

Abstract
Pancreatitis is an inflammatory disorder of pancreas which leads to varying degrees of pancreatic endocrine and exocrine dysfunction and manifests in either acute or chronic forms. Spontaneous pancreatitis in experimental animals has rarely been reported. Here, we found acute to chronic courses of spontaneous pancreatitis in spontaneously hypertensive rats (SHRs), showing the formation of tubular complexes (TCs) and enhanced islet regeneration. We investigated the expression pattern of clusterin in the pancreas of SHRs based on immunohistochemistry (IHC). IHC analysis revealed the strong expression of clusterin in dedifferentiated duct-like cells and regenerative islets of TCs. These results imply that clusterin might be involved in the formation of TCs and parenchymal regeneration during rat pancreatitis.
Keywords: clusterin, pancreatitis, spontaneous hypertensive rat, tubular complex
Pancreatitis is one of the major gastroenteritic diseases whose incidence has been increasing rapidly in advanced countries [1]. Pancreatitis manifests in either acute or chronic forms. Acute pancreatitis models (injection of secretagogue such as caerulein, bile salt infusion, duct obstruction, choline-deficient ethionine-supplemented diet, etc.) in rodents have been widely used to answer each question or test each hypothesis [9]. However, chronic animal models face several challenges when it comes to pathogenesis and clinical relevance, although similar chronic lesions can be induced by repeated injections of caerulein, alcohol or LPS, genetic manipulation, etc [10, 22]. Especially, spontaneous pancreatitis in experimental animals has rarely been reported [13, 15]. Although the etiology remained unclear, it was reported that chronic pancreatitis was sporadically observed in the aged males of Wistar Bonn/Kobori rats, which was accompanied by endocrine-exocrine pancreatic dysfunctions such as hyperglycemia and diabetes [13]. In addition, spontaneously hypertensive rats (SHRs), which are usually used as a model of human hypertension, developed spontaneous pancreatitis with an incidence of 50% until 12 months of age which was accompanied by pancreatic arteriosclerosis [15].
Here, we also found spontaneous pancreatitis from our animal stock of SHR/NCrJ rats (SHRs; Charles River Japan Inc., Yokohama, Japan). We tried to improve our SHRs for a human pancreatitis model by selective breeding of rats showing higher serum values of amylase than WKY rats. Finally, 83.3% of 12-month-old SHRs showed acinar degeneration/atrophy and inflammatory cell infiltration in the pancreatic parenchyma (Table 1). The SHRs fed with a normal diet were maintained as inbred strains by sister-brother mating under SPF conditions. Figure 1A shows representative H&E pictures of the pancreatic changes in 12-month-old SHRs. Wistar-Kyoto rats (WKYs) were used as control rats showing normal pancreatic tissue, because SHRs was bred from WKYs. Multifocal to coalescing chronic pancreatitis was observed in our SHRs. Acinar atrophy and degeneration were frequently found in SHRs (Fig. 1A-1). The parenchymal tissues in SHRs were infiltrated by many lymphocytes, macrophages, and a few plasma cells (Fig. 1A-1). Fat replacement of the parenchyma was occasionally detected (Fig. 1A-1). Severe inflammatory infiltration and fibrosis replaced stromal tissues (Fig. 1A-2). The inflammatory cells consisted of mononuclear cells and few neutrophils. Ductal dilatation and hyperplasia appeared and the dilated lumens of duct were often filled with eosinophilic materials (Fig. 1A-3). The thickness of intimal/medial walls of pancreatic arteries in SHRs was greater than that of WKY rats (Fig. 1A-4). Vascular lesions such as vasculitis and thrombosis in the arterial lumens were occasionally observed in pancreas of SHRs (Fig. 1A-4). As reported in the previous study [15], spontaneous pancreatitis in our SHRs might be attributable to pancreatic ischemia caused by vascular lesions such as arteriosclerosis. However, considering the observation that inflammatory cell infiltration was observed without vascular changes at early stage (Table 1), we need to consider other primary causes such as immunological abnormalities.
