

Abstract
Inflammation is a common symptom in joint disorders such as rheumatoid arthritis, osteoarthritis (OA) and implant aseptic loosening (AL). The sympathetic nervous system is well known to play a critical role in regulating inflammatory conditions, and imbalanced sympathetic activity has been observed in rheumatoid arthritis. In AL it is not clear whether the sympathetic nervous system is altered. In this study we evaluated the systemic and local profile of neuroimmune molecules involved in the interplay between the sympathetic nervous system and the periprosthetic inflammation in hip AL. Our results showed that periprosthetic inflammation does not trigger a systemic response of the sympathetic nervous system, but is mirrored rather by the impairment of the sympathetic activity locally in the hip joint. Moreover, macrophages were identified as key players in the local regulation of inflammation by the sympathetic nervous system in a process that is implant debris-dependent and entails the reduction of both adrenergic and Neuropetide Y (NPY)-ergic activity. Additionally, our results showed a downregulation of semaphorin 3A (SEMA3A) that may be part of the mechanism sustaining the periprosthetic inflammation. Overall, the local sympathetic nervous system emerges as a putative target to mitigate the inflammatory response to debris release and extending the lifespan of orthopedic implants.
Introduction
Osteoarthritis (OA) is one of the most prevalent chronic joint diseases and a major contributor to functional disability and loss of autonomy in older adults1. It is associated with a substantial economic and social burden, which will be even higher in the upcoming years, with the expected aging of the population1. Total joint replacement is considered the actual gold standard for the treatment of patients with severe OA, providing pain relieve, improving joint function and enhancing patients´ quality of life2,3. Unfortunately, total joint replacements can fail, mostly due to periprosthetic inflammation featured by sustained chronic inflammatory response initiated by implant degradation products that shed and accumulate in the neighbor tissue4. This adverse tissue reaction is orchestrated by a large plethora of immune cells of which the macrophage lineage has been shown to be of major relevance4,5. It is well established that macrophage differentiation towards M1 (pro-inflammatory) or M2 (anti-inflammatory) phenotype is of key importance to the inflammation state and/or resolution. In vitro studies have shown that Poly(methyl methacrylate) (PMMA) and ultra-high molecular weight polyethylene (UHMWPE) implant particles can polarize macrophages to pro-inflammatory M1 phenotype6–8. The activation of macrophages and other local cells results in the release of pro-inflammatory factors such as cytokines, chemokines, prostanoids, degradative enzymes and reactive oxygen species5,9. These factors underlie the chronic inflammatory scenario that may lead to painful synovitis, pathologic fracture of the surrounding bone and impaired function, instability and loosening of the implant9.
Over the past decades, accumulating evidence has clearly attributed a pivotal role to the sympathetic nervous system and its neurotransmitters in the regulation of chronic inflammatory conditions10,11. It has been demonstrated that the activation of the sympathetic nervous system in the context of inflammation results in the release of high amounts of sympathetic neurotransmitters known to induce an anti-inflammatory effect in a context-dependent manner11,12. The immunomodulatory effect of sympathetic nervous system can be achieved directly via adrenergic receptors (ADRs) expressed by the immune cells. Two types of ADRs have been characterized, the alpha (A) and beta (B), which were further divided into nine receptors subtypes (A1A, A1B, A1D, A2A, A2B, A2C, B1, B2 and B3)13. Stimulation of ADRB2 is reported to activate anti-inflammatory mechanisms on immune cells, while stimuli via ADRA activates pro-inflammatory mechanisms14. Therefore, the overall result will depend on the ADRs family being activated, which depends on the receptor expression profile and also on the norepinephrine concentration (norepinephrine has a high affinity to ADRA, only binding to ADRB when at high concentrations)14.
The neuropeptide Y (NPY), a neurotransmitter co-released with norepinephrine by the sympathetic nerve fibers, has also been reported to have modulatory effect on the activity of immune cells15. On the context of the immune response, of the five NPY receptors, the Y1 receptor (Y1R) is the most well studied, and has been shown to have a critical role in immunomodulation, as demonstrated by the attenuation of inflammation in Y1R knockout mice16. In healthy human joints, the synovium is highly innervated with both sympathetic and sensory nerve fibers17. In rheumatoid arthritis, data obtained in humans and in animal models revealed a deprivation of neuronal derived neurotransmitters in synovium tissue, due to the loss of sympathetic innervation14,18–20. Moreover, the extent of this deprivation is correlated with the severity of the inflammation. In fact, comparative studies reported a reduction of sympathetic innervation in synovial tissue of rheumatoid arthritis patients while in OA patients this does not occur18. In our previous work, we reported similar absence of sympathetic nerve fibers in periprosthetic tissues from AL patients while, again, in OA patients this does not occur21. In this line of evidence, the sympathetic activity is affected by the intensity of inflammation in the joint. However, it is still unknown whether in debris-associated periprosthetic inflammation occurs a complete shutdown of the sympathetic activity without any rescue mechanisms, and if the observed alterations are restricted to the joint or are also reflected at systemic level.
In this study we evaluated the systemic and the local profile of neuroimmune molecules involved in the interplay between sympathetic nervous system and the inflammatory response to the debris released by hip implants in AL. A comparison with OA was performed.
