug screening need to be improved for better predictability of in vivo outcomes. Microphysiological systems (MPS), or bioengineered 3D microfluidic tissue and organ constructs that mimic physiological and pathological processes in vitro, can be leveraged across preclinical research and clinical trial stages to transform drug development and clinical management for a range of diseases. Here we review the current state-of-the-art in 3D tissue-engineering models developed for cancer research, with a focus on tumor-on-a-chip, or tumor chip, models. From our viewpoint, tumor chip systems can advance innovative medicine to ameliorate the high failure rates in anti-cancer drug development and clinical treatment.
1. Introduction
Cancer is the second leading cause of death in the United States, with 1,500 people dying from the disease every day.1 The high rate of mortality and morbidity for this disease highlights the need for more effective therapies. Despite the high incidence, drug discovery has been slow to translate into clinical benefit for patients and the paucity of effective treatments in oncology is consequent to the high attrition rate during drug development.2 Indeed, oncology has the lowest success rate of any therapeutic area with only 5.1% of anti-cancer drugs entering phase I clinical trials ultimately gaining FDA approval.3 For every 10,000 compounds that proceed through research and development, only 1 will ultimately become FDA-approved for market use.2,4 To bring a new drug to market for any disease indication can take in excess of 10 years and $2.5 billion.5 Despite promising preclinical data, the majority of drugs fail during clinical stages due to issues with efficacy (>50%) and/or safety (>10%).6 One of the main reasons for such a high attrition rate is that current methods for disease modeling and drug screening are poor predictors of human outcomes. Given the fact that about two thirds of drug development cost occurs during clinical trial phases, the ability to more accurately identify lead candidates and eliminate ineffective drugs earlier in the process will save a significant amount of time and resources, reduce human risk, and accelerate the translation of effective therapies to the clinic.7,8
While animal models have advanced our understanding of complex diseases such as cancer and provide essential readouts of organism-level drug effects in vivo, these same models are expensive, time consuming and often fail to predict human responses during clinical trials.9,10 In fact, it is estimated that less than 8% of successful animal trials for cancer drugs translate to successful human clinical trials, primarily due to species-specific differences in physiology and cell biology.8 Furthermore, animal models allow only limited manipulation to study the mechanisms at play during disease progression or therapeutic response.11,12 On the other hand, while standard 2D cell monocultures used for drug screening are cost-efficient and simple to use, such monolayer cultures fail to recapitulate the 3D cellular spatial arrangement and microenvironment of in vivo tissues leading to poor predictive power. Cell growth in 2D versus 3D environments not only promotes changes in cellular morphology, function, response to stimuli, and gene expression patterns, but also leads to drug responses that vary dramatically from the in vivo situation.13 Translation of results obtained from cell culture studies into animal trials during preclinical stages of drug development is difficult because of the inability of these oversimplified in vitro models to simulate the complex and heterogeneous tissue architectures of their in vivo counterparts.
To bridge the translational gap between current preclinical models and clinical outcomes, in vitro platforms that better mimic native tissue physiology are undergoing rapid evolution. Advances in tissue engineering have assisted the development of functional, miniaturized human healthy or diseased organs termed microphysiological systems (MPS, also known as ‘organ-on-a-chip’, organ chip or tissue chip).8,14,15 MPS integrate microfluidics, microfabrication techniques, biomaterials and tissue engineering to create tissue or organ constructs via co-culture of multiple cell types, often embedded in a hydrogel or extracellular matrix (ECM), within a palm-sized device. By leveraging microfluidics technology, physiological relevance can be built into the MPS to model the dynamic microenvironment and inter-cellular interactions of complex tissues or organ-systems. High-fidelity modeling of essentially any tissue in the human body to reproduce corresponding functional units is now possible. For example, microscaled platforms have been developed to model lung16, liver17, brain18, endocrine tissues19, intestine20, kidney21,22, and heart23, among many others. In addition to these micro-organ platforms, MPS technology offers new opportunities for building and applying functional three-dimensional in vitro human tumor models for oncology research.
Besides 3D cellular assembly, the tumor microenvironment consists of a complex combination of ECM, stromal cells and interstitial fluids. This complex composition influences the tumor cell phenotype via mechanical and biochemical factors that ultimately contribute to tumor growth.24 To recreate the tumor microenvironment, tumor chip models have been engineered to incorporate stromal cells such as pericytes25, cancer associated fibroblasts26, smooth muscle cells and myofibroblasts27, mesenchymal stem cells28, as well as endothelial cells29,30 to form a vascular compartment, either self-organized29,31–38, or spatially organized by design.39,40 Rudimentary (natural) immune systems have also been incorporated into tumor chips through the addition of macrophages27, dendritic cells41 and T cells.42,43 Fully autologous systems are still on the horizon. Tumor chips have been arrayed for high throughput drug screening applications44, optimized for cancer metastasis studies such as tumor cell extravasation and micrometastasis generation45, and populated with patient-specific cells for personalized medicine approaches.42 Organotypic tumor chips capable of recapitulating complex organ-level patterns of cancer growth, dissemination and therapeutic response observed in patients are quickly advancing.46,47 Multi-MPS have been generated for the detailed study of drug pharmacokinetics (PK), pharmacodynamics (PD) and toxicity.15,48–50 In particular, tumor chip models allow experimental manipulation and well-controlled real-time study of dynamic interactions among tumor cells, stromal cells and the tumor microenvironment, which is less simple to accomplish using regular tissue culture and animal models.
In this review, we first briefly describe the strengths and limitations of current model systems and highlight critical features of the tumor microenvironment that contribute to disease progression. We then review the current state-of-the-art in 3D tissueengineering models developed for cancer research and outline how the technology is revolutionizing disease modeling, drug screening and personalized medicine for oncology. Within this context, we critically evaluate limitations in current tumor chip models and address challenges in the field by proposing solutions to accelerate the translation of tumor chips into mainstream use.
2. Strengths and Limitations of Standard Preclinical Models
2D Monocultures
While assays derived from 2D monolayers of cell lines grown on plastic are low cost, easy to use and high throughput, these same models have limited predictive capability since they fail to mimic natural human physiology.5,51 Differences in cell morphology, differentiation, proliferation, viability, response to stimuli, metabolism, gene/protein expression and drug sensitivity are observed when cells, previously cultured in 2D, are moved to a 3D environment.52–54 This is not surprising considering that, with few exceptions, human organs develop and maintain their specific functions owing to the 3D structure they adopt. 2D cell cultures have vastly different substrate topography, stiffness, and architecture than in vivo counterparts, and fail to recapitulate the cell-cell and cell-matrix interactions of endogenous tissues.55 Furthermore, 2D culture places a selective pressure on cells that can cause genetic drift and loss of heterogeneity, resulting in substantial changes to their original phenotypes.56
Another major limitation of 2D assays is that artificial in vitro conditions for growing cells on plastic dishes prevents investigation and therapeutic targeting of many cell behaviors that lead to disease progression and treatment failure, such as immune suppression and metastasis.13 Moreover, 2D assay properties may lead to false-positive selection of drugs that have only limited efficacy in vivo, often due to the greater heterogeneity seen in more complex environments, where stem cells and quiescent cells may cycle more slowly and demonstrate resistance to cell cycle arrest.57 Since the tumor microenvironment and tumor-stromal interactions that create a barrier to drug delivery in vivo are not modeled in 2D cell culture, drugs that look promising in cell assays may not be able to reach target cells in vivo.58 Drug screening is typically performed in 2D on cells growing as monocultures, despite evidence that direct association of tumor cells with stroma renders populations of cancer cells resistant to chemotherapeutic drugs. 59–63 The intrinsic limitations of 2D cell culture models have prompted the development of three-dimensional (3D) models that can provide a cellular microenvironment and physiologic context that more closely mimics the microenvironment observed in native tissues. This feature is critical for drug testing since environmental cues can have profound effects on cell functions, which often affect cellular responses to drugs.
3D Spheroids
By maintaining tumor cells in a native 3D conformation, spheroid cultures address certain limitations in 2D cell models. Spheroids develop distinct areas of rapidly dividing cells on the outer layer vs necrotic and slow cycling cells at the center and intermediate layers, respectively.64 In this regard, spheroid cultures more accurately mimic the metabolic gradients and drug resistance of in vivo tumors than standard 2D cultures.65 These models have progressively evolved from the simplest form comprising homogeneous epithelial cell populations to 3D co-cultures that can be embedded in matrix with variable ECM properties and derived from numerous cell sources (such as established cell lines, patient-derived cells, and stem cells). Three-dimensional models provide sound insight into the differences between normal and malignant epithelial cells and serve as an excellent basis for determining the intermediate steps that are responsible for the transition from a normal to a malignant fate.66
While spheroid cultures can recapitulate disease characteristics such as chemo-/radio-resistance67, some aspects of tumor cell heterogeneity and invasive/migratory potential of tumor cells68, there are several limitations in these models. Spheroids are useful models of avascular tumors but lack the structure and complexity seen in vascularized tumors in vivo.69 As a result, spheroid cultures are not able to fully recapitulate the spectrum of cell phenotypes within the tumor milieu. Due to static culture conditions, cells in spheroid models do not experience the same mechanical forces that would be expected in vivo and lack of dynamic flow also prevents long-term culture for drug sensitivity and toxicity studies.70 Thus several important factors of the tumor microenvironment are not reproduced and cannot be studied in these models. Another significant limitation is that many tumor types, especially those with a highly invasive phenotype, do not readily form spheroids and so cannot be assayed in these cultures (e.g. MDA-MB-231 breast cancer cell line). To address these shortcomings, tumor chips represent more sophisticated tissue-based culture models that mimic critical features not represented in traditional monolayer or spheroid cultures.
Animal Models
Animal surrogates of human disease are a necessary component in the drug development pipeline because these in vivo models can emulate physiological complexity at the whole-organism level. Although animal studies have advanced our understanding of complex diseases such as cancer, a major shortcoming of these models is that they often have only limited translatability to humans. This is evidenced by the fact that >90% of drugs that show promise during animal studies fail in clinical trials, suggesting that current animal models fail in critical ways to fully recapitulate the human disease condition.71 Species-specific differences between mouse and human in physiology, and cell biology, variations in the homology of molecular targets, and differences in the number of required key mutations to develop tumors can impede clinical translation of preclinical results.72 Further, immunological and inflammatory response vastly differs in the murine model.73,74 During preclinical drug development, tumor cells are often injected subcutaneously into the flanks of severely immunocompromised mice to generate xenograft tumors for in vivo testing of candidate drugs. This procedure greatly facilitates tumor monitoring by palpation and visual inspection, but is poorly representative of tumor development in the native tissue microenvironment. Less commonly, transplants are generated orthotopically, or in the original site of cancer, to better mimic tumor-specific disease evolution, although these procedures can present technical challenges both in establishing and monitoring the xenograft tumor.75 Moreover, tumors generated from transplantation in mice will inherently contain non-human host cells. In contrast, tumor chips can be composed entirely of human cells and tissue-specific factors of the microenvironment can be readily incorporated into the chip by design to better mimic the organ site of tumor origin.
To better replicate the heterogeneity of human tumors, patient-derived xenograft (PDX) tumor models that are established from transplantation of primary tumors are increasingly being adopted for drug screening, disease modeling and personalized medicine applications.75,76 These models are limited by the small amounts of patient-derived tissue available, thus it can take months to expand and serially transplant PDX to generate sufficient replicates for in vivo drug testing, and many primary tumors simply fail to engraft from the outset. While there have been increasing efforts to use PDX as models to study drug response, recent evidence suggests that PDX may not recapitulate parent tumor characteristics as faithfully as initially assumed.77 Indeed, Ben-David et al56 assessed copy number alterations (CNA) in 1,100 PDX samples derived from 24 cancer types and analyzed PDX genomic stability through the process of serial engraftment, compared to human primary tumors and primary-derived cell lines. Interestingly, individual PDX models often gained or lost CNAs and mutations in cancer-related genes during PDX passage, quickly diverging genomically from the parental tumors from which they derived. These changes in genomic landscape were comparable to those observed in primary-derived cells maintained and passaged in vitro, which included loss of recurrent chromosomal aberrations that are believed to have casual roles in tumor progression and therapy response. These results suggest that primary-derived cells are critically influenced by the amount of time maintained outside of the body, and that MPS can address this limitation by providing an in vivo-like environment amenable to more rapid analysis.
