

Abstract
The metabolic intermediate of acetaminophen (APAP) can cause severe hepatocyte necrosis, which triggers aberrant immune activation of liver non-parenchymal cells (NPC). Overzealous hepatic inflammation determines the morbidity and mortality of APAP-induced liver injury (AILI). Interleukin-1 receptor (IL-1R) signaling has been shown to play a critical role in various inflammatory conditions, but its precise role and underlying mechanism in AILI remain debatable. Herein, we show that NLRP3 inflammasome activation of IL-1β is dispensable to AILI, whereas IL-1α, the other ligand of IL-1R1, accounts for hepatic injury by a lethal dose of APAP. Furthermore, Kupffer cells function as a major source of activated IL-1α in the liver, which is activated by damaged hepatocytes through TLR4/MyD88 signaling. Finally, IL-1α is able to chemoattract and activate CD11b+Gr-1+ myeloid cells, mostly neutrophils and inflammatory monocytes, to amplify deteriorated inflammation in the lesion. Therefore, this work identifies that MyD88-dependent activation of IL-1α in Kupffer cells plays a central role in the immunopathogenesis of AILI and implicates that IL-1α is a promising therapeutic target for AILI treatment.
Keywords: acetaminophen, IL-α, Kupffer cells, sterile immunity, TLR4
Introduction
Interleukin-1 (IL-1) plays a major role in the pathogenesis of various autoimmune, infectious and degenerative diseases.1 IL-1 is produced by two distinct genes, IL-1α and IL-1β, both of which are first produced as 31-kDa peptides and then converted into17-kDa mature forms upon cleavage by specific proteases.2 IL-1α and IL-1β also differ in their cell source and release mechanism.1 The IL-1α precursor is constitutively expressed in the epithelial layers of the gastrointestinal tract, lung, liver, kidney and astrocytes, either as chromatin-associated or membrane-bound.3 The IL-1α precursor is fully active and functions as an alarmin to initiate the early phases of sterile inflammation via direct release from necrotic cells.4 The IL-1α precursor can be cleaved by proteases, such as granzyme B, elastase and calpain-1, leading to a marked enhancement of its bioactivity.5 Secretion of the processed IL-1α is largely restricted to stimulated myeloid cells.6 By contrast, the IL-1β precursor is produced by myeloid cells in response to Toll-like receptors (TLRs), inflammatory cytokines and activated complement components1 and is released into extracellular spaces after the inactive IL-1β precursor is cleaved by caspase-1. Therefore, synthesis and release of IL-1β rely on caspase-1-activating inflammasomes,7 notably the NLR family pyrin domain containing 3 (NLRP3). There is also the involvement of Caspase-1 independent mechanisms for IL-1β mediated inflammation, such as proteinase 3, elastase cleavage of the IL-1β precursor8 and non-canonical inflammasome component caspase-11.9 Intriguingly, although caspase-1 is not involved in IL-1α processing, in certain instances, activation of the inflammasome can promote extracellular release of IL-1α, possibly as a result of inflammasome-induced cell death.6 Both IL-1α and IL-1β engage the same receptor (IL-1R1), with similar biochemical properties.2 However, they have distinct impacts on immune regulation and inflammation, as exhibited in tumorigenesis and metastasis,9 fatty acid-induced vascular response and atherosclerosis,10 and mediation of carbohydrate metabolite-driven inflammation and aerobic glycolysis.11
APAP-induced liver injury (AILI) is caused by an overdose of the over-the-counter analgesic drug acetaminophen (APAP). AILI is characterized by centrilobular hepatic necrosis.12 The initial event of AILI is mediated by the toxic xenobiotic metabolite of APAP, N-acetyl-p-benzoquinone imine (NAPQI), which can be neutralized by administration of N-acetylcysteine.13 Direct drug-induced hepatic damage occurs within the first few hours and partially accounts for the overall injury of AILI.14 Necrotic hepatocytes can release damage-associated molecular patterns (DAMPs) that trigger an irreversible secondary injury mediated by the abnormal activation of the innate immune system.14 DAMPs trigger both local and systemic sterile inflammation,12 of which the IL-1R1/MyD88 signaling pathway is crucially involved.4 Much debate has occurred regarding the requirement and role of the NLRP3 inflammasome and IL-1β in AILI.15,16,17 Moreover, whether18 or not19 the ATP-driven P2X7 purinergic receptor is involved in the assembly of the NLRP3 inflammasome in AILI remains elusive. More recently, several studies have highlighted the critical role of IL-1α, but not IL-1β, in AILI.4,20 It is therefore necessary to determine both the cell source and target of IL-1 within the liver microenvironment to better understand the sterile inflammatory response in AILI.
In this work, we demonstrated that IL-1α, rather than IL-1β, is critically involved in the immunopathogenesis of AILI. Activation of IL-1α depends on Kupffer cells that sense and transduce DAMP signaling through the TLR4/MyD88 pathway. Moreover, CD11b+ Gr1+ neutrophils and monocytes are targeted by IL-1α to amplify the inflammatory signals for AILI.