Table 1. Summary of the incidence of histological lesions in the pancreas of SHRs.
| Month | 4 | 8 | 12 | 16 | 20 | |
|---|---|---|---|---|---|---|
| No. of animals | 6 | 6 | 6 | 2 | 2 | |
| Exocrine | Atrophy/degerneration | 2 (33.3%) | 2 (33.3%) | 5 (83.3%) | 2 (100%) | 2 (100%) |
| Focal or small necrosis | 3 (50%) | 3 (50%) | 3 (50%) | 2 (100%) | 2 (100%) | |
| Duct | Dilatation/hyperplasia | 3 (50%) | 3 (50%) | 2 (33.3%) | 2 (100%) | 2 (100%) |
| Vessel | Arteriosclerosis | 0 (0%) | 0 (0%) | 0 (0%) | 2 (100%) | 2 (100%) |
| Arteritis | 0 (0%) | 0 (0%) | 1 (16.7%) | 2 (100%) | 2 (100%) | |
| Thrombosis | 0 (0%) | 0 (0%) | 1 (16.7%) | 2 (100%) | 0 (0%) | |
| Neovascularization | 0 (0%) | 0 (0%) | 1 (16.7%) | 1 (50%) | 2 (100%) | |
| Stroma(inter-, intralobular) | Hemorrhage | 2 (33.3%) | 2 (33.3%) | 3 (50%) | 0 (0%) | 2 (100%) |
| Fibrin/edema | 3 (50%) | 3 (50%) | 1 (33.3%) | 1 (50%) | 0 (0%) | |
| Hemosiderin | 0 (0%) | 0 (0%) | 1 (16.7%) | 2 (100%) | 2 (100%) | |
| Mononuclear cell Inflammatory cell infiltration | 5 (83.3%) | 5 (83.3%) | 4 (66.7%) | 2 (100%) | 2 (100%) | |
| Fibrosis | 0 (0%) | 0 (0%) | 3 (50%) | 2 (100%) | 2 (100%) | |
| Adipose tissue | Mixed inflammatory cell infiltration | 6 (100%) | 6 (100%) | 6 (100%) | 2 (100%) | 2 (100%) |
Fig. 1.
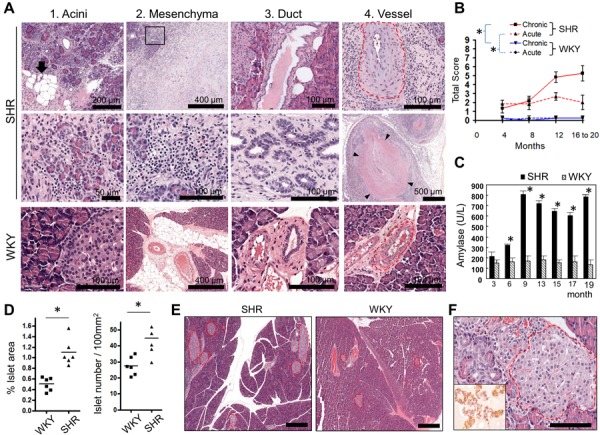
Histopathological features of spontaneous pancreatitis in SHRs. (A) Representative H&E images of pancreatic lesions observed in 12-month-old SHRs. (A-1) Acinar lesions. The arrow indicates fatty replacement of interlobular tissues (top). The middle panel is the high magnification of parenchymal lesions such as degenerative and atrophic acini and severe infiltration of inflammatory cells. The bottom panel is a representative picture of normal pancreatic exocrine and endocrine glands in WKY rats. (A-2) Mesenchymal lesions. Severe fibrosis is observed in stromal tissues (top). The middle panel, a higher magnification of the insert on the top panel, demonstrates the severe infiltration of inflammatory cells consisting of lymphocytes, macrophages, and a few plasma cells. The bottom panel is a representative picture of normal pancreatic mesenchymal tissues in WKY rats. (A-3) Ductal lesions. Pancreatic ducts were irregularly dilated (top) and occasionally hyperplastic (middle). The bottom panel is a representative picture of interlobular pancreatic duct in WKY rats. (A-4) Vascular lesions. Thickened vascular walls and narrow luminal spaces of pancreatic arteries in SHRs (top). Pancreatic vasculitis and thrombus (middle). The artery contained a thrombus whose margin is indicated by arrowheads. The bottom panel is a representative picture of normal pancreatic arteries of WKY rats. Red dotted lines indicate pancreatic interlobular arteries. (B) Pathologic scores for the pancreatitis of SHRs and WKY rats based on H&E staining. (C) Plasma levels of amylase in SHRs and WKY rats at the indicated time points. SHR and WKY group at an indicated time consisted of 4 and 5 mice, respectively. (D) Quantification of total islet areas (left graph) and islet numbers (right graph) in 12-month-old SHRs and WKY rats. The total area of islets was normalized to the entire pancreas area. The area and number were measured by using ImageScope software (Aperio, Vista, CA, U.S.A.). (E) Representative H&E images of the increased number and area of islets in SHRs compared with WKY rats. Islets are marked as red dotted lines. Bar=400 µm. (F) Representative H&E images of regenerative islets in TC of SHRs. Insert, an IHC image for insulin/glucagon expression in regenerative islets of TCs. Bar=100 µm. *, P<0.05, Student’s t-tests. Error bar, mean ± SEM.
