

Abstract
Extensive research during the last decade demonstrated that a single systemic administration of erythropoietin (EPO) lead to significant attenuation of myocardial infarction (MI) induced in animals, mostly small rodents, either by a myocardial ischemia followed by reperfusion or by a permanent ligation of a coronary artery. Both methods are critically reviewed with the aim of helping the reader in appreciating key issues in the translation of experimental results to the clinic. Results of several clinical trials in patients with acute MI completed to date failed to demonstrate beneficial effects of EPO, and thus put into question the validity of results obtained in animal models. Comprehensive review of design and results of animal experiments and clinical trials presented here allowed authors to postulate that therapeutic window for EPO during developing MI is very narrow and was possibly missed in negative clinical trials. This point was illustrated by the negative outcome of experiment in the rat model of MI in which timing of EPO administration was similar to that in clinical trials. The design of future clinical trials should allow for a narrow therapeutic window of EPO. Given current standards for onset-to-door and door-to-balloon time the optimal time for EPO administration should be just prior to PCI.
Keywords: Heart, Cardiac, Ischemia, Ischemia-reperfusion, Cardiomyocytes, Clinical trials
1. Introduction
Chronic Heart Failure (CHF)—a debilitating condition that reached epidemic proportion in the Western society and responsible for probably the largest portion of health care budget predominantly affecting the aging population. The major cause for severity of developing CHF is the extent of myocardial damage during an ischemic episode leading to a myocardial infarction (MI). Thus, one strategic approach to reduce the severity of CHF is to limit the extent of myocardial damage during an ischemic event. In spite of a great progress achieved during the last 20 years to treat an acute MI, mainly using timely revascularization of myocardium, the need for additional therapies to limit myocardial damage remains on the forefront of cardiovascular research.
The first experimental study demonstrating that exogenous recombinant human erythropoietin rhEPO protects myocardium from ischemic damage had been published in 2003. Since then, numerous successful studies in different experimental models had been reported and summarized in several review papers (1–6). Now, almost a decade and several phase I and II clinical trials later, we still failed to translate these highly promising experimental findings into clinical advances. In this review we reassess the wells of experimental data and outcomes of clinical trials and attempt to analyze the reason of a chasm between them. Should we write off the decade of intensive experimental work as yet another example of biological differences between species or is there another, currently overlooked, reason for the negative outcomes of clinical trials? To this effect we systematically review all available in vivo experimental studies, conducted in two major experimental models, MI induced by a permanent ligation of a coronary artery or by an ischemia-reperfusion method. We put an emphasis of this review on the experimental details such as effective doses and therapeutic window. Then, in the light of these experimental details, we reassess the design and clinical outcomes of reported clinical trials.
1.1. Review of published studies
1.1.1. Permanent occlusion of a coronary artery model
Animal experiments in the model of permanent occlusion of a coronary artery are summarized in the Table 1. The first experiment had been published in 2003 (7). Rats were subjected to a surgical occlusion of the left descending coronary artery and were treated either with intraperitoneal injection of 5,000 IU/kg of erythropoietin (type is not reported) or with saline. After 60 min of occlusion rats were sacrificed and apoptosis was assessed in the area at risk via TUNEL staining. The number of TUNEL positive nuclei in the AAR were dramatically reduced (p<0.0001) in EPO-treated rats.
Table 1.
