

Abstract
Continuing introduction of multi-drug resistant, zoonotic pathogens such as methicillin-resistant Staphylococcus aureus (MRSA) in horse clinics challenges the biosafety of employees and animal patients. This study was aimed to determine the occurrence of mobile genetic elements facilitating survival in the early stages of invasive infection in different host species, including humans and horses, in MRSA carried by equine patients admitted to a large horse clinic. A total of 341 equine patients were investigated for carriage of MRSA by hygiene screening directly at hospital admission. MRSA were further investigated by antimicrobial susceptibility testing, whole-genome sequencing and genomic composition, including virulence factors involved in immune evasion and host adaption. From a total of 340 validated specimens from equine nostrils, 3.5% yielded positive results for MRSA. All MRSA were found to be closely related belonging to sequence type (ST) 398_t011 with up to four additional antimicrobial resistances. All MRSA harbored a specific Staphylococcal Pathogenicity Island (SaPIbov5) involved in facilitating survival in ruminant and equine plasma. Moreover, a β-hemolysin (hlb) converting ΦSa3 phage encoding the human-specific Immune Evasion Cluster (IEC) was present in 72% of the isolates. An equid-specific leukotoxin encoded by a further temperate phage (Saeq1) was only rarely detected (22%). Despite the absence of β-hemolysin production for all IEC-positive ST398, a prominent hemolysis zone was demonstrable on sheep blood agar. Thus, IEC might remain undetected among the ST398 lineage, since the presence of IEC is commonly associated with reduction of hemolysis in S. aureus belonging to other genetic backgrounds. Here we describe MRSA-ST398 harboring different mobile genetic elements encoding variants of immune evasion factors and toxins previously shown to contribute to S. aureus invasive diseases in specific host species or ecologic niches. We suggest these combinations contribute to the adaptation of MRSA belonging to ST398 with respect to epidemic spread across different habitats and hosts, and may therefore confer a host “generalist” phenotype.
Keywords: MRSA, multi-drug resistance, horses, one health, host range, ST398
Introduction
Epidemic and zoonotic methicillin-resistant Staphylococcus aureus (MRSA) lineages are characterized by the capacity to colonize and infect a broad host spectrum and circulate within distinct ecological niches (Walther et al., 2009, 2017; Vincze et al., 2014). Recent studies suggested that the dominant European livestock-associated S. aureus lineage belonging to sequence type (ST) 398 could increase its threat to public health by continued acquisition of virulence and antibiotic resistance genes (Diene et al., 2017).
Virulence-associated factors promoting colonization and infection and involved in counteracting the host's innate immune defense are often located on mobile genetic elements (MGE) such as pathogenicity islands (SaPI), temperate phages, and plasmids (Everitt et al., 2014; Diene et al., 2017). Examples include phages such as Saeq1 harboring a leukocidin (LukPQ) with a potent and specific killing activity toward equine neutrophils (Koop et al., 2017), and SaPIbov-encoded variants of von Willebrand factor-binding protein (vWbp) (Viana et al., 2010). Together with coagulase (Coa), chromosomally-encoded vWbp form a macromolecular complex with prothrombin, fibrinogen, factor XIII, and fibronectin in human plasma (McCarthy and Lindsay, 2013). The resulting staphylococcal clots are fibrin barriers against host phagocytes, providing a niche for local population development (Thomer et al., 2013). It has been suggested that genes encoding vWbp (vwb) might be a factor involved in animal adaptation, since the vWbp encoded on SaPIbov4 showed specific activity toward ruminant and equine plasma (Viana et al., 2010).
However, additional host defenses against invading bacteria are the recognition, opsonisation, and elimination functions of the complement system (Foster, 2005). Distinct phylogenetic S. aureus lineages evade host immune responses differently, with adaption to new host environments often accompanied by acquisition of MGEs and the immune evasion protein complexes that they encode (McCarthy and Lindsay, 2013). The immune evasion cluster (IEC) is commonly associated with beta-hemolysin (hlb) converting phages (van Wamel et al., 2006). As a result, IEC-positive S. aureus with a disrupted hlb often lack a prominent hemolysis zone on sheep blood agar plates (Katayama et al., 2013).
IEC are generally regarded as human-specific, since immune-modulating factors such as the complement-protein cleaving Staphylokinase (sak) and the Staphylococcal complement inhibitor (scn) inhibit complement factor C3 activation in the human host (McCarthy and Lindsay, 2013). Additional toxins, adhesins, coagulation- and immunomodulatory factors are frequently found to be encoded by genes within SaPIs (Malachowa and DeLeo, 2010).
Here we report on MRSA-carriage rates recorded for horses at hospital admission, the genetic relationship of these isolates and the presence of genes located on MGEs encoding factors involved in establishment of infection in distinct host species.
Materials and methods
Inclusion criteria for equine specimens
All specimens were obtained from horses by hygiene screening, directly at hospital admission at the Equine Clinic, Surgery and Radiology, Department of Veterinary Medicine, Freie Universität Berlin, as described earlier (Walther et al., 2018), following the regulations and approval of the Landesamt für Gesundheit und Soziales (LAGESO), Berlin (14.07.2014). Inclusion criteria for all equine samples were defined as: sterile cotton swabs with Amies transport medium (Mast Diagnostica, Reinfeld) of both anterior nostrils taken on arrival without delay and fecal samples taken within 120 min after admission (Walther et al., 2018).
Statistical analysis
Data were analyzed using IBM SPSS version 24. Differences in the occurrence of positive nostril swabs between the “colic” group and the “open wound” group were investigated using the chi-square-test. P-values below 0.05 were considered statistically significant.
Microbiological approach and antimicrobial susceptibility testing
Fecal specimens, nostril- and wound swabs were initially cultured on Columbia agar with 5% sheep blood (bioMérieux, Germany), and chromID® MRSA (bioMérieux, Germany) agar plates. Identification of S. aureus was determined by MALDI-TOF MS (Bruker, Germany). Wound swabs were taken immediately upon admission from the “open wound” patients within the reception area. Further information on the complete sampling procedure and local setting has been published elsewhere (Walther et al., 2018). Antimicrobial susceptibility testing (AST) using the VITEK®2 system (BioMérieux, Germany) was performed according to the standards given by the CLSI VET01-A4 and M100-S21 (Clinical Laboratory Standards Institute, 2011, 2013).
