

Abstract
The 7-deazapurine derivatives, 2’-deoxy-7-cyano-7-deazaguanosine (dPreQ0) and 2’-deoxy-7-amido-7-deazaguanosine (dADG) are recently-discovered DNA modifications encoded by the dpd cluster found in a diverse set of bacteria. Here we identify the genes required for formation of dPreQ0 and dADG in DNA and propose a biosynthetic pathway. The preQ0 base is a precursor that in Salmonella Montevideo, is synthesized as an intermediate in the pathway of the tRNA modification queuosine. Of the eleven genes (dpdA - dpdK) found in the S. Montevideo dpd cluster, dpdA and dpdB are necessary and sufficient to synthesize dPreQ0, while dpdC is additionally required for dADG synthesis. Among the rest of the dpd genes, dpdE, dpdG, dpdI, dpdK, dpdD and possibly dpdJ appear to be involved in a restriction-like phenotype. Indirect competition for preQ0 base led to a model for dADG synthesis in which DpdA inserts preQ0 into DNA with the help of DpdB, and then DpdC hydrolyzes dPreQ0 to dADG. The role of DpdB is not entirely clear as it is dispensable in other dpd clusters. Our discovery of a minimal gene set for introducing 7-deazapurine derivatives in DNA provides new tools for biotechnology applications and demonstrates the interplay between the DNA and RNA modification machineries.
Keywords: DNA modification, 7-deazapurine, restriction
Graphical abstract
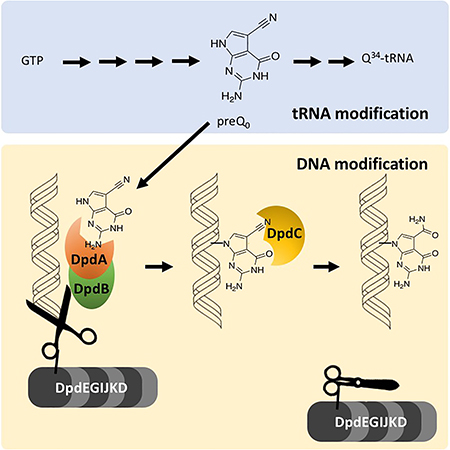
DNA modifications allow Bacteria to recognize self from non self. We describe a novel pathway responsible for the insertion of a complex modification that was thought to be found only in RNA.
Abbreviated Summary
DNA modifications allow Bacteria to recognize self from non self. We describe a novel pathway responsible for the insertion of a complex modification that was thought to be found only in RNA.
Introduction
Although the first discovery of a non-canonical nucleoside in a nucleic acid was the identification of 5-methylcytosine (5mC) in DNA in the early 1950s (Hotchkiss, 1948; Wyatt, 1950), it is in RNA that both the quantity and complexity of nucleic acid modifications have been realized. More than one-hundred RNA modifications have been found to date (Machnicka et al., 2013), mainly in transfer RNA (tRNA) and ribosomal RNA (rRNA) (Grosjean, 2009). In contrast, only twenty modifications have been identified in DNA and most of the known DNA modifications are simple methylated derivatives of canonical nucleosides (Grosjean, 2009; Weigele & Raleigh, 2016). The few structurally complex DNA modifications are mainly found in phages where they serve to evade the host-encoded restriction systems (Warren, 1980; Weigele & Raleigh, 2016). Indeed, numerous bacteria have acquired restriction and modification (R-M) systems to defend against foreign DNA (Vasu & Nagaraja, 2013). Well-studied phage DNA modifications include 5-methylcytosine in phage XP12 (Feng, Tu, & Kuo, 1978; Huang, Farnet, Ehrlich, & Ehrlich, 1982); 5-hydroxymethylcytosine in phage T4gt, β-glucosyl-5-hydroxymethylcytosine in phage T4 (Cao, Huang, Farnet, & Ehrlich, 1983), N6-(1-acetamido)-adenine in bacteriophage Mu (Hattman, 1979) and 5-hydroxymethyluracil in Bacillus SP8 (Vilpo & Vilpo, 1995). In addition, recently discovered 5-(2-aminoethoxy)methyluridine in Salmonella phage VII, 5-(2-aminoethyl)uridine in Pseudomonas phage M6 (Lee et al., 2018) and phosphorothioate (PT) modification of the DNA backbone (Chen et al., 2017; Wang et al., 2007) expanded the list of naturally-occurring DNA modifications.
Until recently, 7-deazapurine modifications were thought to be limited to tRNA. Queuosine (Q, 7-{5-[((1S,4S,5R)-4,5-Dihydroxy-2-cyclopenten-1-yl)amino]methyl}−7-deazaguanosine) is found at the wobble base (position 34) of a subset of bacterial and eukaryotic tRNAs (histidine, asparagine, aspartate and tyrosine) (Fergus, Barnes, Alqasem, & Kelly, 2015). Archaeosine (G+, 7-formamidino-7-deazaguanosine) is found at position 15 in the D-loop of many archaeal tRNAs (Gregson et al., 1993; Jühling et al., 2009; Sprinzl, Horn, Brown, Loudovltch, & Steinberg, 1998). However, this paradigm was changed by the recent discovery of 2’-deoxy-7-amido-7-deazaguanosine (dADG) and 2’-deoxy-7-cyano-7-deazaguanosine (dPreQ0) in bacterial genomic DNA and of 2’-deoxy-G+ (dG+) in phage DNA (Thiaville et al., 2016). [Note that the standard nomenclature uses “Q” to represent the ribonucleoside, “preQ0” and “ADG” to represent the corresponding base, “dPreQ0” and “dADG” to represent the corresponding 2’-deoxyribonucleosides.] In phages such as the Escherichia coli phage 9g, the presence of dG+ confers resistance to many restriction enzymes (Kulikov et al., 2014; Tsai, Corrêa, Xu, & Xu, 2017). The eleven-gene deazapurine-in-DNA (dpd) cluster of Salmonella enterica serovar Montevideo ATCC BAA-710 (S. Montevideo) was shown to be involved in modification of genomic DNA with dPreQ0 and dADG, and in restriction-like activity that caused low transformation efficiency when using unmodified DNA compared with modified DNA. Deletion of the whole cluster in S. Montevideo eliminated both modification and proposed restriction-like activities, and the presence of the cluster in other phylogenetically-diverse bacteria correlated with the presence of dADG in DNA (Thiaville et al., 2016). A R-M system encompassing a 20kb region is unusual, as classical systems are usually much smaller (Roberts, Vincze, Posfai, & Macelis, 2015; Wilson, 1991).
