

Abstract
Individuals infected with human immunodeficiency virus type-1 (HIV-1) usually show a general dysregulation and hyper-activation of the immune system. A direct influence of HIV-1 particles on B-cell phenotypes and functions has been previously described. However, the consequences of B-cell dysregulation are still poorly understood. We evaluated the phenotypic changes in primary B cells after direct contact with HIV-1 particles in comparison with different types of stimuli. The functionality of treated B cells was challenged in co-culture experiments with autologous CD4+ and CD8+ T cells. We demonstrated that HIV-1 induces a phenotypic change in B cells towards a regulatory B-cell phenotype, showing a higher level of IL-10, TGF-β1, EBI3 or IL-12(p35) mRNA expression and acquiring an immunosuppressive profile. The acquisition of a Breg phenotype was confirmed by co-culture experiments where HIV-treated B cells reduced the proliferation and the TNFα production of CD4+ or CD8+ T cells. This suppressive ability of HIV-treated B cells was dependent on cell-to-cell contact between these B cells and effector cells. To our knowledge, these data provide the first evidence that HIV-1 can directly induce a regulatory B cell-like immunosuppressive phenotype, which could have the ability to impair specific immune responses. This dysregulation could constitute one of the mechanisms underlying unsuccessful efforts to develop an efficient vaccine against HIV-1.
Keywords: HIV-1, immune hyper-activation, regulatory B cells
Introduction
Human immunodeficiency virus type-1 (HIV-1) infection is associated with B-cell dysregulation and perturbations in the production of antibodies, which can lead to hypergammaglobulinemia, polyclonal activation of the B-cell compartment, depletion and impaired function of memory B cells. These perturbations might induce B-cell hyperactivity, ineffective recall responses and dysregulation of class switch recombination (CSR), which leads to the production of non-specific IgG, IgE and IgA antibodies.1,2,3 As a consequence, the B-cell compartment becomes exhausted, leading to defective responses to opportunistic pathogens and compromised B-cell responses to vaccination.4,5
Dysregulation of B cells can occur irrespective of T cells. Direct contact between viral particles and B cells, such as the gp120/C-type lectin interaction with B-cell receptor engagement (BCR) in the presence of CD40L at the virion membrane, are signals strong enough to activate the B-cell compartment.6,7,8,9 In our previous work, we investigated the extent of B-cell dysregulation in a T cell-free context.7 We determined that HIV-mediated cell activation, AID mRNA expression and CSR induction were induced by BCR engagement through the c-Jun N-terminal kinase (JNK) and the spleen tyrosine kinase (SYK)/BCR pathways.6 However, even if HIV-mediated dysregulation of B cells has been demonstrated, its functional significance is still poorly understood.
It was previously described that CD21, CD24, CD38 and CD27 expression levels were deeply altered at the surface of B cells during HIV-1 infection.7,10 All of these markers are associated with a relatively new B-cell compartment named regulatory B cells (Bregs). Bregs are formed by different B-cell subsets with regulatory functions and with a pivotal impact on the immune response in infection, cancer or autoimmune diseases.11,12 Several studies have attempted to determine which markers define human Breg cells in peripheral blood. The Breg subsets in humans have alternatively been classified as CD19+CD24hiCD38hi13 or CD19+CD24hiCD27+.14,15 More recently, other B-cell subsets, such as CD19+CD38+CD1d+IgM+CD147+, CD25hiCD71hiCD73lo or circulating CD39+CD73+ B cells, were also classified as Breg cells (reviewed in ref.15). Although a general Breg phenotype is still not well defined, it is widely accepted that the presence of suppressive capacity is the best indicator for the identification of Breg cells. The majority of mechanisms of suppression described to date are related to the ability of Breg cells to produce IL-10 and to inhibit the production of pro-inflammatory cytokines.16,17 However, IL-10-independent mechanisms of suppression have also been described in several studies and in the review written by Ray et al. 18 HIV-1 infection has been associated not only with an increase in IL-10 in peripheral blood but also with an increase in Breg frequency in the early stages of infection, which consequently leads to the inhibition of anti-HIV-1-specific T cells.19,20 The Breg frequency was correlated with the appearance of markers of HIV-1 disease progression and with the loss of anti-HIV-1 CD8+ T-cell function.21 Therefore, the presence of Bregs is potentially related to the lack of a correct immune response in HIV-1 infection. Regarding potential mechanisms, it has been suggested that microbial translocation might be partially responsible for the high frequency of IL-10-producing B cells in untreated HIV-1 individuals.20,22 Microbial translocation in the gut induces the expression of TLR ligands and CD40L in peripheral blood, which are stimuli required to activate Breg cells in vitro. Nevertheless, mechanisms explaining the increase in Breg cells and IL-10 expression in HIV-1-infected individuals have not yet been fully determined. Comprehension of the mechanisms of Breg-cell differentiation, function and expansion in the context of HIV-1 infection could help to better understand the immune dysregulation and to improve the efficacy of vaccines in the generation of neutralizing antibodies.
We describe for the first time the ability of HIV-exposed B cells to develop a Breg-like phenotype in vitro. The increased frequency of a Breg-like phenotype was not due to specific expansion of Breg subsets already present in the pool of cells. Interestingly, HIV-exposed B cells were able to produce IL-10, TGF-β1 and IL-35, which are cytokines associated with the Breg-cell phenotype. Moreover, HIV-exposed B cells were able to block CD4+ and CD8+ T cell proliferation and TNFα production in a cell-to-cell contact interaction in part involving the HIV-1 envelope protein. These results could provide a partial explanation for the increased frequency of Breg cells in HIV-1-infected individuals and the extent of immune system dysregulation in HIV-1 infection.
Materials and methods
Isolation of B cells from peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMCs) were isolated with a Ficoll-Hypaque density gradient (Rafer, Zaragoza, Spain) from buffy coats obtained from the transfusion center of Madrid, following national guidelines. B cells were purified using anti-human CD19 MicroBeads (positive selection, Miltenyi, Bergisch Gladbach, Germany) and the purity was >96%. In several experiments, B cells were also isolated using unaltered CD19 purification Microbeads (negative selection, Miltenyi). PBMCs and isolated B cells were cultured with RPMI 1640 medium (Biochrom, Cambridge, UK) supplemented with 5% heat-inactivated FCS and a mix of antibiotics (125 μg/ml ampicillin, 125 μg/ml cloxacillin and 40 μg/ml gentamicin; Sigma Aldrich, St Louis, MO, USA).
Virus stock production
HIV-1 stocks were produced by infection of activated PBMCs with NL4-3 virus stocks. NL4-3 stocks came from previous transient transfections of pNL4-3 (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH) in 293 T cells (ATCC, LGC Standards). HIV-1, HIV-89.6, HIV-Ba-L and HIV-LAI (BRU) were generated by infecting activated PBMCs with cell-free virus (cat# 1966; 510 and 2522, respectively). HIV-1 89.6 came from Dr Ronald Collman; HIV-1 Ba-L from Dr Suzanne Gartner, Dr Mikulas Popovic and Dr Robert Gallo; and HIV-1 LAI (BRU) from Dr Jean-Marie Bechet and Dr Luc Montagnier. HIV-1 transmitted/founder viruses (T/F), HIV-WITO, HIV-THRO, HIV-CH058 and HIV-CH077, were generated by transfecting infectious molecular clone pWITO.c/2474 (cat# 11739), pTHRO.c/2626 (cat# 11745), pCH058.c/2960 (cat# 11856) and pCH077.t/2627 (cat# 11742) into 293 T cells. T/F plasmids were from Dr John Kappes and Dr Christina Ochsenbauer. All virus isolates (virus or plasmids) were obtained via the AIDS Research and Reference Reagent program, Division of AIDS, NIAID, NIH. The resulting virus stocks were expanded in activated PBMCs. Physical titers of HIV-1 were evaluated by quantification of HIV-1 p24gag by an ELISA kit (Innogenetics, Ghent, Belgium). The 50% tissue culture infectious dose (TCID50) of each isolate was determined in activated PBMCs using the Spearman Karber formula (ACTG Lab Man, 25 May 200423). We calculated the ratio TCID50/ng by dividing TCID50/ml by ng/ml of p24gag obtained for each HIV-1 isolate. For HIV-1NL4-3, which was essentially used in this study, we tested five different viral productions, with a TCID50/ng mean of 290.8±247 (range ±SD: 40–665 TCID50/ng of p24gag). Other viral ratios were as follows: HIV-1-Ba-L (958 TCID50/ng), HIV-1-LAI(BRU) (393 TCID50/ng), HIV-1-WITO (84 TCID50/ng), HIV-1-THRO (67 TCID50/ng), HIV-1-CH058 (90 TCID50/ng) and HIV-1-CH077 (395 TCID50/ng).
Culture and treatment of B cells
B cells were treated for 24 h to quantify intracellular IL-10, and 48 h to analyze gene expression and cytokine production. B cells were treated with HIV-1NL4-3 (25 ng of p24gag/106 B cells) as positive controls. We treated B-cells with CD40L (eBioscience, San Diego, CA, USA; 200 ng/ml)+IL-4 (ImmunoTools, Friesoythe, Germany; 20 ng/ml), which induces B-cell differentiation, or with CpG-B oligodeoxynucleotide-2006 (CpG; Eurogentec SA, Liege, Belgium; 10 μg/ml)+CD40L+LPS (Sigma Aldrich; 1 μg/ml), which induces IL-10-producing Breg cells and a Breg phenotype in vitro.14 Alternatively, B cells were activated with CD40L+IL-4+HIV-1NL4-3 or with CpG+CD40L+LPS+HIV-1NL4-3. A mock condition was defined using supernatant from activated and non-infected PBMC cell culture. A non-treated (NT) condition was defined as B cells cultured only with medium as a negative control.