CAMP test
The CAMP test (originally described by Christie et al., 1944) is commonly used to identify Group B streptococci, which secrete a protein called CAMP factor known to interact with the β-hemolysin of S. aureus (Spellerberg and Brandt, 2015). All isolates were tested for the CAMP phenomenon using a diffusion test on Columbia agar with 5% sheep blood (bioMérieux, Germany), with the quality control strain Streptococcus agalactiae (ATCC12386), and the beta-toxin-producing S. aureus strain ATCC25923 as a positive control. Note that β-hemolysin enhances lysis by δ-hemolysin, but inhibits lysis by α-hemolysin (Traber et al., 2008). After 18 h at 37°C incubation followed by 4 h at 4°C, isolates were inspected for CAMP hemolysis.
Whole genome sequencing of equine MRSA
MRSA isolates were whole-genome sequenced (WGS) using Illumina MiSeq 300 bp paired-end sequencing with an obtained coverage > 90X. Adapter-trimmed reads were used for de novo assembly into contiguous sequences (contigs) and subsequently into scaffolds using SPAdes v3.11. All draft genomes were annotated using Prokka (Seemann, 2014). WGS data were used for genotypic characterization including the determination of the sequence type (ST) with online tools (https://cge.cbs.dtu.dk/services) including MLSTFinder, spaTyper 1.0 and ResFinder 2.1 (threshold: 95% ID, 80% minimum length) for transferable resistance gene detection (Larsen et al., 2012; Zankari et al., 2012; Bartels et al., 2014). Genomic integration sites of mobile genetic elements and amino acid exchanges in resistance-associated genes were investigated using Geneious 10.0.5 (Biomatters Ltd., Australia).
Maximum likelihood tree based on determination of the maximum common genome (MCG)
In order to compare the genomes at high resolution, we used the maximum common genome (MCG) that is defined by those orthologous genes present in all considered genomes (von Mentzer et al., 2014). The coding sequences were clustered based on the parameters of sequence similarity (min. 70%) and coverage (min. 90%). The MCG was defined as those genes that were present in each genome and fulfilled the threshold parameters, yielding 2,346 genes. Allelic variants of these genes were subsequently extracted from all genomes by an in-house developed blast-based pipeline, then aligned individually for each gene and concatenated, resulting in an alignment of 1.981 Mbp for these strains. The alignment was used to generate a maximum likelihood phylogenetic tree using RAxML 8.1 (Stamatakis, 2014), including livestock-associated methicillin-resistant S. aureus strain 08S00974 (accession number CP020019) isolated from a pig on a fattening pig farm in Germany (Makarova et al., 2017) as an outgroup.
The resulting alignment was screened for pairwise single nucleotide polymorphism (SNP) differences and based on these values a SNP-distance matrix was created.
Results
We prospectively investigated equine patients admitted to the equine clinic of the Freie Universität Berlin, Department of Veterinary Medicine, which showed clinical signs associated with either “colic” or “open wounds,” from April through October in 2014, and again in 2015. All horses were screened for colonization/infection with multidrug resistant and zoonotic bacteria to develop hygiene improvement strategies (Walther et al., 2018).
Carriage rates and antibiotic resistance among MRSA from equine patients at hospital admission
MRSA carriage was investigated in 341 equine patients (Table 1). Information on the inclusion/exclusion criteria for horses has been previously described (Walther et al., 2018). One nostril swab and 23 fecal samples were excluded due either to time delays in sampling post-admission or because they were not available.
Table 1.
Results from MRSA screening of equine patients at hospital admission.
| Specimens (admission) | Screening period I & II | Screening period I | Screening period II | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 341 horses | Apr-Oct 2014 (n = 174 horses) | Apr-Oct 2015 (n = 167 horses) | ||||||||||||||||
| Total (I+II) | Colica | Open woundb | Total (I) | Colica | Open woundb | Total (II) | Colica | Open woundb | ||||||||||
| n | % | n | % | n | % | n | % | n | % | n | % | n | % | n | % | n | % | |
| Nostril swabs (valid) | 340 | 100 | 232 | 100 | 108 | 100 | 173 | 100 | 113 | 100 | 60 | 100 | 167 | 100 | 119 | 100 | 48 | 100 |
| MRSA | 12 | 3.5 | 10 | 4.3 | 2 | 1.9 | 5 | 2.9 | 3 | 2.7 | 2 | 3.3 | 7 | 4.2 | 7 | 5.9 | 0 | 0 |
| Feces (valid) | 318 | 100 | 220 | 100 | 98 | 100 | 166 | 100 | 109 | 100 | 57 | 100 | 152 | 100 | 111 | 100 | 41 | 100 |
| MRSA | 2 | 0.6 | 1 | 0.5 | 1 | 1.0 | 2 | 1.2 | 1 | 0.9 | 1 | 1.8 | 0 | 0 | 0 | 0 | 0 | 0 |
| Wound swabs | 108 | 100 | 60 | 100 | 48 | 100 | ||||||||||||
| MRSA | 4 | 3.7 | 3 | 5.0 | 1 | 2.1 | ||||||||||||
n, number; MRSA, methicillin-resistant S. aureus.
horses admitted to the horse clinic with clinical signs associated with the colic complex.
horses admitted to the horse clinic with an open wound.
Out of a total of 340 validated nostril swabs, 3.5% were found positive for MRSA, with a slightly higher rate (4.3%) among the “colic” group in comparison with the “open wound” group (1.9%), but were not statistically significant (p = 0.253, chi-square-test). Four wound swabs taken from horses of the latter group (n = 108) were MRSA-positive (3.7%). Moreover, 318 validated fecal samples were obtained, with an isolation rate of 0.6% MRSA.