Animal experiments (permanent occlusion)
| First author and reference |
Data | Species | EPO, type and doses (IU/kg) |
Time of EPO |
End-points |
||
|---|---|---|---|---|---|---|---|
| Relative to occlusion | Relative to reperfusion |
End-points | Outcomes | ||||
| Tramontano (7) | 2003 | Rat | EPO, 3,000 single Dose |
0 min | NA | At 60 min: apoptosis in the ARR |
↓ p<0.001 |
| Moon (8) | 2003 | Rat | Epoetin alfa, 3,000 Single dose |
0 min | NA | At 24 h: apoptosis in the ARR 8 weeks: MI size LVEDV LVESV LVEF |
↓ 50% p<0.001 ↓ 80% p<0.001 ↓ p<0.002 ↓ p<0.005 ↓ p<0.001 |
| Parsa (13) | 2003 | Rabbits | Epoetin alfa, 5,000 Single dose |
0 min | NA | At 6 h: apoptosis At 4 days: AAR MI size |
↓ p<0.001 ns ↓ p<0.1 |
| Van der Meer (14) |
2005 | Rat | Darbepoetin alfa, 8,000 single dose or repeated |
(1) 0 min or (2) 0 min + every 3 weeks or (3) + 3 weeks + every 3 weeks |
NA | At 9 weeks: MI size Capillary density |
↓ (1) p<0.01 ↓ (2) p<0.01 ns (3) ↑ (1,2,3) p<0.01 |
| Moon (9) | 2005 | Rat | Epoetin alfa, 3,000 or 1,000 or 500 or 150 or 50,Single Dose 3,000 or 150, single Dose |
0 min +4 h or +8 h or +12 h or +24 h |
NA | At 24 h: apoptosis At 4 weeks: MI size |
0 min group: ↓ 3,000 p<0.05 ↓ 150 p<0.05 50 ns 0 min group: ↓ 3,000–150 p<0.05 50 ns Delayed: 3,000: ↓ +4,+8,+12 h 150: ↓ +4h |
| Hale (15) | 2005 | Rat | EPO 5,000 repeated doses |
‒24 h, 0 min,+24 h and 4 more times daily |
NA | At 6 weeks: MI size LV volume LVEF |
ns ns ns |
| Moon (10) | 2006 | Rat | Epoetin alfa, 3,000 single dose and repeated doses |
0 min or 0 min,+24 and 6 more times daily |
NA | At 4 weeks: MI size LV volume LVEF |
Single ↓ p<0.05 Repeated ↓ ns ↓ p<0.05 ↑ p<0.05 |
| Moon (11) | 2006 | Rat | Epoetin alfa, 3,000 single dose |
0 min | NA | At 4 weeks: MI size LVEDV LVESV LVEF |
↓ p<0.05 ↓ p<0.05 ↓ p<0.05 ↑ p<0.05 |
| Hirata (16) | 2006 | Dog | EPO 1,000 single dose |
0 min (o min group) or +6 h (6 h group) or +1 week (1 week group ) |
NA | At 6 h: MI size At 1 week: CD34+ cells At 4 weeks: MI size` LVEF Capillary density |
↓ p<0.05 ↑ p<0.05 ↓ in 0 min group ns in 6 h group and 1 week group ↑ in 0 min and 6 h ns in 1 week ↑ in 0 min and 6 h ns in 1 week |
| Chua (17) | 2011 | Mini- pigs |
EPO 7,500 repeated doses |
+30 min and +24 h | NA | At 14 days: Apoptosis Fibrosis Capillary density MI size LVEF |
↓ p<0.001 ↓ p<0.01 ↓ p<0.05 ↓ p<0.05 ↓ p<0.05 |
| Ahmet (12) | 2011 | Rat | Epoetin alfa, 3,000 single dose |
0 min | NA | At 24 h: Apoptosis Inflammation MI size At 8weeks: MI size LVEDV LVESV LVEF |
↓ (80%) p<0.05 ↓ (30%) p<0.05 ↓ (50%) p<0.05 ↓ p<0.05 ↓ p<0.05 ↓ p<0.05 ↑ p<0.05 |
Two more studies were published at the same time. In one (8) rats were treated with a single systemic (intraperitoneal) injection of Epoetin-alfa (3,000 IU/kg) immediately following a permanent occlusion of an anterior descending coronary artery. Apoptosis in the area at risk (peri-infarct area) was assessed in the subgroup of rats 24 h after coronary occlusion and was found to be reduced by 50%. The rest of the animals were followed up with serial echocardiography during 8 weeks. Experiment was concluded by a histological evaluation of the heart. At the end of experiment left ventricular (LV) volumes were significantly smaller and EF was significantly higher in the EPO treated group comparing with control, untreated animals. Moreover, histologically measured MI size was fourfold smaller in the EPO-treated animals. Interestingly, hematocrit level measured during the first 3 weeks after surgery was not elevated in the EPO-treated group. Results of this first study had been consistently reproduced by the same group later on in 2005, 2006, and 2011 in different experiments in which a single injection of 3,000 IU/kg of EPO immediately after coronary occlusion served to elicit a standard effect to compare with other types of interventions (9–12).
In another study employing a permanent ligation model in rabbits (13) a single dose of 5,000 IU/kg of epoetin-alfa was injected immediately after ligation (the route of injection was not reported) and animals were sacrificed 4 days after surgery. AAR was not different between groups. While the number of TUNEL- positive cells in the AAR at 6 h after ligation was 50% less in the EPO-treated animals, the MI size at day 4 had only a tendency to be smaller (p<0.1).