For the study of cell-surface markers and intracellular IL-10 labeling, B cells were also treated for 48 h with 20 μg/ml HIV-1 IIIB gp120 recombinant protein (cat# 11784, from ImmunoDX, LLC, NIH AIDS Reagent Program). B cells were treated with a combination of 10 μm of zidovudine (AZT, GSK, Madrid, Spain) and 20 μm of enfuvirtide (T20, Roche, Basel, Switzerland) for 1 h before adding virus. Alternatively, HIV-1NL4-3 (25 ng of p24gag/106 B cells) was treated for 1 h with an anti-HIV-1 gp120 monoclonal antibody used at a 1:5 ratio (volume antibody: volume virus; cat# 2343, neutralizing monoclonal antibody clone, ID6, raised against a recombinant LAV-1 gp160, from Dr Kenneth Ugen and Dr David Weiner), with human AB-serum used at a ratio of 1:5 (volume serum: volume virus; Sigma Aldrich), HIV-1 Neutralizing Serum #1 used at a ratio of 1:5 (volume serum: volume virus; cat# 1984, from Dr Luba Vujcic) or with 50 μg/ml of anti-human CD40L (clone 24-31, Biolegend, San Diego, CA, USA) before adding this treated virus to B-cell culture. Anti-HIV-1 gp120 monoclonal antibody, HIV-1 IIIB gp120 recombinant protein and HIV-1 neutralizing serum #1 were obtained via the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. SF162-gp120 monomeric (gp120 m) and gp140 trimeric proteins (gp140t) were obtained from Dr Leo Stamatatos.24 Trimeric gp140 was formed by a fusion site between gp120 and gp41 that has been mutated to result in a fused gp140 soluble protein; most of the proteins exist in a trimeric form. Gp120 m and gp140t were tested at 10 and 50 μg/ml; only results from the 10 μg/ml conditions are shown, as 50 μg/ml treatment with monomeric gp120 and trimeric gp140 was toxic.
Flow cytometry for determination of B-cell phenotype
Cells were stained to verify the purity of isolated B cells or to define B-cell phenotype using anti-CD3, anti-CD4, anti-CD8, anti-CD71, anti-CD20, anti-CD19, anti-CD38, anti-CD24, anti-CD27, anti-CD1d and anti-CD5 (Beckman Coulter, Brea, CA, USA); anti-PD-1, and anti-PD-L1 (BD Biosciences, San Jose, CA, USA) and anti-FasL (Abcam, Cambridge, UK). Viability of B cells was analyzed with 0.5 μg/ml of 7-amino-actinomycin D (7AAD, Sigma Aldrich). For intracellular labeling of anti-Granzyme B (Biolegend), cells were washed, surface-stained, stained for viable cells with Fixable Viability Dye eFluor450 (eBiosciences), and fixed/permeabilized (Cytofix/Cytoperm, BD biosciences). For intracellular labeling of cytokines IL-10 (Miltenyi Biotec) or TNFα (Biolegend), cell cultures were supplemented with PMA (10 ng/ml)+ionomycin (0.25 μg/ml, both from Sigma Aldrich), and GolgiStop (BD biosciences) for the last 5 h of incubation before staining and permeabilization. Cells were then analyzed by flow cytometry by using a Gallios cytometer (Beckman Coulter), and the data were analyzed by using FlowJo 7.6.1 software (Tree Star Inc., Ashland, OR, USA).
B-cell proliferation and determination of B-cell phenotype
Freshly isolated B cells were first labeled with carboxyfluorescein succinimidyl ester (CFSE) (CFSE from Life Technologies, Carlsbad, CA, USA) following the manufacturer’s recommendations. B cells were mock-treated or treated with HIV-1NL4-3 (25 ng of p24gag/106 B cells) for 5 days. B-cell phenotype and intracellular IL-10 were then analyzed by flow cytometry after surface and intracellular marker labeling. Frequencies of proliferating Breg cells or IL-10-producing B cells were determined by flow cytometry following the loss of CFSE signal.
Cytokine mRNA quantification by real-time polymerase chain reaction
RNA was extracted from 2 × 106 non-treated or treated B cells with Trizol LS Reagent (Life Technologies, Santa Clara, CA, USA). Overall, 100–500 ng of total mRNA were reverse transcribed into cDNA with the GoScript Reverse Transcription System (Promega, Madison, WI, USA). cDNA was used in the real-time polymerase chain reaction (RT-PCR) with Brilliant II SYBR Green QPCR Master Mix (Agilent Technologies). The RT-PCR reaction conditions were: 95 °C 30 s, 60 °C 1 min and 72 °C 1 min for 40 cycles. Primers used to amplify cytokine mRNA are described in Supplementary Table 1. The fold-change of gene mRNA expression levels was calculated by the 2−ΔΔCt equation.
Cytokine production quantified by ELISA assays
B cells were NT or treated with CD40L/IL-4, CD40L/IL-4/HIV-1NL4-3, CpG/CD40L/LPS, CpG/CD40L/LPS/HIV-1NL4-3 or mock-treated for 48 h, and IL-10, IL-12 (Diaclone, Besancon, France), TGF-β1 (Cusabio, Hubei, China) and IL-35 (USCN Life Science, Houston, Tx, USA) were quantified from supernatants of cell cultures by ELISA assay.
Proliferation assay
B cells were NT or treated with CD40L/IL-4, CD40L/IL-4/HIV-1NL4-3, CpG/CD40L/LPS, CpG/CD40L/LPS/HIV-1NL4-3 or mock-treated for 48 h. B cells were washed once with PBS and then washed with acid buffer (PBS-glycine, pH 3.2). Briefly, B cells were washed with 0.2 m glycine buffer containing 0.15 m NaCl (pH 3.2), which was employed to remove cell-surface bound virus, followed by PBS washes. Autologous CFSE-labeled CD19-depleted PBMCs (50 000 cells, named ‘effector cells’ or EC) (CFSE from Life Technologies) were co-cultured with HIV-1-exposed B cells or stimulated B cells (100 000 cells) and subsequently stimulated with anti-CD3/anti-CD28-coated magnetic beads (50 000 beads, Life Technologies). After 72 h, the frequencies of proliferation of CD8 and CD4 T cells were determined by flow cytometry, following the loss of CFSE signal to determine the suppressive capacity of the B cells.
TNFα production assay
B cells were NT or treated with several stimuli and/or HIV-1NL4-3 for 48 h. B cells were washed once with PBS and then washed with acid buffer (PBS-glycine, pH 3.2) followed by PBS washes. Autologous CD19-depleted PBMCs (50 000 cells, EC) were co-cultured with HIV-1-exposed B cells or stimulated B cells (100 000 cells) and then stimulated with anti-CD3/anti-CD28-coated magnetic beads (50 000 beads). After 72 h, the frequencies of TNFα-producing CD8 and CD4 T cells were determined by flow cytometry, and the reduction in TNFα expression was calculated as follows: 100−((TNFα expression in EC co-cultured with treated B cells × 100)/TNFα expression in EC co-cultured with mock-treated B cells).
Apoptosis/necrosis assays
Overall, 50 000 EC were co-cultured with HIV-1-exposed B cells or stimulated B cells (100 000 cells) and subsequently stimulated with anti-CD3/anti-CD28-coated magnetic beads. After 72 h, CD3+ T cells were isolated by anti-CD3 magnetic beads (Dynabeads, Thermo Fisher, Waltham, MA, USA) and lysed to detect the lactate dehydrogenase (LDH) activity following the manufacturer’s recommendations (Promega). Semi-quantitative analysis of cleaved caspase-3 and -9 or full-length caspase-3 and -9 proteins in the mock and stimulated B cell conditions were performed by western blotting. Isolated CD3+ T cells from the 72 h co-culture (EC: B cells) were resuspended in 50 μl of lysis buffer (20 mm HEPES pH 7.5, 150 mm NaCl, 2.5 mm MgCl2, 250 mm sucrose, 0.05% NP-40, 0.5% Triton X-100, all from Sigma Aldrich), with a cocktail of proteases inhibitors (Thermo Scientific, Waltham, MA, USA). Antibodies used for western blotting analysis were anti-caspase-3, anti-caspase-9 (Cell Signaling Technology, Danvers, MA, USA) and anti-actin (Sigma Aldrich). The level of protein was revealed using Immun-Star Western C Chemiluminescent Kit (Bio-Rad, Hercules, CA, USA) and measured using ImageJ software (NIH). Quantity of cleaved proteins was quantified as follows: (intensity of cleaved caspase bands × 100/(intensity of full-length band+intensity of cleaved caspase bands)).
Proliferation assay with neutralization of immune suppression markers
A total of 50 000 EC were co-cultured with mock-treated B cells, HIV-1-exposed B cells or stimulated B cells (100 000 cells) and subsequently stimulated with anti-CD3/anti-CD28-coated magnetic beads. Anti-IL-10 (JES3-19F1, Biolegend), anti-IL-35 (IL27-IL-35 EBI3 Subunit Peptide, Novus Biologicals, Abingdon Oxon, UK), anti-PD-1 (EH12.2H7, Biolegend), anti-TGF-β1 (19D8, Biolegend), anti-TRAIL (RIK-2, Biolegend), anti-PD-L1 (29E.2A3, Biolegend); anti-FasL (NOK-1, Biolegend) and their isotype controls, purified Rat IgG1, Rat IgG2b and mouse IgG1 were added. After 72 h, the frequencies of proliferating CD8 and CD4 T cells were determined by flow cytometry by following the loss of CFSE signal to analyze the suppressive capacity of the B cells.
Transwell assay
Transwells of 0.4 μm pore size were used (Millicell, Merck Millipore, Billerica, MA, USA). CFSE-labeled EC (50 000 cells) were added to the lower chambers of 96-well plates and subsequently stimulated with anti-CD3/anti-CD28-coated magnetic beads, whereas non-treated or stimulated B cells (100 000) were added to the upper chambers in the transwells. As controls for the transwell assay, CFSE-labeled EC (50 000) and B cells (100 000 cells) were also co-cultured in the same plate in the lower chambers with anti-CD3/anti-CD28-coated magnetic beads. After 72 h of co-culture/transwell, EC were gathered and EC proliferation was analyzed by flow cytometry following the loss of CFSE signal. Percentage of relative proliferation was quantified as follows: (% of proliferation of EC in HIV-1-treated B cell condition × 100/ % of proliferation of EC in mock-treated B-cell condition).
Statistical analysis
Results are expressed as the mean±s.e.m. The comparisons between the frequency of Breg subsets, proportions of cytokines quantified by ELISA, mRNA between non-treated and treated B cells, B-cell proliferation, suppression of EC proliferation in co-culture experiments and suppression of EC proliferation between transwell and co-culture conditions were done using the non-parametric Wilcoxon test for paired samples. Statistical comparison of apoptosis (caspase-3/-9 cleavage) or necrosis (LDH expression) between different conditions was performed by paired Student's t-test. The statistical correlation between variables was calculated by the Spearman rank correlation analysis. P values <0.05 were considered statistically significant. All analyses were performed using SPSS 17.0 Inc. software (IBM, Armonk, NY, USA).