Antibiotic susceptibility testing results revealed additional resistances to aminoglycosides (gentamicin, kanamycin) and tetracycline for all 18 MRSA tested, with 66.7% of the isolates also showing resistance toward fluoroquinolones (enrofloxacin, marbofloxacin) and 61% to trimethoprim-sulfonamide (Table 2). Whole genome sequence data yielded corresponding resistance genes including blaZ and mecA (penicillin/methicillin resistance) tet(M) for tetracycline resistance, aacA-aphD (aminoglycoside resistance), and the trimethoprim resistance gene dfrK located on transposon Tn559 (Kadlec and Schwarz, 2010). Moreover, all genomes harbored strA (streptomycin resistance). Phenotypic resistance toward fluoroquinolones was associated with amino acid exchanges in the gyrase and topoisomerase IV at the following positions: Ser84Leu in GyrA, Glu182Asp in GyrB, Ser80Phe, Val590Ile and Val656Ile in GrlA and Ile72Thr, Glu422Asp and Glu596Asp in GrlB, matching a profile described only recently for MRSA-ST398-t011 isolated from specimen of equine ocular surfaces (Soimala et al., 2018).
Table 2.
Resistance profiles and virulence characteristics of 18 MRSA-ST398 isolated from horses at hospital admission.
| No.a | IMT _IDb | Date | IECc | lukP/Qd | CAMPe | Resistance phenotypef |
|---|---|---|---|---|---|---|
| 1 | 33368 | May 2014 | pos. | neg. | neg. | GEN, KAN, TET, SXT |
| 2 | 33391 | May 2014 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR |
| 3 | 33826 | Jul 2014 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR, SXT |
| 4 | 33828 | Jul 2014 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR |
| 5 | 33861 | Jul 2014 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR |
| 6 | 33862 | Jul 2014 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR |
| 7 | 33997 | Aug 2014 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR, SXT |
| 8 | 34080 | Aug 2014 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR, SXT |
| 9 | 34209 | Sep 2014 | neg. | neg. | pos. | GEN, KAN, TET, SXT |
| 10 | 34426 | Sep 2014 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR, SXT |
| 11 | 36995 | Jun 2015 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR |
| 12 | 37082 | Jul 2015 | neg. | pos. | pos. | GEN, KAN, TET, SXT |
| 13 | 37264 | Aug 2015 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR |
| 14 | 37277 | Aug 2015 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR |
| 15 | 37325 | Aug 2015 | neg. | pos. | pos. | GEN, KAN, TET, SXT |
| 16 | 37340 | Aug 2015 | neg. | pos. | pos. | GEN, KAN, TET, SXT |
| 17 | 37426 | Sep 2015 | pos. | neg. | neg. | GEN, KAN, TET, ENR, MAR, SXT |
| 18 | 37510 | Sep 2015 | neg. | pos. | pos. | GEN, KAN, TET, SXT |
All MRSA-ST398-t011 of equine origin harbor SaPIbov5 (1–18).
Strain collection number at Institute of Microbiology and Epizootics (IMT), Freie Universität Berlin.
Presence of a beta-hemolysin converting ΦSa3 phage (40 kb) harboring a variant of the Immune Evasion Cluster (Figure 2).
Presence of 45 kb prophage (ΦSaeq1) known to harbor a leukocidin (LukPQ) with potent and specific killing activity toward equine neutrophils.
CAMP test result: enlarged area of hemolysis formed by β-hemolysin production from Staphylococcus aureus (Figure 4).
Results from antimicrobial susceptibility testing (technical appendix) by VITEK®2 system (BioMérieux, Nürtingen, Germany) for non-beta-lactam antibiotics. GEN, gentamicin; KAN, Kanamycin; ENR, enrofloxacin; MAR, marbofloxacin; TET, tetracycline; SXT, trimethoprim-sulfamethoxazole.
Phylogenetic relationship of MRSA ST-398 isolated from equids
All MRSA were submitted for whole-genome sequencing (WGS) using Illumina MiSeq 300 bp paired-end sequencing, and screened for virulence- and resistance factors. The accession numbers of the current strains are listed in Supplemental Table 1. All MRSA belonged to the so-called livestock-associated lineage predominating in Europe (ST398, spa type t011), and harbored a complete SCCmecIV element and SaPIbov5 (Viana et al., 2010).
A phylogenetic tree was generated from the MCG comprising 2,346 orthologues genes including WGS data (CP020019) of a livestock-associated MRSA-ST398 strain (08S00974) as an outgroup. Overall, all 18 equine MRSA strains appeared closely related. However, separation of two distinct clusters was possible based on the core genome phylogeny (Figure 1). Pairwise distance ranges among the MCG were calculated for all equine MRSA isolates (Supplemental Table 2), yielding 0-1 SNPs for the most related strains (cluster A, Figure 1), 0-43 SNPs for the MRSA clustering together within B, and 60-92 SNPs for a singleton, representing the most distantly related strain (IMT34209) in this study. A range of 187-201 SNPs was calculated based on the WGS data of the outgroup strain (08S00974), which is also positive for SAPIbov5 and shares the ST398 background.
Figure 1.

Close relationship of 18 equine MRSA-ST398-t011 isolated from horses at hospital admission. Maximum Likelihood Phylogeny: Phylogenetic tree generated with RAxML, displayed with CLC Genomics Workbench on the basis of 2,346 orthologous genes from 19 ST398 S. aureus WGS, including livestock-associated methicillin-resistant Staphylococcus aureus (ST398) strain 08S00974 (CP020019) as an outgroup. Presence of mobile genetic elements harboring host adaptation factors is marked blue. phiNM3-var., ΦSa3 phage (40 kb) harboring an immune evasion cluster (IEC); Saeq1, 45 kb prophage Φ Saeq1; SaPIbov5, Staphylococcus pathogenicity island SaPIbov5. Two subgroups could be distinguished, cluster (A) and (B).
Of note, four MRSA identified in 2015 were positive for a recently described temperate phage, Saeq1 (Table 2). In 13 equine MRSA-ST398, a ΦSa3 phage of 40 kb length closely related to phiNM3 (DQ530361.1; coverage: 96%, similarity 98%) was found to have a disrupted β-hemolysin gene, hlb (Figure 2). A single strain (IMT34209) lacks both MGEs. The presence/absence of these MGEs matches with the core genome phylogeny (Figure 1).
Figure 2.