Materials and methods
Animals and treatment
C57BL/6N mice were purchased from Vital River Laboratory Animal Tech Co. (Beijing, China). TLR3−/−, TLR4−/−, TLR7/9−/−, MyD88−/−, Trif−/− and MyD88−/−/Trif−/− mice in a C57BL/6N background were purchased from Oriental BioService (kyoto, Japan), while IL-1R1−/− mice (C57BL/6N background) and Caspase-1−/− mice (NOD.129 background) were purchased from The Jackson Laboratory (MI, USA). Caspase-1−/− mice were backcrossed to C57BL/6N for >10 generations before use. NLRP3−/− mice in a C57BL/6N background21 were kindly provided by Dr. Warren Strober (National Institutes of Health, USA). IL-1α−/−and IL-1β−/− mice were from a C57BL/6N background and had been described before.22 All mice used in this study had a comparable pattern of cytochrome P450, family 2, subfamily e, polypeptide 1 gene (Cyp2e1) expression, GSH exhaustion and APAP adduct formation after APAP injection (Supplementary Figure S1). Male age-matched (8–12 week old) mice were used throughout the study. All mice were maintained under specific pathogen-free conditions at the Institute of Biophysics or the Institut Pasteur of Shanghai, CAS. An APAP (Sigma-Aldrich, St Louis, CA, USA) solution (20 mg/ml in PBS) was made fresh for each experiment and was i.p. injected at different doses after mice were starved for 15 h. The animals were killed by ketamine/xylazine injection at the indicated time for collection of sera and liver tissues. Animal experiments were performed in accordance with institutional guidelines approved by the Animal Care and Use Committee of each CAS institute.
Antibodies (200 μg, BioLegend, San Diego, CA, USA) against IL-1R1 (clone JAMA-147), IL-1α (clone ALF-161) and IL-1β (clone B122) were i.v. injected 1 h before APAP treatment. The in vivo blocking activities were reported by others.23,24,25 For myeloid cell depletion, antibodies against Gr-1 (clone RB6-8C5) and Ly6G (clone 1A8) were i.p. injected (200 μg) 24 h before APAP injection. The depletion efficiencies were verified by FACS analysis. To offset any off-target effects by a given antibody, we routinely used isotype-matched immunoglobulin as a control.
Conditioned medium
Primary hepatocytes were isolated from wt mice by a two-step collagenase perfusion protocol. In brief, the hepatic portal vein was ligated and perfused first with HBSS containing 5 mM EDTA without Ca2+ and Mg2+ (Beyotime Co., China), then with HBSS containing 0.025% type IV collagenase (Sigma-Aldrich). After perfusion, the liver was excised and hepatocytes were suspended in serum-free DMEM (Gibco, MD, USA) and passed through a 100-μm strainer. The filtrate was centrifuged (50g for 3 min at 4 °C), and the pelleted hepatocytes were resuspended in WEM containing 10% FBS (Gibco, MD, USA). The cells were plated (2 × 105 cells/ml) and stimulated with 10 mM APAP or DMSO for 16 h. The supernatants were collected, as conditioned media.
FACS analysis of hepatic nonparenchymal cells (NPCs)
Hepatic NPCs were isolated at the indicated time, as previously described.26 All stainings were performed in PBS/2% FBS in the presence of purified anti-CD16/32 (BioLegend) at saturation to block nonspecific staining via FcRII/III. Multi-color flow cytometry was performed on a LSRFortessa (BD Biosciences, NJ, USA) after NPCs (1 × 106) were incubated with the indicated mAb (10 μg/ml). For intracellular staining, isolated NPCs were cultured in RIPI medium (Gibco, MD, USA) with Brefeldin A (1 μl per 106 cells; BioLegend) for 6 h before staining. The FACS antibodies (BioLegend) used were: AF700-NK1.1 (PK136), BV421-CD45R/B220 (RA3-6B2), F4/80-BV605 (BM8), PE/Cy7-CD8α (53-6.7), APC-CD3ɛ (145-2C11), PerCP/Cy5.5-Gr-1(RB6-8C5), FITC-CD146 (ME-9F1), PE-IL1α (ALF-161), APC-CD121 (JAMA-147), APC-Cy7-CD11b(OX-42), FITC-Ly6G (1A8) and Ly6C-APC (HK1.4). 7-AAD (BioLegend) was used to exclude dead cells. Isotype controls were used where needed. The data were analyzed with FlowJo software (TreeStar, OR, USA).
Serology and serum chemistry
Mice were bled retro-orbitally at the indicated time post APAP challenge. Serum (10 μl) cytokines and chemokines were measured with Milliplex MAP kits (Millipore, MA, USA) in the FLEXMAP 3D system (Luminex, Austin, TX, USA). The data were analyzed by xPONENT software (Luminex). Serum (20 μl) ALT and AST were measured in an automated Blood Chemistry Analyzer (ZS-200B, BioSino Biotech, China).
Macrophage depletion, purification and adoptive transfer
For macrophage depletion, mice were i.p. injected with 200 μl of clodronate liposomes, as previously described.27 To elicit peritoneal macrophages, mice of different genetic backgrounds were i.p. injected with 2 ml of a 4% thioglycollate broth (Sigma-Aldrich) for 3 days. To adoptively transfer macrophages, the indicated donor peritoneal macrophages (5 × 106 cells/ml) were i.v. transferred to recipient mice 48 h after liposomal depletion of Kupffer cells. Both the depletion and reconstitution efficiencies were monitored by immunofluorescent staining of liver sections with rat anti-mouse F4/80 mAb (Serotec, Oxford, UK). To improve the survival rates of the recipient mice after adoptive transfer of different cells at 24 hpi, a slightly lower dose of APAP (550 mg/kg) was administered to mice 24 h after macrophage reconstitution.
APAP adducts detection
APAPcysteine (APAP-Cys) adducts were measured by high-pressure liquid chromatography with electrochemical detection according to the method of Miteshkumar et al. 28 Briefly, a 100-mg portion of frozen liver was mixed with 0.2 ml of PBS (pH 7.4) and homogenized. The supernatant was placed on a Nanosep centrifugal device with a membrane weight cutoff of 30 kDa. The protein retained on the membrane was washed and then mixed with a protease E solution (8 units/ml, Pronase E type XIV, Sigma-Aldrich), followed by incubation at 50° for 16 h on a heating block. The digest was transferred to a conditioned Nanosep centrifugal device and centrifuged at 12 000g for 10 min to collect the filtrate. The membrane on the filtration device was rinsed twice with distilled water, and the washings and filtrate were pooled. This solution was injected into the liquid chromatograph. The APAP-Cys standard (C994750, TRC) was dissolved in 1 ml of DMSO at 5 mg/ml.