All bacterial dpd islands include a set of 11 common genes with minor variations. The signature gene for the island, dpdA, encodes a homolog of the archaeal tRNA-guanine transglycosylase (aTGT; EC 2.4.2.48), the enzyme involved in inserting the 7-cyano-7-deazaguanine (preQ0) precursor into archaeal tRNAs (Bai, Fox, Lacy, Van Lanen, & Iwata-Reuyl, 2000; Watanabe et al., 1997) (Fig. 1A). Analysis of DpdA sequences suggests that, like aTGT, these proteins bind preQ0. dpdA is usually surrounded by the dpdB and dpdC genes in a tight physical cluster. dpdB encodes a protein similar to DndB, a DNA binding protein involved in regulating expression of genes involved in phosphorothioate DNA modifications (He et al., 2015), although the exact molecular function of DndB is not clear. The Aravind group has proposed that DpdC might be similar to ArcS, the archaeal amidotransferase that modifies the preQ0 ribonucleoside into G+ (Phillips et al., 2010) (Fig. 1A) based on very weak homologies around the active site (Iyer, Zhang, Maxwell Burroughs, & Aravind, 2013). Further sequence analysis (Alva, Nam, Söding, & Lupas, 2016; Hildebrand, Remmert, Biegert, & Söding, 2009) showed that DpdC harbored a domain of unknown function DUF328 (Thiaville et al., 2016). The dpd clusters encode several putative DNA-binding enzymes, including a member of the DEAD/DEAH box helicase family (dpdJ), a SNF2-type helicase (dpdE), and a RecQ-like Superfamily II DNA helicase (dpdF) (Fig. 1B). dpdK encodes a protein harboring a phospholipase D-like domain. Little information is available about the proteins encoded by dpdD, dpdG and dpdI, but sequence analyses revealed low-confidence similarities of DUF3479 with a small portion of DpdD, of PF09821 with DpdG, and of PF15967 with DpdI (Thiaville et al., 2016).
Figure 1. Biosynthesis of 7-deazaguanine modifications in tRNA and DNA.

(A) The biosynthetic pathways for queuosine and archaeosine in tRNA. Dashed arrows represent proposed reactions. CPH4, 6-carboxytetrahydropterin; bTGT, tRNA-guanine transglycosylase. Enzymes abbrevations are described in text except for QueA, QueG and QueH that are described in (Zallot, Harrison, Kolaczkowski, & de Crécy-Lagard, 2016) in Zallot, Ross, et al., 2017. (B) The gene cluster governing 7-deazapurine biosynthesis in Salmonella enterica subspecies enterica serovar Montevideo (GCA_000238535.2; TgtA5 UniProt ID E7V8J4) (Thiaville et al., 2016). TA, toxin–antitoxin gene pair (ccdA, ccdB); leuX, tRNA-leu gene.
The S. Montevideo dpd cluster has been clearly shown to encode a R-M system (Thiaville et al., 2016), but it is still poorly understood, and the actual function of each gene has yet to be determined. Which genes are required for the formation of the modification in DNA, which genes are required for cleaving unmodified DNA, and why so many genes are involved, are all open questions.
PreQ0 is the common precursor of Q in Bacteria and G+ in Archaea (Fig. 1). It is synthesized from GTP in both kingdoms. The first step of the pathway, catalyzed by GTP cyclohydrolase I (GCHI; EC 3.5.4.16), is shared in Bacteria with the tetrahydrofolate synthesis pathway (Phillips et al., 2008), and produces dihydroneopterin triphosphate (H2NTP). H2NTP is then converted by 6-carboxytetrahydropterin synthase (QueD; EC 4.1.2.50) and 7-carboxy-7-deazaguanine synthase (QueE; EC 4.3.99.3) to give 7-carboxy-7-deazaguanine (CDG) (Mccarty, Lin, Jacobsen, & Bandarian, 2009; Reader, Metzgar, Schimmel, & de Crécy-Lagard, 2004). The last step in preQ0 synthesis is catalyzed by 7-cyano-7-deazaguanine synthase (QueC; EC 6.3.4.20) (Nelp & Bandarian, 2015; Reader, Metzgar, Schimmel, & de Crécy-Lagard, 2004) in two ATP dependent half-reactions. Adenylation of CDG, followed by the addition of ammonia, generates 7-amido-7-deazaguanine (ADG) as an amide intermediate. Subsequently, dehydration of ADG consumes a second equivalent of ATP and produces the nitrile product, preQ0 (Nelp & Bandarian, 2015). The discovery of dADG and dpreQ0 in the DNA of species like S. Montevideo that use preQ0 in Q synthesis in tRNA raises many questions. Do the synthesis pathways for 7-deazapurine production in RNA and DNA share a common precursor? Is there any competition for pathway intermediates? Here we define the necessary genes and propose a synthetic pathway for incorporation of the PreQ0 and ADG bases into DNA.
Results
The S. Montevideo dpd island is composed of two transcription units
The 11 genes of the dpdC-D island are encoded by the same DNA strand, some overlapping, and could hence be in one transcriptional operon. RT-PCR analysis was performed on cDNA synthesized from RNA extracted from the WT S. Montevideo strain. (Fig. S1 and Methods). Each intergenic region was successfully amplified with a unique pair of primers, except the ~580-bp intergenic region between dpdB and dpdE (Fig. S1B). This suggests the presence of two operons: the dpdCAB operon and the dpdEFGHIJKD operon (Fig. S1A).
Promoter and terminator prediction tools reinforce the RT-PCR results. The BPROM promoter prediction tool (Softberry Inc., Solovyev & Salamov, 2011) predicts one promoter ~20 bp upstream the dpdC gene in the S. Montevideo 20 kb island and another ~200 bp upstream dpdE (Fig. S1A). The ARNold terminator prediction tool (Université Paris-Sud web server, Gautheret and Lambert, 2001; Macke et al., 2001) predicted three rho-independent terminators in the dpd gene cluster with both the Erpin and RNAmotif programs. The top-scoring one is located ~8 bp downstream of the dpdD gene. The one with the second-best score is located ~15 bp downstream of dpdB (Fig. S1A).
The dpdEFGHIJKD operon may encode a restriction system inhibited by the dADG modification
We have previously shown that the transformation efficiency of the WT S. Montevideo strain with pUC19 DNA lacking dADG and dPreQ0 was reduced compared to pUC19 DNA containing dADG modifications. The efficiency of transformation was fully recovered when YYF3022, a S. Montevideo derivative with a 21-kb deletion eliminating the entire dpd cluster, was used a recipient (Fig. 5 in Thiaville et al., 2016), suggesting the dpd island encodes a R-M system. Growth rate and yield of the YYF3022 strain did not show significant changes under standard laboratory conditions compared to the WT (Fig. S2).
Figure 5. dADG can be formed from exogenous preQ0 favoring a role of DpdC after preQ0 insertion.

(A) Schematic representation of possible models for the roles of DpdA, DpdB and DpdC in dADG formation. Dashed arrows represent proposed reactions. All molecule abbreviations and protein names are described in the text. (B) pUC19 (10 ng) extracted from the ΔqueC deletion strain was transformed in the S. Montevideo WT host. The donor strain was fed either with DMSO (negative control) or preQ0. The WT and ΔdpdC-dpdD::kan donors are respectively positive and negative control of the experiment. The average of three experiments is shown, with error bars representing SD (**P < 0.01, two-tailed Student’s t-test).
The whole dpdEFGHIJKD operon was deleted as described in the Methods section to give strain YYF3345 (Fig. S3). When unmodified pUC19 DNA was transformed into this strain, a ~100-fold increase in transformation efficiency was observed compared to a WT recipient (Fig. 2A), suggesting that genes required for the restriction-like phenotype are part of the dpdEFGHIJKD operon. Individual deletions of every gene of this operon were then tested. Transformation efficiencies with unmodified pBAD33 DNA were low in the dpdF or dpdH mutants, as in the WT strain (Fig. 2C). On the other hand, deletion of dpdE, dpdG, dpdI, dpdJ, dpdK or dpdD resulted in a significant increase in transformation efficiency that was abolished with the expression of the corresponding gene in trans (with the exception of dpdJ encoding a Lhr-like helicase that we were unable to clone, hence in this case a polar effect cannot be eliminated). We also showed that the none of the dpdEFGHIJKD genes are involved in the synthesis of the modifications, as DNA extracted from the ΔdpdE-dpdD strain still harbors the dADG modification (Fig. 2B and Table 1).