Results
Periprosthetic inflammation does not trigger the activation of the systemic neuroimmune regulatory pathway
Several studies have shown the activation of the systemic sympathetic nervous system in response to pro-inflammatory cytokines as a means to mobilize energy-rich molecules and sustain the inflammatory process14. In order to investigate the impact of periprosthetic inflammation on the systemic sympathetic nervous system activity, the serum levels of norepinephrine, epinephrine and NPY (markers of sympathetic nervous system activity) were measured in AL patients, OA patients, and healthy donors. In addition, the serum levels of cortisol were also assessed in the same groups as an indicator of the Hypothalamus-Pituitary-Adrenal (HPA) axis activity. The HPA axis together with the sympathetic nervous system compose the hormonal pathway through which the central nervous system exerts a regulatory control over inflammation14. No differences were found in the norepinephrine and NPY serum levels between the three groups (Fig. 1a). Epinephrine levels were below the detection limit of the used commercial kit. A tendency of higher cortisol levels was observed in AL patients when compared with healthy donors (p = 0.0658), although no statistically significant differences were found (Fig. 1a).
Figure 1.

The systemic neuroimmune regulatory pathway was not targeted by periprosthetic inflammation in AL patients. The levels of norepinephrine, NPY and cortisol (a) and the levels of IL-6 (b) were evaluated by ELISA in the serum collected pre-operatively from OA and AL patients, and from healthy donors. The ratio serum cortisol/IL-6 was calculated (c). Results are presented as mean ± SEM, n = 13–15 for healthy donors and AO patients and n = 14–20 for AL patients. **p < 0.01; ****p < 0.001.
The serum levels of the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α, key players in the activation of the sympathetic nervous system and HPA axis in response to inflammation14, were also measured in AL and OA patients and in healthy donors. The blood levels of IL-6 were higher in AL and OA patients than in healthy donors (p < 0.001 and p < 0.01, respectively) (Fig. 1b). The levels of IL-1β and TNF-α were observed to be below the ELISA kits’ detection limits in all groups.
Normal cortisol serum levels in the presence of high IL-6 concentrations are indicative of an inadequate cortisol secretion22, and the ratio serum cortisol/IL-6 was shown to be the more suitable indicator of the HPA axis activity23. Therefore, to further evaluate the HPA axis activity, the ratio of serum cortisol / IL-6 was calculated. Results showed no differences in this ratio between the groups, although a not statistically significant trend to lower values was observed in AO patients when compared with healthy donors (p = 0.0517) (Fig. 1c).
Local sympathetic response is impaired in macrophages in periprosthetic tissues from AL patients
Locally, the sympathetic nervous system is known to modulate the inflammatory response throughout adrenergic and NPY-ergic signaling14. In order to explore the effect of the sustained release of debris from implants on the local sympathetic immune-regulation, the expression of tyrosine hydroxylase (TH), ADRA1, ADRA2A, ADRB2, NPY and Y1R was analyzed in macrophages, B and T cells. A comparison between periprosthetic tissues and OA synovial tissues was performed. Macrophages in OA synovial tissues were found to express TH, ADRA1 and ADRB2, but not the macrophages present in periprosthetic tissues (Fig. 2a). ADRA2A was expressed by macrophages in both tissues (Fig. 2a). Macrophages in OA synovial tissues also stained positively for NPY but not macrophages in AL tissues (Fig. 2b).
Figure 2.

Macrophages in AL periprosthetic tissues do not express TH or ADRB2. The expression of TH, ADRA1, ADRA2A and ADRB2 (a) and the expression of NPY and Y1R (b) by macrophages (CD68+) was evaluated in periprosthetic tissues from AL patients and in synovial tissues from OA patients through double immunohistochemistry staining. Macrophages expressing TH, ADRA1, ADRA2A, ADRB2 or NPY are highlighted with triangle head white arrows. Simple head white arrows indicate macrophages and white arrowheads highlight TH, ADRA1, ADRB2, NPY and Y1R staining in cells other than macrophages (positive control). Scale bar = 20 μm.
Interestingly, the activation of ADRB2 in macrophages has been shown to promote a preferential differentiation of macrophages towards anti-inflammatory M2 phenotype over the pro-inflammatory M1 phenotype24. However, to our knowledge, the expression profile of ADRB2 in M1 and M2 macrophages was never described. Here we show that in vitro macrophages display a lower ADRB2 mRNA expression levels in M1 than in M2 phenotypes (p < 0.05) (Fig. 3). The in vitro analysis of ADRA1 and ADRA2 mRNA expression in M1 and M2 macrophages showed very low expression values, which were in some samples even below the detection limit. Still, from the results obtained, no differences were found in the ADRA1 and ADRA2 mRNA expression between M1 and M2 macrophages (data not shown).
Figure 3.
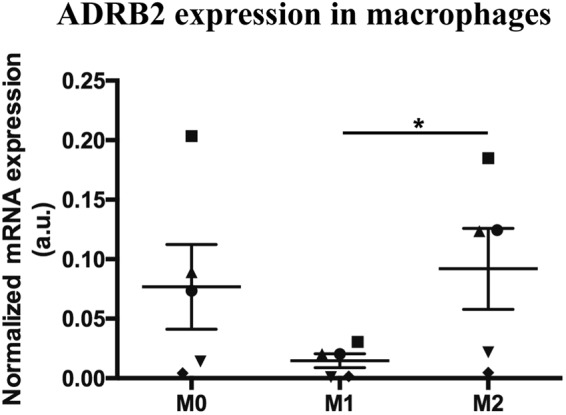
The in vitro expression of ADRB2 is lower in M1 as compared with M2 macrophages. The in vitro mRNA expression of ADRB2 was evaluated in M0, M1 and M2 macrophages phenotypes. Results are represented as mean ± SEM, for n = 5 per group. Each symbol represents macrophages obtained from one specific blood donor. *p < 0.05.
T cells were found to express ADRA1 both in periprosthetic tissues and OA synovial membrane, but not ADRA2A and ADRB2 (Fig. 4a). T cells were also expressing TH in OA but not in AL tissues. B cells were not expressing the adrenergic markers investigated neither in AL nor in OA (Fig. 5a) and the expression of NPY and Y1R was absent from T and B cells in both pathologic conditions (Figs 4b and 5b).