Real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) from liver tissue, PC or NPC. Quantitative RT-PCR (7500, ABI) was carried out using gapdh as the internal control. The primers used were: Cyp2e1-F: , Cyp2e1-R: , IL-1R1-F: , IL-1R1-R: , IL-1α-F: , IL-1α-R: , IL-1β-F: , IL-1β-R: , GAPDH-F: , GAPDH-R: .
Histochemistry
H&E stains of formalin-fixed paraffin-embedded liver tissues (right lobules, slice thickness 5 μm) were performed according to the manufacturer’s instructions (Leica, Germany). The statistical results of the necrosis area were averaged from 20 random view fields of each section (× 200 magnification) using ImageJ software. IHC was carried out with a 1:200 dilution of the primary antibody to F4/80 (Serotec) using a tyramide signal amplification kit (Invitrogen). The images were acquired by a Leica SCN400 digital slide scanner and analyzed with QWin V3 imaging software (Leica, Germany).
Statistics
All in vivo experiments were repeated at least times for confirmation, and the representative results of one experiment are shown. Kaplan–Meier plots and statistical analysis were performed using GraphPad Prism software version 5.01. An unpaired two-tailed Student’s t-test was used to compare groups. A P value of <0.05 was considered significant.
Results
Aberrant innate cell infiltration and activation in response to AILI
Previous studies have shown that mice of different genetic backgrounds respond to APAP rather differently; therefore, we managed to unify all mice used in this study under the C57BL/6N type. To precisely determine the role of IL-1 signaling in AILI, we carefully calibrated lethal doses (LD) of APAP by i.p. administration of various doses of APAP (400–800 mg/kg) in mice. The LD50 at 48 h post injection (hpi) was determined at 600 mg/kg and was used throughout the study, except where specified (Supplementary Figure S2a). Under the regimen of LD50=600 mg/kg, the livers showed typical centrilobular hepatic necrosis and increased ALT and AST in serum (Supplementary Figures S2b and c). The cell counting assay of liver NPC in APAP-treated mice showed that the majority of infiltrated leukocytes were neutrophils (CD11bhighF4/80−Gr-1high) and monocytes (CD11bhighF4/80intGr-1int), in agreement with previous work.29 Neutrophils started infiltrating as early as 6 hpi and peaked at 12 hpi, followed by an influx of monocytes. Liver resident macrophages/Kupffer cells (CD11bintF4/80high), NK/NKT (CD3−NK1.1+/ CD3+ NK1.1+), DC (CD11c+) or T (CD3−NK1.1−) cells were unaltered after APAP administration (Figure 1a; see Supplementary Figure S2d for gating strategies). Furthermore, the measurement of pro-inflammatory cytokines and chemokines (PICC) before and after APAP treatment showed that serum IL-1α, IL-6, KC/CXCL1, MCP-1, G-CSF and MIG were preferentially elevated, in contrast with the undetectable IL-1β, IFN-γ or other PICCs throughout the course of the study (Figure 1b). Up-regulation of KC, MCP-1 and MIG agreed with the hepatic infiltration and activation of neutrophils and monocytes,30 and the increase of IL-6 was indicative of macrophages/Kupffer cells mediating the response to APAP.31
Figure 1.
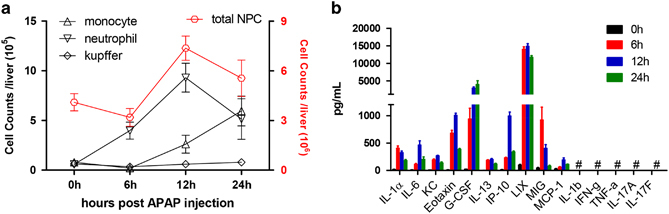
Aberrant innate cell infiltration and activation in response to AILI. Wt mice (n=7 for each time point) were injected with 600 mg/kg APAP, i.p. (a) Intrahepatic leukocytes were counted by FACS at the indicated times. (b) Serum PICCs of mice were measured at the indicated times. # denotes under the detection limit.
IL-1α but not IL-1β exacerbated the immunopathology in AILI
The aforementioned response of IL-1α, but not IL-1β, to APAP stimulation agreed with previous studies,15,16 but contradicted another study.17 Therefore, it was necessary to clarify whether IL-1α or IL-1β participated in AILI and how. Quantitative RT-PCR (qPCR) analysis of liver cells showed that APAP stimulation could weakly activate transcription of IL-1β, primarily in PC cells (Figure 2a). Liver PC cells are known to be ineffective in inflammasome processing and activation.32 Indeed, caspase-1 was hardly activated after i.p. injection of APAP in wt mice (Figure 2b). Ineffective IL-1β activation also suggested that inflammasomes might not be involved in the modulation of AILI. Consistent with this hypothesis, NLRP3−/− and Caspase-1−/− mice showed a similar morbidity and mortality to AILI as the wt littermate controls (Figure 2c). Therefore, inflammasomes, including the NLRP3 inflammasome, would be dispensable for AILI, as suggested by other studies.15,16,19 More importantly, the same panel of experiments revealed a relatively more effective induction of IL-1α transcription in NPC cells (Figure 2a). Therefore, IL-1α and IL-1β responded to APAP differentially, with IL-1α swiftly induced and secreted into circulation. To confirm that IL-1α rather than IL-1β was accountable for AILI, mice were pre-treated with blocking antibodies to IL-1α, IL-1β or an isotype control prior to APAP injection. The results showed that blockade of IL-1α, but not IL-1β, effectively rescued mice from AILI lethality (Figure 2d). Blockade of IL-1α correlated with significantly reduced hepatic necrosis (Figure 2e); serum ALT and AST (Figure 2f); serum levels of inflammatory cytokines, such as IL-6 and KC (Figure 2g); and hepatic infiltration of neutrophils and monocytes (Figure 2h). Taken together, these findings indicated that IL-1α signaling played a critical role in AILI. In contrast, the NLRP3/IL-1β inflammasome and its activation might exhibit only a trivial impact on AILI.