Figure 2. The dpdEFGHIJKD mutant showed WT level transformation efficiency as donor and increased transformation efficiency as host.

(A) Unmodified pUC19 DNA (10 ng) was transformed in the S. Montevideo WT and ΔdpdE-dpdD::kan strains. (B) pUC19 DNA (10 ng) extracted from different mutant donor cells was transformed in S. Montevideo WT. (C) Unmodified pBAD33 DNA (10 ng) was transformed in the WT host and in each individual mutant strain containing pBAD24 or its derivative expressing the corresponding dpd gene in trans (with the exception of dpdJ that could not be cloned). The average of three experiments is shown, with error bars representing SD (*P < 0.05, ***P < 0.001, two-tailed Student’s t-test).
Table 1.
Levels of dADG and dPreQ0 in DNA from strains lacking or expressing various components of the dpd cluster.
| Strains | Genotype | Condition | dADG per 106 nt | dPreQ0 per 106 nt |
|---|---|---|---|---|
| YYF3048 | S. Montevideo WT | 330 | <5† | |
| YYF3022 | ΔdpdC-D::kan | <51 | <5 | |
| YYF3286 | ΔdpdE-dpdD::kan | 308 | <5 | |
| YYF3014 | ΔdpdD::kan | 69 | 10 | |
| YYF3434 | ΔdpdA::kan ΔdpdD | <5 | <5 | |
| YYF3402 | ΔdpdB::kan ΔdpdD | <5 | <5 | |
| YYF3401 | ΔdpdC::kan ΔdpdD | <5 | 28 | |
| YYF3422 | ΔdpdA::kan ΔdpdD pBY279.1::dpdA pUC19 |
500 ng mL−1 aTET | 86 | <5 |
| YYF3416 | ΔdpdB::kan ΔdpdD pBY279.1::dpdB pUC19 |
500 ng mL−1 aTET | 58 | <5 |
| YYF3418 | ΔdpdC::kan ΔdpdD pBY279.1::dpdC pUC19 |
500 ng mL−1 aTET | 475 | <5 |
| YYF3324 |
S. M ΔdpdC-dpdD::kan pBAD33::dpdCAB |
0.4% arabinose | 529 | <5 |
| YYF3425 |
S. M ΔdpdC-dpdD::kan pBY279.1::dpdAB |
500 ng mL-1 aTET | <5 | 334 |
| YYF3346 | ΔqueC::Kan | <5 | <5 | |
| YYF3460 | ΔqueC::kan pBAD24 |
0.4% arabinose | <5 | <5 |
| YYF3461 | ΔqueC::kan pBAD24::queC |
0.4% arabinose | 394 | 130 |
| YYF3346 | S. M ΔqueC::kan | 5 nM preQ0 | 24 | <6 |
| YYF3297 |
E. coli ΔqueC pBAD33 |
5 nM preQ0 | <6 | <6 |
| YYF3297 |
E. coli ΔqueC pBAD33::dpdAB |
5 nM preQ0 | <6 | 75 |
| YYF3303 |
E. coli ΔqueC pBAD33::dpdCAB |
5 nM preQ0 | 76 | <6 |
Limit of detection for both dADG and dPreQ0.
dpdCAB are necessary for ADG insertion in DNA
We were able to delete the dpdA gene only in a ΔdpdD strain (YYF3014). When the pUC19 DNA extracted from the ΔdpdA::kan ΔdpdD strain was used to transform WT cells, transformation efficiencies were low and comparable to the results with unmodified pUC19 DNA extracted from YYF3022 strain lacking the whole dpd cluster. This suggested that DpdA is required for the formation of dADG and/or dPreQ0 modifications (Fig. 2B). This hypothesis was confirmed by mass spectrometric analysis of genomic DNA extracted from the ΔdpdA mutant as both dADG and dPreQ0 levels were below the detection limits (Table 1). Using a similar strategy, deletion of dpdB and dpdC was performed in the ΔdpdD background, to give the YYF3401 (ΔdpdC::kan ΔdpdD) and YYF3402 (ΔdpdB::kan ΔdpdD) strains, respectively. Mass spectrometric analysis of genomic DNA from these strains revealed that they lacked dADG, demonstrating that the dpdB and dpdC genes were also required for ADG formation in DNA. However, dPreQ0 was detected in DNA from the ΔdpdC strain, but not from the ΔdpdA or ΔdpdB strains (Table 1), suggesting that dpdC is dispensable for dPreQ0 formation.
To confirm that all three genes were indeed required for dADG synthesis and our results were not due to polar effects, we first showed that deletion of dpdC or dpdA did not affect transcription of dpdAB or dpdB respectively by RT-PCR (Fig. S4). In parallel, we transformed each individual mutant with a plasmid expressing the deleted gene in trans and analyzed the genomic DNA of the resulting strains LC-MS/MS. As shown in Table 1, the dADG deficiency phenotype was complemented in all three cases. As dPreQ0 levels were low and not consistently detected in WT DNA, its absence in the complemented strains was not really conclusive.
dpdAB are sufficient for dPreQ0 synthesis and dpdC is required for dADG formation
The previous genetic results suggested that only DpdA and DpdB were necessary for the synthesis of dPreQ0 (Table 1, strain YYF3401). To test this hypothesis, the YYF3022 strain that lacks the entire dpd island was transformed with plasmids expressing dpdAB or dpdCAB. Mass spectrometric analysis of the genomic DNA of these strains (Table 1) showed an increased amount of dPreQ0 compared to WT when dpdAB was overexpressed (the YYF3324 strain), while dADG could be detected only in the YYF3425 strain expressing the three genes (dpdCAB).
To address the question of what modified base (preQ0 and/or ADG) conferred protection from the potential restriction activity, we isolated plasmid DNA from a variety of strains and investigated the transformation efficiencies of each of the plasmid samples. Transformation efficiencies were high when pUC19 was extracted from S. Montevideo mutant strain YYF3022 (Fig. 3A), Salmonella enterica serovar Typhimurium LT2 and E. coli K12 (Fig. 3B) expressing dpdCAB in trans. These results suggest that dpdC, dpdA and dpdB represent the minimal set of genes required for ADG insertion. In contrast, the transformation efficiencies were low and comparable to the negative control when pUC19 was extracted from these strains expressing only dpdAB (Fig. 3). Taken together the data support the conclusion that it is dADG, and not dPreQ0, that confers protection from the potential dpd restriction machinery. The data are also consistent with dpdC, dpdA and dpdB comprising the minimal set of genes required for dADG formation in DNA, at least in organisms that synthesize the preQ0 precursor as discussed below.
Figure 3. Expression of dpdCAB genes restores high transformation efficiencies in the WT strain.