Results
Induction of Breg phenotype in vitro in stimulated B cells
We have previously demonstrated that HIV-1 induces changes in the phenotype of B cells7 including modifying the expression of surface markers, such as CD27, CD24 and CD38, which have been associated with a Breg phenotype.13,14,25 We studied the acquisition of two Breg phenotypes in in vitro experiments previously described in the literature, CD19+CD24hiCD38hi and CD19+CD24hiCD27+. Total B cells were cultured for 24 h in the presence of HIV-1NL4-3 or, as a control, in the presence of other stimuli (CD40L/IL-4 and CpG/CD40L/LPS). CD40L/IL-4 induces B-cell differentiation,26 and CpG, CD40L and LPS was shown to induce the differentiation of B cells towards Breg cells in vitro. 14 The B cells were first analyzed for viability, and none of the treatments used in this study induced cell death (Figure 1a). The CD19+CD24hiCD38hi and CD19+CD24hiCD27+ frequencies were markedly modified after stimulation. All stimuli induced a significant increase in CD19+CD24hiCD38hi frequency, and a higher induction was obtained in cells stimulated with HIV-1NL4-3 (7.34±2.13%) or CD40L/IL-4/HIV-1NL4-3 (7.50±2.59%) in comparison to the mock-treated condition (1.69±0.54%, Figure 1b). The frequency of the Breg phenotype CD19+CD24hiCD27+ was significantly decreased when treated with HIV-1NL4-3 (3.36±0.53%), CD40L/IL-4/HIV-1NL4-3 (3.79±0.60%), CpG/CD40L/LPS (1.27±0.29%) and CpG/CD40L/LPS/HIV-1NL4-3 (1.35±0.41%) in comparison to the mock-treated condition (7.64±0.94%, Figure 1c).
Figure 1.
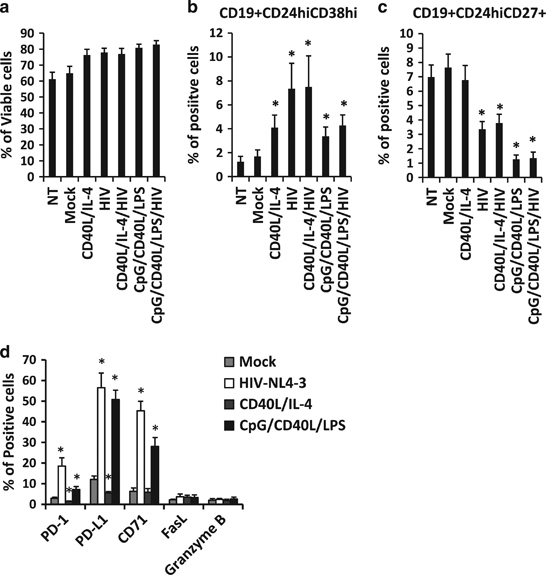
Acquisition of a Breg phenotype in HIV-1-treated B cells. B cells were treated with CD40L/IL-4, HIVNL4-3, CD40L/IL-4/HIVNL4-3, CpG/CD40L/LPS and CpG/CD40L/LPS/HIVNL4-3 for 24 h. B cells were also mock-treated (mock) or not-treated (NT). Twenty-four hours post stimulation, the percentage of (a) viable B cells (7AADneg cells), (b) CD19+CD24hiCD38hi cells, (c) CD19+CD24hiCD27+ cells and (d) B cells expressing PD-1, PD-L1, CD71, FasL and Granzyme B were determined by flow cytometry. Data are shown as the average±s.e.m. of seven experiments for each condition. *P<0.05 when comparing mock-treated condition versus treated conditions.
Breg cells are also defined by other surface markers associated with their suppressive function. Therefore, we analyzed the expression of PD-1, PD-L1, CD71 and FasL at the surface of B cells and Granzyme B intracellularly, which are pivotal for Breg function.27 B cells treated with HIV-1NL4-3 or with Breg-associated stimuli (CpG/CD40L/LPS) induced a significant modification in PD-1, PD-L1 and CD71 expression at the surface of the B cells (Figure 1d). In summary, previously described Breg-associated phenotypes must be challenged when studied in culture since the two subsets presented in this work showed different reactions to stimulation. It is interesting to note that not only the frequency of the CD19+CD24hiCD38hi Breg phenotype showed an increase after B cell stimulation but also the CD71 and PD-1/PD-L1 axis, which takes part in Breg suppressive mechanisms.
IL-10-producing B cells in vitro
IL-10 is the major marker of functional Breg cells. Therefore, we investigated whether the HIV-1-associated dysregulation of B cells is linked to their IL-10 production in vitro. After 24 h of stimulation, we analyzed the production of intracellular IL-10 in the total viable B-cell population (Figure 2a). As expected, CpG/CD40L/LPS, which is the stimulus that steers B cells towards a Breg phenotype, induced a significant increase in the frequency of IL-10-producing B cells in comparison to mock-treated B cells (2.74±1.56% and 0.22±0.06%, respectively, Figure 2b). Surprisingly, HIV-1NL4-3 alone was able to induce a significant increase in the frequency of IL-10-producing B cells (1.53±0.73%, Figure 2b). HIV-1NL4-3 was also able to significantly increase the frequency of IL-10-producing B cells when combined with CD40L/IL-4 or with CpG/CD40L/LPS.
Figure 2.
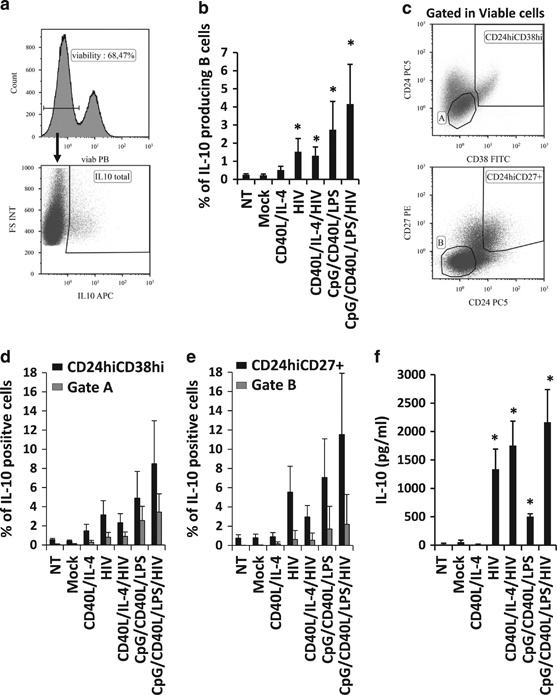
IL-10 production in HIV-1-treated B cells. B cells were treated for 24 h and analyzed by flow cytometry. (a) Total IL-10-producing cells were analyzed within viable cells (histogram) using intracellular labeling (dot plot). (b) Average percentage of IL-10-producing viable B cells was analyzed. Data are shown as the average±s.e.m. of seven experiments for each condition. (c) Gates were established for Breg subsets, such as CD19+CD24hiCD38hi and CD19+CD24hiCD27+, and for non-Breg subsets, gate A (corresponding to CD19+CD24intCD38int) and Gate B (corresponding to CD19+CD24negCD27neg). The percentage of IL-10-positive cells was detected in (d) CD24hiCD38hi or in gate A or in (e) CD24hiCD27+ or in gate B. (f) Production of IL-10 was also analyzed 48 h post stimulation in the supernatant of cell cultures by ELISA (pg/ml). *P<0.05 when comparing mock-treated condition versus treated conditions. Data are shown as the average±s.e.m. of seven experiments.
We also quantified the frequency of IL-10-producing cells in CD19+ cells expressing classical Breg phenotypes (CD24hiCD38hi and CD24hiCD27+, Figure 2c) and in non-Breg populations (see gates A and B in Figure 2c, respectively). As expected, a significantly higher frequency of IL-10-positive cells was found in the two Breg populations in comparison to the non-Breg populations (gates A and B in Figure 2c) in all the conditions tested, with the exception of the CD40L/IL-4 condition (Figures 2d and e). Even if the percentage of CD19+CD24hiCD27+ was decreased when B cells were treated with HIV-1NL4-3 (Figure 1c), they still showed high expression of IL-10 (Figure 2e). Therefore, the loss of the CD27 marker at the surface of B cells already described in HIV-1-infected individuals highlights that even if the phenotype of these cells is perturbed, their function could still be preserved. However, the frequency of IL-10-producing B cells with Breg phenotypes was <20% in all cases, showing that the definition of a Breg phenotype must be refined in the context of HIV-1 infection.
As the percentages of IL-10-producing B cells were low, IL-10 was also quantified in cell-culture supernatants (Figure 2f). HIV-1NL4-3 alone was able to increase the IL-10 production to a higher level than B cells activated with Breg-associated stimuli (Figure 2f).
We analyzed the phenotypic changes observed in B cells in the presence of additional virus isolates, such as dual-tropic HIV-1-89.6, CCR5-tropic HIV-1-Ba-L, CXCR4-tropic HIV-1-LAI(BRU) and four CCR5-tropic transmitted/founder viruses (WITO, THRO, CH058 and CH077). The frequency of CD24hiCD38hiIL-10+ subsets was significantly increased when B cells were treated with HIV-89.6 and HIV-1-Ba-L (Figure 3a), as observed with HIV-1NL4-3 (Figure 2d). However, only HIV-1NL4-3 was able to induce a clear increase in CD24hiCD27+IL-10+ expression (Figure 3b). In addition, percentages of total IL-10-producing B cells were significantly increased when B cells were treated with HIV-1NL4-3, HIV-1-89.6 and HIV-1-BaL (Figure 3c). It was interesting to note that the CXCR4-tropic HIV-1-LAI (BRU) and the transmitted/founder viruses did not induce phenotypic changes in B cells, and we wondered whether a longer incubation time could induce Breg-like phenotype frequency modifications. Nevertheless, changes observed in in vitro experiments were not restricted to the HIV-1NL4-3 isolate; in fact, they were observed with diverse HIV-1 isolates.
Figure 3.