Figure 2.
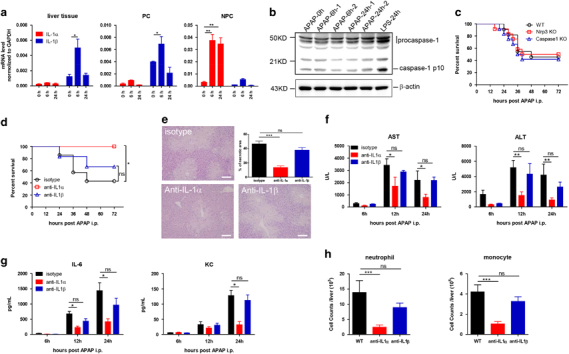
IL-1α, but not IL-1β, exacerbated the immunopathology by APAP, which accounted for the mortality and morbidity of AILI. (a) IL-1α and IL-1β expression before and after APAP injection (600 mg/kg, i.p) in wt mice in liver tissue; PC and NPC were analyzed by qPCR, as indicated, n=7, for each time point. (b) Hepatic caspase-1 expression before and after APAP injection (600 mg/kg, i.p.) in wt mice was detected by immunoblot. Liver samples from LPS-treated mice (250 mg/kg, i.p.) were used as a positive control. (c) Wt, NLRP3−/− or Caspase1−/− mice (n⩾10) were i.p. injected with APAP. Measurement of the survival rates and analysis by the Kaplan-Meier method. (d–h) Wt mice were pre-treated with anti-IL-1α, anti-IL-1β or isotype control antibodies (200 μg/mouse, i.v.) 1 h before APAP injection (n=8 for each group, 600 mg/kg, i.p.). (d) Measurement of the survival rates. (e) Representative hepatic necrosis 24 hpi viewed by H&E staining and statistical analysis. Bar=50 μm. Measurement of the (f) serum ALT and AST levels and (g) serum levels of KC and IL-6 at the indicated time post APAP treatment. (h) Absolute numbers of infiltrated neutrophils and monocytes 24 hpi were analyzed by FACS staining. The data are presented as the mean±s.e.m. Student’s t-test. ***P<0.001; **P<0.01; *P<0.05; ns, no significant difference.
IL-1α secreted by Kupffer cells attributed to AILI
To explore the intrahepatic cell source of IL-1α in response to AILI, hepatic NPCs were isolated after APAP stimulation and assayed for IL-1α by intracellular staining. Flow cytometric analysis (Figure 3a) strikingly showed that over 90% of the IL-1α-staining cells were dominantly Kupffer cells after 6 hpi (CD11bintF4/80high) and not other leukocytes (including Gr-1+, NK, NKT, DC, T or B cells) or liver sinusoidal endothelial cells (LSEC, CD45− CD146+). The proportion of IL-1α-positive cells increased by almost three-fold after APAP stimulation for 6 h (Figure 3a). Kupffer cells have been reported to be pro-inflammatory mediators to initiate and/or amplify hepatic injury in the early phase of AILI.33 To test whether Kupffer cells were a major IL-1α producer, we used clodronate liposomes to deplete Kupffer cells in the liver before APAP injection (Figure 3b). The removal of Kupffer cells significantly improved survival rates early in APAP injection (Figure 3c) and liver injury (Figure 3d), which was concomitant with a reduction in the serum levels of IL-1α, IL-6 and KC (Figure 3e). Adoptive transfer of isolated macrophages rendered those Kupffer cell-depleted mice sensitive to AILI (Figures 3c–e). To further test whether the IL-1α of macrophage origin played a role in AILI, mice depleted of Kupffer cells by clodronate liposomes were adoptively transferred with macrophages isolated from wt, IL-1α−/− or IL-1β−/− mice. Mice reconstituted with IL-1α-deficient macrophages were resistant to AILI, with much less liver injury (Figure 3f) and lower serum PICCs (Figure 3g) than mice reconstituted with wt or IL-1β−/− macrophages (Figures 3f and g). Therefore, Kupffer cells, although not the only cell source, might serve as an important producer of IL-1α in mediating the immunopathogenesis of AILI. Of note, Kupffer cells also play a protective role in liver homeostasis,34 as characterized by the regulation of homeostasis and integrity of LSEC,35 portal vein tolerance36 and prolonging of liver allograft survival.37 This was demonstrated at the recovery stage (after 24 hpi) of AILI, when Kupffer-depleted mice showed more severe liver injury and poorer survival rates (Figures 3c and d). This finding of a dual role of Kupffer cells in AILI, which is deteriorative in the initial phase and protective in the solution phase, is also confirmed by other observations.38
Figure 3.