IEC-positive phiNM3-like phage integrated in hlb gene of equine MRSA-ST398. Gene map of the hlb-encoding phospholipase C (ß-hemolysin) region in equine MRSA-ST398. The colored bar at the top indicates the mean pairwise nucleotide sequence identity in the column: bright green = 100% identity; green-brown = <100% but >30% base pair identity. Prophage genes are colored based on putative or known function. Genomic attachment sites were shown in gray. The integration site (5′-GTATCCGAATTGG-3′) of 40,699 bp or 40,703 bp phiNM3-like phages (coverage: 96%, similarity 98%; DQ530361.1) harboring genes of the Immune Evasion Cluster (scn, sak) within the first 13 genomes is indicated by an orange arrow, resulting in a disrupted hlb gene (red) of 301 bp (left) and 825 bp (right). So far, ΦSa3 integration in hlb of ST-398 was rarely reported (Kraushaar et al., 2017).
All strains harbored two allelic variants of vwb, a chromosomally-encoded gene of 1,515 base pair length sharing 100% identity with vwb of HM240418.1 and a further variant located on SAPIbov5, which is 100% similar to the vwb gene reported for SaPIbov4 (HM211303.1) (Diene et al., 2017).
Hemolysis on sheep blood agar plates
Different bi-component leucocidins including γ-hemolysin, the pore forming toxins α-hemolysin (hla), β-hemolysin and phenol soluble modulins, are among the factors which determine the hemolytic appearance of S. aureus on sheep blood agar (SBA) plates. Despite an explicit hemolysis zone on SBA plates for all MRSA-ST398 isolates reported here (Figure 3), the lack of β-hemolysin production (phospholipase C, a sphingomyelinase) was shown by employing a CAMP cross-streaking test using the CAMP factor produced by Streptococcus agalactiae (ATCC12386) as an indicator for β-hemolysin production. As shown in (Figure 4), the absence of β-hemolysin production was verified by the lack of a CAMP phenomenon in all MRSA-ST398 possessing the ΦSa3 phage, although a relatively large hemolysis zone is apparent. In addition, β-hemolysin production inhibits lysis by α-hemolysin (Traber et al., 2008), resulting in weaker total hemolysis zones for the IEC-negative isolates 9, 12, 15, 16, and 18 (Figures 4B,C).
Figure 3.
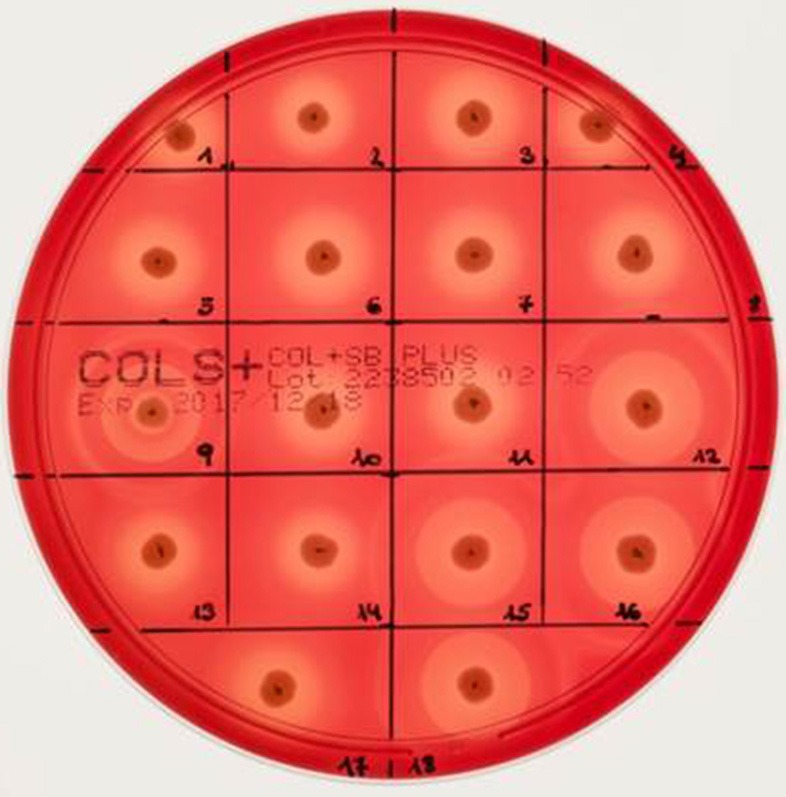
Hemolysis of equine MRSA-ST398 on sheep blood agar plate. Hemolysis zones of closely related MRSAST398-t011 isolated from horses on sheep blood agar plates after 18 h at 37°C incubation followed by 4 h at 4°C. The β-hemolysin activity induced “double zone” hemolysis (halo) was noticed only for isolate 9, 12, 15, 16, and 18. Isolates 1–8, 10, 11, 13, 14, and 17 harbor a β-hemolysin disrupting phage carrying IEC (numbers as indicated in Table 2).
Figure 4.

Lack of β-hemolysin production in MRSA-ST398 harboring IEC. (A–C) CAMP test results for Streptococcus agalactiae (grown as central diameter on each plate) together with 18 MRSA-ST398-t011 isolates of equine origin. grown as central diameter on each plate) together with 18 MRSA-ST398-t011 isolates of equine origin. (A) White arrow: large hemolytic zone irrespectively of CAMP factor presence in 1–8, 10, 11, 13, 14, and 17. These isolates harbor a β-hemolysin disrupting phage carrying IEC (numbers as indicated in Table 2). (B) White arrow: enlarged hemolysis zone induced by synergistic activity of CAMP factor- and β-hemolysin production in 9, 12, 15, 16, and 18. (C) White arrow: β-hemolysin production inhibits lysis by α-hemolysin (Traber et al., 2008), resulting in weaker total hemolysis zones for the IEC-negative isolates 9, 12, 15, 16, and 18. +, positive control S. aureus ATCC 25923.
Discussion
Here we provide evidence not only for the continuing entrance of MRSA into veterinary clinics through colonized and/or infected horses, but also additional molecular evidence for contemporary carriage of MGEs within the so-called “live-stock” MRSA-ST398 lineage— MGEs encoding virulence factors previously found to be involved in adaption to different host species.