In 2005, in experiments on rats van der Meer et al. (14) used 8,000 IU/kg of Darbepoetin alfa, prolong action epoetin, in three different treatment strategies: (1) a single dose of EPO immediately after coronary ligation, (2) repeated doses of EPO every 3 weeks starting the first dose immediately after coronary ligation, and (3) repeated doses of EPO every 3 weeks starting the first dose 3 weeks after coronary ligation. Experiment lasted 9 weeks. At the end of 9 weeks MI size was significantly smaller in groups 1 and 2, i.e., in groups in which treatment was started immediately after coronary ligation. There were no effects on the MI size in group 3 (treatment started 3 weeks after coronary ligation) and there were no differences in the magnitude of effect on the MI size between groups 1 and 2. Capillary density was significantly increased in all three treatment groups and there were no differences in the magnitude of effect.
Study reported by Hale et al. (15) is the only one among studies employing the permanent coronary occlusion model with clearly negative results. 24 h prior coronary ligation rats had been injected subcutaneously with 5,000 IU/kg of Epoetin alfa. The dose had been repeated at the day of ligation (20–25 min before occlusion), and five more times daily. Six weeks after surgery there were no significant effects of treatment either with respect to MI size or with respect to LV volume and function. It was noted, however, that at each given MI size the EF was higher in the EPO treated animals. Notably, after 7 days the hematocrit in the EPO treated rats was elevated to 67%, compared to 45% in untreated animals.
Hirata et al. (16) studied the effect of EPO in the dog experimental model of MI. rhEPO (type of EPO is not reported) had been injected as a single bolus intravenously in three different treatment groups either immediately after permanent coronary occlusion (0 group), or 6 h after occlusion (6 h group), or 1 week after occlusion (1 week group). Six hours after occlusion (0 group) MI was statistically smaller in the EPO treated group vs. control. One week after occlusion the number of progenitor cells was increased in the both EPO treated groups (0 group and 6 h group). Measured 4 weeks after occlusion, the MI was significantly smaller only in the 0 group; EF and capillary density were increased in the 0 group and 6 h group, but not in 1 week group of EPO treated dogs.
Recently (2011) the effect of EPO has been tested in the mini-pig model of MI (17). 7,500 IU/kg of EPO (type is not reported) was injected 30 min following coronary occlusion. The dose had been repeated once, 24 h later. Fourteen days after surgery MI size was smaller and EF was higher in EPO-treated in comparison with placebo-treated animals. In the hearts of the EPO group there were less fibrosis and higher capillary density. Apoptosis in the peri-infarct area was reduced.
Therapeutic window and effective dose of EPO in the rat model of permanent coronary occlusion had been systematically studied by Moon et al. (9). In the first experiment five different doses (3,000, 1,000, 500, 150, and 50 IU/kg) were injected intraperitoneally immediately after coronary ligation. Apoptosis in the AAR 24 h after treatment measured as a number of TUNEL positive nuclei was significantly reduced in all treatment groups except one, treated with the lowest, 50 IU/kg, dose. All doses, but 50 IU/kg were also found effective 4 weeks after coronary ligation with respect to reduction of MI size, LV remodeling and preservation of EF. In the second experiment 3,000 IU/kg had been injected with different delays after coronary ligation (0 min, 4, 8, 12, and 24 h). Four weeks after surgery MI was significantly smaller and EF significantly less reduced in all delayed treatment groups, except when treatment had been delayed by 24 h. In the final experiment the smallest effective dose (150 IU/kg) was injected with the same delays as in the second experiment. Four weeks later it was found that this dose was effective only if injected immediately. Any delay longer than 4 h did not affect the MI size or LV remodeling. In the group where 150 IU/kg treatment was delayed by 4 h, EF was higher and MI was smaller than in untreated group, but with respect to MI size the difference did not reach statistical significance (p<0.07).
Finally, using the rat model of permanent coronary occlusion (10), the effect of a single dose of 3,000 IU/kg injected immediately after coronary ligation had been compared with the effect of treatment when immediate dose of 3,000 IU/kg was supplemented with seven more daily injections at the same dose. Comparing with untreated animals 4 weeks after surgery LV remodeling and the reduction of EF were similarly attenuated in single dose and repeated dose groups. However, the reduction of MI size was statistically significant only in the group treated with a single dose.