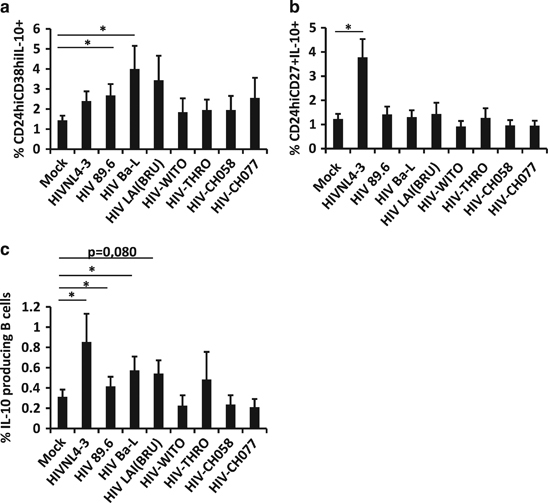
Breg-like phenotype induction when B cells were treated with different virus isolates. B cells were treated for 48 h and analyzed by flow cytometry. (a) Average percentage of CD24hiCD38hiIL-10+, (b) CD24hiCD27+IL-10+ or (c) total IL-10-producing cells were analyzed in viable cells. The average+s.e.m. of nine experiments for mock and HIV-1NL4-3, six experiments for HIV-1-89.6, five experiments for HIV-1-Ba-L and HIV-1-LAI(BRU), and three experiments for T/F viruses (WITO, THRO, CH058 and CH077) are shown. *P<0.05 when comparing mock-treated condition versus HIV-treated conditions.
In summary, even though Breg-associated phenotypes were modified in different ways, these B cells were still the major source of IL-10 production. It is interesting to note that HIV-1 is a strong inducer of IL-10, which is the major marker for Breg-cell function. The combination of HIV-1NL4-3, CpG, CD40L and LPS, which are stimuli that could be frequently found in HIV-1+ individuals who suffer from bacterial translocation or immune activation, would increase this effect, which could constitute the main cause of the hyper-expression of IL-10 in HIV-1+ individuals.
HIV-1NL4-3 induced a Breg phenotype
When B cells were treated with HIV-1NL4-3, we detected an increase in CD19+CD24hiCD38hi and IL-10-producing B-cell subsets; nevertheless, whether these increases were due to the generation of new Breg-related subsets or to an expansion of an already existing Breg population was not clear. Therefore, CFSE-labeled B cells were mock-treated or treated with HIV-1NL4-3 for 5 days, and cellular proliferation was observed following the loss of the CFSE signal. HIV-1-treated B cells showed a higher level of proliferation than mock-treated B cells (Figure 4a), as previously described.7 However, no difference in proliferation frequency was detected between CD19+CD24hiCD38hi and CD19+CD24−CD38− (non-Breg phenotype) in mock-treated or in HIV-1-treated conditions, even though CD19+CD24−CD38− subsets showed a slight increase in proliferation frequency (Figures 4b and c). The viability of CD19+CD24hiCD38hi and CD19+CD24−CD38− subsets was identical (data not shown). In addition, we analyzed the proliferative capacity of IL-10+ or IL-10neg (IL-10-) B-cell subsets when treated with HIV-1NL4-3, and no significant differences were observed between both subsets in mock- or in HIV-1-treated conditions (Figure 4d). Taken together, increased frequency of Breg-associated phenotypes appears not to be due to an expansion of Breg subsets already present in B-cell culture.
Figure 4.
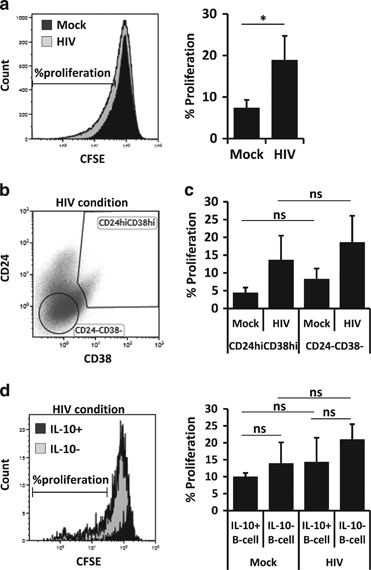
HIV-treated B-cell proliferation. CFSE-labeled B cells were treated for 5 days and analyzed by flow cytometry. (a) Histogram plot of CFSE-labeled mock- and HIVNL4-3-treated cells (left panel). Proliferation was analyzed as the loss of CFSE signal. Average percentage of B-cell proliferation (a, right panel), average percentage±s.e.m. of five experiments. (b) Gates were established for the Breg subset, CD19+CD24hiCD38hi, and for the internal control, CD19+CD24−CD38−. One representative experiment out of four total experiments is shown. (c) Average percentage of B-cell proliferation in CD24hiCD38hi and in CD24−CD38− populations in mock- or HIV-1NL4-3-treated conditions. Average percentage±s.e.m. of four experiments. (d) Histogram plot of proliferation (loss of CFSE signal) gated on IL-10+ and IL-10neg (IL-10−) B-cell when cells were treated with HIVNL4-3 (left panel). Average percentage of B-cell proliferation in IL-10+ and in IL-10− populations in mock- or HIV-1NL4-3-treated conditions were studied. Average percentage ±s.e.m. of four experiments. *P<0.05 when comparing mock-treated versus HIV-treated conditions. ns, non-significant.
Increased frequency of IL-10-producing cells was related to direct contact with viral particles
HIV-1-treated B cells showed a Breg-like phenotype that was related to an increase in IL-10 expression. We studied whether such changes were associated with viral or cellular components present in the viral membrane, including through HIV-1-Env (neutralizing antibody raised against a recombinant LAV-1 gp160 or using recombinant gp120/gp140 derived from IIIB (CXCR4-tropic) or SF162 (CCR5 tropic)), direct B-cell infection or CD40L that could be found at the surface of the viral particles. Therefore, we treated HIV-1NL4-3 with an anti-HIV-1 neutralizing serum (or a human AB-serum as control), with anti-gp120 or with anti-human CD40L for 1 h before adding this treated virus to the B cells. Alternatively, B cells were treated with full-length gp120 IIIB or with AZT+T20 for 1 h before adding HIV-1 to the cells. Breg phenotype (CD19+CD24hiCD38hi or CD19+CD24hiCD27+) and intracellular IL-10 production were analyzed by flow cytometry. As already observed, HIV-1NL4-3 was able to induce an increase in CD19+CD24hiCD38hi frequency and a decrease in CD19+CD24hiCD27+ frequency (Figures 1b and c and 5a and b). None of the treatments were able to reverse the effect of HIV-1NL4-3 on the CD19+CD24hiCD38hi phenotype (Figure 5a). The use of full-length HIV-1-gp120 IIIB was not sufficient to induce the Breg phenotype. However, full-length HIV-1-gp120 IIIB induced an increase in the CD19+CD24hiCD27+ B cell subset (Figure 5b). Treatment of HIV-1NL4-3 with anti-HIV-1 neutralizing serum, anti-gp120 or anti-CD40L was able to partially but significantly reverse the effect of HIV-1NL4-3 on B cells regarding the CD19+CD24hiCD27+ phenotype. These data suggest that HIV-1 Env, CD40L or other HIV-1-derived proteins could be directly implicated in the B-cell phenotype changes (Figure 5b).
Figure 5.
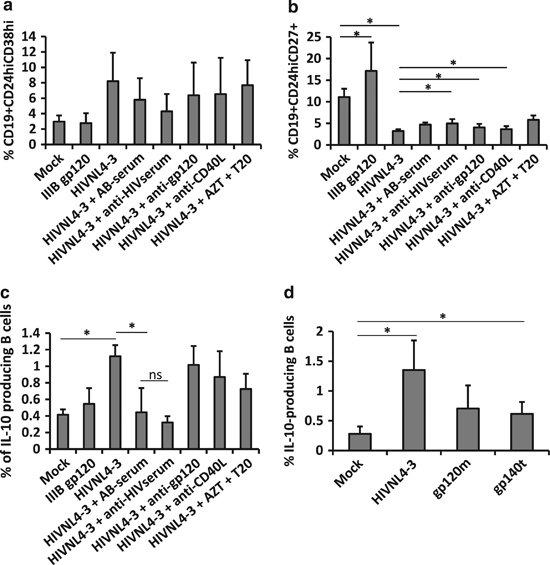
Breg-like phenotype was acquired through direct cell-virus contact but not by direct infection. B cells or virus were treated with various compounds before mixing, and cell phenotype was analyzed after 48 h by flow cytometry (a) Average percentage of CD24hiCD38hi, (b) CD24hiCD27+ or (c) total IL-10-producing cells were analyzed in viable cells. Average±s.e.m. of nine experiments for mock, HIV-1NL4-3 and HIV-1NL4-3+ATZ+T20, and five experiments for gp120 IIIB, HIV-1NL4-3+AB-serum, HIV-1NL4-3+anti-HIV serum, HIV-1NL4-3+anti-gp120 and HIV-1NL4-3+anti-CD40L conditions are shown. (d) Total IL-10-producing cells were analyzed in viable cells after B cell treatment with HIV-1NL4-3 or with 10 μg/ml of monomeric gp120 (gp120m) or trimeric gp140 (gp140t). Average±s.e.m. of four experiments for mock, HIV-1NL4-3, gp120m and gp140t are shown. *P<0.05. ns, non-significant.
As IL-10 expression is a clear marker of phenotypic change in B cells, we analyzed the intracellular IL-10 expression in treated B cells. As expected, HIV-1 treatment induced an increase in the frequency of IL-10-producing B cells, which was only significantly reversed using anti-HIV-1 neutralizing serum. There was no significant difference in the expression of IL-10 in B cells treated either with HIV-1 treated with human AB-serum or with HIV-1 treated with anti-HIV-1 serum, most likely due to the higher variability when using human AB-serum (Figure 5c). To determine whether the viral envelope is related to such IL-10 expression, we treated cells with SF162-gp140 trimeric (gp140t) and SF162-gp120 monomeric (gp120m) proteins. Cells treated with gp120m and gp140t showed increased expression of intracellular IL-10, even if IL-10 expression detected in gp120m-treated B cells was not significantly different from mock-treated cells (Figure 5d). In contrast, gp140t-treated cells showed a significant increase in IL-10 expression in comparison to mock-treated cells, even though this increase was not comparable to the one observed when the cells were treated with HIV-1NL4-3 (Figure 5d). Taken together, envelope proteins and unidentified proteins related to HIV-1 were implicated in IL-10 production by B cells as anti-HIV-1 serum could have various HIV-1 targets. CD40L and HIV-1-envelope might be implicated in Breg phenotype induction (Figure 5b), implying that a direct contact between the virus and a B-cell is required for the B-cell phenotypic changes observed in in vitro experiments. However, direct infection of B cells was not responsible for Breg-like phenotype induction since the use of AZT+T20 was not able to reverse the signal induced by HIV-1. As B cells were isolated by positive selection, which could influence B-cell reactions, we analyzed these results using B cells isolated by negative selection. Similar results were obtained with negatively selected B cells (untouched B cells), as HIV-1NL4-3 treatment increased the frequency of IL-10-producing cells, which was not reversed by the use of anti-CD40L or anti-gp120 (Supplementary Figure 1). As the reversion of the HIV-1 effect upon B cells was not observed with the compounds used in this study, we can assume that different proteins at the surface of the HIV-1 particles (from human or viral origin) must be implicated in this phenomenon.