Figure 4.

T cells express ADRA1 both in AL periprosthetic tissues and OA synovial membrane. The expression of TH, ADRA1, ADRA2A and ADRB2 (a), and the expression of NPY and Y1R (b) in T cells (CD3+) was evaluated in periprosthetic tissues from AL patients and in synovial tissues from OA patients through a double immunohistochemistry staining. T cells expressing TH or ADRA1 are highlighted with triangle head white arrows. Simple head white arrows indicate T cells and white arrowheads highlight ADRA1, ADRA2A, ADRB2, NPY and Y1R staining in cells other than T cells (positive control). Scale bar = 20 μm.
Figure 5.

B cells in AL periprosthetic tissues and OA synovial membrane do not express TH, ADRA1, ADRA2A, ADRB2, NPY or Y1R. The expression of TH, ADRB2, ADRA1 and ADRA2A (a), and the expression of NPY and Y1R (b) in B cells (CD20+) was evaluated in periprosthetic tissues from AL patients and in synovial tissues from OA patients through double immunohistochemistry staining. White arrows indicate B cells and white arrowheads highlight positive staining for TH, ADRA1, ADRA2A, ADRB2 and Y1R in cells other than B cells (positive control). Scale bar = 20 μm.
The absence of sympathetic innervation in periprosthetic tissues from AL patients is not mediated by the classic regulators of the innervation pattern
In our previous work21, using the same samples that were used in this study, we reported an unbalanced innervation pattern in hip periprosthetic tissues, mirrored by the lack of sympathetic nerve fibers. Here, in order to investigate the molecular mechanism underlying this effect, we evaluated the mRNA expression of the classical neurotrophins NGF and BDNF, of the nerve repellent molecule SEMA3A, and of the sympathetic nerve repellent factors SEMA3C and SEMA3F in the AL and OA joint tissues. No differences were found in the mRNA expression levels of NGF and BDNF between periprosthetic tissues and OA synovial membrane (Fig. 6a). Interestingly, the mRNA levels of SEMA3A, SEMA3C and SEMA3F were found to be decreased in periprosthetic tissues when compared with OA synovial tissues (SEMA3A: p < 0.01; SEMA3C and SEMA3F: p < 0.05) (Fig. 6b).
Figure 6.

Neurotrophins and semaphorins expression in AL periprosthetic tissues and OA synovial membrane. The mRNA levels of neurotrophins NGF and BDNF (a), and semaphorins SEMA3A, SEMA3C and SEMA3F (b) were assessed in OA synovial membrane and periprosthetic tissues from AL patients. Results are represented as mean ± SEM, for n = 8–9 per group. **p < 0.01, *p < 0.05.
Discussion
In this study we show that the periprosthetic inflammatory response is not decoded by the sympathetic nervous system at systemic levels, but is mirrored locally in the hip joint by the impairment of adrenergic and NPY-ergic activity in macrophages.
Pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 released by local immune cells were identified as key players in the central activation of sympathetic nervous system14. Moreover, implant debris were shown to promote the in vitro release of pro-inflammatory cytokines by lymphocytes25 and macrophages26, and in OA an increase of TNF-α in synovial membrane and blood, and of IL-6 in synovial fluid and blood was reported27. In this study, the comparison of the IL-1β, IL-6 and TNF-α profile showed an increase in the serum levels of IL-6 in both AL and OA patients (in comparison with healthy donors) but no differences between AL and OA. Such results highlight IL-6 as a main mediator of the systemic inflammatory response in the studied joint disorders. Still, these increased blood levels of IL-6 were not reflected in an increased systemic sympathetic tone neither in AL nor in OA patients, suggesting that the inflammatory response is not being regulated by the systemic sympathetic nervous system, even in scenarios where debris is being released. Moreover, the evaluation of the cortisol blood levels and the analysis of the cortisol/ serum IL-6 ratio, strong indicators of HPA axis activity23, showed no significant differences between the three analyzed groups, suggesting no impact in the HPA axis activity. The combined lack of activation of the systemic sympathetic nervous system and the HPA axis supports the absence of a systemic control of the inflammatory reaction in both OA and AL conditions.
Within the inflammation site, sympathetic nervous system can directly influence immune cells via the ADRs expressed on their cell surface28. The sympathetic immunomodulatory effects are known to be dependent on parameters such as the ADRs profile and the catecholamines concentration. The stimulation of ADRA, sensitive to low concentrations of norepinephrine, activates pro-inflammatory mechanisms, while ADRB2 activation by high concentrations of norepinephrine, triggers anti-inflammatory processes14. The sympathetic influence on immune function can also be exerted by other neurotransmitters co-released with norepinephrine, such as NPY15. In order to understand the impact of the debris release in the local sympathetic control of the inflammatory response we investigated both the adrenergic and the NPY-ergic activity in immune cells. In the follow up of our previous work21, the expression of TH, ADRA1, ADRA2A and ADRB2 (adrenergic markers) and of NPY and Y1R (NPY-ergic markers) in macrophages, B and T cells, the main immune cell types previously identified in AL periprosthetic tissues and in OA synovial membrane21, were investigated by immunohistochemistry.