While severely immunocompromised mice are necessary to allow engraftment of human tumors, such models preclude the study and therapeutic targeting of interactions between adaptive immune cells and tumor cell populations. Humanized mouse models are being developed to address this concern, whereby human immune components are incorporated to partially reconstitute the immune-inflammatory response during disease progression.73,78 Still, appropriate mouse models may not be readily available for certain applications and are impractical for routine drug screening.65,79 Another important limitation of animal models is that only limited experimental manipulation can be performed to interrogate mechanisms of disease progression, due to the complexity of generating knockout animals and difficulty investigating dynamic cell-cell interactions in vivo. Furthermore, spatially random and temporally rapid events such as tumor cell intravasation that can be easily visualized in real-time using tumor chips cannot be easily interrogated using animal models. Transgenic mouse models have been genetically engineered to partially recapitulate aspects of human carcinogenesis in situ, however evidence suggests key differences in the signaling requirements for transformation of mouse and human cells.80,81 Additional considerations for the use of different types of mouse models in cancer research have been reviewed previously.82–84
Although animal models represent a necessary component in the drug development pipeline and have provided useful information on disease processes, these same models require tremendous time and resources and thus represent a low throughput model system. Even so, approximately 27 million vertebrate animals are used for research purposes in the US every year, highlighting the ethical burden associated with these studies.85 Indeed, the US National Research Council recommends that animal model based tests be replaced as soon as possible with an increased emphasis on epidemiology, in silica models and in vitro human cell-based assays, including MPS.10 This is in accordance with federal and ethical guidelines originating from the 3R’s initiative to replace, reduce and refine the use of animals in scientific and medical research.86
3. Overview of the Tumor Microenvironment
To create a realistic tumor model, several key features of an actual tumor must be replicated. A tumor comprises numerous cell types in a dynamic tumor microenvironment wherein a host of biochemical and biophysical cues dictate cellular responses. Although tumor genetic heterogeneity remains a significant barrier to effective cancer eradication, it is now widely recognized that the tumor microenvironment plays an equally critical role in cancer initiation, progression and drug resistance, thus representing an attractive therapeutic target independent of the myriad genomic aberrations unique to each tumor.58,59,81,87,88 The tumor microenvironment serves as a complex ecosystem containing diverse cellular and non-cellular components that modulate the proliferation, function and fate of cancer cells via bi-directional communication.89,90 Cell signaling within the tumor microenvironment occurs through release of soluble factors in the interstitial fluid, cell-cell or cell-ECM adhesion, and mechanical forces. These mechanical forces are generated by fluid forces, shear stresses, interstitial flow and ECM organization, composition and stiffness.70,91,92 The functional association of tumor cells with surrounding tissues leads to the development of a new pathological ‘organ’ that continually changes as malignancy progresses and in response to treatment.90
Like normal tissues, tumors require delivery of oxygen and nutrients, and elimination of metabolic wastes, via the vascular supply. In the absence of new vasculature, central necrosis will develop in a solid tumor due to limited diffusion of oxygen to the tumor core resulting in hypoxia, high acidity and the accumulation of wastes.68 Before undergoing cell death, cells at the core adapt their metabolism and become quiescent in order to maintain homeostasis. Quiescent tumor cells are difficult to eradicate with conventional therapies that target rapidly proliferating cells, such as radiation and chemotherapy.47 Gradients of nutrients, oxygen and cytokines develop as the tumor mass grows, often leading to zonation within the tumor whereby viable, proliferating cells survive at the periphery of the tumor while quiescent and necrotic cells are harbored at the center. Necrotic cells can also release growth-stimulating factors, such as IL-1α, that can directly stimulate neighboring cells to proliferate, thus contributing to disease progression.81 Overexpression of hypoxia inducible factors such as HIF-1 promotes the expression of hypoxia-inducible genes that enhance cell survival, alter glucose metabolism, increase vascular permeability and inflammation, and induce new vessel sprouting via angiogenesis.89 Such genes include phosphoglycerate kinase 1 (PGK-1), glucose transporter 1 (Glut-1), lactate dehydrogenase A (LdhA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), vascular endothelial growth factor (VEGF) and nitric oxide synthetase (iNOS).93 Hypoxia also promotes cancer metastasis of solid tumors via a step-wise, physical process that is heavily influenced by the ECM density and composition of the surrounding tumor microenvironment. Synergistic interactions between malignant cells and the tumor microenvironment lead to active ECM remodeling that further promotes the recruitment of fibroblasts, immune-inflammatory cells, and perivascular cells to facilitate cancer cell dissemination and invasion to distant organs.63
Bissell and colleagues demonstrated the importance of faithfully recapitulating the tissue-specific tumor microenvironment in a series of seminal studies.53,54,94,95 when mammary epithelial cells were cultured on laminin-rich reconstituted basement membrane, the cells self-assembled into spherical structures with a central lumen and produced milk protein in response to stimuli, similar to normal mammary acini. However, when the same mammary luminal epithelial cells were cultured in 3D collagen I gels, the self-assembled spheres failed to form a central lumen, demonstrated inverse cell polarity and did not produce milk protein. Interestingly, the physiological phenotype (i.e. lumen formation and cell polarity) could be restored if the mammary luminal epithelial cells were co-cultured with myoepithelial cells that could deposit the basement membrane in situ, suggesting that the composition of the ECM is a critical determinant of tissue structure and function.63 Further, our work has demonstrated the profound effect of ECM composition and stiffness on cell behavior within the tumor microenvironment. By extracting and comparing ECM from normal human colon tissue and colon tumor metastases, we found differences in protein composition and stiffness between the two reconstituted matrices with overrepresentation of a number of ECM proteins (e.g. collagens IV and XIV, laminin) in the tumor ECM as well as a 3-fold increase in stiffness compared to normal ECM.88 In an in vitro assay of vascularized tumors whereby tumor cells were co-cultured with endothelial cells and fibroblasts in the reconstituted matrices, vascular network formation and tumor growth were significantly increased in the tumor ECM compared to normal ECM, and tumor cells exhibited increased glycolysis. When introduced in vivo, tumor ECM promoted enhanced vascularization to the cancer cells.88 These findings highlight the importance of studying tumor cells in the correct context. A better understanding of interactions within the tumor microenvironment, gained through use of appropriate experimental models such as MPS, will be critical to overcome treatment resistance through the development of successful targeted therapeutics.
4. Microphysiological Systems for Cancer Research
The ability to rapidly screen drugs and study disease mechanisms within a physiologically relevant context is critically important to facilitate clinical translation of preclinical findings. To address current limitations in preclinical models, multiple research groups have focused on innovating MPS that model both normal and pathological human tissue functions in vitro.96,97 By utilizing advanced microfabrication, microfluidic and tissue engineering techniques, physiological relevance can be designed into MPS to emulate the important functions of practically any human tissue or organ in corresponding microscale configurations. Major advantages of on-chip tissue models are that they recapitulate both the 3D organization and multicellular complexity of tissues and at the same time enable enhanced dynamic control over the cellular microenvironment to accommodate systematic experimental intervention.14 Furthermore, organ-on-a-chip platforms are composed exclusively of human cells and require fewer cells and drug volumes that standard preclinical models since assays are performed on a microscale. Microfabrication techniques such as soft lithography and replica molding are often used to manufacture tissue chips based on precise microfluidic designs. These bioengineering approaches allow manipulation of fluids at ultralow volumes (i.e. nanoliter and below) to simulate physiological flow, shear stress, nutrient delivery and drug exposure.70,98–100 Furthermore, on-chip devices enable careful spatiotemporal control over cell growth to better model complex tissue structure and function within micrometer-sized channels. Since fluid flow in microfluidic channels is laminar, it can be easily mathematically modeled, allowing theoretical predictions of complex biological phenomena48 that, when coupled with experimental analysis, provide a robust in vitro system for understanding tissue function and testing promising approaches for treating disease.
Microfluidic devices for biomedical purposes are often fabricated using poly(dimethylsiloxane) (PDMS), an elastic silicone-based polymer that is biocompatible, oxygen-permeable, and optically transparent, allowing for continuous observation of tissue constructs by microscopy for real-time assessment of cell behavior and response to treatment.101 Recent advances have allowed continuous in situ monitoring of biochemical, physical and optical responses via fully integrated sensing platforms on chip.8 Physical properties of individual organs can be modeled on-chip via cyclic deformations (to model breathing or peristaltic motions20,46), mechanical loading (to mimic the weight of the body on the musculoskeletal system102) or contractile forces (such as those important for heart function23). Current on-chip approaches mainly rely on combining pre-differentiated cells in particular ratios, often within an ECM or hydrogel that acts as scaffolding for cell growth, to emulate the native tissue composition.65 Cells are often fluorescently labeled, labeled with dye or immunofluorescently stained to facilitate tracking by fluorescence microscopy, but other sensitive, non-invasive imaging methods have also been applied to these systems.31 By integrating microfluidic assays, advanced microscopy and computational modeling, single events can be investigated with unprecedented temporal and spatial resolution as part of the complex biological processes that define pathophysiological responses. Based on advances in tissue-engineering strategies, individual organ-on-a-chip platforms are now being linked together to generate multi-organ systems for the study of drug pharmacokinetics, pharmacodynamics and toxicity.7,48,50,103–108 With knowledge gained in the field of stem cells, human induced pluripotent stem (iPS) cells or adult tissue resident stem cells can be differentiated into patient-specific cell populations for incorporation into tissue chips to achieve personalized medicine approaches.23,65,109,110 On-chip devices are anticipated to enhance preclinical predictability of drug responses by more accurately mimicking complex tissue- and disease-specific microenvironments than standard models.
4.1. Tumor Chips
Organ-on-a-chip platforms are rapidly evolving as powerful tools for oncology research (see Table 1). By replacing healthy cells and associated ECMs in tissue-specific constructs with those of cancer origins, so called tumor-on-a-chip or tumor chip systems have emerged. Tumor chips can ideally reproduce specific key aspects of the tumor microenvironment, such as biochemical gradients and niche factors, dynamic cell-cell and cell-matrix interactions, and complex tissue structure comprised of tumor and stromal cells. Moreover, tumor chips are able to reproduce cell confinement, a parameter imposed on cell movement in the interstitial space of tissues that is totally absent in 2D assays yet essential for studying the behavior of motile cells such as immune and cancer cells.41
Table 1.
Applications of Tumor Chip Technology
| Disease modeling | Drug Screening | Personalized Medicine |
|---|---|---|
| •Metastasis modeling | •PK-PD modeling | •Incorporation of primary cells |
| •Invasion/intravasation | •Absorption | •Induced pluripotent stem (iPS) cells |
| •Survival/dormaney | •Distribution | •Adult tissue resident stem cells |
| •Extravasation | •Metabolism | •Biopsy-, blood- or tissue-derived cells |
| •Metastatic niche | •Excretion | •Cancer stem cells |
| •Epithelial-mesenchymal transition | •Toxicity | •Patient-derived organoids |
| •Tumor heterogeneity/evolution | •High-throughput designs | •Genetically modified cells |
| •Mechanical forces | •Efficacy testing | •Integration with Big Data |
| •Tumor-ECM interactions | •Novel compound validation | ‘Omics’ signatures/readouts |
| •Tumor-stromal interactions | •Tumor resistance/sensitivity | •Genome wide association studies (GWAS) |
| •Organ-specific microenvironments | •Mechanistic studies | •Computational modeling |
| •Immuno-oncology studies | •Biomarker discovery/validation | |
| •Species-specific models | •Individualized trials-on-chip | |
| •Tailored clinical management |
Vascularized Tumor Chips
Nearly every tissue in the human body, including those of malignant origin, depends for survival on a supply of oxygen and nutrients delivered through blood vessels. Angiogenesis refers to the sprouting of new vessels from pre-existing vasculature, and vasculogenesis occurs when vessels form de novo from progenitor cells. In combination they represent the fundamental processes by which new blood vessels are formed (reviewed in111–113) and are critical during physiological processes such as tissue homeostasis, wound healing, pregnancy and fetal development. However, during malignant progression angiogenesis and, to a lesser degree, vasculogenesis are co-opted to feed the growing tumor mass, while also providing a means for metastasis. Metastasis is a primary reason for therapy failure and accounts for >90% of cancer deaths.114 Thus, tumor-associated vasculature represents an important component of the tumor niche and an attractive therapeutic target. Indeed, anti-angiogenic drugs have been developed extensively for use in cancer but with mixed clinical trial outcomes and oftentimes marginal survival gains.113 Elucidating the factors that contribute to therapy failure will ultimately lead to more effective therapies. Furthermore, approximately 25% of drugs entering clinical trials fail due to pharmacological issues such as lack of absorption or penetration into the tumor.10 High efficacy drug delivery to cancer remains a challenge primarily due to the heterogeneity and complexity of the tumor microenvironment, therefore models that mimic physiological barriers to drug or gene delivery will facilitate translation of in vitro results to in vivo studies.
To advance drug development in this area, our group and others have designed microvascularized tissue constructs on-chip in which vascular and perivascular cells self-organize de novo into a living and perfused vascular network in response to fluid flow and shear stress.31,115–117 Incorporation of these microvascular networks into tumor chips is a breakthrough for several reasons: 1) it better mimics the structure, function and disease processes of a vascularized tumor mass in vivo; 2) it models key steps of metastasis, which involve tumor-endothelial and stromal cell interactions that are poorly understood and difficult to investigate in current preclinical models; 3) it more accurately establishes physiologically selective barriers to nutrient and drug delivery in target tissues allowing for more realistic pharmaceutical screening; and, 4) drugs with anti-angiogenic and anti-metastatic capabilities can be directly assessed in such a system.
For realistic tumor modeling and anti-cancer drug screening, our group has adapted our base vascularized micro-organ (VMO) platform32,33,118 for cancer studies by incorporating tumor cells into the model to generate vascularized micro-tumors, or VMTs (Fig. 1).31,44 We have previously validated our VMO model by demonstrating that perfused, living microvessels that self-assemble within the microfluidic device (Fig. 1a,b) model the physiology of blood vessels in vivo. In response to gravity-driven physiologic flow, VMO microvessels derived from endothelial cells (EC) (Fig. 1c) form tight junctions by day 5 of culture (Fig. 1d). Stromal cells seeded with EC in the tissue chambers acquire a pericyte phenotype (NG2+ , PDGFRβ+) with tight appositions to vessels (Fig. 1e), and once established, vessels rapidly lay down a collagen IV rich basement membrane that increases in density over time. Importantly, the VMO recapitulates the barrier functions of vessels in vivo, with fully perfused and non-leaky vessels that show permeability coefficient values in line with values obtained from capillaries (Fig. 1f). We then created biomimetic VMT models for colorectal cancer (CRC) using HCT116 (Fig. 1g), SW620 and SW480 cell lines, breast cancer (MCF7 and MDA-MB-231) and melanoma (MNT1) by introducing each cancer cell line mixed with stromal cells, EC and ECM into the three tissue chambers of the device. Interestingly, tumor cells showed reproducibly different growth patterns, with SW480 and MCF7 growing as tight spheroids and MDA-MB-231 and MNT1 showing highly diffuse, invasive phenotypes reminiscent of their in vivo behaviors. Differences in growth rate, vessel development, and collagen synthesis suggests that each tumor cell line uniquely remodels the tumor microenvironment within the VMT.
Fig. 1. Vascularized micro-tumor (VMT) model.