Carriage of multi-drug resistant MRSA-ST398-t011 in horses at hospital admission
Similar to the large geographic variations for MRSA detection rates reported for human patients at hospital admission, MRSA (nostril-)screening results for horses at hospital admission have shown large variations in former studies, generally ranging from 2.9 to 10.9% (Tokateloff et al., 2009; Weese and van Duijkeren, 2010). In this study, the average nostril-swab detection rate was 3.5%. However, the colic group yielded more MRSA-positive isolates than the open wound group (4.3 vs. 1.5%), but the difference was not statistically significant. Since horses often suffer from recurrent colic, prior episodes of intensive veterinary care or hospital stays might have an influence on the detection rate for this group. Unfortunately, long-term retrospective data was not available for the horses presented here and should be included in further studies. While S. aureus fecal carriage has been identified as a potential source for nosocomial transmission and a risk factor for disease development in human medicine (Claassen-Weitz et al., 2016), the detection rate of 0.6% for horses at hospital admission seems to be of less importance with respect to equine hospital hygiene. In contrast, in human medicine, intestinal carriage seems of much greater importance and has been assumed to be generally underestimated (Acton et al., 2009).
All MRSA isolates reported here revealed a remarkable multi-drug resistance, comprising up to five different classes of antimicrobials (Table 2). A further study including horses with acute or chronic ocular disorders presented at the horse clinic of the Freie Universität Berlin in 2015/2016 revealed the occurrence of closely related, multi-drug resistant MRSA-ST398-t011 in samples from ocular surfaces as well (Soimala et al., 2018).
While MRSA-ST398 were infrequently reported as cause of human infections (van Duijkeren et al., 2011; Kinnevey et al., 2014; Larsen et al., 2015), hospitals located in pig population-dense regions appear to be a hot-spot for this genotype (Köck et al., 2009, 2013; van Alen et al., 2017).
Phylogenetic relationship of MRSA-ST398 isolated from hospitalized horses
Although the occurrence of different genotypes has previously reported in prior studies (Cuny et al., 2006, 2008; Walther et al., 2009), more recent studies, including this study, revealed an absolute predominance of MRSA-ST398 in samples obtained from horses (Vincze et al., 2014).
In recent years, several reports indicated not only the occurrence of phages harboring the IEC in MRSA-ST398 of equine origin (Cuny et al., 2015; Islam et al., 2017), but also a possible beneficial impact of IEC-carriage in MRSA-ST398 on bacterial survival in the presence of human and equine polymorphic neutrophils (Jung et al., 2017).
Here, we report on WGS data from MRSA isolates of 18 horses sampled directly at hospital admission, before entering the hospital stables or other rooms. The core genome together with the SNP ranges calculated based on the 2,346 bp MCG of these 18 MRSA-ST398-t011 revealed a close phylogenetic relationship, mirrored by range of 0-92 SNPs considering pairwise distance ranges for all 18 equine MRSA (Supplemental Table 2). However, two subgroups (cluster A and B in Figure 1) and a singleton (IMT34209) could be distinguished, yielding SNP ranges from 0 to 1 within cluster A and 0–43 in cluster B. A former study on MRSA from veterinary clinics in the UK revealed that isolates from a particular clinic often clustered together on the phylogenetic tree, including different clades of closely related isolates from a broad range of infections (Harrison et al., 2014). Interestingly, the presence or absence of either Saeq1 or an IEC-carrying phiNM3-like phage, which were not part of the core genome analysis, matches strictly with the phylogenetic tree (Figure 1).
A recent study investigating WGS data of LA-MRSA belonging to clonal complex 398 revealed some evidence of phylogeographic patterns with a majority of European isolates clustering together and forming a unique lineage compared with the non-European isolates (Sharma et al., 2016). The largest European sub-lineage, denominated as EU t011, was further separated into two branches: One group harboring SCCmecIV and the other SCCmecV (Sharma et al., 2016). Interestingly, transferable resistance genes detect in this study were similar to the profiles reported by Sharma et al. for sub-lineage EU t011 SCCmecIV, including blaZ, mecA, tetM, aac(A)-aph(D), dfrK, and strA (Sharma et al., 2016). In addition, this sub-lineage was associated with MRSA strains from a broad range of host species, including horses, cattle and pigs. In contrast to our results, none of the ST-398 isolates investigated by Sharma et al. carried any human-associated virulence genes (Sharma et al., 2016), supporting the idea of ongoing adaptive changes within this host generalist lineage.
In this study, three different homologs of scn were identified within the IEC (scn), SaPIbov5 (scnbov), and Saeq1 (scneq), and three MGEs of a completely different nature and composition. Interestingly, 17/18 MRSA-ST398 of equine origin harbored two different SCIN-encoding variants (Figure 1). Inactivation of the C3 convertase complex in the alternative pathway of complement activation is considered one of the most important immune evasion strategies in S. aureus (Ricklin et al., 2009). A recent study on the biological effects of the equine variant of SCIN encoded by scneq revealed its ability to block activation of the equine complement system, hence interfering with phagocytosis (de Jong et al., 2018).
Strikingly, eqSCIN was identified as a SCIN variant that functions in a much broader range of hosts, including horses, humans, and pigs (de Jong et al., 2018). In addition, our data might indicate a greater importance for contemporary carriage of allelic variants encoding Staphylococcal complement inhibitor (SCIN) for successful S. aureus niche- and host-adaption than previously thought, since complement activation is pivotal for S. aureus killing (Jongerius et al., 2007).
All MRSA ST398 positive for SAPIbov5 harbor two allelic variants encoding the von Willebrand binding protein, a potent activator of the blood prothrombin (Thomer et al., 2013). The SAPIbov5-encoded vwb variant shows specific activity toward equine and ruminant plasma (Viana et al., 2010). The presence of two, different vWbp-encoding genes might indicate a broadening in this genetic background of its flexibility with respect to host range. However, the functional role of the chromosomally-encoded vwb reported here, as well as the interplay and regulation of both variants requires further investigation.
Further research is warranted to track the origin and distribution of MGEs promoting niche adaption within the successful and zoonotic ST398 lineage, including genes facilitating resistance toward antimicrobials and biocides but also virulence and biofilm formation abilities (Feßler et al., 2010; Fessler et al., 2017; Wendlandt et al., 2013).