To evaluate the onset and progression of the pancreatitis in SHRs, scheduled sacrifices were performed at 2, 4, 8, 12, 16 and 20 months of age. Based on histopathological analyses with H&E stain, our SHRs developed acute to chronic courses of pancreatitis (Table 1). Lesions typically observed in acute pancreatitis, such as fibrin/edema/hemorrhage and small focal necrosis of acini, were first found in SHRs at 4 months of age and the frequencies remained steady. The incidence of chronic lesions, such as acinar atrophy/degeneration, fibrosis, and ductal lesions increased with age. Most SHRs of over 4 months showed inflammatory cell infiltration in inter- and intralobular stroma and surrounding adipose tissue. We scored the severity of pancreatitis based on the previously described scoring systems [5, 15, 20]. In brief, the grading is based on three lesions that typically occur in chronic pancreatitis: lymphocytic inflammation (0–3), interstitial fibrosis (0–3), and acinar degeneration (0–3); and two lesions that typically occur in acute pancreatitis: interstitial edema/necrosis of mesenteric fat (0–3) and neutrophilic inflammation (0–3). The total score was calculated by the sum of the scores for each criterion. Pathologic scores for chronic pancreatitis increased steadily in SHRs with age; the scores for acute pancreatitis in SHRs remained higher than those in WKYs, but did not increase with age (Fig. 1B). The chronic lesions were locally and sporadically observed at early ages and extended over the whole pancreas with age.
We examined the plasma levels of amylase and glucose in our SHRs, which were determined by commercial service from Seoul Clinical Laboratory Co. (Seoul, Korea). In line with the early-onset of pancreatitis and an increase in severity during the disease, the levels of serum amylase started to increase in 3-month-old SHRs, continued to increase steadily until 9 months, and peaked until 19 months (Fig. 1C). However, despite pancreatic damages, glucose levels remained unchanged in our SHRs compared with WKY rats (data not shown). Notably, the total area and number of islets were significantly greater in SHRs than in WKY rats (Fig. 1D and 1E). We also frequently found regenerative islets in duct-like tubular structures called tubular complexes (TC) in the exocrine pancreas of SHRs (Fig 1F). The TC formation is considered as one of sources for reactivated and regenerated pancreatic islets after pancreatic injury [19]. Indeed, Insulin/glucagon-positive clusters of islet with irregular shapes were frequently observed in TC of SHRs (Fig. 1F). Collectively, the unchanged glucose levels in our SHRs might be partially attributable to the regeneration and compensation of islets.