Therefore, all published studies but one demonstrated that in the experimental model of MI induced by a permanent occlusion of a coronary artery in rats, dogs, and mini-pigs high dose of rhEPO (3 3,000 IU/kg) applied after coronary ligation effectively protects myocardium from ischemic damage by reducing apoptosis in the area at risk resulting in smaller MI size and attenuated LV remodeling. The effect is clearly dose-depended and has a narrow therapeutic window, which is also dose-dependent. If treatment is applied immediately after occlusion, the effective dose can be as low as 150 IU/kg (9, in rats). 1,000 IU/kg was not effective if treatment was delayed by 6 h (16, dogs). A single dose applied immediately after coronary occlusion is as effective as repeated doses (14). Moreover, there are some indications that repeated doses may reduce the effectiveness of treatment (10). In the only study in which EPO treatment did not improve the outcome, the high dose of EPO was applied seven times resulting in the massive elevation of hematocrit.
1.1.2. Ischemia–Reperfusion Model
A summary of published animal experiments in the in vivo model of temporary occlusion of a coronary artery followed by reperfusion are summarized in Table 2.
Table 2.
Animal experiments with in vivo models of ischemia-reperfusion
| Time of treatment |
End-points |
||||||
|---|---|---|---|---|---|---|---|
| First author and references |
Date | Species | EPO, type and doses (lU/kg) |
Relative to Isch/Rep | Duration of occlusion (min) |
End-points | Outcomes |
| Calvillo (18) | 2003 | Rat | rhEPO, 5,000 i.p. multiple doses |
24 h, 0.5 h before Isch, then daily |
30 | At 7 days: LVEDP Number of CM |
↓ p<0.001 ↑ p<0.01 |
| Parsa (19) | 2004 | Rabbit | EPO, 1,000 or 5,000 single dose i.v. |
12 h before Isch, at Isch or start Rep |
45 | At 45 min: MI size LV function CM apoptosis |
↓ p< 0.005 (all EPO groups) ↑ p<0.05 (only EPO 12 h before or at Isch) ↓ p<0.05 (only EPO 12 h before Isch) |
| Abdelrahman (20) |
2004 | Rat | EPO 300 i.v. | Start of Rep or of resuscitation |
20 | At 2 h: MI size Renal and liver function |
↓ p<0.05 ↑ p<0.05 |
| Lipsic (21) | 2004 | Rat | EPO 5,000 i.p. | 2 h before Isch or at start Isch or right after Rep |
45 | At 24 h: MI size LV function CM apoptosis |
↓ p<0.05 p<0.05 (=NS for 2 h before Isch) ↓ p<0.05(=NS for 2 h before Isch) |
| Rui (22) | 2005 | Mouse | rhEPO 5,000 i.p. | 24 h and 30 min before Isch |
30 | At 4 h: myocardial MPO |
↓ p<0.05 |
| Hirata (23) | 2005 | Dog | EPO 100 to 1,001 i.v. |
10 min before Rep | 90 | At 6 h: MI size VF CM apoptosis |
↓ p<0.05 ↓ p<0.05 ↓ p<0.05 |
| Xu (24) | 2005 | Rat | rhEPO 3,000 i.p. | 24 h before Isch | 30 | At 2 h: MI size Hsp70 NF-kB |
↓ p<0.001 ↑ p<0.001 ↓ p<0.001 |
| Bullard (25) | 2005 | Rat | EPO 5,000 i.v. | Right before Rep | 40 | At 24 h: MI size | ↓ p<0.05 |
| Kristensen (26) | 2005 | Pig | EPO 500 i.v. | 1.5 h or 24 h before Isch |
45 | At 2.5 h: MI size | p=NS |
| Liu (27) | 2006 | Rat | rhEPO 5,000 i.v. | 24 h before Isch | 30 | At 3 h: MI size MPO NF-kB, Ap-1 TNFa |
↓ p<0.001 ↓ p<0.001 ↓ p<0.001 ↓ p<0.001 |
| Nishihara (28) | 2006 | Rat | EPO 5,000 i.v. | 5 min before Rep | 20 | At 2 h: MI size Akt phosphorylation |
↓ p<0.05 ↓ p<0.05 |
| Olea (29) | 2006 | Sheep | rhEPO 3,000 i.v. | Right before Rep, than daily for 3 days |
90 | At 10 weeks: MI size LVEDP LV Volume Septum thickening |
p=NS ↑ p<0.03 ↑ p<0.05 ↓ p<0.04 |
| Liu (30) | 2006 | Rat | rhEPO 5,000 i.p. | 24 h before Isch | 30 | At 24 h: MI size | ↓ p<0.01 |