(A) pBAD33 and pUC19 DNA (20 ng of total) extracted from S. Montevideo strains carrying empty pBAD33 vector, pBAD33::dpdAB or pBAD33::dpdCAB was transformed in the WT strain. (B) pBAD33 and pUC19 DNA (20 ng of total) extracted from the indicated strains was transformed in the S. Montevideo WT strain. The average of three experiments is shown, with error bars representing SD (*P < 0.05, **P < 0.01, two-tailed Student’s t-test).
PreQ0 is the precursor of the dADG modification
PreQ0 and ADG are intermediates in the tRNA Q modification pathway (Fig. 1), with QueC converting CDG to preQ0 through the ADG intermediate (Nelp & Bandarian, 2015). S. Montevideo possesses all of the genes involved in the Q pathway (Fig. 1) (Reader, Metzgar, Schimmel, & de Crecy-Lagard, 2004; Thiaville et al., 2016) and we have previously shown that Q is present in tRNA extracted from this organism (Thiaville et al., 2016). The parsimonious hypothesis was therefore that S. Montevideo QueC synthesizes the precursors needed for the formation of dPreQ0 and dADG in DNA as well as Q in tRNA. We were therefore surprised that deletion of the S. Montevideo queC in a WT background produced the viable strain YYF3346. Indeed, if this gene is essential to the modification, then it should be impossible to delete queC without also deleting the potential restriction system, since the S. Montevideo putative restriction system would cleave its own DNA.
Using a gel-based Q detection method, we showed that this strain lacked Q in tRNAAspGUC (Fig. 4A, left), consistent with disruption of the Q pathway, and that this phenotype was complemented by expressing the queC gene in trans (Fig. 4A, middle, see methods). To determine the modification status of DNA in the queC mutant pUC19 was extracted from this strain and used to transform WT host cells (Fig. 4B). The low efficiency of transformation suggested that the DNA lacked the dADG modification, and subsequent mass spectrometric analysis confirmed that neither dADG or dPreQ0 were detected in DNA from the ΔqueC strain, but that both modifications were present in the complemented strain YYF3461 (Table 1). This demonstrates that queC, and the other genes required for preQ0 biosynthesis, are required for the synthesis of the dADG/dPreQ0 modifications.
Figure 4. The queC deletion strain show deficiency in Q modification and dADG-dependent modification and increased transformation efficiency with unmodified DNA.

(A) Q− phenotype in the queC deletion strain can be complemented by preQ0 feeding. Q incorporation into tRNAAspGUC tRNA was detected in total tRNA extracted at 0, 20, 40 and 60 min after supplementing S. Montevideo culture medium with 5 nM preQ0 or DMSO in M9 broth containing 0.4% arabinose. tRNAASPGUC was visualized by hybridization with a specific biotinylated probe. tRNAs modified with Q (upper bands) migrate slower than the unmodified tRNA (lower bands) (Igloi & Kossel, 1985). (B) pUC19 DNA (10 ng) extracted from different S. Montevideo mutant donor strains was transformed in the WT host and YYF3022. (C) Modified and unmodified pUC19 DNA (10 ng) was transformed in different S. Montevideo mutant host cells. The average of three experiments is shown, with error bars representing SD (***P < 0.001, two-tailed Student’s t-test).
The fact that YYF3346 was viable despite the absence of dADG suggested that the queC mutant might also be deficient in restriction. Testing the restriction-like phenotype confirmed this suspicion (Fig. 4C), explaining the viability of the strain. The restriction deficiency was not complemented by expressing the queC gene in trans (Fig. 4C), suggesting that this phenotype was due to second-site mutations that inactivated the restriction machinery. However, none of the dpd genes we tested complemented the restriction deficiency phenotype (Fig. S5), leading us to sequence the dpdJ gene (4518 bp), the only gene in the cluster that we could not clone. Sequencing showed a G-to-C mutation at position 272 (Proline to Arginine) in YYF3346 compared with the WT strain, indicating that a second-site mutation had occurred in the dpdJ gene.
Role of DpdC in dADG synthesis
While the previous results showed that DpdC is required for dADG synthesis, several models for its role in the pathway can be proposed. DpdC could be channeling ADG away from QueC or inhibiting part of the enzyme (Fig. 5A and 5B), or it could be converting preQ0 to ADG before or after its insertion in DNA (Fig. 5C and 5D). Providing the ΔqueC strain (YYF3346) with exogenous preQ0 suppressed the Q− phenotype (Fig. 4A, right), which demonstrates that, like E. coli, S. Montevideo can transport exogenous preQ0 (Zallot, Yuan, & de Crécy-Lagard, 2017). pUC19 extracted from the ΔqueC strain exhibited higher transformation frequencies in WT cells when the ΔqueC mutant was provided with exogenous preQ0 (Fig. 5E). This was consistent with the presence of dADG, which was confirmed by mass spectrometric analysis (Table 1). These results suggests that DpdC can synthesizes dADG directly from preQ0 and does not require prior ADG synthesis by QueC, hence disproving the two models shown in Figure 5A and 5B.
To confirm DpdC produces ADG from preQ0, we used an indirect assay in a heterologous system. We postulated that if DpdC converted preQ0 to ADG before the incorporation of preQ0 into DNA, then DpdC should compete with QueF for the available preQ0 and the formation of Q in tRNA would be reduced when dpdC is overexpressed. To detect this potential competition, conditions of preQ0 limitation had to be engineered. This is the case in an E. coli ΔqueC strain when 10 nM of exogenous preQ0 is added and cells are harvested shortly after supplementation. When dpdC was overexpressed, we saw a reduction of Q formation in tRNA (Fig. 6A), suggesting that DpdC competes with QueF for the available preQ0. This reduction was not observed when dpdA or dpdB were overproduced individually (Fig. 6B). However, when both dpdA and dpdB were overexpressed, Q formation in tRNA was lower than when dpdC was overexpressed (Fig. 6A), consistent with the DpdAB complex competing more effectively for free preQ0 than DpdC. The fact that mass spectrometric analyses have shown that dADG, not dPreQ0, is the predominant form found in S. Montevideo DNA (Table 1) suggests that the conversion of ADG from preQ0 might occur after incorporation of preQ0 in DNA, as depicted in the model in Figure 5D.
Figure 6. Overexpression of DpdC in E. coli K12 reduces the rate of Q formation in tRNAASP.

(A) (Left) The formation rate of Q was estimated by visualizing tRNAASPGUC extracted at different time points (0, 20, 40 and 60 min) after supplementing with preQ0. tRNA extracted from WT and Δtgt strains were used as control. (Right) The density of the bands was measured by ImageJ (Rueden et al., 2017). The percentage of Q modified tRNAASPGUC was plotted. The average of three experiments is shown, with error bars representing SD. (B) The effect of expression of DpdA and DpdB on the Q formation rate was estimated with the same approach.