Our immunostaining results showed that TH, ADRA1, ADRB2 and NPY were expressed by macrophages in OA synovial tissues but are absent in macrophages from periprosthetic tissues. These results show that both the adrenergic and the NPY-ergic arms are implicated in the local regulation of joint inflammation by the sympathetic nervous system in a debris-dependent manner and reinforce macrophages as key cellular players in the process. Moreover, the absence of TH and ADRB2 in AL macrophages suggests a lack of sympathetic-mediated anti-inflammatory mechanisms in response to periprostetic inflammation. It was demonstrated that the stimulation of ADRB2 in macrophages promotes their differentiation towards an M2 anti-inflammatory profile and serves as a mean to prevent hyper-inflammation24. In addition, the pro-inflammatory M1 macrophages are increased in AL periprosthetic tissues when compared with OA synovial membrane6, and in vitro studies showed that PMMA and UHMWPE implant particles promoted macrophages polarization towards a M1 pro-inflammatory phenotype6–8. In our study we observed that only OA macrophages, and not AL macrophages expressed ADRB2. Hence, we decided to assess whether macrophages polarization in M1 or M2 could impact the ADRB2 expression profile. For this aim, an in vitro experiment exploring ADRB2 expression in M1 and M2 macrophages was performed. The results showed that the expression of ADRB2 is lower in M1 pro-inflammatory phenotype than in M2 anti-inflammatory phenotype macrophages, suggesting that the lack of ADRB2 observed in macrophages in AL tissues might be involved in the harshness of the periprosthetic inflammation.
ADRA were also found to be differentially expressed in OA and AL macrophages (in OA both ADRA1 and ADRA2A are present while in AL only ADRA2A were found) suggesting a debris-dependent activity of ADRA-mediated macrophages pro-inflammatory mechanisms. Very limited research is available on ADRA expression profile and function in macrophages29. Whether such different expression profile is a direct consequence of the presence of debris, and/or conditions the different inflammatory outcomes is not known and should be further investigated.
Regarding NPY-ergic regulation, NPY produced by immune cells during inflammation is known to modulate the cellular activity through autocrine or paracrine actions15,30. NPY has been shown to decrease the expression of the pro-inflammatory TNF-α by macrophages after stimulation with LPS31 and to increase the expression of the anti-inflammatory TGF-β132, contributing to ameliorate the inflammatory response. Moreover, it was recently suggested that NPY promotes the differentiation of macrophages towards a M2 anti-inflammatory phenotype33. Based on this information, and in the same way to what was found for ADRB2, the observed lack of NPY expression in AL macrophages may induce a preferential differentiation towards a pro-inflammatory phenotype. Such increase in the M1/M2 ratio may underlie the perpetuation of inflammation and increased osteolysis leading to implant failure. Although several immunomodulatory roles of NPY were suggested to occur through Y1R15, the expression of this receptor was not observed neither in macrophages in periprosthetic tissues nor in OA synovial membrane, hence, other NPY receptors (namely Y2R and Y5R) might be involved in the NPY regulation of macrophages activity30,34.
In regard to the B and T cells, we have previously shown that they are present in low number in both AL and OA tissues, and no differences in their population densities between the two joint conditions were found21. The activity of B and T cells has been reported to be modulated by the sympathetic nervous system, mostly through the ADRB235. The sympathetic modulatory effects seem to be dependent on (i) the activation and differentiation state of the cells, on (ii) the molecular signalling pathway activated, and on (iii) the cytokine microenvironment35. In this study, we found that T cells and B cells were not expressing ADRB2 neither in AL nor in OA tissues, suggesting an absence of ADRB2 modulation of these cells populations. Moreover, B cells were also not expressing any of the investigated ADRA neither in OA nor in AL tissues indicating that these receptors are not involved in the regulation of these cells activity by the sympathetic nervous system. It was found that ADRA1 is expressed in T cells, both in OA and AL. This receptor has been described as an inhibitor of T cells proliferation36. Through our results, that show the expression of ADRA1 by mature T cells (CD3+) in both AL and OA tissues, a different putative role of ADRA1 in T cells-mediated inflammation, other than the inhibition of proliferation, which is independent of particles released by the implants, is suggested. As observed in macrophages, T cells were found to express TH in OA but not in AL tissues, supporting the higher ability of non-neuronal synthesis of catecholamines in OA synovial membrane when compared to AL periprosthetic tissues.
Evidence showed a repulsion of sympathetic nerve fibers from inflamed tissues that cause the loss of anti-inflammatory neurotransmitters and allow the establishment of a privileged pro-inflammatory area18,37–39. In a previous work, performed in the same samples used in the present study, we have also reported the repulsion of sympathetic innervation from periprosthetic tissues in AL patient but not from synovial membrane in OA patients21. Here, we investigate the putative involvement of well-studied neurotrophic factors, NGF and BDNF, of the nerve repellent molecule SEMA3A, and of the sympathetic nerve repellent factors SEMA3C and SEMA3F in this specific innervation pattern. No differences were found in the expression levels of NGF and BDNF between AL periprosthetic tissues and OA synovial membrane. The expression of the chemorepellent molecule SEMA3A, SEMA3C and SEMA3F was lower in periprosthetic tissues when compared with OA synovial membrane. Such results indicate that the most well studied molecules involved in the modulation of the innervation pattern were not responsible for the repulsion of the sympathetic innervation from periprosthetic tissues of AL patients. Considering that SEMA3A has also been described as having an immunomodulatory role40–42, the observed decrease SEMA3A expression may be part of the mechanism underlying the different inflammatory profile between OA synovial membrane and periprosthetic tissues. Actually, decreased levels of SEMA3A were observed in synovial tissues from rheumatoid arthritis patients compared to synovial tissues in OA patients, and the reduction of SEMA3A expression has been correlated with rheumatoid arthritis exacerbation43. Moreover, overexpression of SEMA3A reduced inflammation in a mouse model of collagen-induced arthritis44, further supporting SEMA3A as an important immunomodulatory molecule.