| Gao (31) | 2007 | Rat | Darbepoetin 2,7, 11,30 mg/kg i.v. |
Start Isch, 1 or 24 h after Rep |
30 | At 3 h: MI size Cardiac reserve CM apoptosis |
↓ p<0.05−0.01 ↑ p<0.05−0.001 ↓ p<0.05 |
| Baker (32) | 2007 | Rat | Darbepetin 2.5 mg/kg i.v. |
15 min before Isch or start Rep, or 24 h post Rep |
30 | At 2 h: MI size cTnI |
↓ p<0.05 ↓ p<0.05 |
| Toma (33) | 2007 | Pig | Darbepetin 30 mg/kg i.v. |
At Rep | 60 | At 2 weeks: MI size Fibrosis Capillaries LV wall motion |
p=NS ↓ p<0.02 ↓ p<0.003 ↑ p<0.05 |
| Singh (34) | 2007 | Rat | rhEPO 5,000 intra-right atrium |
15 min before VF or at 10 min VF, start of chest compression |
10 min cardiac arrest with VF |
At resuscitation: Aortic press Cardioc index |
↑ p<0.05 ↑ p<0.05 only after 10 min VF |
| Boucher (35) | 2008 | Rat | Darbepetin 30 mg/kg i.v. |
Start of Isch | 30 | At 3 days: MI size CM apoptosis LVEF echo |
↓ p<0.05 ↓ p<0.05 ↑ p<0.05 |
| Prunier (36) | 2009 | Rat | Darbepoetin 5,000, or 5,000 and 300 (∼1.5 mg/kg) i.p |
Start Rep+-once-a- week |
45 | At 8 weeks: MI size Thrombosis |
↓ p<0.05 p<0.05 |
| Shan (37) | 2008 | Mouse | EPO 1,000 | Start Rep | 45 | At 4 h: MI size LVEF CM apoptosis |
↓ p<0.05 ↑ p<0.05 ↓ p=NS |
| Doue (38) | 2008 | Rat | EPO 200 i.v. | Start Rep | 20 | At 30 min: Annexin V area At 2 and 4 weeks: LVEDd LVFS |
↓ p<0.001 ↓ p<0.01 ↑ p<0.01 |
| Huang (39) | 2008 | Mouse | EPO 1,000,3,000, 5,000 i.v. |
3 min after resuscita tion from asphyxia- induced cardiac arrest |
6.5 or 9.5 | At 3 days: Survival dP/dt |
↑ p<0.003 ↑ p<0.003 (only at EPO 5,000 U/kg) |
| Burger (40) | 2009 | Mouse | rhEPO 2,500 i.v. | 24 h before Isch or 5 min after Isch start |
45 | At 3 or 6 h: MI size CM apoptosis |
↓ p<0.05 ↓ p<0.05 (no effect pf EPO in HO-1−/− mice) |
| Burger (41) | 2009 | Mouse | rhEPO 2,500 | 24 and 0.5 h before Isch or 5 min after Isch start |
45 | At 45 min: PVCs and VT At 6 h: MI size |
↓ p<0.05 ↓ p<0.003 (only for EPO pretreatment) |
| Frence (42) | 2009 | Mouse | EPO 10,000 i.p. | Before Isch or right afrter Rep |
3 h | At 72 h: MI size Echo LV FS |
p=NS p=NS |
| Schlecht-Bauer (44) |
2009 | Rat | Darbepoetin 3–30 mg/kg i.v. |
Pre Isch | 40 | At 3 or 28 days: MI size LVEF |
↓ p<0.05 ↑ p<0.05 |
| Tamareille (44) | 2009 | Rat | EPO 10,000 i.p. | Right before Rep | 45 | At 24 h: MI size CM apoptosis |
↓ p<0.05 ↓ p<0.05 |
| Miki (45) | 2009 | Rat | EPO 5,000 route not reported |
1 min before Isch | 20 | At 2 h: MI size | ↓ p<0.01, EPO in control rats p=NS, EPO in type 2 diabetic rats |
| Hotta (46) | 2010 | Rat | EPO 5,000 route not reported |
15 min before Isch | 20 | At 2 h: MI size | ↓ p<0.01, EPO in control rats p=NS, EPO in type 2 diabetic rats |
| Treguer (47) | 2010 | Rat | Darbepoetin 1,000 i.v. |
Right before Rep | 45 | At 7 days: MI size Transmural MI |
↓ p<0.01 ↓ p<0.01 (no benefit on LV function by EPO with conventional echo) |
| Shen (48) | 2010 | Rat | rhEPO 100–5,000 i.v. |
30 min before Isch | 30 | At 2 and 4 h: Ventricular arrhythmias TNFa, IL6 |
↓ p<0.05 ↓ p<0.05 |
| Strin (49) | 2010 | Nude rat | rhEPO 200 U ± hEPCs intramyocardial |
At Rep | 30 | At 4 weeks: MI size Regional LV wall motion LVEF Vessel density EPC apoptosis |
Only EPC + EPO p=NS ↑ p<0.05 p=NS ↑ p<0.05 ↓ p<0.05 |
| Yano (50) | 2011 | Stroke- prone rats (SHR-SP) |
EPO 5,000 i.v. | 15 min before Isch | 20 | At 2 h: MI size | ↓ p<0.05 in WKY p=NS in SHR-SP |
Only 1.5 dose