Clusterin, encoded by CLU, was first identified as a secretory molecule related to the process of aggregation and maturation of sperm cells of ram rete testes [2]. Clusterin has been identified in many cell types, including epithelial cells, and is thought to be involved in diverse pathophysiological processes including cell death and tissue remodeling/regeneration [17]. Clusterin was transiently expressed in developing and differentiating exocrine/endocrine cells during pancreatic development and regeneration in rats [4, 11, 12]. It was reported that clusterin plays important roles to promote exocrine and endocrine regeneration as well as differentiation of stem cells in ducts and islet cells [4, 18, 20]. Clusterin-deficient mice showed a more severe caerulein-induced acute pancreatitis with enhanced apoptosis in acinar cells [18]. Clusterin-deficient mice also showed a poor formation of regenerating lobule after pancreatectomy [9]. Taken together, these previous studies suggest the involvement of clusterin in the process of regeneration during pancreatitis and led us to evaluate the expression of clusterin in pancreas of our SHRs.
The TC formation has been observed in pathogenic conditions related to pancreatic damages such as caerulein-induced pancreatitis and pancreatectomy, and diabetes-prone rats [16, 19]. TC formation in the pancreas is regarded as the result of acinar to duct dedifferentiation [3, 6, 19]. TCs were identified based on dedifferentiated duct-like acinar cells showing reduced zymogen granules. In our SHRs, zymogen granules of duct-like acinar cells in TCs decreased as acinar cells gradually lost their differentiation (Fig. 2A–C). Based on immunohistochemistry (IHC), clusterin was highly expressed in duct-like dedifferentiated cells and regenerative islets of TC in our SHRs (Fig. 2D), whereas the immunoreactivity of clusterin was only found in the cytoplasm of periphery regions of islets and weakly detected in the nuclei of acinar cells in WKY rats (Fig. 2E).
Fig. 2.
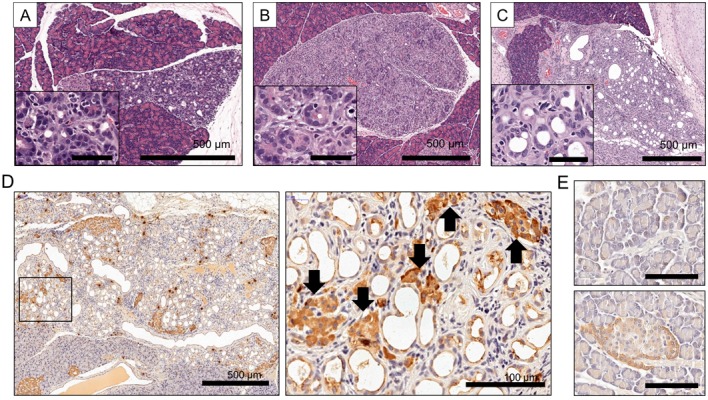
High expression of clusterin in TCs of SHRs. (A–C) Representative H&E images of TCs in SHRs. An entire lobule was affected by TC formation. Inserts are magnified images for TCs of (A), (B), and (C). Bar in inserts=50 µm. (A and B) Duct-like acinar cells showed reduced zymogen granules. (C) Dedifferentiated duct-like acinar cells were characterized by complete loss of zymogen granules. (D) Representative IHC pictures for clusterin expression in TCs. The right panel presents a higher magnification of the boxed area on the left panel. Arrows indicate the strong clusterin expression in regenerative islets of TCs. The paraffin sections of the pancreas were immunostained with a primary antibody against clusterin (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) according to the avidin-biotin complex methods (ABC technique), using the commercial kit (Vectastain Elite ABC kit, Vector Laboratories, Burlingame, CA, U.S.A.). (E) Representative IHC pictures for clusterin in pancreatic exocrine (top) and endocrine (bottom) glands of 9-month-old WKY rats. Bar=50 µm.