Interactions of Dpd proteins with DNA
To further address the specific functions of the Dpd protein in dADG synthesis, we investigated the DNA binding properties of the recombinant DpdA, DpdB, and DpdC proteins in as set of preliminary experiments as we have yet to identify the target sequence. The dpdABC genes were each subcloned into E. coli expression vectors to allow over-production of recombinant C-terminal His6-affinity-tagged proteins. The cleaved recombinant proteins (Fig. S6), which retained a C-terminal Factor Xa sequence (IGER), were incubated at a range of concentrations with linearized pUC19 plasmid DNA followed by electrophoresis on 0.5% agarose gels. As can be seen (Fig. 7), all three of the proteins clearly exhibit binding to the DNA relative to the BSA negative control (Fig. 7A), with DpdA exhibiting the strongest interaction (Fig. 7B). DpdB, which on the basis of the genetic experiments we hypothesized might provide binding specificity for DpdA, exhibited the weakest interaction (Fig. 7C), and did not appear to enhance binding of DpdA or DpdC when present together with either of them (Fig. 7E and 7F, respectively). While initially surprising, this result is not inconsistent with the proposal that DpdB determines DpdA location if DpdA possesses a non-specific affinity for DNA and requires DpdB for targeting to the specific sites where preQ0 is to be inserted. Indeed, while it is tempting to interpret the high affinity of DpdA binding to the potential formation of the covalent intermediate in the base exchange reaction, the requirement for both DpdA and DpdB for the incorporation of preQ0 into DNA argues against this interpretation and suggests that the binding is non-specific. Notably, the distinct DNA binding exhibited by DpdC is consistent with the results of the competition experiment (vide supra), which suggested that DpdC catalyzes hydrolysis of the preQ0 nitrile group in the context of preQ0-modified DNA (Fig. 5D), not the free base.
Figure 7. Dpd protein/DNA binding assays.

Gel mobility shift analysis of Dpd proteins with DNA. In each panel the lanes correspond to the following: lane 1 (far left), kb ladder; lane 2, linear pUC19 alone; lane 3, protein at 0.1 μM; lane 4, protein at 0.2 μM; lane 5, protein at 0.5 μM; lane 6, protein at 1.0 μM; lane 7, protein at 2.0 μM; lane 8, protein at 4.0 μM. The proteins in each panel are (A) BSA negative control, (B) DpdA, (C) DpdB, (D) DpdC, (E) DpdA/B, and (F) DpdB/C. pUC19 is present at 0.5 μg in each lane.
Discussion
Here we have demonstrated that preQ0 base is a shared intermediate in the tRNA and DNA 7-deazapurine derivative modification pathways, and that only three genes of the S. Montevideo dpd islands, part of the dpdABC operon, are required for the formation of preQ0 and ADG in DNA. Based on these results, the model we favor is that the DpdAB complex inserts preQ0 into DNA as the first step in modifying DNA, followed by DpdC-catalyzed hydration to dADG, although the alternate model in which DpdC acts at the level of the free base before ADG insertion in DNA by DpdAB cannot be rigorously ruled out at this time. The fact that in our indirect competition assay the DpdAB complex, and not DpdA or DpdB alone, effectively competes for preQ0 was surprising, since DpdA, the TGT paralog predicted to contain a preQ0 binding pocket, is the logical candidate for the enzyme responsible for the base exchange (Thiaville et al., 2016). However, the results are consistent with our genetic data, which demonstrated that the base exchange reaction in DNA only occurs in the presence of both DpdA and DpdB, at least in the S. Montevideo system. Indeed, DpdB might not always be required, as we identified fused DpdCA proteins in several organisms in which dpdB is missing, such as Meiothermus chliarophilus DSM 9957, Spirosoma spitsbergense DSM 19989, Haloarcula sinaiiensis ATCC 33800, Herbaspirillum sp. JC206 and Paenibacillus polymyxa SC2 (Thiaville et al., 2016, Table. S1). Only when the dADG insertion system can be reconstituted in vitro will we be able to fully confirm the predicted biochemical roles of the three DpdABC proteins. Trials are ongoing, the results of which will be reported in due course.
Results from the transformation assays with both unmodified DNA and DNA containing preQ0 or ADG show that only ADG confers resistance to the predicted restriction activity encoded by the dpd cluster. Unlike dG+ in 9g phage (Tsai et al., 2017), the presence ADG in pUC19 does not confer resistance to the Type II restriction enzymes we tested (Fig. S7). However we do know that the 9g phage is heavily modified while the plasmid carries only a few modified bases (Thiaville et al., 2016). Further work is needed to explore if high densities of dADG can like dG+ protect from Type II restriction enzyme cleavage.
Genetic analyses suggest that at least six of the eight genes of the dpdEFGHIJKD operon are part of a nuclease system that restricts DNA lacking ADG. Here again, biochemical studies will be required to provide a definitive confirmation and to elucidate the specific role(s) of each protein. Though the classification scheme for R-M systems was initially designed specifically for methylations, the dpd system would be most similar to a type I RM system (Loenen & Raleigh, 2014). Indeed, we identified a “modification complex” (DpdABC), composed of a potential DNA sequence specificity protein and two modification proteins, and a “restriction complex” (potentially DpdEFGHIJKD) that contains SNF2 helicase homologs (see Thiaville et al., 2016 for description). However, the complexity of this system, with at least six different proteins involved in restriction, and the novelty of the modification, could warrant a new RM system category.
Experimental Procedures
Bioinformatics
Sequences were acquired from the PubSEED subsystem “dpd cluster”. Promoter prediction was performed by BPROM online server (Softberry Inc., Solovyev & Salamov, 2011). Terminator prediction was performed by ARNold web server (Université Paris-Sud web server, Gautheret and Lambert, 2001; Macke et al., 2001). For other sequence analyses, the BLAST tools (Altschul, Gish, Miller, Myers, & Lipman, 1990) and resources at NCBI (http://www.ncbi.nlm.nih.gov/) were routinely used. The protein sequences were retrieved from the NCBI using the following accession numbers: DpdA, AHW12286.1; DpdB, AHW12287.1; DpdC, AHW12295.1 and AHW12296.1; DpdD, AHW12295.1; DpdE, AHW12288.1; DpdF, AHW12289.1; DpdG, AHW12290.1; DpdH, AHW12291.1; DpdI, AHW12292.1; DpdJ, AHW12293.1; DpdK, AHW12294.1; QueC, AHW08582.1.