Based on experimental evidence that EPO attenuates both primary (apoptotic) and secondary (inflammatory) causes of cell death, Calvillo et al. (18) administered 5,000 U/kg i.p. of recombinant human EPO (rhEPO) to rats that underwent 30 min ischemia followed by reperfusion. rhEPO was given 24 and 0.5 h before coronary ligation, then daily for 7 days after coronary ligation rhEPO reduced cardiomyocyte loss by approximately 50%, and accordingly normalized hemodynamic function within 1 week after reperfusion. RhEPO markedly prevented the apoptosis of cultured adult rat cardiac myocytes subjected to 28 h of hypoxia (approximately 3% normal oxygen). This in vitro observation supported the hypothesis that cardiac protective effects of EPO were independent of hematopoiesis.
Parsa et al. (19) evaluated the in vivo protective potential of erythropoietin in the reperfused rabbit heart following 30 min ventricular ischemia followed by 45 min of reperfusion. They showed that 5,000 U/kg i.v. EPO given 12 h before ischemia or at time of coronary ligation limited infarct size and improved global LV function. However, when EPO was administered at the low dose of 1,000 U/kg at any time and/or at the time of reperfusion, it did not improve cardiac function even if significantly reduced infarct size and incidence of apoptosis in cardiac myocytes. EPO activated cell survival pathways in myocardial tissue in vivo and in adult rabbit cardiac fibroblasts in vitro. These pathways, activated by EPO in both whole hearts and cardiac fibroblasts, are also activated acutely by ischemia–reperfusion injury.
As part of a study on the effects of EPO on the tissue/organ injury caused by hemorrhagic shock, endotoxic shock, Abdelrahman et al. (20) showed that 300 U/kg i.v. of EPO after 20 min myocardial ischemia right before reperfusion significantly reduced infarct size in anesthetized rats at 2 h. The administration of the same dose of EPO before resuscitation abolished the renal dysfunction and liver injury in hemorrhagic, but not endotoxic, shock at 4–6 h.
Lipsic et al. (21) studied the effect of timing of EPO administration on cardio protection during cardiac ischemia–reperfusion. Male Sprague-Dawley rats were subjected to 45 min of coronary occlusion, followed by 24 h of reperfusion. Animals were randomized to receive saline or single dose of EPO (5,000 IU/kg i.p.) either 2 h before ischemia–reperfusion, at the start of ischemia, or after the onset of reperfusion. Administration of EPO at all three time points resulted in a significant 19–23% reduction in the infarct area/area at risk after24-h reperfusion, which was accompanied by a trend toward better left ventricular hemodynamic parameters only in EPO at start of ischemia and right after reperfusion groups. The same two experimental groups showed a significant decrease in apoptosis, while the decrease was not statistically significant in the group pretreated with EPO. These results suggest that cardiac protection by EPO does not need pretreatment with EPO.
Rui et al. (22) showed that EPO can ameliorate the myocardial inflammatory response in both in vitro and in vivo murine models of ischemia-reperfusion, in agreement with what shown in central nervous system injury. In vivo, in wild type mice, 5,000 U/kg i.p. of rhEPO given 24 h before 30 min ischemia followed by 2 h reperfusion prevented ischemia–reperfusion-induced increase in myocardial myeloperoxidase activity (indicative of polymorphonuclear cells infiltration). With the help of in vitro experiments, the authors attribute this beneficial effect of EPO to the production of NO by eNOS via aPI3-kinase-dependent activation of the nuclear transcription factor AP-1.