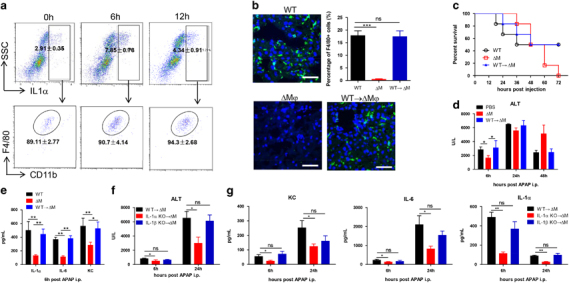
Kupffer cells were the main source of activated IL-1α. (a) NPCs isolated from mice (n=6 for each time point) at 0 or 6 h post APAP injection (600 mg/kg, i.p.) were analyzed by FACS for cell types that produced IL-1α. (b–e) Mice were pre-treated with control empty liposomes (PBS, WT), clodronate liposomes (CL2MBP, ΔM), or clodronate liposomes and then reconstituted with macrophages (WT→ΔM) enriched from wt donor mice (2 × 106 cells in 400 μl of PBS for each mouse, i.v.). Subsequently, APAP (550 mg/kg) was i.p. injected into these mice (n=11 for each group). (b) Macrophage depletion and reconstitution efficiencies. Immunofluorescence staining of frozen liver sections by the F4/80 antibody (green). The nuclei were counterstained with DAPI (blue). Bar=50 μm. Survival rates (c), serum ALT (d) and serum cytokines (e) were analyzed 24 hpi. (f and g) Mice depleted of macrophages by clodronate liposomes (200 μl/mouse, 3 d before APAP injection) were adoptively transferred with macrophages enriched from indicated donor mice (2 × 106 cells in 400 μl PBS for each mouse, i.v.). Two days later, APAP (550 mg/kg) was i.p. injected into these mice (n=8 for each group). Measurement of serum ALT (f) and serum levels of KC, IL-6 and IL-1α (g) at the indicated time post APAP treatment. The data were averaged and are presented as the mean±s.e.m. Student’s t-test. **P<0.01; *P<0.05.
TLR4/MyD88 signaling mediated IL-1α activation in Kupffer cells
TLRs have long been suggested to play a role in the sterile inflammatory response to AILI. For example, TLR2,39,40 TLR3,41 TLR4(refs 33,42,43,44 ) and TLR9(ref.17 ) have been reported to contribute to AILI. In agreement with these previous studies, TLR3−/−, TLR4−/− and TLR7/9−/− mice all showed improved survival rates (Supplementary Figure S3a), as well as decreased hepatic necrosis (Supplementary Figure S3b) and serum ALT and AST (Supplementary Figure S3c) after APAP challenge. Notably, alleviation of TLR4 gave rise to profound protection and significant reduction in serum IL-1α and other PICC levels (Supplementary Figure S3d).
To test whether the TLR4 signaling pathway regulates IL-1α production in macrophages, mice depleted of Kupffer cells by clodronate liposomes were adoptively transferred with macrophages isolated from wt, TLR3−/−, TLR4−/−, TLR7/9−/−, MyD88−/−, Trif−/− or MyD88−/−/Trif−/− mice. Mice reconstituted with macrophages of TLR3−/−, Trif−/− or TLR7/9−/−origin became sensitive to AILI, with liver necrosis (Figure 4a), injury (Figure 4b), serum IL-1α and other PICCs elevated to similar levels as wt mice or mice reconstituted with wt macrophages (Figure 4c). By contrast, supplementation with TLR4−/− or MyD88−/−macrophages failed to sensitize mice to AILI (Figures 4a–c). These results therefore suggested that TLR4/MyD88 activation in Kupffer cells accounted for the immunopathogenesis of AILI. To substantiate this notion, we added conditioned medium prepared from APAP-treated primary hepatocytes (APAP CM; IL-1α and KC undetectable, data not shown) to peritoneal macrophages of different genetic backgrounds. First, IL-1α and KC expression in macrophages was activated by APAP CM and the conditioned medium prepared from heat-killed hepatocytes (Figure 4d). By contrast, APAP by itself or conditioned medium from DMSO-treated hepatocytes (DMSO CM) failed to activate macrophages. Second, detection of IL-1α and KC in supernatants showed that macrophages from wt, TLR3−/−, Trif−/−or TLR7/9−/− mice, but not from TLR4−/−, MyD88−/− or MyD88−/−/Trif−/− mice, could be activated by DAMPs released from necrotic hepatocytes (Figure 4e). Taken together, these results strongly indicated that TLR4/MyD88 signaling in Kupffer cells was involved in IL-1α-mediated AILI. Because MyD88 is the adapter for both IL-1R1 and TLR4 signaling, it is intriguing to speculate that MyD88 in Kupffer cells could be simultaneously engaged by two pro-inflammatory signals, one being the DAMPs released from necrotic hepatocytes, such as HMGB1 on TLR4,45 and the other the potentiation of IL-1α signaling on IL-1R1 via an autocrine mode.46
Figure 4.
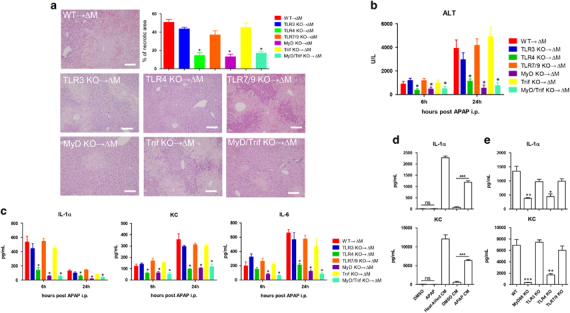
IL-1α activation depended on TLR4/MyD88 signaling in macrophages. Mice depleted of macrophages by clodronate liposomes (200 μl/mouse, 3 d before APAP injection) were adoptively transferred with macrophages enriched from indicated donor mice (2 × 106 cells in 400 μl of PBS for each mouse, i.v.). Two days later, APAP (550 mg/kg) was i.p. injected into these mice (n=8 for each group). (a) Representative hepatic necrosis and statistical analysis were performed 24 h after APAP injection. Bar=50 μm. (b) Measurement of serum ALT in macrophage reconstituted mice at the indicated time post APAP treatment. (c) Serum levels of KC, IL-6 and IL-1α at the indicated time post APAP treatment. (d) Wt macrophages were stimulated with DMSO, APAP, conditioned medium from heat-killed hepatocytes (Heat-killed CM), conditioned medium from DMSO-treated hepatocytes (DMSO CM) or conditioned medium from APAP-treated hepatocytes (APAP CM) for 16 h. Indicated cytokines in the supernatants were measured by LUMINEX assays. (e) Macrophages enriched from wt, MyD88, TLR3−/−, TLR4−/− or TLR7/9−/− mice were stimulated with 100 μl of conditioned medium (described in the Methods section) for 16 h. Indicated cytokines in the supernatants were measured by LUMINEX assays. The data are presented as the mean±s.e.m. Student’s t-test. ***P<0.001; **P<0.01; *P<0.05.