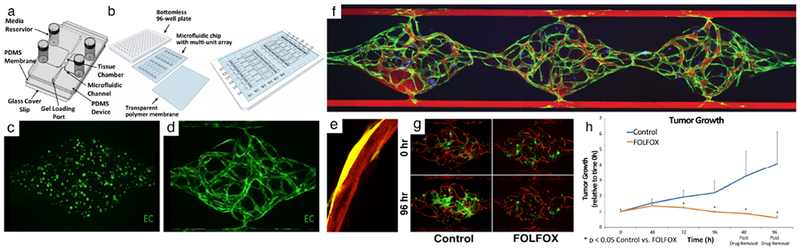
(a) A schematic of the microfluidic platform with a single unit. Three tissue chambers (1 mm × 1 mm × 0.1 mm) constitute 1 unit. Different levels of medium in the four vials drive flow. (b) A schematic of the microfluidic platform with 12 units/plate. (c) GFP+ EC at day 0. (d) A fully-formed vascular network at day 7. (e) A vessel (mCherry, red) wrapped by a pericyte (YFP, yellow) (f) 70 kDa rhodamine dextran flowing through the capillary network (green) formed within the three tissue chambers showing tight barrier function. Tumor cells are labeled in blue. (g) HCT116 colorectal cancer cells (GFP) with vessels (mCherry) are either non-treated (control) or treated with FOLFOX standard chemotherapy on day 7 (0 hour) and imaged every 24 hours. (h) Quantitation of FOLFOX treatment in HCT116 VMT. Tumor significantly regresses with treatment compared to control. Reproduced from Sobrino et al31 and Phan et al18 with permission from Nature Publishing Group and the Royal Society of Chemistry.
We next performed drug screening to test VMT response to FDA-approved chemotherapeutics and small molecule receptor tyrosine kinase (RTK) inhibitors representing both anti-cancer and anti-angiogenic drugs, including the standard-of-care therapies indicated for specific tumor types.31 By treating VMTs with physiologically-relevant doses, we demonstrated that the IC50 in our VMT model is higher than for 2D cultures (i.e. cancer cells are more resistant to treatment in VMTs) and better representative of the IC50 observed in vivo or in patients based on effective plasma concentration dose. This indicates that 2D models fail to accurately model certain critical features of in vivo tumors and that certain survival-signaling pathways essential for tumor progression in vivo are not activated in 2D culture. In response to FOLFOX, a chemotherapeutic regimen indicated for CRC, HCT116 tumors displayed marked regression after 48 hours of treatment vs control, whereas the already established vasculature remained intact (Fig. 1g,h). Tumor regression continued even 96 hours post-drug removal, confirming the cytotoxic effect of FOLFOX treatment. The anti-angiogenic multi-kinase inhibitors sorafenib and pazopanib were also tested in the VMT model and induced marked vascular regression in response to treatment. Both drugs target VEGFR2 and PDGFRβ with similar efficacy, but pazopanib additionally targets VEGFR1 and caused a greater degree of vascular regression in the platform, whereas sorafenib induced greater tumor regression due to its targeting of RAF. Additional drug screening results indicate that the VMT robustly recapitulates anticipated drug response based on mechanism of action, animal studies and clinical trial results.31 We have now arrayed our platform for high-throughput experiments to facilitate drug-screening studies as well as to enable downstream molecular biology techniques that are difficult to perform using standard microfluidic platforms.44
Recent contributions by Kamm and colleagues35,116,117,119 have provided an unprecedented, high-resolution view of tumor cell extravasation through microvessels formed in a microfluidic device (Fig. 2). The authors employed a de novo vascularized platform to study the process of tumor cell extravasation from within in vitro microvessels and were able to track each step in real-time. Suspended human umbilical vein endothelial cells (HUVEC) and normal human lung fibroblasts (NHLF) (Fig. 2a) cultured under dynamic gravity-driven flow conditions in the two-gel channel device (Fig 2b) self-organize to form stable, functional and perfused microvessels via paracrine signaling across the central media channel (Fig 2c–e). The microvessels formed tight cell-cell endothelial junctions, deposited basement membrane and demonstrated physiologic vascular permeability. Breast cancer cells (MDA-MB-231) were introduced into the device and high-resolution time-lapse microscopy revealed the highly dynamic nature of extravasation events (Fig 2f). The cancer cells first penetrated the EC barrier by extending thin fillipodial protrusions that continued to increase and branch out while the remaining body on the apical side of the lumen maintained its sphericity, even as the nucleus traversed the vessel. Throughout the process, tumor cells underwent significant shape changes as the cell body extruded through a gap in the microvessel of subnuclear dimensions. Interestingly, staining for VE-cadherin revealed that EC cell-cell junctions remained largely intact. Employing this assay, the authors found that TNFα stimulation impaired endothelial barrier function and increased tumor cell extravasation efficiency, and noted positive correlations between the metastatic potentials of MDA-MB-231, HT1080 and MCFlOa cancer cells and their extravasation capabilities. These results indicate that human tumor cells exhibiting different metastatic potentials exit the vascular system with different efficiencies, and that the platform possesses the sensitivity to detect such variations.
Fig. 2. Tumor cell extravasation from in vitro microvessels.
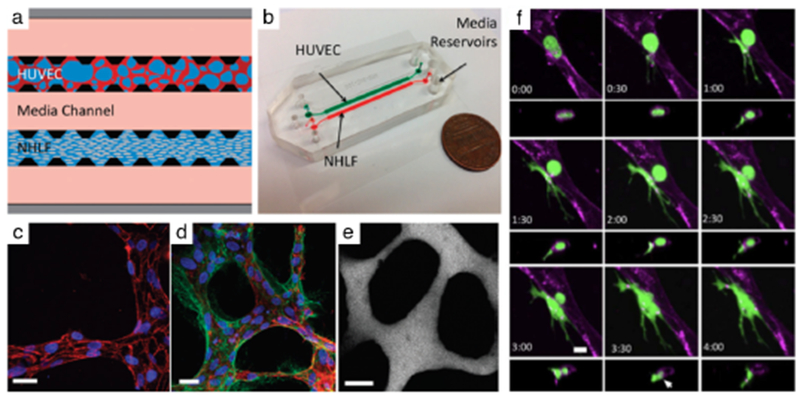
(a) A schematic of a microfluidic device and cell-seeding configuration. Suspended HUVECs form microvascular networks in a gel matrix via paracrine signaling with NHLFs across the central media channel. (b) Photograph of 2-channel microfluidic device. (c) Visualization of VE-cadherin (red) at 60X reveals continuous cell-cell junctions. (d) Collagen IV basement membrane deposition (green) around the lumen (red) and in the perivascular space suggests vessel maturation. (e) Perfusion of vessels with 70 kDa dextran reveals patent lumens void of local leaks. Scale bars are 20 μm. (f) High resolution time-lapse confocal microscopy (40X) of an extravasating entrapped MDA-MB-231 (green). Lumens were labeled with a far-red plasma membrane stain (purple). Tumor cells transmigrate through the endothelium and into the 3D matrix over a period of 4 h. The white arrow at 3:30 h indicates the location of a vessel opening at the site of tumor cell extravasation. Reproduced from Chen et al45 with permission from the Royal Society of Chemistry.
Cancer Type Modeling On-Chip
Cancer progresses via dynamic organ- and tissue-specific interactions; therefore, accurately modeling tissue-specific factors of the tumor microenvironment is crucial to creating physiologically and clinically relevant in vitro platforms for cancer research. In a recent study, Hassell et al46 created a sophisticated in vitro human orthotopic model of non-small cell lung cancer (NSCLC) using a biomimetic microsystem of the alveolar-capillary interface in the human lung (Fig 3a).16,120 The lung alveolar chip consists of two closely-apposed upper and lower channels separated by a thin, flexible and porous ECM-coated PDMS membrane that serves as the alveolar-capillary interface, or can serve as an airway-capillary interface. NSCLC tumor cells were cultured on the upper surface of the membrane containing primary human lung alveolar or small airway epithelial cells and human lung microvascular EC lined all four walls of the lower channel, forming a hollow vascular lumen. Physiological breathing motions were mimicked on-chip by applying cyclic suction to two parallel side chambers that rhythmically deformed the adherent lung tissues. Interestingly, NSCLC cells proliferated rapidly when cultured in the human alveolus chip, yet displayed a relatively dormant phenotype when cultured in the airway chip, reflecting organ microenvironment-specific lung cancer growth observed in human patients in vivo. Surprisingly, when NSCLC cells were cultured in the presence of cyclic mechanical strain to mimic physiological breathing motions, lung cancer growth was significantly suppressed by >50%. Without breathing motions on-chip, the tumor cells expanded to replace large regions of the epithelium and invaded into the epithelial layer, whereas the same tumor cells remained limited to smaller localized regions of the epithelium when grown with cyclic deformation. The authors note the possibility that in vivo obstruction of lung motion due to filling of alveolar spaces by growing cancer cells or other causes could produce a positive feedback loop to further enhance tumor growth. NSCLC cells also displayed decreased sensitivity to tyrosine kinase inhibitors (TKI) when treated under mechanical stimulation due to significantly reduced EGFR expression and phosphorylation, as well as an increase in both expression and phosphorylation of c-Met. Thus, it was concluded that mechanical breathing motions may suppress NSCLC cell response to TKI therapy by altering signaling pathway activation. These studies reveal that mechanical cues within the tumor microenvironment can significantly influence NSCLC growth, drug sensitivity and tumor dormancy in vitro to mimic unique NSCLC tumor growth patterns that are observed in human patients.
Fig. 3. Cancer type-specific modeling on-chip.
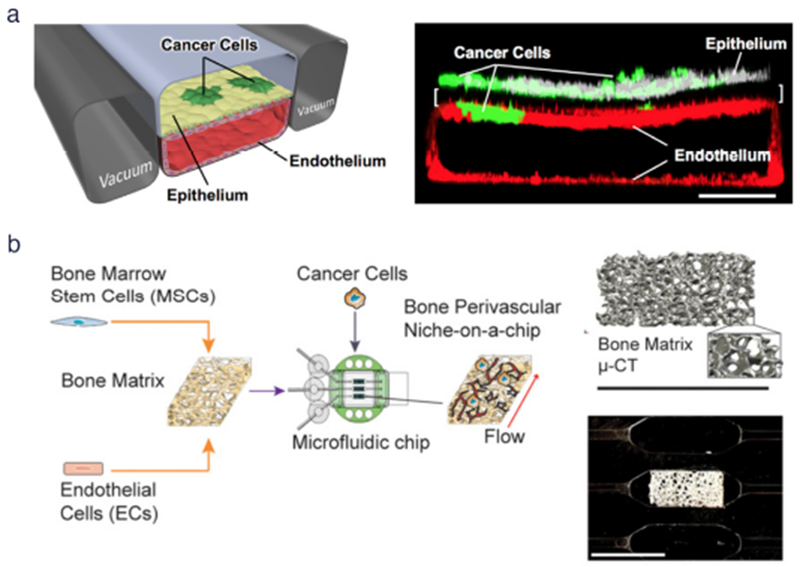
(a) (Left) Schematic diagram of a cross-section through 2-channel microfluidic lung-on-a-chip device. (Right) Confocal fluorescence micrograph of a cross-section of the two central cell-lined channels of an alveolus chip. NSCLC cells are labeled with GFP and endothelium with RFP, as shown. Reproduced from Hassell et al46 with permission from Cell Press, (b) (Left) Workflow for generating bone perivascular (BoPV) niche for studies of breast cancer colonization. (Right Top) Bone tissue reconstruction based on micro-computed tomography (μ-CT) data. (Right Bottom) Rectangular-shaped bone matrix in microfluidic chip. Reproduced from Marturano-Kruik et al47 with permission from the National Academy of Sciences.
Metastases can arise months or even years after a patient is treated for primary disease due to residual disseminated tumor cells that enter dormancy and evade therapies.121 Tissue-specific experimental models of the metastatic niche are needed to identify critical factors leading to metastatic-cell homing and colonization at distant sites, as well as tumor latency and resistance to treatment. Tumor chips are uniquely primed to elucidate the roles of stromal cells, secreted factors, exosomes and ECM proteins in niche priming and reveal therapeutic targets that may prevent metastatic progression.122 To investigate breast cancer metastatic colonization and drug resistance, Marturano-Kruik et al47 developed a functional human triculture that formed stable vascular networks within a 3D native bone matrix cultured on a microfluidic chip, termed the bone perivascular (BoPV) niche-on-a-chip (Fig 3b). Human endothelial and bone marrow-derived mesenchymal stem cells were seeded into decellularized bone and exposed on-chip to physiologically relevant interstitial flow and oxygen gradients. Recreation of these niche factors on-chip allowed the long-term maintenance of self-assembled microvascular networks without continuous addition of angiogenic factors. Interestingly, breast cancer cells that were introduced into the BoPV niche-on-a-chip under physiologic flow conditions showed a 4-fold reduction in cell numbers after 1 week in culture compared to static BoPV niche-on-a-chip cultures. Furthermore, treatment with the RTK inhibitor sunitinib, which is commonly used to target proliferative cancer cells and vasculature, was effective only in the static BoPV niche-on-a-chip. Cancer cell growth was inhibited by 50% following drug treatment in the absence of flow, whereas slowly-proliferating cancer cells in the perfused niche showed no response to sunitinib treatment. This study reveals the importance of recapitulating key cancer-specific characteristics of the tumor microenvironment to reveal physiologic mechanisms of drug resistance in vitro. It also demonstrates how tumor chips provide the unique capability to manipulate the host microenvironment to study the contribution of each niche component to tumor cell dormancy and drug sensitivity.