Pro- or anti-inflammatory cytokine mRNA expression
As shown in Figure 2, B-cell stimulation by HIV-1 induced a marked increase in IL-10 production. Thus, we analyzed and quantified TGF-β1 and IL-35 anti-inflammatory cytokines by ELISA assay, but quantification of these cytokines was either heterogenic (TGF-β1, Supplementary Figure 2a) or undetectable (IL-35, data not shown). Therefore, total mRNA from stimulated B cells was extracted 48 h post stimulation, and IL-10, TGF-β1, IL-21, IL-35 (composed by EBI3 and p35), IL-12 (composed by EBI3 and p40) and IL-27 (composed by p28 and EBI3) transcripts were then quantified by quantitative PCR (Figure 6f). IL-27 (measured as p28 expression) and IL-21 were not detectable by quantitative PCR (data not shown).
Figure 6.
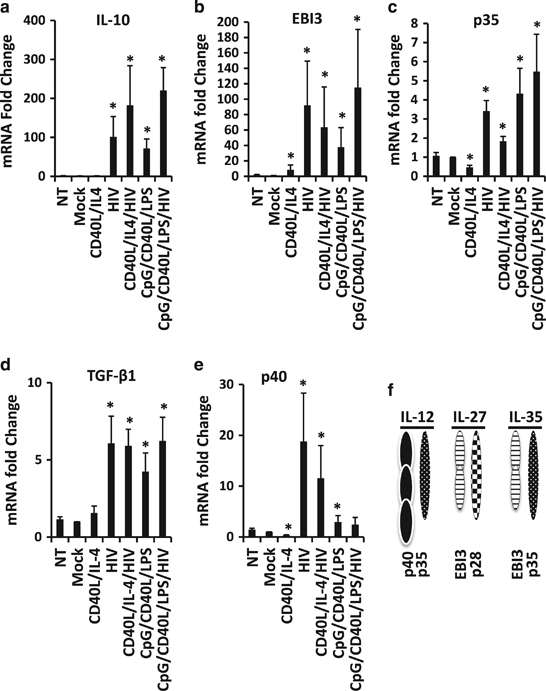
mRNA expression levels of cytokines in HIV-1-treated B cells. B cells were treated with CD40L/IL-4, HIV-1NL43, CD40L/IL-4/HIV-1NL4-3, CpG/CD40L/LPS and CpG/CD40L/LPS/HIV-1NL4-3 for 48 h. Cells were also mock-treated or not-treated (NT). Forty-eight hours post stimulation, mRNA was extracted from NT or treated B cells, and expression of IL-10 (a), EBI3 (b), p35 (c), TGF-β1 (d) or p40 (e) mRNA was determined by real-time PCR. *P<0.05 when comparing mock-treated condition versus treated conditions. Average±s.e.m. of six experiments for each condition are shown. (f) Scheme of the constituent subunits of IL-12 (p40 and p35), IL-27 (EBI3 and p28) and IL-35 (p35 and EBI3).
As expected, in the presence of CpG/CD40L/LPS, a significant increase in IL-10, TGF-β1, EBI3 and p35 transcripts was detected (Figures 6a–d). HIV-1NL4-3 alone was also able to induce a very strong increase in IL-10, TGF-β1, EBI3 and p35 transcript expression, confirming the induction of the Breg-like phenotype observed in Figures 1 and 2. The induction of anti-inflammatory cytokine expression was identical regardless of whether the B cells were treated with HIV-1NL4-3 or with the Breg-associated stimuli (CpG/CD40L/LPS; Figures 6a–d).
Expression of p40 transcripts was also upregulated when cells were treated with HIV-1NL4-3 alone or in combination with CD40L/IL-4 in B cells (Figure 6e). Altogether, p40 and p35 are the two subunits that form IL-12 (Figure 6f). We quantified the amount of IL-12 produced in the supernatant of cell cultures by ELISA assay. IL-12 was hardly detectable in the majority of conditions analyzed, but it was detectable when B cells were treated with HIV-1NL4-3 alone or in combination with CD40L/IL-4 (Supplementary Figure 2B). Taken together, HIV-1-stimulated B cells were able to express not only transcripts that encode for anti-inflammatory cytokines (IL-10, IL-35 and TGF-β1) but also for p40 (IL-12). These data could partially explain the dysregulation of B cells associated with HIV-1 infection.
HIV-1-stimulated B cells acquired a functional suppressive capacity
As B cells exposed to HIV-1 acquire a regulatory phenotype (CD19+CD38hiCD24hi phenotype and the expression of IL-10, IL-35 and TGF-β1 transcripts), we studied whether these cells could also acquire a regulatory function. We analyzed the suppressive capacity of these cells by measuring their ability to block the proliferation and TNFα production of EC. EC are autologous CD19-depleted PBMCs; thus, they include CD4+ and CD8+ T cells. First, we measured the proliferation of autologous EC labeled with CFSE and co-cultured with non-treated, mock-treated or stimulated B cells. The treated B cells were washed with acid buffer, which was employed to remove cell-surface bound virus, before co-culture. EC were labeled for CD4 and CD8, and their ability to proliferate following the loss of CFSE signal was analyzed by flow cytometry (Supplementary Figure 3). The results showed that NT-, mock- or CD40L/IL-4-treated B cells did not change the capacity of CFSE-labeled CD4+ or CD8+ T cells to proliferate in co-culture assays (Supplementary Figure 3B and Figure 7), indicating an absence of suppressive function of these B cells. As expected, CpG/CD40L/LPS-treated B cells were able to significantly diminish the proliferation of EC when compared with mock-treated B cells (Figure 7). B cells treated with HIV-1NL4-3 alone were also able to diminish markedly the proliferation of EC, with a mean percentage of inhibition of 57.25±11.91% for CD4+ T cells and 57.64±9.92% for CD8+ T cells when compared with mock-treated B cells (Figure 7). It is interesting to note that the addition of HIV-1NL4-3 together with CpG/CD40L/LPS induced greater suppression on EC proliferation than these stimuli alone (Figure 7).
Figure 7.
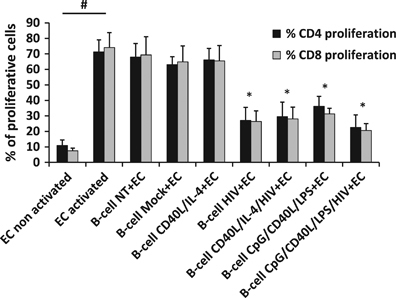
Anti-proliferative function of HIV-1-treated B cells. Proliferation of autologous effector cells (EC) treated with CFSE and co-cultured with not-treated (NT) or treated B cells (ratio Breg:EC of 2:1) at day 3 post co-culture. As a control, EC were also cultured alone without B cells (EC non-activated and EC activated). Average±s.e.m. of the percentage of proliferative CD4+ and CD8+ T cells measured in anti-CD3/anti-CD28-stimulated EC in the presence of B cells. Values from five different donors are shown. *P<0.05 when comparing mock-treated condition versus treated conditions and # P<0.05 when comparing activated and non-activated EC cells.
We also studied the ability of the treated B cells to block TNFα production in EC (Supplementary Figure 4). We measured the frequency of TNFα-producing cells in autologous EC co-cultured with non-treated, mock-treated or stimulated B cells. Only three out of six experiments performed (one of which is shown in Supplementary Figure 4) showed a marked reduction of TNFα when EC were co-cultured with HIV-1NL4-3-, CD40L/IL-4/HIV-1NL4-3-, CpG/CD40L/LPS- or CpG/CD40L/LPS/HIV-1NL4-3-treated B cells in comparison to the TNFα values quantified in co-culture EC+ mock-treated B cells. In the other three experiments, we were not able to detect suppression of TNFα expression for any of the conditions tested.
TNFα is associated with inflammation, immune system hyper-activation and senescence in HIV-1+ individuals. Another parameter recently described (the CD4/CD8 ratio) was found to be associated with an increased immune activation, an immune-senescent phenotype, and a greater risk of morbidity/mortality in HIV-1+ individuals.28,29 Because of the importance of the CD4/CD8 ratio in immune activation/inflammation in HIV-1 disease progression, we correlated the percentage of reduction in TNFα production by EC when co-cultured with stimulated B-cells in comparison to EC co-cultured with mock-treated B cells and the CD4/CD8 ratio. HIV-1NL4-3-, CpG/CD40L/LPS- and CpG/CD40L/LPS/HIV-1NL4-3-treated B cells were able to decrease the TNFα production of EC when the ratio of CD4/CD8 was ~2 (normal value of CD4/CD8 in healthy individuals). A strong correlation between the CD4/CD8 ratio and the percentage of reduction in TNFα production in EC (Figures 8a, c and d) when EC were co-cultured with B cells treated with stimuli that induced a Breg phenotype was found. This correlation was not detected when B cells were treated with CD40L/IL-4 in co-culture experiments (Figure 8b), showing that the reduction in TNFα production in EC was dependent on the regulatory state of the treated B cells. In HIV-1-infected individuals treated with active anti-retroviral therapy, which recover a normal value of the CD4/CD8 ratio, HIV-1-induced Breg phenotype could have a role in the decreasing TNFα levels observed in these treated HIV-1+ individuals.30,31 In summary, we describe for the first time that the presence of HIV-1 induces a Breg-like phenotype in B cells, which acquire regulatory functions showing a suppressive capacity on activated CD4+ or CD8+ T cells.
Figure 8.