Strains, Media, and Growth Conditions
Strains used in this study are listed in (Table S1). S. Montevideo strains were routinely grown in LB (Tryptone 10 g L−1, yeast extract 5 g L−1, NaCl 5 g L−1) at 37 °C. S. Montevideo deletion constructs were made using the linear recombination method described by Datsenko and Wanner (Datsenko & Wanner, 2000). Antibiotic resistance gene was eliminated also as described by Datsenko and Wanner (Datsenko & Wanner, 2000). Oligonucleotides used for RT-PCR (Fig. 2), mutants construction (Fig. 3 and Fig. S2) and plasmids construction are listed in Table S2. Primers SM_deletion_dpdA_F and SM_deletion_dpdA_R were used for the introduction of the dpdA deletion; SM_deletion_dpdB_F and SM_deletion_dpdB_R for dpdB deletion; SM_deletion_dpdC_F and SM_deletion_dpdC_R for dpdC deletion; SM_deletion_dpdD_F and SM_deletion_dpdD_R for dpdD deletion; SM_deletion_dpdE_F and SM_deletion_dpdE_R for dpdE deletion; SM_deletion_dpdF_F and SM_deletion_dpdF_R for dpdF deletion; SM_deletion_dpdG_F and SM_deletion_dpdG_R for dpdG deletion; SM_deletion_dpdH_F and SM_deletion_dpdH_R for dpdH deletion; SM_deletion_dpdI_F and SM_deletion_dpdI_R for dpdI deletion; SM_deletion_dpdJ_F and SM_deletion_dpdJ_R for dpdJ deletion; SM_deletion_dpdK_F and SM_deletion_dpdK_R for dpdK deletion. SM_deletion_dpdE_F and SM_deletion_dpdD_R were used for the introduction of the dpdE-D deletion. S. Montevideo dpdA was cloned into the sites SbfI and HindIII of pBAD24 using primers GO36 and GO37 following standard procedures. dpdC and dpdB was cloned into the same sites with GO34 and GO35 and GO38 and GO39 respectively. dpdAB was cloned with GO36 and GO39. S. Montevideo dpdCAB was cloned into the sites NheI and HindIII of pBAD33 using primers Sm-NheI-RBS-dpdC-F and Sm-dpdB-HindIII-R. S. Montevideo queC was cloned into the sites EcoRI and HindIII of pBAD24 using primers Sm-EcoRI-RBS-queC-F and Sm-HindIII-queC-R. dpdE was cloned into the sites SbfI of pBAD24 using primers GO136 and GO82. dpdD, dpdF, dpdG, dpdH, dpdI, and dpdK were cloned into the SalI and SbfI sites of pBAD24 using primers GO72 and GO73, GO83 and GO84, GO85 and GO86, GO87 and GO88, GO89 and GO90, and GO137 and GO106, respectively.
For recombinant protein production the dpdABC genes were subcloned from the relevant pBAD constructs into the NdeI/NotI sites of pET30-Xa to give C-terminal His6-affinity tagged proteins with a Factor Xa site preceding the affinity tag. The cloning utilized the primers Sm-TGTa5-F and SM-TGTa5-R to generate pAGI-42.1 (dpdA), Sm-DpdB-F and Sm-DpdB-R to generate pAGI-42.2 (dpdB), and Sm-DpdC-F and Sm-DpdC-R to generate pAGI-42.3 (dpdC). Expression and purification of the recombinant proteins followed standard protocols.
Plasmid Restriction Test
Restriction of the pUC19 plasmid was tested using a modified transformation efficiency (TE) method (Xu, Yao, Zhou, Deng, & You, 2010). TE reflects the number of transformants obtained per microgram of DNA and is calculated by dividing the number of transformants by the amount of DNA used in the electroporation. The pUC19 plasmid or the mix of pUC19 plus pBAD33 plasmid was propagated through S. Montevideo WT, YYF3022 (ΔdpdC-dpdD::kan) and other strains, and isolated from each strain using QIAprep Spin Miniprep Kit (Qiagen). 0.4% arabinose was added 30 min before plasmid extraction to induce the PBAD promoter. The presence or absence of the modification on pUC19 was confirmed as described (Thiaville et al., 2016). Plasmid (modified or unmodified) was transformed by electroporation into freshly prepared electrocompetent recipient strains. Transformants were recovered in 1 mL LB for 1 h at 37 °C and dilutions were plated onto LB agar plates with or without antibiotic selection for the transforming DNA: ampicillin (100 μg mL−1). Transformation efficiency was determined by calculating the number of transformants per 106 viable CFU and dividing by the ng of DNA transformed.
DNA Preparation
Total DNA was extracted from bacteria with phenol-chloroform followed by alcohol precipitation as described (Thiaville et al., 2016). A 400 mL culture of each strain was grown in the proper conditions described above. 0.4% arabinose and 500 ng mL−1 aTET was added 30 minutes before harvest to induce the PBAD and PTET promoter respectively. Cells were harvested by centrifugation at 4,500 rpm for 20 min. Pellets were washed once in 40 mL TEN buffer (100 mM Tris, pH 8.0; 100 nM NaCl; 10mM EDTA, pH 8.0) and centrifuged again. Pellets were suspended in 20 mL TEN buffer with 600 μg mL−1 lysozyme, 1 % SDS, 6 mAU mL−1 Proteinase K (Qiagen), and incubated at 56 °C for 1 h. Following incubation, an equal volume of Tris-buffered phenol, pH 7 was added to each sample and incubated with shaking at room temperature overnight. The samples were centrifuged at 4,000 × g at room temperature for 10 min and the aqueous phase was transferred to the new tubes. The extractions were repeated with the mixture of 25:24:1 phenol:chloroform:isoamyl alcohol followed by a chloroform extraction. After the final extraction, the aqueous phase was treated with 200 μg mL−1 RNAse A for 30 min at 37 °C. RNAse was removed by another round of phenol:chloroform extraction. DNA was precipitated with 0.1 volume sodium acetate and 1 volume isopropanol and spooled with a glass rod. After washing briefly in 70% ethanol, the DNA was suspended in sterile deionized water.
DNA Modification Analysis
Quantification of dADG and dPreQ0 was achieved by liquid chromatography-coupled triple quadrupole mass spectrometry (LC-MS/MS) (Thiaville et al., 2016). DNA (100 μg) was hydrolyzed in 10 mM Tris-HCl (pH 7.9) with 1 mM MgCl2 with Benzonase (20 U), DNase I (4 U), calf intestine phosphatase (17 U) and phosphodiesterase (0.2 U) for 16 h at ambient temperature. Following passage through a 10 kDa filter to remove proteins, the filtrate was lyophilized and resuspended to a final concentration of 2 μg μL−1 (based on initial DNA quantity); a final concentration of 0.2 μg μL−1 was used for dADG quantification. Aliquots of DNA hydrolysates (2 μg) were injected on an Agilent 1290 series HPLC equipped with a Kinetex EVO C18 column (150 mm × 2.1 mm, 2.6 μm particle size, 100Å pore size; Phenomenex) and a diode array detector (DAD). The column was heated to 40 °C and eluted at 0.5 mL min−1 with 100% solvent A (5 mM ammonium acetate, pH 5.3) for 2 min followed by a linear gradient to 10% solvent B (acetonitrile) over 8 min, 60% solvent B at 9 min and held for 1 min. The column eluent was monitored by UV absorbance at 260 nm to determine the retention times of canonical 2’-deoxynucleosides (dC, 1.2 min; dG, 3.8 min; dT, 4.2 min; dA, 5.5 min). The HPLC column was coupled to an Agilent 6430 triple quadruple mass spectrometer with an electrospray ion source in positive ion mode and parameters as follows: gas temperature 300 °C, gas flow 12 L min−1, nebulizer pressure 40 psi, and capillary voltage 3800 V. 2’-Deoxynucleosides were quantified using selected reaction monitoring (SRM) for the following HPLC retention times and transitions involving loss of 2-deoxyribose: 6.7 min, m/z 292→176 for dPreQ0; and 6.4 min, m/z 310→194 for dADG. Quantification of dPreQ0 and dADG was achieved using external calibration curves, with standards synthesized as described elsewhere (Thiaville et al., 2016). Nine calibration solutions were prepared with the lowest concentrations of 2 μM dPreQ0 and 200 pM dADG. Calibration samples (10 μL) were interspersed with DNA hydrolysate samples. The limit of detection was determined to 1 fmol for dADG and dPreQ0, which corresponds to ~5 modifications per 106 nucleotides in 2 μg of injected DNA digest.