Onco-Immuno Chips
Cancer is well recognized as an immunogenic disease that stimulates complex immune responses via activation of both immune-inflammatory and immune-suppressive signaling pathways. Cancer cells hijack immune checkpoint signals (including upregulation of PDL1 and PDL2) to evade immune surveillance, while concurrently recruiting immune-inflammatory tumor-associated immunosuppressive cells that actively contribute to malignant progression and metastasis.79 The tumor microenvironment itself plays a critical role in influencing tumor-immune interactions and response to immunotherapy.123 For example, desmoplastic stroma within the tumor microenvironment can function as a barrier to T cell infiltration to foster a permissive, tumor-promoting environment.123 By mobilizing the host immune system against malignant cells, cancer immunotherapy induces long-term remissions in a subset of patients with metastatic disease and has become a clinically validated treatment for many cancers.79,124 However, despite the remarkable success of cancer immunotherapy, overcoming treatment resistance and the variable responses among patients remain major challenges. The interactions between the immune system and cancer cells are dynamic and constantly evolving within each individual patient – from the initial establishment of cancer to the progression to metastatic disease, which is dependent on immune evasion.124 Immunotherapy efficacy relies on this tumor-immune crosstalk within the tumor microenvironment. Given the emerging importance of the tumor microenvironment in modulating immune cell function, more sophisticated tumor models that incorporate features of the tumor microenvironment are needed to elucidate mechanisms of response and resistance to immunotherapies. Broadening the clinical applicability of onco-immunotherapies requires an improved understanding of the mechanisms limiting therapeutic response in order to derive actionable strategies to overcome them. Ex vivo systems such as tumor chips that model the dynamic interactions between the immune system and cancer cells may facilitate efforts in precision immune-oncology and the development of effective combination therapies.125 Onco-immuno chip models, incorporating both malignant and immune components, fed through a vasculature are well positioned for these studies since they are derived entirely from human cells and the delivery of patient-based antibody therapies to the tumor occurs via tumor-associated vessels. Immune and tumor response can subsequently be assessed in real-time within the tumor microenvironment.
Microfluidic technologies hold many advantages for studying tumor-immune cell interactions since they capture the essential features of multiple cell type interactions while allowing tight control of the microenvironment and real-time monitoring. Hsu et al27 developed a microfluidic platform that allowed cancer cells, myofibroblasts and macrophages to be cultured in each of three separate chambers connected by a Y-shaped channel equipped with microvalves that, when opened, allowed release of cell-conditioned media (CM) from myofibroblasts and/or macrophages to the cancer cells. Employing the platform, it was observed that CM from myofibroblasts and macrophages increased migration of cancer cells and that TNFα secretion by macrophages counteracted the migration-promoting effects of myofibroblasts. This study provides insights into the crosstalk between tumor and stromal cells and highlights the importance of modeling these aspects of the tumor microenvironment in disease models. In a recent contribution by Parlato et al41, a novel platform was used to monitor behavior of patient-derived interferon-α-conditioned DCs (IFN-DCs) toward SW620 colorectal cancer cells, either untreated or exposed to an innovative anti-tumor combined treatment (termed RI). IFN-DCs moved through the microfluidic device toward RI-treated cancer cells, rather than the untreated counterparts, which was facilitated by CXCR4/CXCL12 dendritic cell-cancer cell signaling. RI-treatment of SW620 resulted in a significant increase in phagocytosis of SW620 cells as IFN-DCs modified their motion within the platform to take up more tumor antigens from drug treated cancer cells. The microfluidic device allowed real-time visualization of the dynamic tumor-immune interactions involved in disease progression and treatment response, while further revealing that cancer treatment with the novel dual therapy RI facilitated anti-tumor immune function.
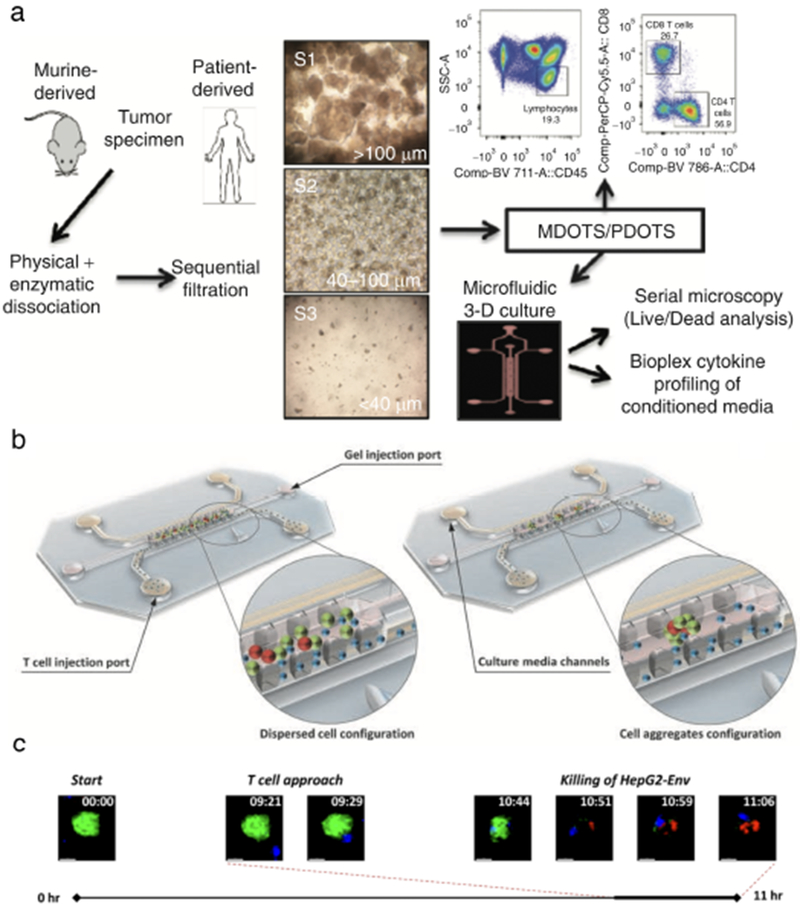
Similarly, in a recent publication by Jenkins et al42, ex vivo response to immune checkpoint blockade (ICB) was interrogated using Murine- and Patient-Derived Organotypic Tumor Spheroids (MDOTS/PDOTS) cultured in a microfluidic device (Fig 4a) and validated against in vivo data. Primary spheroids isolated from mouse and human tumors retained autologous lymphoid and myeloid cell populations that recapitulated response and resistance to ICB within the tumor chips. Profiling of MDOTS with in the tumor chips revealed that TBK1/IKKε inhibition enhanced response to PD-1 blockade, which effectively predicted tumor response in vivo. Systematic profiling of secreted cytokines in PDOTS captured key features associated with response and resistance to PD-1 blockade, such as increased CCL19 and CXCL13 that facilitate recruitment of immune cells to sites of chronic inflammation to coordinate anti-tumor response. These findings were further confirmed in paired biopsy specimens collected from patients with melanoma both before and after ICB treatment. Thus, MDOTS/PDOTS profiling represents a novel platform to evaluate ICB using established murine models as well as clinically relevant patient specimens. A 2017 study by Pavesi et al43 describes a microfluidic model to test the antitumor efficacy of TCR-engineered T cells wherein cancer cells and T cells interact in a 3D collagen matrix on-chip (Fig 4b). Effector T cells introduced into medium-filled side channels encountered a central collagen-filled region (i.e. the “tissue”) containing hepatocellular carcinoma (HepG2-Env) cells expressing both hepatitis B virus (HBV) envelope protein (HBsAg) and GFP. Only T cells engineered via retroviral transduction to express a specific TCR recognizing the complex HLA-A0201 molecule and hepatitis B Envl83-191 epitope (TCRe-T cells) engaged and killed the target cells upon migration into the gel. This was observed in real-time via a high resolution, 11-hour time-lapse video (Fig 4c).
Fig. 4. Examples of onco-immuno chips.
(a) Schematic for preparation and analysis of MDOTS/PDOTS from murine or patient-derived tumor specimens. Reproduced from Jenkins et al42 with permission from the American Association of Cancer Research. (b) 3D rendering of onco-immuno devices from Pavesi et al43. (c) Time-lapse video showing TCR-eT cell killing of HepG2-Env cells on-chip. Reproduced with permission from the American Society for Clinical Investigation.
Future Opportunities
Given their versatility and wide range of capabilities, microfluidic tumor chip assays could be exploited for more complex modeling of immune-cancer cell interactions by using additional cell types in 3D.125 Stromal cells such as cancer-associated fibroblasts and macrophages should be included in future studies because of their critical role in modulating the immune response and facilitating cancer cell dissemination and invasion to distant organs.55,63,81 Incorporation of a functional vascular network is also necessary to model physical barriers to drug or cell delivery, cell homing to distinct microenvironments, and trans-endothelial migration of tumor, stromal and immune cells. 126,127 An important limitation to such studies is that incorporation of adaptive immune cells (e.g. T and B cells) would require an entirely autologous, HLA-compatible vascularized tumor chip model since human endothelial cells are highly immunogenic antigen presenting cells.128 However, one can envision the development of patient-specific onco-immuno chips, incorporating tumor, stroma, and immune components for studying critical initiating steps in metastasis and developing novel immune-modulating therapies.125 These individualized tumor chips are now becoming feasible with advanced cell culturing and genetic engineering techniques. 129,130
While modeling of blood cancers-on-chip is still in its infancy, a recent study by Bruce et al131 demonstrates a reductionist bone marrow-on-a-chip for study of acute lymphoblastic leukemia. It was shown that co-culture of primary human bone marrow stromal cells, osteoblasts and human leukemic cells in a 3D collagen matrix under dynamic flow conditions within the platform conferred enhanced cell viability and chemoresistance to the cancer cells compared to 2D and 3D static models. In a study by Torisawa et al132, an in vivo engineered organotypic bone marrow-on-a-chip that recapitulates the structural, cellular and physical complexity of the hematopoietic niche in vitro is described. Bone marrow-on-a-chip can be used not only for study of toxicity/drug response and solid tumor colonization within the metastatic niche, these models can also be developed to study leukemia, myeloma, myeloproliferative neoplasms and lymphoma. In addition to endothelial cell self-assembly into microvessels on-chip in response to flow, lymphatic microvasculature133 and primitive lymph node models have also been described.121,134 These microphysiological elements can be further integrated into novel models of blood cancers, metastatic disease and onco-immune responses to increase physiological relevance, faithfully recreate steps of cancer progression in vitro and interrogate mechanisms of drug resistance.
4.2. Integrated Tumor-Organ-Chip Systems: Toxicity, PK-PD Modeling, and Metastasis Studies
Cancer is a heterogeneous and individualized disease involving multiple systems. Each cancer is both patient-specific and tissue-specific – i.e. breast cancer is not bone cancer is not lymphoma; all will behave differently, even between individuals with the same type. Yet, all cancers require integration of multiple tissue components to create a complex tumor microenvironment for tumorigenesis to proceed. This cannot be modeled with overly reductionist models like 2D or 3D models that lack tissue structure, or in immunocompromised non-human animal models. MPS represent a physiological, humanized model that allows for customization to specific cancers and integration of multiple systems. Integration of tumors into multi-MPS platforms will allow truly predictive in vitro modeling of human pathophysiology. These coupled tumor-organ-chip systems can enable comprehensive studies of drug toxicity, PK-PD parameters, and complex systemic cell-cell interactions such as those influencing the establishment of distant metastases. The importance of recapitulating orthotopic tumor growth and metastasis further necessitates the use of integrated tumor-organ-chip systems. Metastasis is a physical process by which mobile cancer cells break away from the primary site, travel into lymph vessels to seed nearby lymph nodes or squeeze through blood vessel walls to gain access to the systemic circulation. Metastatic cells can then invade distant tissues by extravasating through the resident vasculature and establishing a secondary tumor at the new site.126,127 Coupling of organ systems holds tremendous potential for modeling metastasis of tumor cells from primary organs to distant sites in the body, especially if the organs are interconnected by microfluidic channels lined with living, perfused vessels to mimic the blood flow pattern in the human body.108
One of the reasons for high rates of attrition during drug development is unforeseen drug toxicity that is not revealed until the later stages of testing when the drug has progressed to clinical trials. Many drugs have even been FDA-approved and available on the market before being recalled by the FDA for unanticipated side effects.135 Current preclinical and computational models are unable to fully reproduce the pharmacokinetics-pharmacodynamics (PK-PD) of drugs in vivo, underscoring the need for clinically relevant model systems capable of revealing safety issues prior to testing in humans. The majority of therapeutic failures attributed to toxicity in cancer treatment are accounted by cardiotoxicity, hepatotoxicity and bone marrow toxicity, although nearly every other organ/system of the body (including gastrointestinal tract, kidneys, nervous system, skeletal muscles and gonads) has been reported to have anti-cancer drug-related pathologies.52 To date, each of these organs have been recreated on-chip and could potentially be leveraged into tumor-organ-chip systems. While single tissue chips are useful for many applications, organs and tissues in the human body are not isolated but instead highly interconnected via the vasculature, receiving biochemical signals from cells at distant sites and functioning in concert to dictate the absorption, distribution, metabolism and excretion of drugs. Since the liver is the major site of drug metabolism, tumor chips can be linked to human liver chips to emulate in vivo PK-PD for simultaneous preclinical efficacy and safety testing within a single platform. Toxic effects on other organs can be assessed by incorporating additional functional units onto the platform.
Organ chip systems that integrate multiple human tissues have been described extensively in the literature7,105,136,137. Yet there are fewer integrated systems in which malignant cell populations have been incorporated to study drug response or cancer progression, suggesting that multi-organ systems are still in the developmental phases of physiologic modeling. In 2010, Sung et al48 published a pumpless system containing liver, bone marrow and colon cancer cells that, upon treatment with the chemotherapy 5-fluorouracil (5-FU), recreated anticipated drug actions showing that metabolites generated in the liver recirculated to achieve expected outcomes in all 3 tissue compartments. While a ‘human surrogate’ on-chip has not yet been fully realized, recreating the entire body may not be necessary or even practical for drug screening. When considering the increased costs, time, resources, sophisticated engineering and complex experimental procedures required for establishing such body-chip systems, the advantages of these systems may be insufficient compared to simpler on-chip models.138 Furthermore, many biological and technical challenges remain, including the need to: 1) scale organs and their associated blood supplies to produce outputs that are physiologically accurate compared to other organs 108,139; 2) control oxygen gradients to create areas of inter- and intra-organ zonation; 3) create a systemic and interconnected circulatory system; and, 4) feed the tissues with a universal blood surrogate providing all the necessary nutrients, growth factors and proteins in a physiologically relevant manner.107 Therefore, simplicity must be weighed against physiological relevance and platform usability when considering the appropriate level of model complexity important to reconstitute tissue functions, disease processes or drug responses for a given application.