The molecular mechanism of clusterin regulation is not clear as of yet. However, the detection of clusterin in duct-like cells in TCs might be explained by the upregulation as an embryonic factor during dedifferentiation of acinar cells to duct-like neogenetic cells [14]. In addition, high clusterin expression in regenerative islet cells in TCs could be partially explained by the fact that clusterin can induce differentiation of pancreatic ductal cells into insulin-secreting β-cells [7, 8]. Cytoplasmic clusterin expression was dominant in TCs of SHRs. Two isoforms of Clusterin protein have been reported as the cytoprotective secreted-clusterin and a pro-death factor nuclear-clusterin [21]. Thus, we assume that the induction of cytoplasmic clusterin in our SHRs may have played cell-protective roles in response to pancreatic damage. Taken together, we suggest that clusterin might be involved in dedifferentiation of pancreatic cells and regeneration of islets in TCs of SHRs.
In summary, we found rats that spontaneously developed acute to chronic courses of pancreatitis from our animal stock of SHRs. We assume that pancreatitis in SHRs may be induced by recurrent bouts of acute damage, such as ischemic injury induced by vascular lesions. We found the compensation of islets and TCs with undifferentiated duct-like cells and regenerative islets in our SHRs, partially explaining unchanged glucose levels despite parenchymal damages. TCs showed strong expression of clusterin, implying that clusterin might be involved in the formation of TCs and parenchymal regeneration during pancreatitis.
Acknowledgments
This work was supported by grants from the Brain Korea 21 Program for Veterinary Science and the Research Institute for Veterinary Science of Seoul National University and Korea Mouse Phenotyping Project (2013M3A9D5072550) of the Ministry of Science, ICT and Future Planning through the National Research Foundation. The funders had no role in study design, data collection and analysis, or preparation of the manuscript.
REFERENCES
- 1.Banks P. A., Conwell D. L., Toskes P. P.2010. The management of acute and chronic pancreatitis. Gastroenterol. Hepatol. (N.Y.) 6Suppl 3: 1–16. [PMC free article] [PubMed] [Google Scholar]
- 2.Blaschuk O., Burdzy K., Fritz I. B.1983. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 258: 7714–7720. [PubMed] [Google Scholar]
- 3.Bouwens L.1998. Transdifferentiation versus stem cell hypothesis for the regeneration of islet beta-cells in the pancreas. Microsc. Res. Tech. 43: 332–336. doi: [DOI] [PubMed] [Google Scholar]
- 4.Calvo E. L., Mallo G. V., Fiedler F., Malka D., Vaccaro M. I., Keim V., Morisset J., Dagorn J. C., Iovanna J. L.1998. Clusterin overexpression in rat pancreas during the acute phase of pancreatitis and pancreatic development. Eur. J. Biochem. 254: 282–289. doi: 10.1046/j.1432-1327.1998.2540282.x [DOI] [PubMed] [Google Scholar]
- 5.De Cock H. E., Forman M. A., Farver T. B., Marks S. L.2007. Prevalence and histopathologic characteristics of pancreatitis in cats. Vet. Pathol. 44: 39–49. doi: 10.1354/vp.44-1-39 [DOI] [PubMed] [Google Scholar]
- 6.Iovanna J. L.1996. Redifferentiation and apoptosis of pancreatic cells during acute pancreatitis. Int. J. Pancreatol. 20: 77–84. [DOI] [PubMed] [Google Scholar]
- 7.Kim B. M., Kim S. Y., Lee S., Shin Y. J., Min B. H., Bendayan M., Park I. S.2006. Clusterin induces differentiation of pancreatic duct cells into insulin-secreting cells. Diabetologia 49: 311–320. doi: 10.1007/s00125-005-0106-2 [DOI] [PubMed] [Google Scholar]