Exogenous PreQ0 feeding
Cells were cultured in M9 defined media with 1% glycerol. Arabinose was added to 0.4% after cells reached an optical density (A600nm) of 0.1 to induce the PBAD promoter. PreQ0 (from Ark Pharm, Libertyville, IL, USA) and DMSO was added respectively after cells reached an optical density (A600nm) of 0.2 to 0.3. The transport reaction was stopped at time points of 0, 20, 40 and 60 min after supplementing with preQ0 and DMSO by placing samples on melting ice, and then centrifuging, followed by tRNA extraction.
tRNA Extraction
This extraction method was proven efficient for the purification of tRNA enriched fraction (Zallot, Yuan, et al., 2017). Cells were harvested by centrifugation at 16,000 × g for 2 min at 4 °C. Cell pellets were immediately resuspended in 1 mL of Trizol (Life). Small RNAs were extracted using Purelink miRNA Isolation kit (Life) according to manufacturer protocol. The purified RNAs were eluted in 50 μL of RNase free water.
Detection of queuosine in bulk tRNA
This method of detection of Q in tRNA was originally developed by Igloi and Kossel (Igloi & Kössel, 1985) and adapted by Zaborske et al. (Zaborske et al., 2014). For each sample, bulk tRNAs were deacylated by incubation in 50 mM Tris-HCl (pH 9) for 30 min at 37°C. Deacylated tRNAs were precipitated at 16,000 xg at 4°C after incubation in ammonium acetate, isopropanol, and linear polyacrylamide overnight at −20 °C (Gaillard & Strauss, 1990). The pellet was washed with 70% ethanol and dissolved in RNase-free water (Life). Prepared tRNAs were quantified using a Nanodrop 1000 spectrophotometer. For each well, 150 ng of tRNAs were resuspended in 2× RNA Loading Dye (NEB) and loaded onto a denaturing 8 M urea, 8% polyacrylamide gel containing 0.5% 3-(acrylamido)phenylboronic acid (Sigma-Aldrich). The migration was performed at 4°C in 1× TAE. Migrated tRNAs were transferred onto a Biodyne B precut Nylon membrane (Thermo Scientific) using a wet transfer apparatus in 1× TAE at 150 mA at 4°C for 90 min. After the transfer, the membrane was baked in an oven for 30 min at 60°C and then UV irradiated in a UV Crosslinker (Fisher FB-UVXL-1000) at a preset UV energy dosage of 120 mJ cm−2. tRNAASP was detected with the North2South Chemiluminescent Hybridization and Detection Kit (Thermo). The initial membrane blocking was realized with DIG Easy Hyb (Roche), because it drastically limits the background noise, compared to the membrane blocking buffer supplied with the North2South kit. Hybridization was done at 60°C, with using the specific biotinylated primer for tRNAASPGUC (Phillips et al., 2012) (5’-biotin-CCCTCGGTGACAGGCAGG 3’) at 50 ng mL−1 final. The blot was exposed to X-Ray film (Thermo Scientific, CL-X Posure Film). The film was developed using a Film Processor (Konica QX-60A).
DNA binding experiments
Purified plasmid pUC19 was linearized by cutting at the single Sal I site using FastDigest Sal I (Thermo) following manufacturer recommended protocols, purified on a 1% agarose gel, and isolated using a GeneJET gel extraction kit (Fermentas). A portion (0.5 μg) of linearized pUC19 was incubated in a 20 μL reaction at 37 °C for 10 min in the presence of the relevant Dpd protein(s) at a concentration of either 100 nM, 200 nM, 500 nM, 1 μM, 2 μM, or 4 μM, along with 20 mM HEPES (pH 7.5), 50 mM KCl, 10 mM MgCl2, and 2 mM 2-mercaptoethanol (BME). In experiments containing two proteins the total protein concentration was the same as in single protein experiments, each present at half. The incubations where then subjected to electrophoresis on a 0.5% agarose gel containing 0.33 μg/mL ethidium bromide made with a TBE buffer containing 89 mM Tris (pH 8.3), 89 mM boric acid, and 2 mM EDTA. The gels were run in the same TBE buffer for 40 min at 107 V.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (R01 GM70641 to V dC-L) and the National Research Foundation of Singapore through its Singapore-MIT Alliance for Research and Technology Antimicriobial Resistance IRG (to PCD). A portion of the mass spectrometric analyses were performed in the Bioanalytical Facilities Core of the MIT Center for Environmental Health Sciences, which is supported by a grant from the National Institute for Environmental Health Sciences (ES002109).
Footnotes
The authors declare no conflict of interest.
References
- Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. [DOI] [PubMed] [Google Scholar]
- Alva V, Nam S-Z, Söding J, and Lupas AN (2016). The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research, 44(W1), W410–W415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Fox DT, Lacy JA, Van Lanen SG, and Iwata-Reuyl D (2000). Hypermodification of tRNA in Thermophilic archaea. Cloning, overexpression, and characterization of tRNA-guanine transglycosylase from Methanococcus jannaschii. Journal of Biological Chemistry, 275(37), 28731–28738. [DOI] [PubMed] [Google Scholar]
- Cao X-M, Huang L-H, Farnet CM, and Ehrlich M (1983). Ligation of highly modified bacteriophage DNA. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression, 741(2), 237–243. [DOI] [PubMed] [Google Scholar]
- Chen C, Wang L, Chen S, Wu X, Gu M, Chen X, et al. (2017). Convergence of DNA methylation and phosphorothioation epigenetics in bacterial genomes. Proceedings of the National Academy of Sciences USA, 114(17), 4501–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences USA, 97(12), 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, Tu J, and Kuo T-T (1978). Characterization of deoxycytidylate methyltransferase in Xanthomonas oryzae infected with Bacteriophage Xp12. European Journal of Biochemistry, 87(1), 29–36. [DOI] [PubMed] [Google Scholar]
- Fergus C, Barnes D, Alqasem M, and Kelly V (2015). The Queuine micronutrient: charting a course from microbe to man. Nutrients, 7(4), 2897–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard C, and Strauss F (1990). Ethanol precipitation of DNA with linear polyacrylamide as carrier. Nucleic Acids Research, 18(2), 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheret D, and Lambert A (2001). Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles11Edited by J. Doudna. Journal of Molecular Biology, 313(5), 1003–1011. [DOI] [PubMed] [Google Scholar]
- Gregson JM, Crain PF, Edmonds CG, Gupta R, Hashizume T, Phillipson DW, and McCloskey JA (1993). Structure of Archaeal transfer RNA nucleoside G*−15 (2-Amino-4,7-dihydro-4-oxo-7-b-D-ribofuranosyl-1H-pyrrolo[2,3-d]pyrimidine-5-carboximidamide (Archaeosine)). Journal of Biological Chemistry, 268(14), 10076–10086. [PubMed] [Google Scholar]
- Grosjean H (2009). Nucleic Acids are not boring long polymers of only four types of nucleotides In Grosjean H (Ed.), DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution (pp. 1–18). Landes Bioscience. [Google Scholar]
- Hattman S (1979). Unusual modification of bacteriophage Mu DNA. Journal of Virology, 32(2), 468–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Huang T, Tang Y, Liu Y, Wu X, Chen S, et al. (2015). Regulation of DNA phosphorothioate modification in Salmonella enterica by DndB. Scientific Reports, 5, 12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand A, Remmert M, Biegert A, and Söding J (2009). Fast and accurate automatic structure prediction with HHpred. Proteins: Structure, Function, and Bioinformatics, 77(S9), 128–132. [DOI] [PubMed] [Google Scholar]
- Hotchkiss D (1948). The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. The Journal of Biological Chemistry, 175(1), 315–332. [PubMed] [Google Scholar]