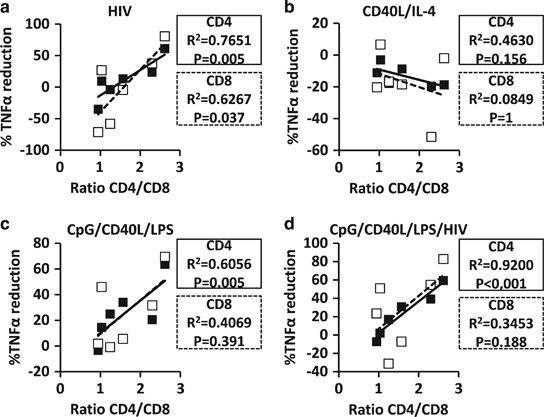
Reduction of TNFα expression in CD4+ or CD8+ T cells after co-culture with HIV-1-exposed B cells. Intracellular expression of TNFα by CD4+ and CD8+ T cells was analyzed after co-culture of effector cells (EC) and non-treated (NT) or treated B cells (ratio Breg:EC=2:1). Correlation between the ratio of CD4/CD8 and the percentage of reduction in TNFα expression in co-culture experiments when (a) HIV-1NL4-3-exposed, (b) CD40L/IL-4-exposed, (c) CpG/CD40L/LPS-exposed and (d) CpG/CD40L/LPS/HIV-1NL4-3-exposed B cells were calculated considering the mock-treated B-cell co-culture as the baseline reference level. Black squares and solid lines represent the results from CD4+ T cells, and open squares and dotted black lines represent the results from CD8+ T cells. Each square represents one experiment.
HIV-1-treated B-cell suppressive capacity was not related to cell death
One can argue that such a suppressive capacity might be related to an increase in T cell death, leading to a lack of EC proliferation or lack of TNFα production. Moreover, induced cell death of effector T cells is one of the suppressive mechanisms of Breg function already described.27 Therefore, we quantified T-cell death by different assays after co-culturing non-treated or treated B cells with CFSE-labeled EC. HIV-1NL4-3- and CpG/CD40L/LPS-treated B cells induced a significant reduction of CFSE-labeled CD4+ and CD8+ T cell frequency after 3 days of co-culture (Figures 7 and 9a). This reduction could be either due to a limited proliferation of these cells or due to specific cell death. We isolated CD3+ T cells using anti-CD3 magnetic beads from the co-culture and quantified the lactate dehydrogenase (LDH), a method for the detection of necrosis, liberated by these cells after 3 days. When cells were co-cultured with mock-treated B cells or with stimulated B cells, no difference in LDH liberation was detected, suggesting that HIV-1NL4-3- or CpG/CD40L/LPS-treated B cells did not induce necrosis of CD3+ T cells (Figure 9b). As caspases are a family of proteases having crucial roles in programmed cell death, essentially apoptosis, CD3+ T cells were isolated from the B cell/EC co-culture, and the expression of cleaved caspase-3 and caspase-9 was analyzed by western blotting in the CD3+ T-cell fraction after 3 days of co-culture (Figure 9c). No changes were observed in EC co-cultured with mock-treated B cells or stimulated B cells; only in the condition where EC were co-cultured with HIV-1NL4-3-treated B cells did we observe a significant decrease in the detection of cleaved caspase-3 (there was no significant reduction for the caspase-9 signal, Figure 9d). Taken together, we demonstrate that HIV-1NL4-3- or CpG/CD40L/LPS-treated B cells neither induce necrosis nor apoptosis in EC in co-culture. The low frequency of EC detected in these co-cultures was related to a suppression of T cell proliferation.
Figure 9.
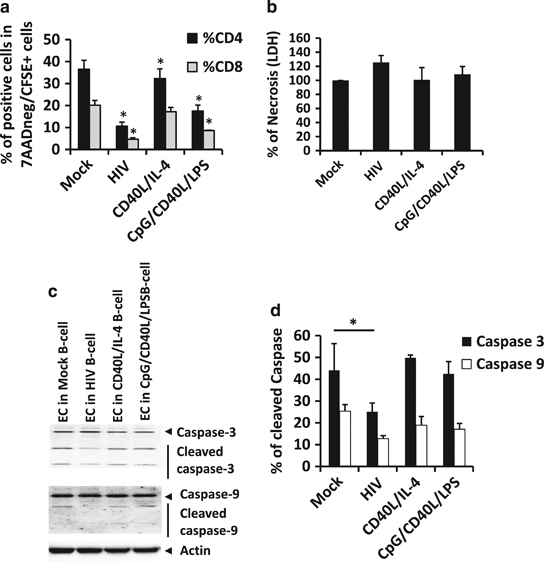
Effector cell (EC) death in co-culture with treated B cells. B cells were treated with CD40L/IL-4, HIV-1NL4-3 or with CpG/CD40L/LPS for 48 h. Cells were also mock-treated (mock). Forty-eight hours post stimulation, cells were co-cultured with autologous CFSE-labeled-EC for 3 days. (a) Frequency of CD4+ or CD8+ T cells in co-culture was analyzed by flow cytometry in total viable cells (7AADneg CFSE-EC). Average±s.e.m. of six experiments for each condition are shown. *P<0.05 when comparing mock-treated condition versus treated conditions. (b) After 3 days of co-culture, CD3+ T cells were isolated by magnetic beads, and LDH release from 100 000 cells was analyzed. Percentage of necrosis was calculated as follows: (LDH released by CD3+ T cells from co-culture with treated B cells × 100/LDH released by CD3+ T cells from co-culture with mock-treated B cells). Average±s.e.m. of six experiments for each condition are shown. LDH released in mock-treated B cell co-culture was considered as baseline-level=100%. (c) Isolated CD3+ T cells from co-culture were also lysed and analyzed for detection of cleaved caspase-3, caspase-9 and actin by western blotting. One representative experiment (out of 3) is shown. (d) Intensity of cleaved and non-cleaved caspase-3 and caspase-9 was quantified, and the percentage of cleaved caspase was calculated as follows: (intensity of cleaved caspase × 100/(intensity of cleaved+intensity of non-cleaved caspase)). Average±s.e.m. of three experiments for each condition are shown. *P<0.05 when comparing mock-treated condition versus treated conditions.
HIV-1-treated B cell suppressive mechanism is acting through cell-to-cell contact
HIV-1-treated B cells showed a Breg phenotype and suppressive function that was not related to the T-cell death induction (Figure 9). We studied whether such actions could be associated with an already known suppressive mechanism through IL-10, TGF-β, IL-35, PD-1 or PD-L1. We co-cultured mock-treated B cells or HIV-1-treated B cells with CFSE-labeled EC in combination with various neutralizing antibodies or peptides, such as anti-IL-10, anti-IL-35 (subunit peptide derived from EBI3), anti-PD-1, anti-TGF-β, anti-TRAIL, anti-PD-L1 or anti-FasL. Subsequently, CD4+ or CD8+ T-cell proliferation was analyzed by flow cytometry after 3 days of co-culture. EC non-activated (NA) and EC activated, cultured alone were used as proliferation controls (Figure 10a). As previously shown, mock-treated B cells did not induce suppression of CD4+ or CD8+ T cells proliferation (Figure 7). HIV-1-treated B cells induced the suppression of T-cell proliferation, but none of the neutralizing antibodies studied were able to reverse such suppression (Figure 10a). The suppressive mechanism of HIV-1-treated B cells was not mediated by the expression of IL-10, IL-35, PD-L1, PD-1, TGF-β, FasL or TRAIL. With the aim of knowing whether these factors inducing suppression could act in combination, we co-cultured mock-treated B cells or HIV-1-treated B cells with CFSE-EC with anti-IL-10, anti-PD-1, anti-PD-L1 and anti-FasL, alone or in combination. CD4+ or CD8+ T cell proliferation was analyzed by flow cytometry after 3 days, and no changes were observed in EC proliferation when co-cultured with HIV-1-treated B cells alone or with antibody combinations (we show only the CD4+ T cell population, Figure 10b). In summary, HIV-1-treated B cells had a suppressive function over EC that was independent of IL-10, PD-L1/PD-1 and FasL alone or in combination.
Figure 10.
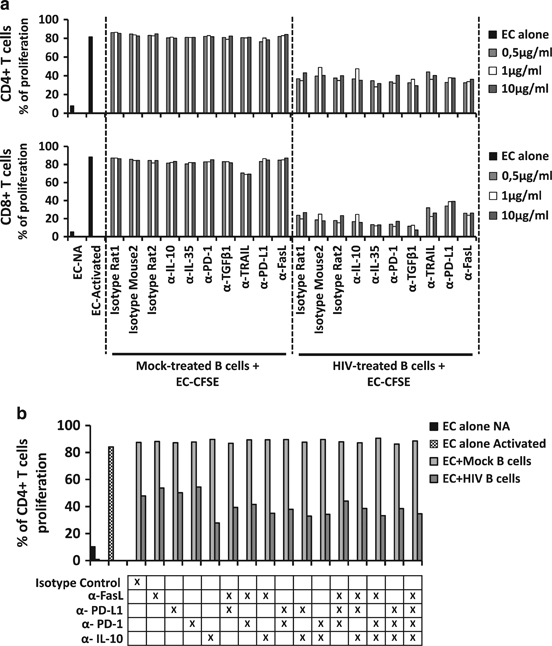
Effector cell (EC) proliferation and neutralizing assay. Proliferation of autologous EC treated with CFSE and co-cultured with mock- or HIV-1NL4-3-B cells (ratio Breg:EC of 2:1) at day 3 post co-culture. As a control, EC were also cultured alone without B cells (EC non-activated (NA) and EC activated). (a) Co-cultures were performed in the presence of isotype controls or neutralizing antibodies or peptide against IL-10 (α-IL-10), IL-35 (subunit peptide anti-IL-35), PD-1 (α-PD-1), TGF-β1 (α-TGF-β1), PD-L1 (α-PD-L1), TRAIL (α-TRAIL) or FasL (α-FasL) at different concentrations (0.5; 1 and 10 μg/ml). At day 3 post co-culture, EC cells were labeled with anti-CD4 and anti-CD8, and the proliferation of CD4+ and CD8+ T cells was measured by flow cytometry as the loss of CFSE signal. One representative experiment is shown. (b) Co-cultures were performed in the presence of isotype controls or a combination of neutralizing antibodies against IL-10 (α-IL-10), PD-1 (α-PD-1), PD-L1 (α-PD-L1) or FasL (α-FasL) (10 μg/ml each). At day 3 post co-culture, proliferation of CD4+ T cells was measured in anti-CD3/anti-CD28 stimulated EC in the presence of B cells by flow cytometry. One representative experiment is shown.
Transwell experiments were performed to examine whether the suppressive mechanism induced by treated B cells over EC required direct cell-to-cell contact. Our results indicate that separation of the two cell types by a 0.4 μm pore size membrane compromised the suppressive activity of the stimulated B cells, supporting a cell-to-cell contact dependence as previously observed (Figure 5). Co-culture experiments in the same plate were performed as a control for the suppressive ability of HIV-1NL4-3-treated B cells. When cells were co-cultured in a transwell system comparing mock-treated B cells or HIV-1-treated B cells, no change in EC proliferation was observed (Figure 11). Even though we could not determine the specific markers responsible for the suppressive function of the HIV-1NL4-3-treated B cells, cell-to-cell contact seems to be essential for these cells to suppress EC function.