Hirata et al. (23) occluded the canine left anterior descending coronary artery for 90 min followed by 6 h of reperfusion. EPO administered (i.v. 100–1,000 IU/kg) just before reperfusion reduced in a dose-dependent way infarct size and the incidence of ventricular fibrillation via the PI3 kinase-dependent pathway in canine hearts. In fact these effects of EPO were abolished by the treatment with wortmannin, an inhibitor of PI3-kinase.
Xu et al. (24) used a rat model of 30 min ischemia followed by 2 h of reperfusion to assess mechanisms of the anti–inflammatory action of recombinant human EPO (rhEPO). A single i.p. injection of 3,000 IU/kg of rhEPO 24 h pre-ligation enhanced the expression of Heat shock protein 70 (Hsp70) and diminished the expression of NF-kB in rat myocardium, and reduced markedly myocardial infarct size, compared to control rats.
Bullard et al. (25) administered 5,000 IU/kg i.v. at the start of reperfusion to rats after 40 min ischemia. Infarct size at 24 h was significantly attenuated in EPO-treated animals. Likewise, wortmannin abrogated EPO-mediated protection, thus suggesting that the protective action of EPO is mediated by ERK and PI3K.
Kristensen et al. (26) tested the effects of rhEPO in a closed chest, catheter-based, porcine coronary occlusion model (45 min of occlusion of the left anterior descending artery). EPO was given i.v. 500 U/kg either 90 min or 24 h and 90 min before coronary occlusion. Infarct size as a proportion of the ischemic area was measured in vivo by myocardial perfusion imaging (MPI) and postmortem by a histochemical procedure (at 150 min of reperfusion). Infarct size relative to the area at risk did not differ significantly between control and EPO groups. These negative results in the pig contrast with all positive results obtained in small rodents, even if the authors cannot exclude possible beneficial effects at later times on apoptosis and/or on left ventricular remodeling.
Liu et al. (27) further investigated mechanisms of the anti-inflammatory action of EPO in a rat model of cardiac 30 min ischemia and 3 h reperfusion. Injection of 5,000 U/kg of rhEPO 24 h before insult remarkably reduced infarct size and neutrophil infiltration. EPO greatly attenuated ischemia–reperfusion-induced NF-kB and AP-1 activation with decreased TNF-alpha, IL-6, and Il-CAM-1 production and enhanced the production of the anti-inflammatory cytokine IL-10.
Nishihara et al. (28) showed that both preconditioning (PC) with 5-min ischemia–5-min reperfusion and EPO (5,000 U/kg i.v.), given 5 min before reperfusion reduced infarct size after 20-min ischemia in rat hearts in situ followed 2 h of reperfusion. The results suggest that EPO and PC afford additive infarct size-limiting effects by additive phosphorylation of GSK-3beta at the time of reperfusion by Akt-dependent and -independent mechanisms.
While high-dose EPO has been found to be cardioprotective in experimental acute MI in small rodents, in large mammals, however, results are controversial and long-term follow-up data are lacking. Olea et al. (29) assessed the long-term effects of high-dose EPO on left ventricular infarct size and function in an ovine model of reperfused MI. After 90 min of coronary occlusion, at the onset of reperfusion, sheep received recombinant human erythropoietin (rhEPO) 3,000 U/kg on 3 consecutive days (rhEPO group) or vehicle (placebo group). Infarct size, evaluated as percent fibrotic myocardium (morphometry) and by hydroxyproline quantification, was similar in both groups. Left ventricular contractile function was decreased in rhEPO group, compared to controls, left ventricle was more dilated, and left ventricular end-diastolic pressure was increased 10 weeks after coronary occlusion.
Liu et al. (30) studied the possible role of cyclooxygenase (COX)-2 in the delayed phase of the cardioprotection induced by recombinant human erythropoietin (rhEPO). Rats were given 5,000 U/kg i.p. of rhEPO 24 h before 30 min of ischemia followed by 3 h of reperfusion. Infarct size reduction in rhEPO group was abolished by the COX-2 inhibitor celecoxib given i.p. at the dose of 5 mg/kg 24 h before ischemia. It appears that COX-2 plays an essential part in cardioprotection of the delayed phase of EPO preconditioning in rats, which might be related to actions of PGE(2) or PGI(2), or both.