- Huang LH, Farnet CM, Ehrlich KC, and Ehrlich M (1982). Digestion of highly modified bacteriophage DNA by restriction endonucleases. Nucleic Acids Research, 10(5), 1579–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igloi GL, and Kössel H (1985). Affinity electrophoresis for monitoring terminal phosphorylation and the presence of queuosine in RNA. Application of polyacrylamide containing a covalently bound boronic acid. Nucleic Acids Research, 13(19), 6881–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Zhang D, Maxwell Burroughs A, and Aravind L (2013). Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Research, 41(16), 7635–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jühling F, Mörl M, Hartmann RK, Sprinzl M, Stadler PF, and Pütz J (2009). tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Research, 37(Database issue), D159–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikov EE, Golomidova AK, Letarova MA, Kostryukova ES, Zelenin AS, Prokhorov NS, and Letarov AV (2014). Genomic sequencing and biological characteristics of a novel Escherichia coli bacteriophage 9g, a putative representative of a new Siphoviridae genus. Viruses, 6(12), 5077–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-J, Dai N, Walsh SE, Müller S, Fraser ME, Kauffman KM, et al. (2018). Identification and biosynthesis of thymidine hypermodifications in the genomic DNA of widespread bacterial viruses. Proceedings of the National Academy of Sciences USA, 115(14), E3116–E3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenen WAM, and Raleigh EA (2014). The other face of restriction: Modification-dependent enzymes. Nucleic Acids Research, 42(1), 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Oglou OO, Purta E, Kurkowska M, Olchowik A, et al. (2013). MODOMICS: A database of RNA modification pathways - 2013 update. Nucleic Acids Research, 41(D1), 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, and Sampath R (2001). RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Research, 29(22), 4724–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccarty RM, Lin G, Jacobsen NE, and Bandarian V (2009). The deazapurine biosynthetic pathway revealed : in vitro Enzymatic Synthesis of PreQ0 from Guanosine-5’-triphosphate in four steps. Biochemistry, 48(18), 3847–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelp MT, and Bandarian V (2015). A single enzyme transforms a carboxylic acid into a nitrile through an amide intermediate. Angewandte Chemie International Edition, 54(36), 10627–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips G, Chikwana VM, Maxwell A, El-Yacoubi B, Swairjo MA, Iwata-Reuyl D, and de Crecy-Lagard V (2010). Discovery and characterization of an amidinotransferase involved in the modification of archaeal tRNA. The Journal of Biological Chemistry, 285(17), 12706–12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips G, El Yacoubi B, Lyons B, Alvarez S, Iwata-Reuyl D, and de Crécy-Lagard V (2008). Biosynthesis of 7-deazaguanosine-modified tRNA nucleosides: a new role for GTP Cyclohydrolase I. Journal of Bacteriology, 190(24), 7876–7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips G, Swairjo MA, Gaston KW, Bailly M, Limbach PA, Iwata-Reuyl D, and de Crécy Lagard V (2012). Diversity of archaeosine synthesis in crenarchaeota. ACS Chemical Biology, 7(2), 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reader JS, Metzgar D, Schimmel P, and de Crécy-Lagard V (2004). Identification of four genes necessary for biosynthesis of the modified nucleoside queuosine. The Journal of Biological Chemistry, 279(8), 6280–6285. [DOI] [PubMed] [Google Scholar]
- Roberts RJ, Vincze T, Posfai J, and Macelis D (2015). REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Research, 43(Database issue), D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BioMed Central Bioinformatics, 18(1), 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V, and Salamov A (2011). Automatic Annotation of Microbial Genomes and Metagenomic Sequences In: Li RW (Ed). Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies. (pp. 61–78). Hauppauge: New York: Nova Science Publishers. [Google Scholar]
- Sprinzl M, Horn C, Brown M, Loudovltch A, and Steinberg S (1998). Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Research, 26(1), 148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiaville JJ, Kellner SM, Yuan Y, Hutinet G, Thiaville PC, Jumpathong W, et al. (2016). Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proceedings of the National Academy of Sciences USA, 113(11), E1452–E1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai R, Corrêa IR, Xu MY, and Xu S (2017). Restriction and modification of deoxyarchaeosine (dG+)-containing phage 9 g DNA. Scientific Reports, 7, 8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasu K, and Nagaraja V (2013). Diverse functions of restriction-modification systems in addition to cellular defense. Microbiology and Molecular Biology Reviews, 77(1), 53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilpo JA, and Vilpo LM (1995). Restriction, methylation and ligation of 5-hydroxymethyluracil-containing DNA. Mutation Research, 316(3),123–31. [DOI] [PubMed] [Google Scholar]
- Wang L, Chen S, Xu T, Taghizadeh K, Wishnok JS, Zhou X, et al. (2007). Phosphorothioation of DNA in bacteria by dnd genes. Nature Chemical Biology, 3(11), 709–710. [DOI] [PubMed] [Google Scholar]
- Warren RA (1980). Modified bases in bacteriophage DNAs. Annual Review of Microbiology, 34, 137–58. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Matsuo M, Tanaka S, Akimoto H, Asahi S, Nishimura S, et al. (1997). Biosynthesis of archaeosine, a novel derivative of 7-deazaguanosine specific to Archaeal tRNA, proceeds via a pathway involving base replacement of the tRNA polynucleotide chain. The Journal of Biological Chemistry, 272(32), 20146–20151. [DOI] [PubMed] [Google Scholar]
- Weigele P, and Raleigh EA (2016). Biosynthesis and function of modified bases in Bacteria and their viruses. Chemical Reviews, 116(20), 12655–12687. [DOI] [PubMed] [Google Scholar]
- Wilson GG (1991). Organization of restriction-modification systems. Nucleic Acids Research, 19(10), 2539–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt GR (1950). Occurrence of 5-methyl-cytosine in nucleic acids. Nature, 166, 237–238. [DOI] [PubMed] [Google Scholar]
- Xu T, Yao F, Zhou X, Deng Z, and You D (2010). A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Research, 38(20), 7133–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborske JM, DuMont VL, Wallace EWJ, Pan T, Aquadro CF, Drummond DA, et al. (2014). A nutrient-driven tRNA modification alters translational fidelity and genome-wide protein coding across an animal genus. PLoS Biology, 12(12), e1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R, Harrison K, Kolaczkowski B, and de Crécy-Lagard V (2016). Functional annotations of paralogs: a blessing and a curse. Life, 6(3), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R, Ross R, Chen WH, Bruner SD, Limbach PA, and De Crécy-Lagard V (2017). Identification of a novel epoxyqueuosine reductase family by comparative genomics. American Chemical Society Chemical Biology, 12(3):844–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R, Yuan Y, and de Crécy-Lagard V (2017). The Escherichia coli COG1738 Member YhhQ is involved in 7-cyanodeazaguanine (preQ0) transport. Biomolecules, 7(1), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.