Overall, in this work we show that periprosthetic inflammation in AL does not trigger a systemic response of the sympathetic nervous system but impairs the local sympathetic activity as a putative mean to enable the perpetuation of the inflammatory state. Macrophages were highlighted as key cellular players in the local regulation of inflammation by the sympathetic nervous system, in a process that is implant debris-dependent and entails the reduction of the activity of both the adrenergic and the NPY-ergic systems. The absence of TH, ADRB2 and NPY expression in macrophages in periprosthetic tissues from AL patients may underlie a preferential differentiation of macrophages towards a M1 pro-inflammatory phenotype, promoting inflammation and increased osteolysis that may lead to implant failure. The local sympathetic nervous system emerges, therefore, as a putative target to mitigate the inflammatory response to debris release and extending the lifespan of orthopedic implants.
Methods
Samples
Biological samples were collected from three groups of patients. Group 1 was constituted by twenty patients (20 hips) undergoing hip revision surgery due to AL, after exclusion of infection, recurrent dislocation and periprosthetic fractures. All patients had a metal-on-polyethylene coupling and eleven prosthesis were cemented. All acetabular components were revised and in five of those the femoral stem was also exchanged. In four patients metallosis was observed and the mean time to revision was 120.05 ± 65.8 months. Group 2 was constituted by fifteen patients (15 hips), submitted to primary hip replacement surgery for primary OA, after excluding patients with a known history of inflammatory or neoplastic diseases. Group 3 was constituted by 15 healthy volunteers (i.e. without known osteoarticular or systemic diseases). Demographics from Groups 1–3 are presented in Table 1.
Table 1.
Demographic data on included patients.
| N | Age (years) | Gender | |||
|---|---|---|---|---|---|
| Mean (±SD) | p | N of male/female | p | ||
| Group 1 Revision Surgery due to AL | 20 | 70.35 ± 11.4 | 0.119 | 6/14 | 0.537 |
| Group 2 Primary Surgery due to OA | 15 | 63.33 ± 14.5 | 6/9 | ||
| Group 3 Healthy Donors | 15 | 32.87 ± 3.4 | < 0.001* | 9/6 | |
AL- Aseptic loosening; OA- Osteoarthritis. *Comparing Group 3 with both Group 1 and Group 2.
Serum was obtained from the blood collected, during the morning, previously to surgery in Groups 1 and 2, and in Group 3, and kept at −80 °C until analyses. Synovial tissue was removed during the surgical procedure in Group 2 patients, and periprosthetic tissue was collected in Group 1.
This study was approved by the Ethics Committee of Centro Hospitalar São João and all patients signed an informed consent to the use of their samples for research purposes. All the procedures were in accordance with the Helsinki Declaration of 1975, as revised in 2000.
ELISA
The levels of Epinephrine/Norepinephrine, NPY and Cortisol were measured in the serum obtained from AL patients, OA patients and healthy donors, using ELISA kits from Abcam plc (Cambridge, UK; Catalog Number: AB108665), Merck KGaA (Darmstadt, Germany; Catalog Number: EZHNPY-25K) and Abnova (Taoyuan, Taiwan; Catalog Number: KA1877) respectively, according to the manufacturers’ protocols. The serum levels of interleukin (IL)-1β, IL-6 and Tumor necrosis factor alpha (TNF-α) were also measured in the same groups, using ELISA kits from BioLegend (CA, USA; Catalog Number: 430507).
Monocyte isolation and macrophage differentiation
Human monocytes were isolated from healthy blood donors and differentiated into macrophages45. Briefly, 106 monocytes/mL/3,8 cm2 were cultured for 10 days in RPMI1640 medium, supplemented with 10% Fetal bovine serum (FBS) and 100 U/mL penicillin and 100 μg/mL streptomycin, in the absence of Macrophages colony-stimulating factor (M-CSF) or other exogenous factors. 10 ng/mL LPS (Sigma-Aldrich) or IL-10 (ImmunoTools, Friesoythe, Germany) were added, for additional 72 h, to polarize macrophages towards M1 or M2 phenotype, respectively. Unstimulated macrophages (M0) were maintained with renewed medium.
Double immunofluorescence staining
The expression of markers of the sympathetic nervous system by immune cells (macrophages, B cells and T cells) was evaluated through a double immunofluorescence staining analysis. The collected tissues were formaldehyde-fixed and processed for paraffin embedding, and cross-sections of 3 μm thickness were cut in the microtome (RM2255, Leica Biosystems). Deparaffinized and dehydrated sections were placed in antigen retrieval for 20 min at 97 °C (10 mM citrate buffer, pH 6.0). After quenching endogenous fluorescence with 0.1% NaBH4 and 100 mM NH4Cl, sections were incubated with blocking buffer (10% FBS, 1% Bovine serum albumin (BSA), 0.2% Triton X-100).
Simultaneous incubation of each primary antibody against immune cells (antibodies mouse anti-CD68 (clone 514H12, dilution 1:100, Novocastra, UK), anti-CD20 (clone L26, dilution 1:100, Cell Marque, USA), anti-CD3 (clone PS1, dilution 1:100, Biocare Medical, USA)) with each primary antibody against sympathetic markers (antibodies rabbit anti-TH (dilution 1:100, Merck KGaA, Darmstadt, Germany), anti-ADRA1 (dilution 1:100, Abcam, USA), anti-ADRA2A (dilution 1:200, Abcam, USA), anti-ADRB2 (dilution 1:100, Proteintech, USA), anti-NPY (dilution 1:1000, Sigma-Aldrich, USA) or anti-Y1R (dilution 1:500, Immunostar, USA)) were performed overnight at 4 °C.