- 8.Kim S. Y., Lee S., Min B. H., Park I. S.2007. Functional association of the morphogenic factors with the clusterin for the pancreatic beta-cell differentiation. Diabetes Res. Clin. Pract. 77Suppl 1: S122–S126. doi: 10.1016/j.diabres.2007.01.045 [DOI] [PubMed] [Google Scholar]
- 9.Lee S., Hong S. W., Min B. H., Shim Y. J., Lee K. U., Lee I. K., Bendayan M., Aronow B. J., Park I. S.2011. Essential role of clusterin in pancreas regeneration. Dev. Dyn. 240: 605–615. doi: 10.1002/dvdy.22556 [DOI] [PubMed] [Google Scholar]
- 10.Lerch M. M., Gorelick F. S.2013. Models of acute and chronic pancreatitis. Gastroenterology 144: 1180–1193. doi: 10.1053/j.gastro.2012.12.043 [DOI] [PubMed] [Google Scholar]
- 11.Min B. H., Jeong S. Y., Kang S. W., Crabo B. G., Foster D. N., Chun B. G., Bendayan M., Park I. S.1998. Transient expression of clusterin (sulfated glycoprotein-2) during development of rat pancreas. J. Endocrinol. 158: 43–52. doi: 10.1677/joe.0.1580043 [DOI] [PubMed] [Google Scholar]
- 12.Min B. H., Kim B. M., Lee S. H., Kang S. W., Bendayan M., Park I. S.2003. Clusterin expression in the early process of pancreas regeneration in the pancreatectomized rat. J. Histochem. Cytochem. 51: 1355–1365. doi: 10.1177/002215540305101012 [DOI] [PubMed] [Google Scholar]
- 13.Mori Y., Yokoyama J., Nishimura M., Kurata H., Miura J., Ikeda Y.1990. Diabetic strain (WBN/Kob) of rat characterized by endocrine-exocrine pancreatic impairment due to distinct fibrosis. Pancreas 5: 452–459. doi: 10.1097/00006676-199007000-00013 [DOI] [PubMed] [Google Scholar]
- 14.Morris J. P., 4th, Cano D. A., Sekine S., Wang S. C., Hebrok M.2010. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J. Clin. Invest. 120: 508–520. doi: 10.1172/JCI40045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onizuka S., Ito M., Sekine I., Tsunoda T., Eto T.1994. Spontaneous pancreatitis in spontaneously hypertensive rats. Pancreas 9: 54–61. doi: 10.1097/00006676-199401000-00008 [DOI] [PubMed] [Google Scholar]
- 16.Reid L. E., Walker N. I.1999. Acinar cell apoptosis and the origin of tubular complexes in caerulein-induced pancreatitis. Int. J. Exp. Pathol. 80: 205–215. doi: 10.1046/j.1365-2613.1999.00116.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg M. E., Silkensen J.1995. Clusterin: physiologic and pathophysiologic considerations. Int. J. Biochem. Cell Biol. 27: 633–645. doi: 10.1016/1357-2725(95)00027-M [DOI] [PubMed] [Google Scholar]
- 18.Savković V., Gantzer H., Reiser U., Selig L., Gaiser S., Sack U., Klöppel G., Mössner J., Keim V., Horn F., Bödeker H.2007. Clusterin is protective in pancreatitis through anti-apoptotic and anti-inflammatory properties. Biochem. Biophys. Res. Commun. 356: 431–437. doi: 10.1016/j.bbrc.2007.02.148 [DOI] [PubMed] [Google Scholar]
- 19.Wang G. S., Rosenberg L., Scott F. W.2005. Tubular complexes as a source for islet neogenesis in the pancreas of diabetes-prone BB rats. Lab. Invest. 85: 675–688. doi: 10.1038/labinvest.3700259 [DOI] [PubMed] [Google Scholar]
- 20.Xie M. J., Motoo Y., Su S. B., Sawabu N.2001. Expression of clusterin in pancreatic acinar cell injuries in vivo and in vitro. Pancreas 22: 126–134. doi: 10.1097/00006676-200103000-00004 [DOI] [PubMed] [Google Scholar]
- 21.Xiu P., Dong X. F., Li X. P., Li J.2015. Clusterin: Review of research progress and looking ahead to direction in hepatocellular carcinoma. World J. Gastroenterol. 21: 8262–8270. doi: 10.3748/wjg.v21.i27.8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan X., Wang F., Bi Y., Ji B.2016. Animal models of gastrointestinal and liver diseases. Animal models of acute and chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 311: G343–G355. doi: 10.1152/ajpgi.00372.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]