For screening purposes, we feel that tumor chips are best positioned for drug efficacy studies because the majority of drugs fail during development due to lack of efficacy. The predictive power of tumor chips in toxicology studies may well be limited as there is a multitude of ways a drug could manifest toxicity, in contrast to efficacy, which is a clearly-delineated endpoint. Certain aspects of organ-specific toxicity may be tested in well-designed multi-organ chips containing malignant cells, however these studies will need to be supplemented with mathematical models, in vivo study, and careful cross-species and drug design considerations. We envision that the greatest commercial and clinical potential for tumor chips resides in efficacy testing, since this application requires only a single tissue unit and output for ease of production, faster readouts and higher throughput screening. The less-complicated, user-friendlier tumor units with fewer barriers to use will likely see greater and faster adoption into drug development and clinical diagnostics. On the other hand, well-designed toxicity studies, with clear hypotheses and defined endpoints, should readily yield useful data from organ chips.
4.3. Personalized Medicine Applications
The goal of personalized medicine (precision medicine) is to choose the most efficacious, and least toxic, therapy for each individual patient. Yet many cancer drugs fail to demonstrate clinical activity due to an inability to identify patients that are most likely to respond.140 Instead, drugs are prescribed by an empirical, one-size-fits-all approach that yields limited efficacy and significant side effects. Lack of efficacy is a significant barrier to FDA-approval for oncology drugs, and the majority of drugs that are approved confer only marginal survival gains. Current standards for precision medicine involve performing molecular testing on patient-derived tumor tissue to identify the specific genomic aberrations present and then categorizing the subtype of cancer based on population-level genetics.141 Patients are then treated as an ‘average patient’ based on the cohort with which their tumor characteristics most segregate – we term this one-way personalized medicine. Although these tests are routinely performed in an effort to guide targeted treatment decisions, the full promise of genome-wide association studies has not yet been realized, as tumor recurrence and high variability in disease patterns remain an important problem. Consequently, there is a lack of clinically validated and actionable biomarkers for which to guide treatment decisions.
Each individual cancer is characterized by considerable inter- and intra-tumor heterogeneity that encompasses genetic, molecular, cellular and microenvironment diversity that evolves over time.142 These defining aspects of actual tumors are insufficiently modeled during preclinical drug development, leading to falsepositive selection of drug candidates that look promising until they reach human efficacy testing in phase II and III clinical trials.5 Part of the problem is that established cancer cell lines are routinely used for drug testing in 2D assays and animal studies, and since these cells have been maintained in culture for decades they are no longer representative of the disease from which they were derived. Even short-term culturing can induce marked genomic changes: cells adapt to growth in 2D monolayers and thus lose the intrinsic heterogeneity of the original tumor through genetic drift, as clonal populations that cannot survive in the culture flask are lost.56 Thus, current preclinical models based on these immortalized cell lines are oversimplified and misrepresentative models of human tumors in vivo. To develop more effective targeted therapy for personalized treatment, understanding of human pathophysiology is critical and requires an investigational cancer model with high clinical predictive value. Tumor chips are now emerging as promising personalized model systems for preclinical drug development and, in the future, may serve as diagnostic tools to inform tailored clinical management.
Cell Sources for Tumor Chips
In order to achieve precision medicine approaches, it is necessary to use human primary cells for disease modeling and drug testing applications to more faithfully recapitulate the heterogeneity inherent to human disease. Primary cells can be derived from surgical resections, biopsies, aspirates and blood specimens.130 In culture, samples can be further enriched for adult resident stem or progenitor cells, or reprogrammed into iPS cells, and subsequently differentiated into mature cell types.109 Ideally, matched healthy and cancerous cells can be derived from a single source and integrated into tumor chips to allow patient-specific disease modeling, drug efficacy and toxicity studies. Tumor chips populated with tissue samples obtained from sites of resistance, matched primary and metastatic lesions or autopsy specimens are valuable resources to study tumor evolution and guide the development of treatment strategies that address tumor heterogeneity. Cutting-edge bioengineering approaches to capture patient- and disease-specific characteristics on-chip through incorporation of renewable cell sources are described below.
Organoid Technology
Advancements in stem cell biology have facilitated the development of patient-derived 3D stem cell cultures termed organoids. Since organoids give rise to multiple lineages that self-organize to reconstitute the cellular hierarchy, heterogeneity and structure of native tissues, these stem cell-derived models are increasingly being integrated into MPS for enhanced tissue fidelity and clinical relevance.143,144 Robust methods that are now established to derive long-term organoid cultures from virtually any matched normal and malignant tissue, such as those originating from intestine, pancreas, liver, prostate, and breast, may also be suitable for MPS applications.145–148 Organoids derived from epithelial tumors, also known as tumoroids, have been shown to be representative of distinct molecular disease subtypes by retaining the genetic heterogeneity of the parent tumors and, consequently, are able to recapitulate some aspects of in vivo treatment responses.146,149,150 However, physiologic relevance is limited in these models because organoid cultures lack key anatomical and functional features that contribute to cancer progression and drug resistance, such as a tissue-tissue interface, stromal cells, a vascular compartment, dynamic fluid flow and mechanical forces.69 Adaption of organoids into tumor chips to replace commonly used established cell lines may facilitate the long-term support of cellular heterogeneity within a physiological context, since the convergence of the two technologies has been previously shown to better model organ-specific structures and in vivo gene expression signatures than either model alone.151 Alternatively, cancer stem cells can be initially positively selected in specialized stem cell media and then differentiated on-chip to reconstitute the native clonal cell populations.152 By exploiting organoid technology for tumor chip applications, both healthy and tumor tissue can be readily generated from the same individual and tested for drugs that specifically target tumor cells while leaving healthy cells unharmed.153 A major limitation to this approach, however, is that cells must be harvested from each represented organ to generate the healthy organoids to be used for drug toxicity screening on-chip. While it may be more reasonable to collect cells from certain tissues than for others, it is neither practical nor ethical to obtain numerous biopsies from patients. Instead, patient-specific iPS cells can be generated.
iPS Cells
iPS cells are derived from primary human somatic cells via reprogramming by transient exogenous expression of a set of transcription factors.154 Two unique properties define iPS cells: they can be maintained in culture in a self-sustaining pluripotent state, and they can be directed to differentiate into virtually any cell type of the human body, although generating fully mature cells with full functionality is still a challenge for most tissue types.130 As a result, iPS cells have made it possible to engineer patient-specific human in vitro systems by deriving every cell type needed in the system from a single iPS cell. This is particularly useful for personalized toxicity screening and to detect variants that alter response to treatment. For example, patients with a genetic variation that reduces their ability to metabolize thiopurines, the drugs most commonly used to treat acute lymphoblastic leukemia, will build up toxic metabolites with standard treatment unless the variant is detected in the clinic and they administered a lower dose.155 In a recent study, cardiomyocytes derived from iPS cells from patients with breast cancer were shown to model the doxorubicin-induced cardiotoxicity manifested in the patients156, demonstrating the potential for iPS cell-derived models to faithfully recapitulate patient-specific disease outcomes in vitro.
To model population-wide variation necessary to detect disease variants, large and diverse collections of iPS cells capturing a range of cancer types, genotypes and patient-specific factors such as age and gender will need to be assembled and tested on-chip. Such studies could streamline the drug development process by accurately representing population variation at preclinical and early clinical trial stages, reducing the attrition rate of promising drugs and averting human risk by informing patient selection for clinical trials.15 Gene editing tools, such as CRISPR-Cas, have enabled researchers to introduce disease-associated mutations into iPS cells to create isogenic models of human disease and then compare them with the original, unedited cell lines.157 These studies will allow better tractability of results from preclinical to clinical stages and will be necessary to reveal novel drug targets and biomarkers of disease progression or prognosis. While iPS cells can theoretically provide an unlimited supply of once-inaccessible human tissues for research, these models are not without limitations. Not all cell lineages can be effectively derived from the same iPS cell line, and can vary between batches.130 In general, iPS cell-derived lineages lose epigenetic markers during derivation and, as noted above, are immature.107,158 To address these limitations, the development of robust protocols for iPS cell differentiation and maturation are ongoing.
Individualized Trials-On-Chip
To circumvent current barriers to personalized medicine, we suggest using one or more approaches outlined above to grow a patient’s own cells within tumor chip systems. Drugs can be tested, in a high throughput manner, directly on this ‘individual-on-a-chip’ in order to better predict the individual response to many therapies at once. Results derived from these tumor chip models can then correctly guide an oncologist to treatments that are most likely to be of clinical benefit to that patient – an approach we term two-way personalized medicine. The goal is to determine which therapeutic regimen will most effectively target their particular cancer before they receive any treatments. This represents a truly personal drug screening methodology whereby tumor chips can be directly used in the clinic as a diagnostic tool. Individualized tumors-on-chip can also be used for ‘micro’-clinical trials in which many unique patient-specific tumor chips are established using patient-derived cells and material.159 Such in vitro trials would allow testing of multiple drug dosing and schedule regimens to inform treatment timing and sequence, guide rational combination therapies, and facilitate discovery of novel agents. Drugs that are currently only approved for treatment of specific cancer types or compounds that are not traditionally prescribed for cancer can be readily tested on tumor chips in vitro and repurposed for additional disease indications based on the results. This may increase the number of treatment options available for patients while facilitating our understanding of cancer type-specific resistance mechanisms to inform development of clinical biomarkers. Individualized trials-on-chip could revolutionize drug development and clinical management in oncology by allowing patient stratification based on individual characteristics without the need for large-scale clinical trials, thereby reducing human risk. Such an approach would also allow therapeutic agents to be screened on tumor chips derived from patient populations unfit to participate in standard clinical trial designs, such as patients with co-morbidities, rare cancers, recurrent or heavily pre-treated disease, and pediatric patients. High-throughput experiments combined with molecular testing could elucidate gene-drug interactions, facilitate tumor subtype stratification in response to drugs and inform clinical trials. Such an approach will allow more accurate patient stratification based on additional tumor characteristics such as the microenvironment milieu.160
Translational Study Design
In order to validate tumor chip models as translational tools, studies utilizing these models must be designed with translation in mind.161 In a recent study47, for example, Vunjak-Novakovic and colleagues validate results from a bone-mimetic tumor chip model against clinical data to unravel the contribution of mechanical strain to treatment resistance in Ewing sarcoma (ES), the second most frequent bone tumor in children and young adults. A bioengineered model of ES was generated by preparing porous 3D scaffolds from collagen I and hyaluronic acid solutions using a freeze-drying technique and then seeding the matrix with established ES cell lines or patient-derived ES xenograft tumor cells. ES-mimetic bone constructs were then placed in a bioreactor and subjected to dynamic compressive loading to model the mechanical stresses generated by body weight and muscle tension on bone. Compared to unstimulated control and 2D culture, bone-like loads increased the expression of RUNX2, a transcription factor that mediates cancer cell proliferation, survival and drug resistance of tumors that reside in the bone. Indeed, mechanical stimulation of the tumor chips led to enhanced ES resistance to treatment with clinically relevant doses of RTK inhibitors. In accordance with clinical trial data and immunohistochemical staining of ES tumor biopsies, patient-derived ES cells demonstrated increased RUNX2 expression and exhibited an enhanced degree of treatment resistance than established ES cell lines, indicating that patient-derived tumor cells better retained characteristics of the native tumor. By analyzing publicly available databases for RUNX2 expression, it was revealed that patients with tumors over-expressing RUNX2 had poorer outcomes and decreased survival. Based on these findings, the authors suggest targeting the RUNX2 downstream effectors ERK1/2 as a rational therapeutic strategy for ES.
The vast numbers of clinically annotated ‘omics’ (genomics, transcriptomics, metabolomics, and proteomics) databases now available constitute a powerful set of research tools to identify high frequency cancer-related biological variations. However, despite significant advancements arising from these association studies, it has proven very difficult to assign causal roles to the identified “cancer-associated” variants.157,162 To successfully verify biomarkers of cancer progression or treatment response, realistic human cell-based cancer models that can be experimentally queried in a reproducible manner are needed. Tumor chips offer unprecedented opportunities to model cancer in a robust, physiologically relevant context that allows repeated experimentation to probe the mechanistic and functional underpinnings of ‘omics’ cancer-associated signatures. Further, tumor chips can reveal novel targets by phenotype-based drug screening.