Both neutrophils and monocytes were target cells of IL-1α in AILI
IL-1α serves as an alarmin to activate an array of PICC in the early phase of sterile inflammation. The important role of IL-1R1 or activation of IL-1R1 by IL-1α, but not IL-1β, in potentiating AILI has been shown by two recent studies.4,20 To determine the potential effector cells of IL-1α signaling in AILI, IL-1R1 expression in liver cells was analyzed. IL-1R1 expression was elevated preferably in NPC but not PC cells post APAP injection, as measured by qPCR analysis (Figure 5a). Thus, IL-1R1 deficiency (Supplementary Figures S4a–c) or antibody blocking of IL-1R-expressing cells (i.v, 200 μg/mouse, Supplementary Figures S4d–f) effectively protected mice from AILI compared with control mice. FACS analysis of NPC cells further indicated that IL-1R1 was only up-regulated in LSEC and Gr-1+ cells in response to APAP (Figure 5b, Supplementary Figure S5 for FACS data). To substantiate that Gr-1+ myeloid cells play a more important role in AILI, we used an anti-Gr1 antibody to deplete both neutrophils and monocytes and an anti-Ly6G antibody to deplete only neutrophils (depletion efficacy in Supplementary Figure S6a). As expected, mice lacking neutrophils and/or monocytes showed improved survival (Supplementary Figure S6b) and reduced serum ALT and AST levels (Supplementary Figure S6c), with depletion of both cells being most protective. Although the Gr1 antibody may protect AILI by activation of metallothionein,47,48 protection from neutropenia by the Ly6G antibody would support the deleterious role of neutrophils in AILI.29,49 Intriguingly, removal of neutrophils or monocytes showed little effect on PICC activation in response to APAP (Supplementary Figure S6d), implying that these myeloid cells function downstream of AILI-associated PICCs. How these activated myeloid cells inflict hepatic damage remains to be determined. Taken together, these results indicated that Gr-1+ myeloid cells could be targeted by IL-1α to orchestrate pathological alteration in AILI.
Figure 5.
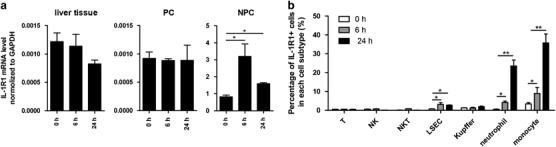
Upregulated IL-1R in non-parenchymal cells attributed to AILI. (a) IL-1R1 expression before and after APAP injection (600 mg/kg, i.p) in wt mice in liver tissue; PC and NPC were analyzed by qPCR, as indicated, n=7, for each time point. (b) IL-1R1 expression before and after APAP injection (600 mg/kg, i.p) in wt mice on NPC was analyzed by FACS, n=7 for each time point. The data are presented as the mean±s.e.m. Student’s t-test. **P<0.01; *P<0.05.
Discussion
AILI is recognized as a ‘two-hit’ process: the first hit is caused by the intracellular toxic signaling of the APAP metabolites, while the second hit is the result of the acute necrotic inflammatory response. The underlying mechanisms of the second hit are still under debate. This work attempts to propose that DAMPs, which are released from necrotic hepatocytes by the first hit, would engage TLR4/MyD88 signaling in Kupffer cells to activate and secrete IL-1α. IL-1α then activates IL-1R+ cells, for example, neutrophils, via a paracrine loop to promote hepatic injury during the early phase of AILI. The central role of TLR signaling in the immunopathogenesis of AILI is supported by the recent findings that several ligands for TLR4, such as HMGB143,50,51,52 and extracellular histones,39 can promote APAP-induced liver inflammation. This work further shows that TLR4 in Kupffer cells might play a critical role in the production and amplification of IL-1α.
Both IL-1α and IL-1β can be activated by TLR signaling and have similar properties through IL-1R1 in the mediation of sterile inflammation.12 However, the dichotomy of IL-1 activities, as shown in this work and others,1 suggests that their precise roles may vary under different physiological and pathological conditions. As an alarmin, IL-1α could be released by necrotic cells directly,53,54 dependent on the intracellular decoy IL-1R2 and caspase-155 or by immune cells as a secondary signaling molecule.4,56 Importantly, this study managed to show that necrotic hepatocytes did not secrete IL-1α or KC, whereas macrophages were the major cell source for IL-1α in vivo. Therefore, it is highly likely that during AILI, IL-1α is predominantly secreted by Kupffer cells in response to DAMPs released from necrotic hepatocytes.
The roles of Gr-1+ myeloid cells in the regulation of AILI are still under debate.57 The controversy surrounding the role of neutrophils in AILI, either as irrelevant58,59 or damage-promoting,43,60 is probably related to the experimental settings. Neutrophils enriched and sustained in the liver after acute injury may also facilitate injury resolution.61 By using antibodies that differentiated neutrophils from monocytes, this work was able to show that neutrophils were the major target cells of IL-1α in AILI. Whether other IL-1R+ cells, such as LSEC, as shown in Figure 5b, might also be involved in this process remains to be determined. Moreover, before this work, monocytes had not been separated from liver-infiltrated macrophages, which has also caused a conflicting interpretation of the role of macrophages in AILI as being either deteriorative62 or protective.30,63,64 In contrast, this work has provided a definitive answer regarding the bipartite role of Kupffer cells in the different phases of AILI progression. Kupffer cells were deteriorative during the acute phase of AILI but protective during the recovery phase of AILI.