Genomic integration site of phiNM3-like phages in equine MRSA-ST398 and hemolysis
The precise hlb integration site (Figure 2) of all phiNM3-like phages (sizes: 40,699 or 40,703 bp) in our study (5′-GTATCCGAATTGG-3′) has been described with respect to the ability of ΦSa3-like phages to integrate into hlb of ST398, an event which has rarely been reported (Kraushaar et al., 2017). A further recent retrospective study on human derived S. aureus isolates sharing the clonal complex 398 revealed 12 different phage types ranging in size from 40,712 bp up to 44,003 bp and four novel genomic integration sites for ΦSa3 phages (van Alen et al., 2018).
Interestingly, a prior study reported that isolates sharing an ST398 background were commonly found to have a significantly higher hla expression compared to other lineages, especially hospital-associated MRSA (Tavares et al., 2014). Notably, a strong expression of α-hemolysin on sheep blood agar might mask the lack of β-hemolysin production as a result of hlb disruption by ΦSa3 phages, a fact that might lead to overlooking an IEC-positive ST398 isolate in routine diagnostics and research (Figure 3).
Conclusion
In equine MRSA-ST398, the contemporary presence of MGEs carrying genes known to encode virulence factors which have been associated with a certain degree of host-specificity, such as SAPIbov5 and β-hemolysin converting phages possessing an IEC variant, most likely mirrors the capacity to survive the initial stages of an invasive infection in different host species.
Further functional analyses are clearly needed to reveal the contribution of scn homologs and other immune modulating factors to host adaption, since the potential of equine MRSA-ST398 to cause severe infectious disease is clearly present.
Consequently, the spread of multi-drug resistant equine MRSA in veterinary clinics must by contained to enhance biosecurity for both veterinary staff and equine patients. This particular aspect of the “One Health” idea requires more emphasis in the development of targeted infection control strategies.
Author contributions
BW and AL-B conceived and designed the experiments. K-SK, A-KB, and HG collected the data and samples. CH and FM performed laboratory analysis. BW, TS, RM, AL-B, KT, and HG analyzed the data. BW, KT AL-B, and HG wrote the article. All authors have read and approved the final draft of the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Christel Simon, Aline Poppe, Angela Schellin, Andrea Schmidt, Vanessa Catharina Johanns, Monika Feldhan, and ZBS-4 photo lab of the Robert Koch Institute for their individual contribution to this project.
Footnotes
Funding. This work was supported by the Federal Ministry of Education and Research [MedVet-Staph, Grant no. 01KI1301F and InfectControl 2020 (IRMRESS) no. 03ZZ0805A] and by Federal Ministry of Food and Agriculture, VetMAB; Grant no. 2817600112. TS was supported by grant 01KI1703B (JPIAMR project HECTOR) from the German Federal Ministry of Education and Research (BMBF).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02516/full#supplementary-material
Whole genome sequences: accession numbers. Single nucleotide polymorphism (SNP) ranges for 18 MRSA-ST398-t011-SCCmecIV of equine origin. As a reference (Ref.), WGS data (CP020019) from an MRSA-ST398 (08S00974) isolated from a pig was included.
Single nucleotide polymorphism-distance matrix.
References
- Acton D. S., Plat-Sinnige M. J. T., van Wamel W., de Groot N., van Belkum A. (2009). Intestinal carriage of Staphylococcus aureus: how does its frequency compare with that of nasal carriage and what is its clinical impact? Eur. J. Clin. Microbiol. Infect Dis. 28, 115–127. 10.1007/s10096-008-0602-7 [DOI] [PubMed] [Google Scholar]
- Bartels M. D., Petersen A., Worning P., Nielsen J. B., Larner-Svensson H., Johansen H. K., et al. (2014). Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 52, 4305–4308. 10.1128/JCM.01979-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie R., Atkins N. E., Munch-Petersen E. (1944). A note on a lytic phenomenon shown by group B streptococci. Aust. J. Exp. Biol. 22, 197–200. [DOI] [PubMed] [Google Scholar]
- Claassen-Weitz S., Shittu A. O., Ngwarai M. R., Thabane L., Nicol M. P., Kaba M. (2016). Fecal carriage of Staphylococcus aureus in the hospital and community setting: a systematic review. Front. Microbiol. 7:449. 10.3389/fmicb.2016.00449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinical and Laboratory Standards Institute (2011). Performance Standards for Antimicrobial Susceptibility Testing: Twenty-first Informational Supplement M100-S21. Wayne, PA. [Google Scholar]
- Clinical and Laboratory Standards Institute (2013). Vet01-S2 Performance Standards for Antimicrobial Disk and Dilution Susceptibility for Bacteria isolated from Animals. Second International Supplement, Wayne, PA. [Google Scholar]
- Cuny C., Abdelbary M., Layer F., Werner G., Witte W. (2015). Prevalence of the immune evasion gene cluster in Staphylococcus aureus CC398. Vet. Microbiol. 177, 219–223. 10.1016/j.vetmic.2015.02.031 [DOI] [PubMed] [Google Scholar]
- Cuny C., Kuemmerle J., Stanek C., Willey B., Strommenger B., Witte W. (2006). Emergence of MRSA infections in horses in a veterinary hospital: strain characterisation and comparison with MRSA from humans. Euro Surveill. 11, 44–47. 10.2807/esm.11.01.00595-en [DOI] [PubMed] [Google Scholar]
- Cuny C., Strommenger B., Witte W., Stanek C. (2008). Clusters of infections in horses with MRSA ST1, ST254, and ST398 in a veterinary hospital. Microb. Drug Resist. 14, 307–310. 10.1089/mdr.2008.0845 [DOI] [PubMed] [Google Scholar]