Considerations for Personalized Medicine
Tumor chip systems are uniquely suited to facilitate the development of personalized medicine because few cells are needed from the patient and rapid, automated results can be obtained within a clinically actionable timeframe. However, several obstacles to the use and study of patient-derived tissues include: 1) the regulatory burdens and logistical issues associated with such research; 2) challenges in successful generation of primary cultures; 3) the scarce amount of viable patient-derived cells available for use after pathological analyses; and, 4) the need to form homogeneous replicates representing the functional heterogeneity of the tumor in order to compare responses under various conditions (e.g. replicates to test different compounds, concentrations and even timing or tissue location). If tumor chip models are to be embraced for precision medicine applications, they must provide reliable results despite these challenges. Close collaboration between primary investigators, physicians, and regulatory agencies is necessary to move the field forward. Biobanks of human primary tissues can facilitate these studies, as well as collaborative efforts among independent investigators to integrate the most critical components for modeling tumor biology on-chip. Phenotypic and genotypic profiling is necessary to demonstrate that tumor chips preserve a high degree of similarity to the original patient tumors. Moreover, it has yet to be demonstrated that tumor chips accurately replicate patients’ clinical outcomes. Co-clinical trials, whereby drug responses of patient-derived tumor chips are compared directly with patient drug response, must be performed to determine the clinical predictive utility of tumor chips.140 Pharmaceutical companies and academic research institutions can design clinical trials to include in vitro studies performed in parallel using patient-derived biopsy tissue to complement in vivo findings. These studies will reveal the value of tumor chips in maximizing drug development success and patient benefit from anticancer treatment.140
4.4. Design of Tumor Chips
Oxygen Control
To create biomimetic 3D models that better recapitulate tissue or organ function during physiological or pathological processes such as cancer, several important factors of the tumor microenvironment must be considered. Oxygen tension is a critical regulator of cell behavior, and in some organs (e.g. liver), tight regulation of oxygen levels across the tissue is required for function. Most adult tissues within the body typically experience 3%–8% O2, with highly oxygenated tissues in the lungs reaching 14% O2, capillaries around 4% O2 and poorly perfused tumors frequently experiencing chronic or intermittent hypoxia (i.e. 1% O2) within the tumor microenvironment.163 In contrast, in vitro studies are usually performed under atmospheric oxygen conditions (20% O2) that do not represent in vivo conditions. Consequently, the results from these studies may be misleading. In cancer, hypoxia is associated with poor prognosis since it promotes treatment resistance and is a major contributor to malignant progression via metastasis.164 In vitro models of metastasis must further expose migrating tumor cells to varying microenvironments and oxygen gradients mimicking what occurs during the intravasation process in vivo.165
Tumor chip systems have several advantages over traditional preclinical models to facilitate experimental manipulation and interrogation of molecular mechanisms of tumorigenesis. Microfluidic devices can be designed to achieve oxygen gradients with high spatial and temporal resolution for modeling physiologically relevant effects of oxygen on tumor progression and metastasis. Such control can be implemented by perfusing oxygen scavengers or carriers, or pumping oxygen or nitrogen, directly into microfluidic channels adjacent to tissue constructs or by exploiting cellular consumption of oxygen in single- or multi-layer devices or in multicellular aggregates.164,166–168 However, some of these designs require cumbersome external apparatuses and pumps to deliver gas or chemicals into the chip, or require multi-layer arrangements that increase fabrication complexity and impair high-resolution microscopic observations.101 Tumor chips that incorporate living, perfused vasculature can deliver oxygen and nutrients to the tissue construct via flow of oxygenated media through the vessels, much as it would occur in vivo. Other options to control O2 delivery in tumor chips include placing microfluidic platforms in controlled gas tissue incubators for simulating chronic hypoxia, or fabricating devices with oxygen-impermeable materials, such as thermoplastics, in place of PDMS, which has high oxygen permeability.166
Fabrication Material Properties
PDMS is traditionally used for fabrication of biomedical microfluidic devices since it is relatively inexpensive, can bond reversibly and irreversibly to itself or other materials, and is elastic, allowing for easy removal from delicate silicon molds for feature replication as well as enabling specialized designs on-chip, such as valves or thin, flexible membranes.101 However, drug and small molecule absorption into PDMS is a major concern since hydrophobic compounds readily diffuse into or adhere to PDMS, reducing the intended drug exposure and causing undesirable mixing between adjacent channels that can confound interpretation of experimental results.168 The hydrophobic surface properties of PDMS can be tuned to a more hydrophilic state prior to drug testing to help prevent absorption/adsorption of drugs to PDMS.169 This is accomplished by oxidizing the PDMS to create a barrier of silicon dioxide on the surface through plasma treating or coating with proteins. Alternatively, devices can be fabricated with a different material altogether although additional considerations must then be given to the fabrication process. Polystyrene is routinely used for cell culture and thus represents a rational alternative to PDMS. Mathematical modeling approaches have also been developed to account for absorption.15,101
Tumor Chip Readouts
Multiple modes of analysis (e.g. biological, electrophysiological, chemical and mechanical) can now be integrated on-chip for dynamic studies of tissue responses.107 The majority of on-chip studies exploit the optical clarity of PDMS devices to perform functional assays on live tissues using microscopy techniques. Morphological tissue features can also be visualized by in situ imaging of fluorescently tagged or unlabeled cells, immunofluorescent or immunohistochemical staining. Sensitive, noninvasive imaging and confocal microscopy techniques can be performed on intact tissue-chips to reveal distinct histological features, cellular populations or tumor characteristics.170 While these tumor chip applications rely on time-consuming and involved microscopy imaging and analyses, which limit the devices in terms of automation, streamlined methods are increasingly reported.8,171 To further characterize cells grown in tumor chips, RNA-expression analysis is now routinely used in combination with imaging.170 However, preparation of the cells for genomic assays is a manual and inefficient process that may be improved by designing tissue chips compatible with the workflow of these assays. Molecular assays such as ELISA or LC/MS based techniques can be coupled to microfluidic cell culture by means of dedicated outlets to collect and analyze effluents from individual chambers and detect secretion of compounds, such as tumor-derived exosomes or pro-inflammatory cytokines.43,138,170 The increased sophistication of analytical techniques applied to MPS studies warrants a critical assessment of bioengineering designs to enhance tumor chip usability, throughput and compatibility with existing technologies.170 Ideally, designs are considered for automation and direct translation into industry settings.107 Data derived from large-scale tumor chip studies can then be compared directly to the in vivo situation to establish correlations of tumor heterogeneity and treatment response.
5. Future Considerations
Advances in microfluidic technologies and tissue engineering have made tumor chips attractive candidates to replace traditional experimental approaches by allowing precise control over the tumor microenvironment, the ability to interrogate dynamic cell-cell and cell-ECM interactions in real time and at high resolution, and the ability to acquire rapid, automated results using high throughput arrays. Depending on tumor chip design, virtually any type of drug or drug delivery system can be tested for efficacy and toxicity within a fully humanized system, including but not limited to: cell-based therapies43,46, chemotherapy, anti-angiogenic treatment, targeted therapy31, antibodies or small molecules, radiation, nanomedicine106,172 and electric field therapy.43 Yet despite their tremendous experimental potential, the use of microfluidic tumor chip assays has mostly been restricted to academic research settings and they have not yet been widely adopted in the pharmaceutical industry or clinic. One of the major reasons for this is that many tumor chip technologies have not yet been rigorously validated against in vivo data, either mouse or human.
Currently, many microfluidic device studies are still considered ‘proof-of-concept’.101 Regulatory hurdles to FDA-approval for tumor chips mean that the burden for preliminary data indicating proof-of-superiority to traditional models lies mostly with the academic labs that developed the platforms. Yet global research initiatives to support and advance this cutting-edge technology are ongoing. In the US, the NIH National Center for Advancing Translational Sciences (NCATS), in collaboration with other NIH Institutes and Centers, the Defense Advanced Research Projects Agency (DARPA), and the FDA, is leading the Tissue Chip Drug Screening program. Under this program, three Tissue Chip Testing Centers have been established to test and validate tissue chip platforms, including tumor chips, independently. The goal of this program is multifaceted: to ensure wide-ranging availability of tissue chip technology, particularly for regulatory agencies and pharmaceutical companies, and to promote adoption of this technology by the broad research community.
Much work still remains before tumor chips, or organ chip models generally, are adopted into healthcare settings and mainstream drug R&D, and predominately hinges on demonstrating the functionality, reproducibility, robustness and reliability of each tissue chip platform. The current trajectory, however, is promising. For example, blood-vessel-on-chip devices have already been implemented into the clinic for the diagnosis of sickle cell disease173, and pharmaceutical companies are now readily forming collaborations with academic centers that develop on-chip technologies. Similarly, start-up companies based on these technologies and founded by the inventors are now gaining commercial recognition as well as support from both public and private entities. Microfluidic technologies have clear potential to advance cancer research, the success of which depends on how effectively the engineering and the biology can be integrated to create a clinically relevant in vitro tumor system.
6. Closing Remarks
Rapid technological advances have led to the development of tumor chip platforms for monitoring in real time the events linked to cancer development, progression and response to therapy. Given their close mimicry of human biology and physiology, tumor chips are anticipated to bridge the enormous gap between in vitro and in vivo preclinical studies as well as serve as a clinical diagnostic for personalized medicine applications. By surpassing the predictive accuracy of conventional 2D cell culture models, tumor chips can reduce reliance on animal models in line with the 3R’s initiative and eliminate false-positive selection of ineffective or toxic drugs earlier in the drug development pipeline, thereby saving time and resources. Most importantly, better predictability of human drug response will reduce human risk and improve patient outcomes. We anticipate a paradigm shift in drug development, disease modeling and precision medicine as tumor chip models become widely adopted in academia, industry and healthcare.
8. Acknowledgements
This research was supported in part by NIH grants UH3 TR000481 and R01 PDQ5-CA180122.
Footnotes
7 Conflicts
CCWH is co-founder of Kino Biosciences Inc., a company that develops MPS.
Notes and references
- 1.Siegel RL, Miller KD and Jemal A, CA. Cancer J. Clin, 2018, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Kola I and Landis J, Nat. Rev. Drug Discov, 2004, 3, 711–716. [DOI] [PubMed] [Google Scholar]
- 3.Smietana K, Siatkowski M and Møller M, Nat. Rev. Drug Discov, 2016, 15, 379–380. [DOI] [PubMed] [Google Scholar]
- 4.Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD,Wang J, Wallace O and Weir A, Nat. Rev. Drug Discov, 2015, 14, 475–486. [DOI] [PubMed] [Google Scholar]
- 5.DiMasi JA, Grabowski HG and Hansen RW, J. Health Econ, 2016, 47, 20–33. [DOI] [PubMed] [Google Scholar]
- 6.Kimmelman J and Federico C, Nature, 2017, 542, 25–27. [DOI] [PubMed] [Google Scholar]
- 7.Wang YI, Oleaga C, Long CJ, Esch MB, McAleer CW, Miller PG, Hickman JJ and Shuler ML, Exp. Biol. Med, 2017, 242, 1701–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang B and Radisic M, Lab Chip, 2017, 17, 2395–2420. [DOI] [PubMed] [Google Scholar]