Last, N-acetylcysteine is commonly administered to treat APAP overdose-induced acute liver failure, which has considerable limitations, such as a narrow therapeutic window and adverse effects.65 This work suggests that IL-1α antagonism might be a promising treatment to buffer the second hit of AILI, considering that IL-1α antagonists are already in use or in clinical trials for the treatment of arthritis and other acute inflammatory syndromes.66
Electronic supplementary material
Acknowledgements
We thank Dr. Yang Liu (Children’s Research Institute, DC) for his brilliant comments and critical reading of the manuscript, Xudong Zhao and Su Liu (Protein Science Core Facility, Chinese Academy of Sciences) for their technical support on serum chemistry analysis and Min Wang (Beijing Institute of Radiation Medicine) for technical support on the isolation and culture of primary hepatocytes. This work was supported by grants from the National Science Foundation of China (31030031 and 81220108018) and the Ministry of Science and Technology of China (2011CB946104) to HT.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.22
References
- 1.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Ann Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 3.Werman A, Werman-Venkert R, White R, Lee JK, Werman B, Krelin Y, et al. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci USA. 2004;101:2434–2439. doi: 10.1073/pnas.0308705101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 5.Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Luthi AU, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell. 2011;44:265–278. doi: 10.1016/j.molcel.2011.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 7.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 8.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 10.Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M, et al. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat Immunol. 2013;14:1045–1053. doi: 10.1038/ni.2704. [DOI] [PubMed] [Google Scholar]
- 11.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos: Biol Fate Chem. 2003;31:1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 14.Maher JJ. DAMPs ramp up drug toxicity. J Clin Investig. 2009;119:246–249. doi: 10.1172/JCI38178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams CD, Farhood A, Jaeschke H. Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol Appl Pharmacol. 2010;247:169–178. doi: 10.1016/j.taap.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams CD, Antoine DJ, Shaw PJ, Benson C, Farhood A, Williams DP, et al. Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol Appl Pharmacol. 2011;252:289–297. doi: 10.1016/j.taap.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. The J Clin Investig. 2009;119:305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoque R, Sohail MA, Salhanick S, Malik AF, Ghani A, Robson SC, et al. P2X7 receptor-mediated purinergic signaling promotes liver injury in acetaminophen hepatotoxicity in mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1171–G1179. doi: 10.1152/ajpgi.00352.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Y, Williams CD, McGill MR, Lebofsky M, Ramachandran A, Jaeschke H. Purinergic receptor antagonist A438079 protects against acetaminophen-induced liver injury by inhibiting p450 isoenzymes, not by inflammasome activation. Toxicol Sci: Off J Soc Toxicol. 2013;131:325–335. doi: 10.1093/toxsci/kfs283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lukens JR, Vogel P, Johnson GR, Kelliher MA, Iwakura Y, Lamkanfi M, et al. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature. 2013;498:224–227. doi: 10.1038/nature12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013;23:201–212. doi: 10.1038/cr.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, et al. Production of mice deficient in genes for interleukin (IL)-1α, IL-1β, IL-1α/β, and IL1 receptor antagonist shows that IL-1β is crucial in turpentine induced fever development and glucocorticoid secretion. J Exp Med. 1998;187:1463–1475. doi: 10.1084/jem.187.9.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers HW, Sheehan KC, Brunt LM, Dower SK, Unanue ER, Schreiber RD. Interleukin 1 participates in the development of anti-Listeria responses in normal and SCID mice. Proc Natl Acad Sci USA. 1992;89:1011–1015. doi: 10.1073/pnas.89.3.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yazdi AS, Guarda G, Riteau N, Drexler SK, Tardivel A, Couillin I, et al. Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1alpha and IL-1beta. Proc Natl Acad Sci USA. 2010;107:19449–19454. doi: 10.1073/pnas.1008155107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu JY, Sadri N, Schneider RJ. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev. 2006;20:3174–3184. doi: 10.1101/gad.1467606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe H, Ohtsuka K, Kimura M, Ikarashi Y, Ohmori K, Kusumi A, et al. Details of an isolation method for hepatic lymphocytes in mice. J Immunol Methods. 1992;146:145–154. doi: 10.1016/0022-1759(92)90223-G. [DOI] [PubMed] [Google Scholar]
- 27.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 28.Acharya M, Lau-Cam CA. Simple reversed-phase HPLC method with spectrophotometric detection for measuring acetaminophen-protein adducts in rat liver samples. Sci World J. 2012;2012:145651. doi: 10.1100/2012/145651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43:1220–1230. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- 30.Ishida Y, Kondo T, Kimura A, Tsuneyama K, Takayasu T, Mukaida N. Opposite roles of neutrophils and macrophages in the pathogenesis of acetaminophen-induced acute liver injury. Eur J Immunol. 2006;36:1028–1038. doi: 10.1002/eji.200535261. [DOI] [PubMed] [Google Scholar]
- 31.Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, Klaunig JE. Role of the Kupffer cell in mediating hepatic toxicity and carcinogenesis. Toxicol Sci: Off J Soc Toxicol. 2007;96:2–15. doi: 10.1093/toxsci/kfl173. [DOI] [PubMed] [Google Scholar]
- 32.Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642–654. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 33.Fisher JE, McKenzie TJ, Lillegard JB, Yu Y, Juskewitch JE, Nedredal GI, et al. Role of Kupffer cells and toll-like receptor 4 in acetaminophen-induced acute liver failure. J Surg Res. 2013;180:147–155. doi: 10.1016/j.jss.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, et al. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 2002;15:1504–1513. doi: 10.1021/tx0255976. [DOI] [PubMed] [Google Scholar]