Figure 11.
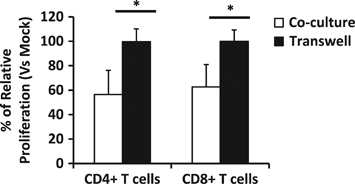
Cell-to-cell contact assay. Proliferation of autologous EC treated with CFSE was analyzed by flow cytometry when either co-cultured or cultured in a transwell plate with mock- or HIV-1NL4-3-B cells (ratio Breg: EC of 2:1) at day 3 post co-culture. EC-CFSE were directly co-cultured or cultured in a transwell system with mock-treated or HIVNL4-3-treated B cells. The percentage of relative proliferation was calculated as follows: (% of CD4+ or CD8+ T cell proliferation in HIV-1NL4-3-treated condition*100 / % of CD4+ or CD8+ T cell proliferation in mock-treated condition) in co-culture or in transwell assay. Average±s.e.m. of five experiments for each condition is shown. *P<0.05 when comparing Co-culture versus Transwell conditions.
Discussion
To our knowledge, we have observed, for the first time, that B cells treated with HIV-1 particles developed the following: (1) the CD19+CD24hiCD38hi Breg phenotype; (2) the expression of immunosuppressive cytokines (IL-10, TGF-β1 and IL-35); (3) Breg-like functions, such as the reduction of T-cell proliferation; and (4) a reduction in TNFα production in T cells through cell-to-cell contact in vitro.
Previously, we described a loss of CD27 and CD24hi, and an increase in CD38hi expression at the surface of HIV-1-treated B cells, which can explain the opposite effect of HIV-1 in the current results on Breg phenotypes CD19+CD24hiCD38hi and CD19+CD24hiCD27+.7 The loss of CD27 in B cells in the presence of HIV-1 provides an explanation for previous articles showing a loss of CD27+ memory B cells and a major presence of CD27 (naive)-B cells in HIV-1-infected individuals.10,32 Considering the doubts about the phenotype that correctly identifies Breg cells, whether the acquisition of a CD19+CD24hiCD38hi phenotype is directly responsible for the increase in suppressive Breg-like cell function observed in this work must be further established. Although the suppressive capacity of CD19+CD24hiCD38hi B cells was previously shown to be related to IL-10 production but not to TGF-β production,13 in our work neither IL-10 nor TGF-β were essential for the functionality of HIV-1-induced Breg-like cells, which makes us hypothesize that, in our model, the induction of this phenotype might not be necessarily related to the regulatory function.
One important finding of this work is that the presence of HIV-1 induces B cells to secrete IL-10, even if the suppressive effect in our culture system seems to be independent of IL-10 production. However, we can hypothesize that the suppressive or regulatory effect of this cytokine might act on a number of immune subsets that could affect immune mechanisms impairing HIV-1-specific immune responses in vivo. Because we have already shown that HIV-1 particles were able to deregulate B cells by BCR engagement through the c-Jun N-terminal kinase (JNK) and the spleen tyrosine kinase (SYK)/BCR pathways,6 and considering that inhibition of Src family kinases such as SYK reduces constitutive IL-10 production by B cells,33 we hypothesize that direct contact between B cells and HIV-1 could induce the production of IL-10 via the SYK/BCR engagement. Further experiments would be necessary to confirm this hypothesis.
Regarding IL-10, its effects on the pathogenesis of HIV-1 are quite controversial. Indeed, several studies have shown that low serum levels of IL-10 are linked to increased susceptibility to HIV-1 infection and accelerated progression to AIDS, particularly in the later stages of the disease.34,35 Other studies have shown that IL-10 is upregulated during viremic HIV-1 infection and that low levels of IL-10 are associated with a protective role in acute HIV-1 infection.19,36,37 High IL-10 levels may promote viral replication by suppressing innate and adaptive immune responses. In addition, in this study we also detected high TGF-β1 expression levels in HIV-1-exposed B cells, which was determined to be more frequent among individuals with higher plasma viral loads. TGF-β1 stimulates the expression of CTLA-4 on CD4+ T cells, which correlates with disease progression and loss of HIV-1-specific CD4+ T cellular functions.37,38 In addition, we demonstrated for the first time that HIV-1-treated B cells expressed mRNA encoding IL-35 subunits. It was recently demonstrated that IL-35 could convert human B-cells into Breg cells,39,40 favoring even more the switch to a Breg phenotype, thus fostering innate and adaptive immune system dysregulation.
HIV-1-related effects on phenotypes and cytokine profiles of B cells were correlated with a clear reduction in the proliferation of effector cells, showing that HIV-1-treated B cells acquire a Breg-like suppressive function. In addition to the anti-proliferative effects, HIV-1-exposed B cells also modulated TNFα production by effector cells. This ability is lost when the CD4/CD8 ratio is decreased. We hypothesized that in HIV-1-infected individuals who suffer from immune system deterioration marked by CD4 lymphopenia, even though B cells were induced towards a Breg-like phenotype, such a regulatory phenotype would not be strong enough to prevent the production of TNFα. This could explain the high levels of TNFα in plasma associated with the depletion of CD4+ T cells. Moreover, in treated HIV-1+-individuals who recover a normal value of the CD4/CD8 ratio, the HIV-1-induced Breg phenotype could have a role in the decrease in TNFα levels. All these results are crucial to understand the extent of dysregulation of the immune system ongoing in HIV-1 infection, and provide a rationale for the impairment of immune responses, in particular in the case of HIV-1+ individuals who suffer from bacterial translocation. In these individuals, the levels of TLR ligands and CD40L in the peripheral blood are high; regarding our results, their presence associated with HIV-1 particles would increase their effects on B cells, which could constitute the main source of the hyper-expression of IL-10 observed in HIV-1-infected individuals.
Despite the increased production of IL-10, IL-35 and TGF-β and the increased expression of surface markers PD-1 and PD-L1 observed on B cells, we did not demonstrate that the suppressive function of HIV-1-treated B cells was related to these factors. However, these markers are well described in the literature to be part of IL-10-dependent and IL-10-independent Breg mechanisms of action.18 None of the blocking antibodies for the factors studied in this work, IL-10, IL-35, TGF-β, PD-1, PD-L1, TRAIL or FasL, alone or in combination, were able to reverse the suppressive function of the HIV-1-treated B cells, although we determined that it was related to cell-to-cell contact between B and effector cells. The use of anti-HIV-1 neutralizing serum in addition to the use of anti-retroviral drugs (AZT and T20) showed that the increased frequency of IL-10-producing B cells and the induction of the Breg phenotype CD19+CD24hiCD38hi were not related to direct infection of the cells, but were related to a mix of viral or/and human components currently undefined but present at the virion surface. As monomeric gp120 and particularly trimeric gp140 were able to increase the frequency of IL-10-producing cells, even though they did not achieve the level detected when cells were treated by full-length viral particles, we hypothesized that the viral envelope is implicated in Breg-like phenotype induction, but that other viral or human proteins currently undefined but present at the viral surface might be implicated in heightened Breg signal induction.
During HIV-1 infection, several soluble factors have been implicated in HIV-1 disease progression and B-cell dysregulation. One of these factors is the B cell-activating factor (BAFF).41 High levels of BAFF have been directly associated with high levels of IL-10 and high frequencies of IL-10-producing B-cells in viremic HIV-1-infected individuals.20,42,43 Remarkably, their levels in aviremic slow progressors/elite controllers were comparable to those detected in healthy donors.44 It is interesting to note that the presence of IL-10 in association with BAFF has the ability to induce the expression of the mannose C-type lectin receptor and mannose receptor at the surface of B cells, receptors that can bind HIV-1 gp120. This linkage triggers the immunoglobulin CSR that elicits the production of specific and non-specific polyclonal IgG, IgA and IgE responses.8 Therefore, the presence of high levels of BAFF and IL-10, even in treated individuals, may be related to chronic activation of B cells, which could lead to immune exhaustion and non-specific Ig production.43 Our work adds a new piece to the puzzle of B-cell dysregulation, and we hypothesized that HIV-1 particles could directly participate through the viral envelope and potentially through other undefined proteins present at the virion’s surface in association with previously described microbial compounds and/or soluble factors (such as BAFF) in B-cell dysregulation. It would be very interesting to further investigate whether the association of HIV-1 particles with BAFF or microbial translocation could induce a higher frequency of Breg-like phenotypes in HIV-1+ individuals.
In summary, HIV-1 particles induced a Breg-like phenotype and suppressive capacities in the B-cell population. As it was previously described that HIV-1 particles could be found attached to the surface of B cells,45 and thus potentially transported to lymph nodes, the results presented here are essential to partially explain the profound dysregulation and inefficiency of the immune responses against HIV-1. An increase in Breg frequency and IL-10 secretion, artificially induced by the direct contact between HIV-1 particles and B cells, could provoke suppression of HIV-specific effector cells. Owing to the importance of an adequate immune response and homeostasis of the HIV-1 infection, this mechanism of immune dysregulation must be taken into consideration and further studied.