- 9.Perel P, Roberts I, Sena E, Wheble P, Briscoe C, Sandercock P, Macleod M, Mignini LE, Jayaram P and Khan KS, Br. Med. J, 2007, 334, 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mak IW, Evaniew N and Ghert M, Am. J. Transl. Res, 2014, 6, 114–8. [PMC free article] [PubMed] [Google Scholar]
- 11.Skardal A, Devarasetty M, Forsythe S, Atala A and Soker S, Biotechnol. Bioeng, 2016, 113, 2020–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai H-F, Trubelja A, Shen AQ and Bao G, J. R. Soc. Interface, 2017, 14, 20170137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stock K, Estrada MF, Vidic S, Gjerde K, Rudisch A, Santo VE, Barbier M, Blom S, Arundkar SC, Selvam I, Osswald A, Stein Y, Gruenewald S, Brito C, Van Weerden W, Rotter V, Boghaert E, Oren M, Sommergruber W, Chong Y, De Hoogt R and Graeser R, Sci. Rep, 2016, 6, 28951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhatia SN and Ingber DE, Nat. Biotechnol, 2014, 32, 760–772. [DOI] [PubMed] [Google Scholar]
- 15.Low LA and Tagle DA, Lab Chip, 2017, 17, 3026–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY and Ingber DE, Science (80-.)., 2010, 328, 1662–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beckwitt CH, Clark AM, Wheeler S, Taylor DL, Stolz DB, Griffith L and Wells A, Exp. Cell Res, 2018, 363, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phan DT, Bender RHF, Andrejecsk JW, Sobrino A, Hachey SJ, George SC and Hughes CC, Exp. Biol. Med, 2017, 242, 1669–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao S, Coppeta JR, Rogers HB, Isenberg BC, Zhu J, Olalekan SA, McKinnon KE, Dokic D, Rashedi AS, Haisenleder DJ, Malpani SS, Arnold-Murray CA, Chen K, Jiang M, Bai L, Nguyen CT, Zhang J, Laronda MM, Hope TJ, Maniar KP, Pavone ME, Avram MJ, Sefton EC, Getsios S, Burdette JE, Kim JJ, Borenstein JT and Woodruff TK, Nat. Commun, 2017, 8, 14584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HJ, Huh D, Hamilton G and Ingber DE, Lab Chip, 2012, 12, 2165. [DOI] [PubMed] [Google Scholar]
- 21.Wilmer MJ, Ng CP, Lanz HL, Vulto P, Suter-Dick L and Masereeuw R, Trends Biotechnol, 2016, 34, 156–170. [DOI] [PubMed] [Google Scholar]
- 22.Jang M, Koh I, Lee SJ, Cheong J-H and Kim P, Sci. Rep, 2017, 7, 41541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huebsch N, Loskill P, Deveshwar N, Spencer CI, Judge LM, Mandegar MA, Fox CB, Mohamed TM, Ma Z, Mathur A, Sheehan AM, Truong A, Saxton M, Yoo J, Srivastava D, Desai TA, So P-L, Healy KE and Conklin BR, Sci. Rep, 2016, 6, 24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruppen J, Wildhaber FD, Strub C, Hall SRR, Schmid RA, Geiser T and Guenat OT, Lab Chip, 2015, 15, 3076–3085. [DOI] [PubMed] [Google Scholar]
- 25.Kim J, Chung M, Kim S, Jo DH, Kim JH and Jeon NL, PLoS One, 2015, 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeong SY, Lee JH, Shin Y, Chung S and Kuh HJ, PLoS One, 2016, 11, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu T-H, Kao Y-L, Lin W-L, Xiao J-L, Kuo P-L, Wu C-W, Liao W-Y and Lee C-H, Integr. Biol. (Camb)., 2012, 4, 177–82. [DOI] [PubMed] [Google Scholar]
- 28.Devarasetty M, Wang E, Soker S and Skardal A, Biofabrication, 2017, 9, 21002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belair DG, Whisler JA, Valdez J, Velazquez J, Molenda JA, Vickerman V, Lewis R, Daigh C, Hansen TD, Mann DA, Thomson JA, Griffith LG, Kamm RD, Schwartz MP and Murphy WL, Stem Cell Rev. Reports, 2015, 11, 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen D-HT, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA and Chen CS, Proc. Natl. Acad. ScL, 2013, 110, 6712–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sobrino A, Phan DTT, Datta R, Wang X, Hachey SJ, Romero-López M, Gratton E, Lee AP, George SC and Hughes CCW, Sci. Rep, 2016, 6, 31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu Y-H, Moya ML, Hughes CCW, George SC, Lee AP, George C and Lee AP, Lab Chip, 2013, 13, 2990–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moya ML, Hsu Y-H, Lee AP, Hughes CC and George SC, Tissue Eng. Part C Methods, 2013, 19, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB and Kamm RD, Proc. Natl. Acad. Sci, 2012, 109, 13515–13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan JM, Zervantonakis IK, Rimchala T, Polacheck WJ, Whisler J and Kamm RD, PLoS One, 2012, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen MB, Whisler JA, Fröse J, Yu C, Shin Y and Kamm RD, Nat. Protoc, 2017, 12, 865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Phan DTT, George SC, Hughes CCW and Lee AP, 3D Cell Culture - Methods and Protocols, Springer; New York, New York, NY, 2017, vol. 1612, pp. 325–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurokawa YK, Yin RT, Shang MR, Shirure VS, Moya ML and George SC, Tissue Eng. Part C Methods, 2017, 23, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan L, Skoko J, Yu J, Leduc PR and Neumann CA, Sci. Rep, 2017, 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caballero D, Blackburn SM, de Pablo M, Samitier J and Albertazzi L, Lab Chip, 2017, 17, 3760–3771. [DOI] [PubMed] [Google Scholar]
- 41.Parlato S, De Ninno A, Molfetta R, Toschi E, Salerno D, Mencattini A, Romagnoli G, Fragale A, Roccazzello L, Buoncervello M, Canini I, Bentivegna E, Falchi M, Bertani FR, Gerardino A, Martinelli E, Natale C, Paolini R, Businaro L and Gabriele L, Sci. Rep, 2017, 7, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, He MX, Walker W, Zhang G, Tian T, Cheng C, Wei Z, Palakurthi S, Bittinger M, Vitzthum H, Kim JW, Merlino A, Quinn M, Venkataramani C, Kaplan JA, Portell A, Gokhale PC, Phillips A, Smart A, Rotem A, Jones RE, Keogh L, Anguiano M, Stapleton L, Jia Z, Barzily-Rokni M, Cañadas I, Thai TC, Hammond MR, Vlahos R, Wang ES, Zhang H, Li S, Hanna GJ, Huang W, Hoang MP, Piris A, Eliane J-P, Stemmer-Rachamimov AO, Cameron L, Su M-J, Shah P, Izar B, Thakuria M, LeBoeuf NR, Rabinowits G, Gunda V, Parangi S, Cleary JM, Miller BC, Kitajima S, Thummalapalli R, Miao B, Barbie TU, Sivathanu V, Wong J, Richards WG, Bueno R, Yoon CH, Miret J, Herlyn M, Garraway LA, Van Allen EM, Freeman GJ, Kirschmeier PT, Lorch JH, Ott PA, Hodi FS, Flaherty KT, Kamm RD, Boland GM, Wong K-K, Dornan D, Paweletz CP and Barbie DA, Cancer Discov, 2018, 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavesi A, Adriani G, Tay A, Warkiani ME, Yeap WH, Wong SC and Kamm RD, Sci. Rep, 2016, 6, 26584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phan DTT, Wang X, Craver BM, Sobrino A, Zhao D, Chen JC, Lee LYN, George SC, Lee AP and Hughes CCW, Lab Chip, 2017, 17, 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen MB, Whisler JA, Jeon JS and Kamm RD, Integr. Biol, 2013, 5, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hassell BA, Goyal G, Lee E, Sontheimer-Phelps A, Levy O, Chen CS and Ingber DE, Cell Rep, 2017, 21, 508–516. [DOI] [PubMed] [Google Scholar]
- 47.Marturano-Kruik A, Nava MM, Yeager K, Chramiec A, Hao L, Robinson S, Guo E, Raimondi MT and Vunjak-Novakovic G, Proceedings of the National Academy of Sciences, 2018, 115, 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sung JH, Kam C and Shuler ML, Lab Chip, 2010, 10, 446. [DOI] [PubMed] [Google Scholar]
- 49.Shuler ML, Lab Chip, 2017, 17, 2345–2346. [DOI] [PubMed] [Google Scholar]
- 50.Tsamandouras N, Chen WLK, Edington CD, Stokes CL, Griffith LG and Cirit M, AAPS J, 2017, 19, 1499–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edmondson R, Broglie JJ, Adcock AF and Yang L, ASSAY and Drug Development Technologies, 2014, 12, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heylman C, Sobrino A, Shirure VS, Hughes CCW and George SC, Exp. Biol. Med, 2014, 239, 1240–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bissell MJ, Rizki A and Mian IS, Curr. Opin. Cell Biol, 2003, 15, 753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li ML, Aggeler J, Farson DA, Hatier C, Hassell J and Bissell MJ, Proc. Natl. Acad. Sci. U. S. A, 1987, 84, 136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bissell MJ and Radisky D, Nat. Rev. Cancer, 2001, 1, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, McFarland JM, Wong B, Boehm JS, Beroukhim R and Golub TR, Nat. Genet, 2017, 49, 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yeung TM, Gandhi SC, Wilding JL, Muschel R and Bodmer WF, Proc. Natl. Acad. Sci, 2010, 107, 3722–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, Ribas A, Lo RS, Flaherty KT, Ogino S, Wargo JA and Golub TR, Nature, 2012, 487, 500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z and Guo A, J. Cancer, 2017, 8, 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams CB, Yeh ES and Soloff AC, NPJ breast cancer, 2016, 2, 15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asano K, Nelson CM, Nandadasa S, Aramaki-Hattori N, Lindner DJ, Alban T, Inagaki J, Ohtsuki T, Oohashi T, Apte SS and Hirohata S, Sci. Rep, 2017, 7, 17225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tauriello DVF and Batlle E, Trends in cancer, 2016, 2, 495–504. [DOI] [PubMed] [Google Scholar]
- 63.Correia AL and Bissell MJ, Drug Resist. Updat, 2012, 15, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sutherland RM, Mccredie JA and Inch WR, J. Natl. Cancer Inst, 1971, 46, 113–120. [PubMed] [Google Scholar]
- 65.Takebe T, Zhang B and Radisic M, Cell Stem Cell, 2017, 21, 297–300. [DOI] [PubMed] [Google Scholar]
- 66.Elliott NT and Yuan F, J. Pharm. Sci, 2011, 100, 59–74. [DOI] [PubMed] [Google Scholar]
- 67.Takai A, Fako V, Dang H, Forgues M, Yu Z, Budhu A and Wang XW, Sci. Rep, 2016, 6, 21174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Verjans E-T, Doijen J, Luyten W, Landuyt B and Schoofs L, J. Cell. Physiol, 2018, 233, 2993–3003. [DOI] [PubMed] [Google Scholar]
- 69.Friedl P and Alexander S, Cell, 2011, 147, 992–1009. [DOI] [PubMed] [Google Scholar]
- 70.Polacheck WJ, German AE, Mammoto A, Ingber DE and Kamm RD, Proc. Natl. Acad. Sci, 2014, 111, 2447–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Esch EW, Bahinski A and Huh D, Nat. Rev. Drug Discov, 2015, 14, 248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Odom DT, Dowell RD, Jacobsen ES, Gordon W, Danford TW, Maclsaac KD, Rolfe PA, Conboy CM, Gifford DK and Fraenkel E, Nat. Genet, 2007, 39, 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mestas J and Hughes CCW, J. Immunol., 2004, 172, 2731–2738. [DOI] [PubMed] [Google Scholar]
- 74.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W and Tompkins RG, Proc. Natl. Acad. Sci, 2013, 110, 3507– 3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruggeri BA, Camp F and Miknyoczki S, Biochem. Pharmacol, 2014, 87, 150–161. [DOI] [PubMed] [Google Scholar]
- 76.Puig I, Chicote I, Tenbaum SP, Arqués O, Herance JR, Gispert JD, Jimenez J, Landolfi S, Caci K, Allende H, Mendizabal L, Moreno D, Charco R, Espín E, Prat A, Elez ME, Argilés G, Vivancos A, Tabernero J, Rojas S and Palmer HG, Clin. Cancer Res, 2013, 19, 6787–6801. [DOI] [PubMed] [Google Scholar]
- 77.Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, Gelmon K, Chia S, Mar C, Wan A, Laks E, Biele J, Shumansky K, Rosner J, McPherson A, Nielsen C, Roth AJ, Lefebvre C, Bashashati A, De Souza C, Siu C, Aniba R, Brimhall J, Oloumi A, Osako T, Bruna A, Sandoval JL, Algara T, Greenwood W, Leung K, Cheng H, Xue H, Wang Y, Lin D, Mungall AJ, Moore R, Zhao Y, Lorette J, Nguyen L, Huntsman D, Eaves CJ, Hansen C, Marra MA, Caldas C, Shah SP and Aparicio S, Nature, 2015, 518, 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zitvogel L, Pitt JM, Daillère R, Smyth MJ and Kroemer G, Nat. Rev. Cancer, 2016, 16, 759–773. [DOI] [PubMed] [Google Scholar]
- 79.Farkona S, Diamandis EP and Blasutig IM, BMC Med, 2016, 14, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rangarajan A and Weinberg RA, Nat. Rev. Cancer, 2003, 3, 952–959. [DOI] [PubMed] [Google Scholar]
- 81.Hanahan D and Weinberg RA, Cell, 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 82.Byrne AT, Alférez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A, Budinská E, Caldas C, Chang DK, Clarke RB, Clevers H, Coukos G, Dangles-Marie V, Gail Eckhardt S, Gonzalez-Suarez E, Hermans E, Hidalgo M, Jarzabek MA, De Jong S, Jonkers J, Kemper K, Lanfrancone L, Mælandsmo GM, Marangoni E, Marine JC, Medico E, Norum JH, Palmer HG, Peeper DS, Pelicci PG, Piris-Gimenez A, Roman-Roman S, Rueda OM, Seoane J, Serra V, Soucek L, Vanhecke D, Villanueva A, Vinolo E, Bertotti A and Trusolino L, Nature Reviews Cancer, 2017, 17, 254–268. [DOI] [PubMed] [Google Scholar]
- 83.Clohessy JG and Pandolfi PP, Nature Reviews Clinical Oncology, 2015, 12, 491–498. [DOI] [PubMed] [Google Scholar]
- 84.Landgraf M, McGovern JA, Friedl P and Hutmacher DW, Trends in Biotechnology, 2018, 36, year. [DOI] [PubMed] [Google Scholar]
- 85.Andersen M, Anderson H, Bailar JC, Boekelheide K, Brent R, Charnley G, Cheung VG, Green S, Kelsey KT, Kerkvliet NI, Li A. a., McCray L, Meyer O, Patterson RD, Pennie W, Scala R. a., Solomon GM, Stephens M, Yager J, Zeise L, Krewski Daniel, Acosta Daniel Jr., Andersen Melvin, Anderson Henry, Bailar John C. III, Boekelheide Kim, Brent Robert, Charnley Gail, Cheung Vivian G., Green Sidney Jr., Kelsey Karl T., Kerkvliet Nancy I., Li Abby A., McCray Lawrence and Zeise L, Natl. Acad. Sci, 2007, 13, 51–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tannenbaum J and Bennett BT, J. Am. Assoc. Lab. Anim. Sci, 2015, 54, 120–32. [PMC free article] [PubMed] [Google Scholar]
- 87.Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, Lees DM, Wong P-P, Alexopoulou A, Elia G, Clear A, Ledoux A, Hunter J, Perkins N, Gribben JG and Hodivala-Dilke KM, Nature, 2014, 514, 112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Romero-López M, Trinh AL, Sobrino A, Hatch MM, Keating MT, Fimbres C, Lewis DE, Gershon PD, Botvinick EL, Digman M, Lowengrub JS and Hughes CC, Biomaterials, 2017, 116, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Portillo-Lara R and Annabi N, Lab Chip, 2016, 16, 4063–4081. [DOI] [PubMed] [Google Scholar]