For signal detection, tissue sections were incubated for 1 hour at room temperature (RT) with a mixture of anti-rabbit Alexa Fluor 568 antibody and anti-mouse Alexa Fluor 488 antibody (1:1000 dilution, Life Technologies, USA), incubated with DAPI and then mounted with Fluoroshield Mounting Medium (Abcam, USA). Immunostaining images were acquired on the confocal Leica TCS SP2 AOBS (Leica Microsystems, Germany) and Leica TCS SP5 microscope (Leica Microsystems, Germany).
Gene expression analysis
Synovial tissues were homogenized in liquid nitrogen using a mortar and pestle to preserve RNA integrity. RNA from synovial tissues and from macrophages was extracted and purified using TRIzol (Invitrogen, UK) and Direct-zol™ RNA MiniPrep (ZYMO Research, USA), according to the manufacturers’ instructions.
RNA purity was estimated from the ratio of absorbance readings at 260 and 280 nm and only ratio between 1.8 and 2 were accepted. RNA quality was verified in agarose gel and RNA concentration was determined in a NanoDrop spectrophotometer (NanoDrop™ 1000 Spectrophotometer, Thermo Fisher Scientific, Wilmington, Delaware, USA NanoDrop). RNA was reverse transcribed using the SuperScriptTM First-Strand Synthesis System for reverse transcription-polymerase chain reaction (RT-PCR) (Invitrogen, Carlsbad, CA, USA).
The transcriptional levels of ADRA1A, ADRA1B, ADRA1D, ADRA2A and ADRB2 in macrophages and neurotrophins (Nerve growth factor (NGF) and Brain-derived neurotrophic factor (BDNF)) and semaphorins (SEMA3A, SEMA3C and SEMA3F) in the AL and OA tissues were evaluated by quantitative real time PCR (qRT-PCR) in the CFX96 Touch Detection System (Bio-rad, USA). β2 microglobulin (B2M) was used as reference gene for internal normalization. The primers used were as follows: ADRA1A sense primer: 5′-TCATGTACTGCCGCGTCTAC-3′; ADRA1A antisense primer: 5′-GGGCGTTTTTCCGATGGATG-3′; ADRA1B sense primer: 5′-CTCTACCGCTTGGCTCCTTGT-3′; ADRA1B antisense primer: 5′-GGAGCATGGGTAGATGATGGG-3′; ADRA1D sense primer: 5′-TCTCCCGTGAGAAGAAAGCG-3′; ADRA1D antisense primer: 5′-CGGGAACAAGGAGCCGAG-3′; ADRA2A sense primer: 5′-ATCCTGGCCTTGGGAGAGAT-3′; ADRA2A antisense primer: 5′-TCTCAAAGCAGGTCCGTGTC-3′; ADRB2 sense primer: 5′GGACTTCCATTGATGTGCTGT-3′; ADRB2 antisense primer: 5′-GTCAGCAGGCTCTGGTACTTG-3′; NGF sense primer: 5′-AGCGCAGCGAGTTTTGG-3′; NGF antisense primer: 5′-GCTGCTCCCTTGGTAAACTG-3′; BDNF sense primer: 5′- GATGCTCAGTAGTCAAGTGCC-3′; BDNF antisense primer: 5′-GCCGTTACCCACTCACTAATAC-3′; SEMA3A sense primer: 5′CAGCCATGTACAACCCAGTG-3′; SEMA3A antisense primer: 5′-ACGGTTCCAACATCTGTTCC-3′; SEMA3C sense primer: 5′-ATCCGGTCCTGATCTTCATC-3′; SEMA3C antisense primer: 5′-CAGCCCCAAGCAAGAGTTTA-3′; SEMA3F sense primer: 5′-CCAACTACCAGTGGATGCCC-3′; SEMA3F antisense primer: 5′-GTACACGGCCTGGTACATGA-3′; B2M sense primer: 5′-CCAGCGTACTCCAAAGATTCAG-3′; B2M antisense primer: 5′- AGTCAACTTCAATGTCGGATGG-3′. Relative transcription levels were calculated by comparative threshold cycle quantification (ΔCt method) using B2M as reference gene.
Statistical analyses
All data were assessed for normal distribution and non-parametric analyses were performed whenever normal distribution was not followed. Hormones and IL-6 levels, and the cortisol/IL-6 ratio were analyzed by Kruskal-Wallis test followed by Dunn’s multiple comparison test. mRNA expression of neutrophins and semaphorins in the synovial tissues was analyzed by Mann-Whitney test and ADRs mRNA expression in macrophages by the Repeated Measures ANOVA followed by Holm-Sidak’s multiple comparisons test. Differences were considered at the significant level of p < 0.05. All data are expressed as mean ± SEM. Statistical analyses were performed using the software Prism 6 (GraphPad software, San Diego, CA, USA).
Acknowledgements
This article is a result of the project NORTE-01-0145-FEDER-000012, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF). CJA was supported by the project NORTE-01-0145-FEDER-000008 financed by NORTE 2020, under the PORTUGAL 2020 Partnership Agreement, through the ERDF. DMV and ISA were supported by Ph.D. and postdoc fellowships SFRH/BD/87516/2012 and SFRH/BPD/75285/2010, respectively. The authors would like to thank orthopaedic surgeons Dr. Rui Pinto, Dr. Manuel Seara, Dr. Silva Pereira, Dr. Carlos Dopico, Dr. Artur Antunes, Dr. João Duarte Silva and Dra. Mariana Cunha Ferreira and nurses from Serviço de Ortopedia e Traumatologia from Centro Hospitalar de S. João and Dr. Francisco Costa Almeida from Serviço de Ortopedia e Traumatologia from Centro Hospitalar V.N.Gaia/Espinho, for the collaboration in samples retrieval.