Electronic supplementary material
Acknowledgements
We acknowledge the centers of transfusion of Madrid for providing the buffy coats. We acknowledge Dr Laura Díaz from the Cytometry unit of IiSGM for sample analysis and Dr Maribel Clemente from the Cell Culture unit of IiSGM. This work was partially supported by Ministry of Economy and Competitiveness ISCIII-FIS grants PI12/01763, PI12/00934 and PI15/00923, and co-financed by ERDF (FEDER) Funds from the European Commission, ‘A way of making Europe’. JL-A was supported by a Grant from IiSGM. AP-S was supported by the Youth Employment Program co-financed by the Madrid community and FEDER Founds. RC-R was supported by the ‘Miguel Servet’ program (CPII13/00033), and MP by the Spanish MICINN through the Ramón y Cajal (RYC-2009-05486). All work was performed in the Laboratorio de Inmuno-Biología Molecular, HGUGM (IiSGM) led by M-AM-F.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.48
References
- 1.De Milito A, Nilsson A, Titanji K, Thorstensson R, Reizenstein E, Narita M, et al. Mechanisms of hypergammaglobulinemia and impaired antigen-specific humoral immunity in HIV-1 infection. Blood. 2004;103:2180–2186. doi: 10.1182/blood-2003-07-2375. [DOI] [PubMed] [Google Scholar]
- 2.Titanji K, De Milito A, Cagigi A, Thorstensson R, Grutzmeier S, Atlas A, et al. Loss of memory B cells impairs maintenance of long-term serologic memory during HIV-1 infection. Blood. 2006;108:1580–1587. doi: 10.1182/blood-2005-11-013383. [DOI] [PubMed] [Google Scholar]
- 3.Moir S, Malaspina A, Ogwaro KM, Donoghue ET, Hallahan CW, Ehler LA, et al. HIV-1 induces phenotypic and functional perturbations of B cells in chronically infected individuals. Proc Natl Acad Sci USA. 2001;98:10362–10367. doi: 10.1073/pnas.181347898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hart M, Steel A, Clark SA, Moyle G, Nelson M, Henderson DC, et al. Loss of discrete memory B cell subsets is associated with impaired immunization responses in HIV-1 infection and may be a risk factor for invasive pneumococcal disease. J Immunol. 2007;178:8212–8220. doi: 10.4049/jimmunol.178.12.8212. [DOI] [PubMed] [Google Scholar]
- 5.Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O'Shea MA, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med. 2008;205:1797–1805. doi: 10.1084/jem.20072683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perise-Barrios AJ, Correa-Rocha R, Alvarez S, Munoz-Fernandez MA, Pion M. HIV-1 induces B-cell activation and class switch recombination via spleen tyrosine kinase and c-Jun N-terminal kinase pathways. AIDS. 2014;28:2365–2374. doi: 10.1097/QAD.0000000000000442. [DOI] [PubMed] [Google Scholar]
- 7.Perise-Barrios AJ, Munoz-Fernandez MA, Pion M. Direct phenotypical and functional dysregulation of primary human B cells by human immunodeficiency virus (HIV) type 1 in vitro. PLoS ONE. 2012;7:e39472. doi: 10.1371/journal.pone.0039472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He B, Qiao X, Klasse PJ, Chiu A, Chadburn A, Knowles DM, et al. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol. 2006;176:3931–3941. doi: 10.4049/jimmunol.176.7.3931. [DOI] [PubMed] [Google Scholar]
- 9.Martin G, Roy J, Barat C, Ouellet M, Gilbert C, Tremblay MJ. Human immunodeficiency virus type 1-associated CD40 ligand transactivates B lymphocytes and promotes infection of CD4+ T cells. J Virol. 2007;81:5872–5881. doi: 10.1128/JVI.02542-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Milito A, Morch C, Sonnerborg A, Chiodi F. Loss of memory (CD27) B lymphocytes in HIV-1 infection. AIDS. 2001;15:957–964. doi: 10.1097/00002030-200105250-00003. [DOI] [PubMed] [Google Scholar]
- 11.Balkwill F, Montfort A, Capasso M. B regulatory cells in cancer. Trends Immunol. 2013;34:169–173. doi: 10.1016/j.it.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Das A, Ellis G, Pallant C, Lopes AR, Khanna P, Peppa D, et al. IL-10-producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J Immunol. 2012;189:3925–3935. doi: 10.4049/jimmunol.1103139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32:129–140. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011;117:530–541. doi: 10.1182/blood-2010-07-294249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol. 2015;27:479–486. doi: 10.1093/intimm/dxv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 17.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 18.Ray A, Wang L, Dittel BN. IL-10-independent regulatory B-cell subsets and mechanisms of action. Int Immunol. 2015;27:531–536. doi: 10.1093/intimm/dxv033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, Sela J, et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood. 2009;114:346–356. doi: 10.1182/blood-2008-12-191296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Zhan W, Kim CJ, Clayton K, Zhao H, Lee E, et al. IL-10-producing B cells are induced early in HIV-1 infection and suppress HIV-1-specific T cell responses. PLoS ONE. 2014;9:e89236. doi: 10.1371/journal.pone.0089236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siewe B, Stapleton JT, Martinson J, Keshavarzian A, Kazmi N, Demarais PM, et al. Regulatory B cell frequency correlates with markers of HIV disease progression and attenuates anti-HIV CD8(+) T cell function in vitro. J Leukoc Biol. 2013;93:811–818. doi: 10.1189/jlb.0912436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 23.ACTG. TCID50 Determination of Viable HIV-1. ATCG Laboratory Technologist Committee. ACTG Laboratory Manual 2004.
- 24.Stamatatos L, Lim M, Cheng-Mayer C. Generation and structural analysis of soluble oligomeric gp140 envelope proteins derived from neutralization-resistant and neutralization-susceptible primary HIV type 1 isolates. AIDS Res Hum Retroviruses. 2000;16:981–994. doi: 10.1089/08892220050058407. [DOI] [PubMed] [Google Scholar]
- 25.Kessel A, Haj T, Peri R, Snir A, Melamed D, Sabo E, et al. Human CD19(+)CD25(high) B regulatory cells suppress proliferation of CD4(+) T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun Rev. 2012;11:670–677. doi: 10.1016/j.autrev.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Cerutti A, Zan H, Schaffer A, Bergsagel L, Harindranath N, Max EE, et al. CD40 ligand and appropriate cytokines induce switching to IgG, IgA, and IgE and coordinated germinal center and plasmacytoid phenotypic differentiation in a human monoclonal IgM+IgD+ B cell line. J Immunol. 1998;160:2145–2157. [PMC free article] [PubMed] [Google Scholar]
- 27.Rincon-Arevalo H, Sanchez-Parra CC, Castano D, Yassin L, Vasquez G. Regulatory B cells and mechanisms. Int Rev Immunol. 2016;35:156–176. doi: 10.3109/08830185.2015.1015719. [DOI] [PubMed] [Google Scholar]
- 28.Serrano-Villar S, Sainz T, Lee SA, Hunt PW, Sinclair E, Shacklett BL, et al. HIV-infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non-AIDS morbidity and mortality. PLoS Pathog. 2014;10:e1004078. doi: 10.1371/journal.ppat.1004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sainz T, Serrano-Villar S, Diaz L, Gonzalez Tome MI, Gurbindo MD, de Jose MI, et al. The CD4/CD8 ratio as a marker T-cell activation, senescence and activation/exhaustion in treated HIV-infected children and young adults. AIDS. 2013;27:1513–1516. doi: 10.1097/QAD.0b013e32835faa72. [DOI] [PubMed] [Google Scholar]
- 30.Serrano-Villar S, Gutierrez C, Vallejo A, Hernandez-Novoa B, Diaz L, Abad Fernandez M, et al. The CD4/CD8 ratio in HIV-infected subjects is independently associated with T-cell activation despite long-term viral suppression. J Infect. 2013;66:57–66. doi: 10.1016/j.jinf.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 31.Wada NI, Jacobson LP, Margolick JB, Breen EC, Macatangay B, Penugonda S, et al. The effect of HAART-induced HIV suppression on circulating markers of inflammation and immune activation. AIDS. 2015;29:463–471. doi: 10.1097/QAD.0000000000000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chong Y, Ikematsu H, Yamamoto M, Murata M, Yamaji K, Nishimura M, et al. Increased frequency of CD27- (naive) B cells and their phenotypic alteration in HIV type 1-infected patients. AIDS Res Hum Retroviruses. 2004;20:621–629. doi: 10.1089/0889222041217455. [DOI] [PubMed] [Google Scholar]
- 33.Alhakeem SS, Sindhava VJ, McKenna MK, Gachuki BW, Byrd JC, Muthusamy N, et al. Role of B cell receptor signaling in IL-10 production by normal and malignant B-1 cells. Ann NY Acad Sci. 2015;1362:239–249. doi: 10.1111/nyas.12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vasilescu A, Heath SC, Ivanova R, Hendel H, Do H, Mazoyer A, et al. Genomic analysis of Th1-Th2 cytokine genes in an AIDS cohort: identification of IL4 and IL10 haplotypes associated with the disease progression. Genes Immun. 2003;4:441–449. doi: 10.1038/sj.gene.6363983. [DOI] [PubMed] [Google Scholar]
- 35.Oleksyk TK, Shrestha S, Truelove AL, Goedert JJ, Donfield SM, Phair J, et al. Extended IL10 haplotypes and their association with HIV progression to AIDS. Genes Immun. 2009;10:309–322. doi: 10.1038/gene.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naicker DD, Werner L, Kormuth E, Passmore JA, Mlisana K, Karim SA, et al. Interleukin-10 promoter polymorphisms influence HIV-1 susceptibility and primary HIV-1 pathogenesis. J Infect Dis. 2009;200:448–452. doi: 10.1086/600072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freitas FB, Lima SS, Feitosa RN, Azevedo VN, Ishak Mde O, Ishak R, et al. Polymorphisms in the IFNgamma, IL-10, and TGFbeta genes may be associated with HIV-1 infection. Dis Markers. 2015;2015:248571. doi: 10.1155/2015/248571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol. 2007;8:1246–1254. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 39.Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. 2014;20:633–641. doi: 10.1038/nm.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen P, Roch T, Lampropoulou V, O'Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507:366–370. doi: 10.1038/nature12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomez AM, Ouellet M, Tremblay MJ. HIV-1-triggered release of type I IFN by plasmacytoid dendritic cells induces BAFF production in monocytes. J Immunol. 2015;194:2300–2308. doi: 10.4049/jimmunol.1402147. [DOI] [PubMed] [Google Scholar]
- 42.Fontaine J, Chagnon-Choquet J, Valcke HS, Poudrier J, Roger M. High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIV-related B-cell disease progression in humans. Blood. 2011;117:145–155. doi: 10.1182/blood-2010-08-301887. [DOI] [PubMed] [Google Scholar]
- 43.Yehudai D, Snir A, Peri R, Halasz K, Haj T, Odeh M, et al. B cell-activating factor enhances interleukin-6 and interleukin-10 production by ODN-activated human B cells. Scand J Immunol. 2012;76:371–377. doi: 10.1111/j.1365-3083.2012.02752.x. [DOI] [PubMed] [Google Scholar]
- 44.Chagnon-Choquet J, Fontaine J, Poudrier J, Roger M. IL-10 and lymphotoxin-alpha expression profiles within marginal zone-like B-cell populations are associated with control of HIV-1 disease progression. PLoS ONE. 2014;9:e101949. doi: 10.1371/journal.pone.0101949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moir S, Malaspina A, Li Y, Chun TW, Lowe T, Adelsberger J, et al. B cells of HIV-1-infected patients bind virions through CD21-complement interactions and transmit infectious virus to activated T cells. J Exp Med. 2000;192:637–646. doi: 10.1084/jem.192.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.