- 35.Holt MP, Yin H, Ju C. Exacerbation of acetaminophen-induced disturbances of liver sinusoidal endothelial cells in the absence of Kupffer cells in mice. Toxicol Lett. 2010;194:34–41. doi: 10.1016/j.toxlet.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 36.Callery MP, Kamei T, Flye MW. Kupffer cell blockade inhibits induction of tolerance by the portal venous route. Transplantation. 1989;47:1092–1094. doi: 10.1097/00007890-198906000-00041. [DOI] [PubMed] [Google Scholar]
- 37.Sato K, Yabuki K, Haba T, Maekawa T. Role of Kupffer cells in the induction of tolerance after liver transplantation. J Surg Res. 1996;63:433–438. doi: 10.1006/jsre.1996.0288. [DOI] [PubMed] [Google Scholar]
- 38.Yang R, Zou X, Koskinen ML, Tenhunen J. Ethyl pyruvate reduces liver injury at early phase but impairs regeneration at late phase in acetaminophen overdose. Crit care. 2012;16:R9. doi: 10.1186/cc11149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626–2631. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. 2013;60:782–791. doi: 10.1016/j.jhep.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cavassani KA, Moreira AP, Habiel D, Ito T, Coelho AL, Allen RM, et al. Toll like receptor 3 plays a critical role in the progression and severity of acetaminophen-induced hepatotoxicity. PLoS ONE. 2013;8:e65899. doi: 10.1371/journal.pone.0065899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yohe HC, O'Hara KA, Hunt JA, Kitzmiller TJ, Wood SG, Bement JL, et al. Involvement of Toll-like receptor 4 in acetaminophen hepatotoxicity. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1269–G1279. doi: 10.1152/ajpgi.00239.2005. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Sun R, Wei H, Tian Z. High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: Interaction of gammadelta T cells with macrophages. Hepatology. 2013;57:373–384. doi: 10.1002/hep.25982. [DOI] [PubMed] [Google Scholar]
- 44.Shah N, Montes de Oca M, Jover-Cobos M, Tanamoto K, Muroi M, Sugiyama K, et al. Role of toll-like receptor 4 in mediating multiorgan dysfunction in mice with acetaminophen induced acute liver failure. Liver Transplant: Off Publ Am Assoc Study Liver Dis Int Liver Transplant Soc. 2013;19:751–761. doi: 10.1002/lt.23655. [DOI] [PubMed] [Google Scholar]
- 45.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Arnandis I, Guillen MI, Gomar F, Pelletier JP, Martel-Pelletier J, Alcaraz MJ. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1beta in osteoarthritic synoviocytes. Arthritis Res Ther. 2010;12:R165. doi: 10.1186/ar3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jaeschke H, Liu J. Neutrophil depletion protects against murine acetaminophen hepatotoxicity: another perspective. Hepatology. 2007;45:1588–1589. doi: 10.1002/hep.21549. [DOI] [PubMed] [Google Scholar]
- 48.Saito C, Yan HM, Artigues A, Villar MT, Farhood A, Jaeschke H. Mechanism of protection by metallothionein against acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2010;242:182–190. doi: 10.1016/j.taap.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Therap. 1973;187:185–194. [PubMed] [Google Scholar]
- 50.Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK. Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Mol Med. 2010;16:479–490. doi: 10.2119/molmed.2010.00126. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Martin-Murphy BV, Holt MP, Ju C. The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol Lett. 2010;192:387–394. doi: 10.1016/j.toxlet.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323:1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eigenbrod T, Park JH, Harder J, Iwakura Y, Nunez G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol. 2008;181:8194–8198. doi: 10.4049/jimmunol.181.12.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci USA. 2010;107:2574–2579. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity. 2013;38:285–295. doi: 10.1016/j.immuni.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kono H, Karmarkar D, Iwakura Y, Rock KL. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol. 2010;184:4470–4478. doi: 10.4049/jimmunol.0902485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jaeschke H, Williams CD, Ramachandran A, Bajt ML. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver Int. 2012;32:8–20. doi: 10.1111/j.1478-3231.2011.02501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams CD, Bajt ML, Farhood A, Jaeschke H. Acetaminophen-induced hepatic neutrophil accumulation and inflammatory liver injury in CD18-deficient mice. Liver Int: Off J Int Assoc Study Liver. 2010;30:1280–1292. doi: 10.1111/j.1478-3231.2010.02284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cover C, Liu J, Farhood A, Malle E, Waalkes MP, Bajt ML, et al. Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2006;216:98–107. doi: 10.1016/j.taap.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 60.Marques PE, Oliveira AG, Pereira RV, David BA, Gomides LF, Saraiva AM, et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology. 2015;61:348–360. doi: 10.1002/hep.27216. [DOI] [PubMed] [Google Scholar]
- 61.Williams CD, Bajt ML, Sharpe MR, McGill MR, Farhood A, Jaeschke H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol Appl Pharmacol. 2014;275:122–133. doi: 10.1016/j.taap.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi DY, Ban JO, Kim SC, Hong JT. CCR5 knockout mice with C57BL6 background are resistant to acetaminophen-mediated hepatotoxicity due to decreased macrophages migration into the liver. Arch Toxicol. 2014;89:211–220. doi: 10.1007/s00204-014-1253-3. [DOI] [PubMed] [Google Scholar]
- 63.Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol. 2008;84:1410–1421. doi: 10.1189/jlb.0308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.You Q, Holt M, Yin H, Li G, Hu CJ, Ju C. Role of hepatic resident and infiltrating macrophages in liver repair after acute injury. Biochem Pharmacol. 2013;86:836–843. doi: 10.1016/j.bcp.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yin H, Cheng L, Holt M, Hail N, Maclaren R, Ju C. Lactoferrin protects against acetaminophen-induced liver injury in mice. Hepatology. 2010;51:1007–1016. doi: 10.1002/hep.23476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dinarello CA. Therapeutic strategies to reduce IL-1 activity in treating local and systemic inflammation. Curr Opin Pharmacol. 2004;4:378–385. doi: 10.1016/j.coph.2004.03.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.