- de Jong N. W. M., Vrieling M., Garcia B. L., Koop G., Brettmann M., Aerts P. C., et al. (2018). Identification of a staphylococcal complement inhibitor with broad host specificity in equid S. aureus strains. J. Biol. Chem. 293, 4468–4477. 10.1074/jbc.RA117.000599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diene S. M., Corvaglia A. R., Francois P., van der Mee-Marquet N., Regional Infection Control Group of the Centre Region . (2017). Prophages and adaptation of Staphylococcus aureus ST398 to the human clinic. BMC Genomics 18:133. 10.1186/s12864-017-3516-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt R. G., Didelot X., Batty E. M., Miller R. R., Knox K., Young B. C., et al. (2014). Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus. Nat. Commun. 5:3956. 10.1038/ncomms4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feßler A., Scott C., Kadlec K., Ehricht R., Monecke S., Schwarz S. (2010). Characterization of methicillin-resistant Staphylococcus aureus ST398 from cases of bovine mastitis. J. Antimicrob. Chemother. 65, 619–625. 10.1093/jac/dkq021 [DOI] [PubMed] [Google Scholar]
- Fessler A. T., Zhao Q., Schoenfelder S., Kadlec K., Brenner Michael G., Wang Y., et al. (2017). Complete sequence of a plasmid from a bovine methicillin-resistant Staphylococcus aureus harbouring a novel ica-like gene cluster in addition to antimicrobial and heavy metal resistance genes. Vet. Microbiol. 200, 95–100. 10.1016/j.vetmic.2016.07.010 [DOI] [PubMed] [Google Scholar]
- Foster T. J. (2005). Immune evasion by staphylococci. Nat. Rev. Microbiol. 3, 948–958. 10.1038/nrmicro1289 [DOI] [PubMed] [Google Scholar]
- Harrison E. M., Weinert L. A., Holden M. T., Welch J. J., Wilson K., Morgan F. J., et al. (2014). A shared population of epidemic methicillin-resistant Staphylococcus aureus 15 circulates in humans and companion animals. MBio 5:e00985–e00913. 10.1128/mBio.00985-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. Z., Espinosa-Gongora C., Damborg P., Sieber R. N., Munk R., Husted L., et al. (2017). Horses in Denmark are a reservoir of diverse clones of methicillin-resistant and -susceptible Staphylococcus aureus. Front. Microbiol. 8:543. 10.3389/fmicb.2017.00543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongerius I., Kohl J., Pandey M. K., Ruyken M., van Kessel K. P., van Strijp J. A., et al. (2007). Staphylococcal complement evasion by various convertase-blocking molecules. J. Exp. Med. 204, 2461–2471. 10.1084/jem.20070818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung P., Abdelbary M. M., Kraushaar B., Fetsch A., Geisel J., Herrmann M., et al. (2017). Impact of bacteriophage Saint3 carriage on the immune evasion capacity and hemolytic potential of Staphylococcus aureus CC398. Vet. Microbiol. 200, 46–51. 10.1016/j.vetmic.2016.02.015 [DOI] [PubMed] [Google Scholar]
- Kadlec K., Schwarz S. (2010). Identification of the novel dfrK-carrying transposon Tn559 in a porcine methicillin-susceptible Staphylococcus aureus ST398 strain. Antimicrob Agents Chemother. 54, 3475–3477. 10.1128/AAC.00464-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y., Baba T., Sekine M., Fukuda M., Hiramatsu K. (2013). Beta-hemolysin promotes skin colonization by Staphylococcus aureus. J. Bacteriol. 195, 1194–1203. 10.1128/JB.01786-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnevey P. M., Shore A. C., Brennan G. I., Sullivan D. J., Ehricht R., Monecke S., et al. (2014). Extensive genetic diversity identified among sporadic methicillin-resistant Staphylococcus aureus isolates recovered in Irish hospitals between 2000 and 2012. Antimic. Agents Chemoth. 58, 1907–1917. 10.1128/AAC.02653-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köck R., Harlizius J., Bressan N., Laerberg R., Wieler L. H., Witte W., et al. (2009). Prevalence and molecular characteristics of methicillin-resistant Staphylococcus aureus (MRSA) among pigs on German farms and import of livestock-related MRSA into hospitals. Eur. J. Clin. Microbiol. Infect. Dis. 28, 1375–1382. 10.1007/s10096-009-0795-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köck R., Schaumburg F., Mellmann A., Köksal M, Jurke A., Becker K., Friedrich A.W. (2013).Livestock-associated methicillin-resistant Staphylococcus aureus (MRSA) as causes of human infection and colonization in Germany. PLoS ONE 8:e0055040. 10.1371/journal.pone.0055040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koop G., Vrieling M., Storisteanu D. M., Lok L. S., Monie T., van Wigcheren G., et al. (2017). Identification of LukPQ, a novel, equid-adapted leukocidin of Staphylococcus aureus. Sci. Rep. 7:40660. 10.1038/srep40660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar B., Hammerl J. A., Kienol M., Heinig M. L., Sperling N., Dinh Thanh M., et al. (2017). Acquisition of virulence factors in livestock-associated MRSA: Lysogenic conversion of CC398 strains by virulence gene-containing phages. Sci. Rep. 7:2004. 10.1038/s41598-017-02175-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J., Petersen A., Sorum M., Stegger M., van Alphen L., Valentiner-Branth P., et al. (2015). Meticillin-resistant Staphylococcus aureus CC398 is an increasing cause of disease in people with no livestock contact in Denmark, 1999 to 2011. Euro. Surv. 20, 5–13. 10.2807/1560-7917.ES.2015.20.37.30021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. V., Cosentino S., Rasmussen S., Friis C., Hasman H., Marvig R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. 10.1128/JCM.06094-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova O., Johnston P., Walther B., Rolff J., Roesler U. (2017). Complete genome sequence of the livestock-associated methicillin-resistant strain Staphylococcus aureus subsp. aureus 08S00974 (Sequence Type 398). Genome Announc 5 e00294–17. 10.1128/genomeA.00294-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malachowa N., DeLeo F. R. (2010). Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 67, 3057–3071. 10.1007/s00018-010-0389-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy A. J., Lindsay J. A. (2013). Staphylococcus aureus innate immune evasion is lineage-specific: a bioinfomatics study. Infect. Genet. Evol. 19, 7–14. 10.1016/j.meegid.2013.06.012 [DOI] [PubMed] [Google Scholar]