Author Contributions
M.R.S., D.M.V., N.N. and D.L. carried out the retrieval of synovial tissues and blood samples. M.R.S., C.J.A. and D.M.V. performed ELISA quantifications. C.J.A., M.R.S. and R.H. performed co-staining analyses. C.J.A. and M.R.S. executed gene expression analysis and all statistical analyses. M.J.O. performed monocytes isolation and differentiation into macrophages. M.R.S., C.J.A., I.S.A., G.C. and M.L. conceived of the study, and participated in its design and coordination. C.J.A. and M.R.S. prepared the first manuscript draft. M.L. and I.S.A. worked in the final manuscript, and all authors read and approved it.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
M. Lamghari and C. J. Alves contributed equally.
References
- 1.OARSI. Osteoarthritis: A Serious Disease, Submitted to the U.S. Food and Drug Administration (2016).
- 2.Mancuso CA, Ranawat CS, Esdaile JM, Johanson NA, Charlson ME. Indications for total hip and total knee arthroplasties. Results of orthopaedic surveys. J Arthroplasty. 1996;11:34–46. doi: 10.1016/S0883-5403(96)80159-8. [DOI] [PubMed] [Google Scholar]
- 3.Dieppe P, et al. Knee replacement surgery for osteoarthritis: effectiveness, practice variations, indications and possible determinants of utilization. Rheumatology (Oxford) 1999;38:73–83. doi: 10.1093/rheumatology/38.1.73. [DOI] [PubMed] [Google Scholar]
- 4.Abu-Amer Y, Darwech I, Clohisy JC. Aseptic loosening of total joint replacements: mechanisms underlying osteolysis and potential therapies. Arthritis Res Ther. 2007;9(Suppl 1):S6. doi: 10.1186/ar2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Athanasou NA. The pathobiology and pathology of aseptic implant failure. Bone Joint Res. 2016;5:162–168. doi: 10.1302/2046-3758.55.BJR-2016-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao AJ, et al. Revision joint replacement, wear particles, and macrophage polarization. Acta Biomater. 2012;8:2815–2823. doi: 10.1016/j.actbio.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antonios JK, Yao Z, Li C, Rao AJ, Goodman SB. Macrophage polarization in response to wear particles in vitro. Cell Mol Immunol. 2013;10:471–482. doi: 10.1038/cmi.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang J, et al. Macrophage Polarization in IL-10 Treatment of Particle-Induced Inflammation and Osteolysis. Am J Pathol. 2016;186:57–66. doi: 10.1016/j.ajpath.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Goodman SB, et al. Novel biological strategies for treatment of wear particle-induced periprosthetic osteolysis of orthopaedic implants for joint replacement. J R Soc Interface. 2014;11:20130962. doi: 10.1098/rsif.2013.0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dekkers JC, Geenen R, Godaert GL, Bijlsma JW, van Doornen LJ. Elevated sympathetic nervous system activity in patients with recently diagnosed rheumatoid arthritis with active disease. Clin Exp Rheumatol. 2004;22:63–70. [PubMed] [Google Scholar]
- 11.Harle P, et al. Increase of sympathetic outflow measured by neuropeptide Y and decrease of the hypothalamic-pituitary-adrenal axis tone in patients with systemic lupus erythematosus and rheumatoid arthritis: another example of uncoupling of response systems. Ann Rheum Dis. 2006;65:51–56. doi: 10.1136/ard.2005.038059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Straub RH, Harle P. Sympathetic neurotransmitters in joint inflammation. Rheum Dis Clin North Am. 2005;31:43–59, viii. doi: 10.1016/j.rdc.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Guimaraes S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev. 2001;53:319–356. [PubMed] [Google Scholar]
- 14.Pongratz G, Straub RH. The sympathetic nervous response in inflammation. Arthritis Res Ther. 2014;16:504. doi: 10.1186/s13075-014-0504-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheway J, Herzog H, Mackay F. NPY and receptors in immune and inflammatory diseases. Curr Top Med Chem. 2007;7:1743–1752. doi: 10.2174/156802607782341046. [DOI] [PubMed] [Google Scholar]
- 16.Hassani H, Lucas G, Rozell B, Ernfors P. Attenuation of acute experimental colitis by preventing NPY Y1 receptor signaling. Am J Physiol Gastrointest Liver Physiol. 2005;288:G550–556. doi: 10.1152/ajpgi.00182.2004. [DOI] [PubMed] [Google Scholar]
- 17.Eitner A, Pester J, Nietzsche S, Hofmann GO, Schaible HG. The innervation of synovium of human osteoarthritic joints in comparison with normal rat and sheep synovium. Osteoarthritis Cartilage. 2013;21:1383–1391. doi: 10.1016/j.joca.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 18.Miller LE, Justen HP, Scholmerich J, Straub RH. The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J. 2000;14:2097–2107. doi: 10.1096/fj.99-1082com. [DOI] [PubMed] [Google Scholar]
- 19.Mapp PI. Innervation of the synovium. Ann Rheum Dis. 1995;54:398–403. doi: 10.1136/ard.54.5.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorton D, Lubahn C, Bellinger DL. Potential use of drugs that target neural-immune pathways in the treatment of rheumatoid arthritis and other autoimmune diseases. Curr Drug Targets Inflamm Allergy. 2003;2:1–30. doi: 10.2174/1568010033344499. [DOI] [PubMed] [Google Scholar]