- 90.Maley CC, Aktipis A, Graham TA, Sottoriva A, Boddy AM, Janiszewska M, Silva AS, Gerlinger M, Yuan Y, Pienta KJ, Anderson KS, Gatenby R, Swanton C, Posada D, Wu CI, Schiffman JD, Hwang ES, Polyak K, Anderson AR, Brown JS, Greaves M and Shibata D, Nature Reviews Cancer, 2017, 17, 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vining KH and Mooney DJ, Nat. Rev. Mol. Cell Biol, 2017, 18, 728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Langley RR and Fidler IJ, Endocr. Rev, 2007, 28, 297–321. [DOI] [PubMed] [Google Scholar]
- 93.Semenza GL, Curr. Opin. Cell Biol, 2001, 13, 167–171. [DOI] [PubMed] [Google Scholar]
- 94.Lee GY, Kenny P. a., Lee EH and Bissell MJ, Nat. Methods, 2007, 4, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Inman JL, Robertson C, Mott JD and Bissell MJ, Development, 2015, 142, 1028–1042. [DOI] [PubMed] [Google Scholar]
- 96.Baker M, Nature, 2011, 471, 661–665. [DOI] [PubMed] [Google Scholar]
- 97.Ingber DE, Cell, 2016, 164, 1105–1109. [DOI] [PubMed] [Google Scholar]
- 98.Song JW and Munn LL, Proc. Natl. Acad. Sci, 2011, 108, 15342–15347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Piotrowski-Daspit AS, Simi AK, Pang M-F, Tien J and Nelson CM, Mammary Gland Dev. Methods Protoc, Springer New York, New York, NY, 2017, pp. 245–257. [DOI] [PubMed] [Google Scholar]
- 100.Shirure VS, Lezia A, Tao A, Alonzo LF and George SC, Angiogenesis, 2017, 20, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sackmann EK, Fulton AL and Beebe DJ, Nature, 2014, 507, 181–189. [DOI] [PubMed] [Google Scholar]
- 102.Marturano-Kruik A, Villasante A, Yaeger K, Ambati SR, Chramiec A, Raimondi MT and Vunjak-Novakovic G, Bio-materials, 2018, 150, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Skardal A, Murphy SV, Devarasetty M, Mead I, Kang H-W, Seol Y-J, Shrike Zhang Y, Shin S-R, Zhao L, Aleman J, Hall AR, Shupe TD, Kleensang A, Dokmeci MR, Jin Lee S, Jackson JD, Yoo JJ, Hartung T, Khademhosseini A, Soker S, Bishop CE and Atala A, Sci. Rep, 2017, 7, 8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bhise NS, Ribas J, Manoharan V, Zhang YS, Polini A, Massa S, Dokmeci MR and Khademhosseini A, J. Control. Release, 2014, 190, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Edington CD, Chen WLK, Geishecker E, Kassis T, Soenksen LR, Bhushan BM, Freake D, Kirschner J, Maass C, Tsamandouras N, Valdez J, Cook CD, Parent T, Snyder S, Yu JJ, Suter E, Shockley M, Velazquez J, Velazquez JJ, Stockdale L, Papps JP, Lee I, Vann N, Gamboa M, LaBarge ME, Zhong Z, Wang X, Boyer LA, Lauffenburger DA, Carrier RL, Communal C, Tannenbaum SR, Stokes CL, Hughes DJ, Rohatgi G, Trumper DL, Cirit M and Griffith LG, Sci. Rep, 2018, 8, 4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang YS, Zhang YN and Zhang W, Drug Discov. Today, 2017, 22, 1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ronaldson-Bouchard K and Vunjak-Novakovic G, Cell Stem Cell, 2018, 22, 310–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sung JH, Esch MB, Prot J-M, Long CJ, Smith A, Hickman JJ and Shuler ML, Lab on a Chip, 2013, 13, 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mathur A, Loskill P, Hong S, Lee JY, Marcus SG, Dumont L, Conklin BR, Willenbring H, Lee LP and Healy KE, Stem Cell Res. Ther, 2013, 4 Suppl 1, S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mathur A, Loskill P, Shao K, Huebsch N, Hong S, Marcus SG, Marks N, Mandegar M, Conklin BR, Lee LP and Healy KE, Sci. Rep, 2015, 5, 8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Folkman J, Nat. Rev. Drug Discov, 2007, 6, 273–286. [DOI] [PubMed] [Google Scholar]
- 112.Chung AS, Lee J and Ferrara N, Nat. Rev. Cancer, 2010, 10, 505–514. [DOI] [PubMed] [Google Scholar]
- 113.Potente M, Gerhardt H and Carmeliet P, Cell, 2011, 146, 873–887. [DOI] [PubMed] [Google Scholar]
- 114.Khazali AS, Clark AM and Wells A, Stem Cell Rev. Reports, 2017, 13, 364–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim S, Lee H, Chung M and Jeon NL, Lab Chip, 2013, 13, 1489. [DOI] [PubMed] [Google Scholar]
- 116.Kim C, Kasuya J, Jeon J, Chung S and Kamm RD, Lab Chip, 2015, 15, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Whisler JA, Chen MB and Kamm RD, Tissue Eng. Part C Methods, 2014, 20, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen X, Aledia AS, Popson S. a., Him L, Hughes CC and George SC, Tissue Eng. Part A, 2010, 16, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jeon JS, Bersini S, Whisler JA, Chen MB, Dubini G, Charest JL, Moretti M and Kamm RD, Integr. Biol. (Camb)., 2014, 6, 555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton G. a., Thorneloe KS, Mcalexander MA and Ingber DE, Sci. Transl. Med, 2012, 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sosa MS, Bragado P and Aguirre-Ghiso JA, Nature Reviews Cancer, 2014, 14, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Aguado BA, Bushnell GG, Rao SS, Jeruss JS and Shea LD., Nature Biomedical Engineering, 2017, 1, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A and DeNardo DG, Nat. Med, 2016, 22, 851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sharma P, Hu-Lieskovan S, Wargo JA and Ribas A, Cell, 2017, 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Boussommier-Calleja A, Li R, Chen MB, Wong SC and Kamm RD, Trends in cancer, 2016, 2, 6–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lamouille S, Xu J and Derynck R, Nat. Rev. Mol. Cell Biol, 2014, 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Marcucci F, Stassi G and De Maria R, Nat. Rev. Drug Discov, 2016, 15, 311–325. [DOI] [PubMed] [Google Scholar]
- 128.Hughes CC, Savage CO and Pober JS, Immunol. Rev, 1990, 117, 85–102. [DOI] [PubMed] [Google Scholar]
- 129.Fellmann C, Gowen BG, Lin PC, Doudna JA and Corn JE, Nature Reviews Drug Discovery, 2017, 16, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Papapetrou EP, Nat. Med, 2016, 22, 1392–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bruce A, Evans R, Mezan R, Shi L, Moses BS, Martin KH, Gibson LF and Yang Y, PLoS ONE, 2015, 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Torisawa YS, Spina CS, Mammoto T, Mammoto A, Weaver JC, Tat T, Collins JJ and Ingber DE, Nature Methods, 2014, 11, 663–669. [DOI] [PubMed] [Google Scholar]
- 133.Knezevic L, Schaupper M, Mühleder S, Schimek K, Hasenberg T, Marx U, Priglinger E, Redl H and Holnthoner W, Frontiers in Bioengineering and Biotechnology, 2017, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Giese C, Lubitz A, Demmler CD, Reuschel J, Bergner K and Marx U, Journal of Biotechnology, 2010, 148, 38–45. [DOI] [PubMed] [Google Scholar]
- 135.Skardal A, Shupe T and Atala A, Drug Discov. Today, 2016, 21, 1399–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang YI, Carmona C, Hickman JJ and Shuler ML, Adv. Healthc. Mater, 2017, 1701000, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Prantil-Baun R, Novak R, Das D, Somayaji MR, Przekwas A and Ingber DE, Annu. Rev. Pharmacol. Toxicol, 2016, 58, 37–64. [DOI] [PubMed] [Google Scholar]
- 138.Marx U, Andersson TB, Bahinski A, Beilmann M, Beken S, Cassee FR, Cirit M, Daneshian M, Fitzpatrick S, Frey O, Gaertner C, Giese C, Griffith L, Hartung T, Heringa MB, Hoeng J, De Jong WH, Kojima H, Kuehnl J, Leist M, Luch A, Maschmeyer I, Sakharov D, Sips AJ, Steger-Hartmann T, Tagle DA, Tonevitsky A, Tralau T, Tsyb S, Van De Stolpe A, Vandebriel R, Vulto P, Wang J, Wiest J, Rodenburg M and Roth A, Altex, 2016, 33, 272–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wikswo JP, Curtis EL, Eagleton ZE, Evans BC, Kole A, Hofmeister LH and Matloff WJ, Lab Chip, 2013, 13, 3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, Ben-Zvi I, Stebbing J, McGuire W, Harris W, Maki R, Gaya A, Bedi A, Zacharoulis S, Ravi R, Wexler LH, Hoque MO, Rodriguez-Galindo C, Pass H, Peled N, Davies A, Morris R, Hidalgo M and Sidransky D, Annals of Oncology, 2017, 28, 2595–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ashley EA, Nat. Rev. Genet, 2016, 17, 507–522. [DOI] [PubMed] [Google Scholar]
- 142.Astolfi M, Péant B, Lateef MA, Rousset N, Kendall-Dupont J, Carmona E, Monet F, Saad F, Provencher D, Mes-Masson A-M and Gervais T, Lab Chip, 2016, 16, 312–325. [DOI] [PubMed] [Google Scholar]
- 143.Wang Y, Wang L, Zhu Y and Qin J, Lab on a Chip, 2018, 18, 851–860. [DOI] [PubMed] [Google Scholar]
- 144.Kim HJ, Li H, Collins JJ and Ingber DE, Proceedings of the National Academy of Sciences, 2016, 113, E7–E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rios AC and Clevers H, Nat. Methods, 2018, 15, 24–26. [DOI] [PubMed] [Google Scholar]
- 146.Van De Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, Van Houdt W, Van Gorp J, Taylor-Weiner A, Kester L, McLaren-Douglas A, Blokker J, Jaksani S, Bartfeld S, Volckman R, Van Sluis P, Li VS, Seepo S, Sekhar Pedamallu C, Cibulskis K, Carter SL, McKenna A, Lawrence MS, Lichtenstein L, Stewart C, Koster J, Versteeg R, Van Oudenaarden A, Saez-Rodriguez J, Vries RG, Getz G, Wessels L, Stratton MR, McDermott U, Meyerson M, Garnett MJ and Clevers H, Cell, 2015, 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, Pogrebniak K, Sandoval J, Cassidy JW, Tufegdzic-Vidakovic A, Sammut SJ, Jones L, Provenzano E, Baird R, Eirew P, Hadfleld J, Eldridge M, McLaren-Douglas A, Barthorpe A, Lightfoot H, O’Connor MJ, Gray J, Cortes J, Baselga J, Marangoni E, Welm AL, Aparicio S, Serra V, Garnett MJ and Caldas C, Cell, 2016, 167, 260–274.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lancaster MA and Knoblich JA, Science, 2014, 345, 1247125–1247125. [DOI] [PubMed] [Google Scholar]
- 149.Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, Rata M, Koh D.-m., Tunariu N, Collins D, Hulkki-Wilson S, Ragulan C, Spiteri I, Moorcraft SY, Chau I, Rao S, Watkins D, Fotiadis N, Bali M, Darvish-Damavandi M, Lote H, Eltahir Z, Smyth EC, Begum R, Clarke PA, Hahne JC, Dowsett M, de Bono J, Workman P, Sadanandam A, Fassan M, Sansom OJ, Eccles S, Starling N, Braconi C, Sottoriva A, Robinson SP, Cunningham D and Valeri N, Science, 2018, 359, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, Chua CW, Barlow LMJ, Kandoth C, Williams AB, Bergren SK, Pietzak EJ, Anderson CB, Benson MC, Coleman JA, Taylor BS, Abate-Shen C, McKiernan JM, Al-Ahmadie H, Solit DB and Shen MM, Cell, 2018, 173, 515–528. el7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kasendra M, Tovaglieri A, Sontheimer-Phelps A, Jalili-Firoozinezhad S, Bein A, Chalkiadaki A, Scholl W, Zhang C, Rickner H, Richmond CA, Li H, Breault DT and Ingber DE, Scientific Reports, 2018, 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Srinivasan T, Walters J, Bu P, Than EB, Tung KL, Chen KY, Panarelli N, Milsom J, Augenlicht L, Lipkin SM and Shen X, Cancer Res, 2016, 76, 3411–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Drost J and Clevers H, Nat. Rev. Cancer, 2018, 18, 407–418. [DOI] [PubMed] [Google Scholar]
- 154.Takahashi K and Yamanaka S, Development, 2013, 140, 2457–2461. [DOI] [PubMed] [Google Scholar]
- 155.Drew L, Nature, 2016, 537, S60–S62. [DOI] [PubMed] [Google Scholar]
- 156.Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmström A, Chang AC, Coronado MJ, Ebert AD, Knowles JW, Telli ML, Witteles RM, Blau HM, Bernstein D, Altman RB and Wu JC, Nature Medicine, 2016, 22, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Low LA and Tagle DA, Expert Review of Precision Medicine and Drug Development, 2018, 3, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fong AH, Romero-López M, Heylman CM, Keating M, Tran D, Sobrino A, Tran AQ, Pham HH, Fimbres C, Gershon PD, Botvinick EL, George SC and Hughes CC, Tissue Engineering Part A, 2016, 22, 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kang L, Chung BG, Langer R and Khademhosseini A, Drug Discov. Today, 2008, 13, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Caballero D, Kaushik S, Correlo V, Oliveira J, Reis R and Kundu S, Biomatenals, 2017, 149, 98–115. [DOI] [PubMed] [Google Scholar]
- 161.Cirit M and Stokes CL, Lab on a Chip, 2018, 18, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Van De Stolpe A and Kauffmann RH, RSC Advances, 2015, 5, 18451–18453. [Google Scholar]
- 163.Carreau A, Hafny-Rahbi BE, Matejuk A, Grillon C and Kieda C, J. Cell. Mol. Med, 2011, 15, 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Acosta MA, Jiang X, Huang P-K, Cutler KB, Grant CS, Walker GM and Gamcsik MP, Biomicrofluidics, 2014, 8, 054117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ehsan SM, Welch-Reardon KM, Waterman ML, Hughes CCW and George SC, Integr. Biol, 2014, 6, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chen Y-A, King AD, Shih H-C, Peng C-C, Wu C-Y, Liao W-H and Tung Y-C, Lab Chip, 2011, 11, 3626. [DOI] [PubMed] [Google Scholar]
- 167.Brennan MD, Rexius-Hall ML, Elgass LJ and Eddington DT, Lab Chip, 2014, 14, 4305–18. [DOI] [PubMed] [Google Scholar]
- 168.Ehsan SM and George SC, J. Biosci. Bioeng., 2015, 120, 347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bashir S, Bashir M, Casadevall i Solvas X, Rees JM and Zimmerman WB, Micromachines, 2015, 6, 1445–1458. [Google Scholar]
- 170.Junaid A, Mashaghi A, Hankemeier T and Vulto P, Current Opinion in Bioinedical Engineering, 2017, 1, 15–22. [Google Scholar]
- 171.Urban G, Bache KM, Phan D, Sobrino A, Shmakov AK, Hachey SJ, Hughes C and Baldi P, IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2018, 1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ghaemmaghami AM, Hancock MJ, Harrington H, Kaji H and Khademhosseini A, Drug Discov. Today, 2012, 17, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wood DK, Soriano A, Mahadevan L, Higgins JM and Bhatia SN, Sci. Transl. Med, 2012, 4, 123ra26–123ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]