- Ricklin D., Tzekou A., Garcia B. L., Hammel M., McWhorter W. J., Sfyroera G., et al. (2009). A molecular insight into complement evasion by the staphylococcal complement inhibitor protein family. J. Immunol. 183, 2565–2574. 10.4049/jimmunol.0901443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Sharma M., Nunez-Garcia J., Kearns A. M., Doumith M., Butaye P. R., Argudin M. A., et al. (2016). Livestock-associated methicillin resistant Staphylococcus aureus (LA-MRSA) Clonal Complex (CC) 398 Isolated from UK Animals belong to European Lineages. Front. Microbiol. 7:1741. 10.3389/fmicb.2016.01741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soimala T., Lübke-Becker A., Schwarz S., Feßler A. T., Huber C., Semmler T., et al. (2018). Occurrence and molecular composition of methicillin-resistant Staphylococcus aureus isolated from ocular surfaces of horses presented with ophthalmologic disease. Vet. Microbiol. 222, 1–6. 10.1016/j.vetmic.2018.06.009 [DOI] [PubMed] [Google Scholar]
- Spellerberg B., Brandt C. (2015). Streptococcus, in Manual of Clinical Microbiology, 11th Edn, eds Pfaller M., Jorgensen J., Carroll K., Funke G., Landry M., Richter S., Warnock D. (Washington, DC:ASM Press; ), 391–392. [Google Scholar]
- Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares A., Nielsen J. B., Boye K., Rohde S., Paulo A. C., Westh H., et al. (2014). Insights into Alpha-Hemolysin (Hla) Evolution and expression among Staphylococcus aureus clones with hospital and community origin. PLoS ONE 9:e0098634. 10.1371/journal.pone.0098634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomer L., Schneewind O., Missiakas D. (2013). Multiple ligands of von Willebrand factor-binding protein (vWbp) promote Staphylococcus aureus clot formation in human plasma. J. Biol. Chem. 288, 28283–28292. 10.1074/jbc.M113.493122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokateloff N., Manning S. T., Weese J. S., Campbell J., Rothenburger J., Stephen C., et al. (2009). Prevalence of methicillin-resistant Staphylococcus aureus colonization in horses in Saskatchewan, Alberta, and British Columbia. Can. Vet. J. 50, 1177–1180. [PMC free article] [PubMed] [Google Scholar]
- Traber K. E., Lee E., Benson S., Corrigan R., Cantera M., Shopsin B., et al. (2008). agr function in clinical Staphylococcus aureus isolates. Microbiology 154(Pt 8), 2265–2274. 10.1099/mic.0.2007/011874-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Alen S., Ballhausen B., Kaspar U., Koeck R., Becker K. (2018). Prevalence and genomic structure of bacteriophage phi3 in human derived livestock-associated MRSA from 2000 to 2015. J. Clin. Microbiol. 56, e00140–e00148. 10.1128/JCM.00140-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Alen S., Ballhausen B., Peters G., Friedrich A. W., Mellmann A., Koeck R., et al. (2017). In the centre of an epidemic: fifteen years of LA-MRSA CC398 at the University Hospital Munster. Vet. Microbiol. 200, 19–24. 10.1016/j.vetmic.2016.01.021 [DOI] [PubMed] [Google Scholar]
- van Duijkeren E., Ten Horn L., Wagenaar J. A., de Bruijn M., Laarhoven L., Verstappen K., et al. (2011). Suspected horse-to-human transmission of MRSA ST398. Emerg. Infect. Dis. 17, 1137–1139. 10.3201/eid1706.101330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wamel W. J., Rooijakkers S. H., Ruyken M., van Kessel K. P., van Strijp J. A. (2006). The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315. 10.1128/JB.188.4.1310-1315.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana D., Blanco J., Tormo-Mas M. A., Selva L., Guinane C. M., Baselga R., et al. (2010). Adaptation of Staphylococcus aureus to ruminant and equine hosts involves SaPI-carried variants of von Willebrand factor-binding protein. Mol. Microbiol. 77, 1583–1594. 10.1111/j.1365-2958.2010.07312.x [DOI] [PubMed] [Google Scholar]
- Vincze S., Stamm I., Kopp P. A., Hermes J., Adlhoch C., Semmler T., et al. (2014). Alarming proportions of methicillin-resistant Staphylococcus aureus (MRSA) in wound samples from companion animals, Germany 2010-2012. PLoS ONE 9:e85656. 10.1371/journal.pone.0085656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mentzer A., Connor T. R., Wieler L. H., Semmler T., Iguchi A., Thomson N. R., et al. (2014). Identification of enterotoxigenic Escherichia coli (ETEC) clades with long-term global distribution. Nat. Genet. 46, 1321–1326. 10.1038/ng.3145 [DOI] [PubMed] [Google Scholar]
- Walther B., Klein K. S., Barton A. K., Semmler T., Huber C., Wolf S. A., et al. (2018). Extended-spectrum beta-lactamase (ESBL)-producing Escherichia coli and Acinetobacter baumannii among horses entering a veterinary teaching hospital: the contemporary “Trojan Horse”. PLoS ONE 13:e0191873. 10.1371/journal.pone.0191873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther B., Monecke S., Ruscher C., Friedrich A. W., Ehricht R., Slickers P., et al. (2009). Comparative molecular analysis substantiates a zoonotic potential of equine Methicillin- resistant Staphylococcus aureus (MRSA). J. Clin. Microbiol. 47, 704–710. 10.1128/JCM.01626-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther B., Tedin K., Luebke-Becker A. (2017). Multidrug-resistant opportunistic pathogens challenging veterinary infection control. Vet. Microbiol. 200, 71–78. 10.1016/j.vetmic.2016.05.017 [DOI] [PubMed] [Google Scholar]
- Weese J. S., van Duijkeren E. (2010). Methicillin-resistant Staphylococcus aureus and Staphylococcus pseudintermedius in veterinary medicine. Vet. Microbiol. 140, 418–429. 10.1016/j.vetmic.2009.01.039 [DOI] [PubMed] [Google Scholar]
- Wendlandt S., Fessler A. T., Monecke S., Ehricht R., Schwarz S., Kadlec K. (2013). The diversity of antimicrobial resistance genes among staphylococci of animal origin. Int. J. Med. Microbiol. 303, 338–349. 10.1016/j.ijmm.2013.02.006 [DOI] [PubMed] [Google Scholar]
- Zankari E., Hasman H., Cosentino S., Vestergaard M., Rasmussen S., Lund O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. 10.1093/jac/dks261 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Whole genome sequences: accession numbers. Single nucleotide polymorphism (SNP) ranges for 18 MRSA-ST398-t011-SCCmecIV of equine origin. As a reference (Ref.), WGS data (CP020019) from an MRSA-ST398 (08S00974) isolated from a pig was included.
Single nucleotide polymorphism-distance matrix.