- 21.Vasconcelos DM, et al. Immune response and innervation signatures in aseptic hip implant loosening. J Transl Med. 2016;14:205. doi: 10.1186/s12967-016-0950-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Straub Rainer H., Cutolo Maurizio. Involvement of the hypothalamic-pituitary-adrenal/gonadal axis and the peripheral nervous system in rheumatoid arthritis: Viewpoint based on a systemic pathogenetic role. Arthritis & Rheumatism. 2001;44(3):493–507. doi: 10.1002/1529-0131(200103)44:3<493::AID-ANR95>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 23.Straub RH, Paimela L, Peltomaa R, Scholmerich J, Leirisalo-Repo M. Inadequately low serum levels of steroid hormones in relation to interleukin-6 and tumor necrosis factor in untreated patients with early rheumatoid arthritis and reactive arthritis. Arthritis Rheum. 2002;46:654–662. doi: 10.1002/art.10177. [DOI] [PubMed] [Google Scholar]
- 24.Grailer JJ, Haggadone MD, Sarma JV, Zetoune FS, Ward PA. Induction of M2 regulatory macrophages through the beta2-adrenergic receptor with protection during endotoxemia and acute lung injury. J Innate Immun. 2014;6:607–618. doi: 10.1159/000358524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Podzimek S, et al. Influence of metals on cytokines production in connection with successful implantation therapy in dentistry. Neuro Endocrinol Lett. 2010;31:657–662. [PubMed] [Google Scholar]
- 26.Nich C, Goodman SB. Role of macrophages in the biological reaction to wear debris from joint replacements. J Long Term Eff Med Implants. 2014;24:259–265. doi: 10.1615/JLongTermEffMedImplants.2014010562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.da Silva MR, et al. Neuroimmune expression in hip osteoarthritis: a systematic review. BMC Musculoskelet Disord. 2017;18:394. doi: 10.1186/s12891-017-1755-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007) Brain Behav Immun. 2007;21:736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scanzano A, Cosentino M. Adrenergic regulation of innate immunity: a review. Front Pharmacol. 2015;6:171. doi: 10.3389/fphar.2015.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimitrijevic M, Stanojevic S. The intriguing mission of neuropeptide Y in the immune system. Amino Acids. 2013;45:41–53. doi: 10.1007/s00726-011-1185-7. [DOI] [PubMed] [Google Scholar]
- 31.Puerto M, Guayerbas N, Alvarez P, De la Fuente M. Modulation of neuropeptide Y and norepinephrine on several leucocyte functions in adult, old and very old mice. J Neuroimmunol. 2005;165:33–40. doi: 10.1016/j.jneuroim.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 32.Zhou JR, Xu Z, Jiang CL. Neuropeptide Y promotes TGF-beta1 production in RAW264.7 cells by activating PI3K pathway via Y1 receptor. Neurosci Bull. 2008;24:155–159. doi: 10.1007/s12264-008-0130-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buttari B, et al. Neuropeptide Y as Regulator of Macrophage Phenotype and Functions: A Neuroimmune Cue in Atherosclerosis Regression? Atherosclerosis. 2017;263:E2–E2. doi: 10.1016/j.atherosclerosis.2017.06.035. [DOI] [Google Scholar]
- 34.Dimitrijevic M, Stanojevic S, Vujic V, Beck-Sickinger A, von Horsten S. Neuropeptide Y and its receptor subtypes specifically modulate rat peritoneal macrophage functions in vitro: counter regulation through Y1 and Y2/5 receptors. Regul Pept. 2005;124:163–172. doi: 10.1016/j.regpep.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 35.Sanders VM. The beta2-adrenergic receptor on T and B lymphocytes: do we understand it yet? Brain Behav Immun. 2012;26:195–200. doi: 10.1016/j.bbi.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grisanti LA, Perez DM, Porter JE. Modulation of immune cell function by alpha(1)-adrenergic receptor activation. Curr Top Membr. 2011;67:113–138. doi: 10.1016/B978-0-12-384921-2.00006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fassold A, et al. Soluble neuropilin-2, a nerve repellent receptor, is increased in rheumatoid arthritis synovium and aggravates sympathetic fiber repulsion and arthritis. Arthritis Rheum. 2009;60:2892–2901. doi: 10.1002/art.24860. [DOI] [PubMed] [Google Scholar]
- 38.Lorton D, et al. Differences in the injury/sprouting response of splenic noradrenergic nerves in Lewis rats with adjuvant-induced arthritis compared with rats treated with 6-hydroxydopamine. Brain Behav Immun. 2009;23:276–285. doi: 10.1016/j.bbi.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Straub RH, et al. Anti-inflammatory role of sympathetic nerves in chronic intestinal inflammation. Gut. 2008;57:911–921. doi: 10.1136/gut.2007.125401. [DOI] [PubMed] [Google Scholar]
- 40.Rienks M, et al. Sema3A promotes the resolution of cardiac inflammation after myocardial infarction. Basic Res Cardiol. 2017;112:42. doi: 10.1007/s00395-017-0630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka J, et al. Semaphorin 3A controls allergic and inflammatory responses in experimental allergic conjunctivitis. Int J Ophthalmol. 2015;8:1–10. doi: 10.3980/j.issn.2222-3959.2015.01.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida Y, et al. Semaphorin 4D Contributes to Rheumatoid Arthritis by Inducing Inflammatory Cytokine Production: Pathogenic and Therapeutic Implications. Arthritis Rheumatol. 2015;67:1481–1490. doi: 10.1002/art.39086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takagawa S, et al. Decreased semaphorin3A expression correlates with disease activity and histological features of rheumatoid arthritis. BMC Musculoskelet Disord. 2013;14:40. doi: 10.1186/1471-2474-14-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catalano A. The neuroimmune semaphorin-3A reduces inflammation and progression of experimental autoimmune arthritis. J Immunol. 2010;185:6373–6383. doi: 10.4049/jimmunol.0903527. [DOI] [PubMed] [Google Scholar]
- 45.Cardoso AP, et al. Matrix metalloproteases as maestros for the dual role of LPS- and IL-10-stimulated macrophages in cancer cell behaviour. BMC Cancer. 2015;15:456. doi: 10.1186/s12885-015-1466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]