In Gao et al. (31) dose–response study, darbepoetin (DA) 2,7,11, and 30 m/kg or vehicle was administered as a single i.v. bolus at the start of 30-min ischemia. To determine the time window of potential cardioprotection, a single high dose of DA (30 mg/kg) was given at either the initiation or the end of ischemia or at 1 or 24 h after reperfusion. After 3 days, cardiac function and infarct size were assessed. DA significantly reduced infarct size in a dose-dependent manner. Treatment with DA up to 24 h after the beginning of reperfusion still demonstrated a significant reduction in infarct size. Consistent with infarction data, DA improved in vivo cardiac reserve compared with vehicle, as assessed by left ventricular dP/dt at increasing doses of isoproterenol. Finally, DA significantly decreased myocyte apoptosis and caspase-3 activity after ischemia-reperfusion.
Baker et al. (32), found that a single intravenous darbepoetin treatment immediately before 30 min of regional ischemia in the rat reduced myocardial necrosis following 120 min of reperfusion in a dose-dependent manner. Optimal protection with darbepoetin-alfa against MI was manifest at a dose of 2.5 mg/kg among doses ranging from 0.25 to 30 mg/kg· Darbepoetin was cardioprotective when administered after the onset of ischemia and at the start of reperfusion. Darbepoetin-alfa (2.5 mg/kg) also reduced infarct size and Troponin I leakage 24 h after reperfusion. Inhibition of p42/44 MAPK (PD98059), p38 MAPK (SB203580), mitochondrial ATP-dependent potassium (KATP) channels (5-HD), sarcolemmal KATP channels (HMR 1098), but not phosphatidylinositol-3 (PI3) kinase/Akt (Wortmannin and LY 294002) abolished darbepoetin-alfa-induced cardioprotection.
Toma et al. (33) randomized to darbepoetin 30 mg/kg i-v. or saline at the time of reperfusion after 60 min ischemia 16 domestic pigs. Ischemia was induced by inflating an angioplasty balloon in the proximal left circumflex artery. Darbepoetin did not reduce infarct size, but a limited decrease in interstitial fibrosis, increased capillary area and regional functional improvement in darbepoetin-treated animals was observed. However, this did not translate to improved wall thickening of the left ventricle.
Singh et al. (34) induced ventricular fibrillation electrically and maintained it, untreated, for 10 min. Chest compression and ventilation were then started and electrical defibrillation was attempted 8 min later. Rats were randomized to receive rhEPO (5,000 U/kg) in the right atrium at baseline, 15 min before induction of VF (rhEPOBL −15-min), or at 10 min of VF, immediately before the start of chest compression (rhEPOVF 10-min), or to receive saline (control). Post-resuscitation, rats in the rhEPOVF 10-min group displayed higher mean aortic pressure associated with numerically higher cardiac index, stroke work index, and systemic vascular resistance index. In this model of short lasting global in vivo cardiac ischemia followed by reperfusion, rhEPO may rapidly induce myocardial protection.
Boucher et al. (35) subjected rats to ischemia–reperfusion and treated them with IGF-1, DA, and a combination of IGF-1 and DA 30 mg/kg i.v., or vehicle at the start of 30 min ischemia. Both IGF-1 and DA reduced infarct size and improved cardiac function 3 days after ischemia–reperfusion compared to vehicle. In the reperfused heart, apoptosis was reduced with either or both IGF-1 and DA treatments as measured by reduced TUNEL staining and caspase-3 activity.
Prunier et al. (36) examined the thrombogenic effects of a chronic EPO therapy after MI. Rats underwent coronary occlusion followed by reperfusion. They were assigned to one of the following groups: EPO-A, single i.p. injection of EPO 5,000 U/kg at the time of reperfusion; EPO-C, injection of EPO 5,000 U/kg at the time of reperfusion followed by 300 U/kg/week; PBS-C, injection of vehicle only. After 8 weeks of treatment they were exposed to a prethrombotic test based on partial stenosis of the inferior vena cava. As compared to the rats receiving vehicle only, the rats treated with EPO exhibited a significant reduction in MI size, the hematocrit was significantly increased in EPO-C, but the proportion of rats in which a thrombus occurred was similar in all groups.
Shan et al. (37) subjected mice to 45 min ischemia followed by 4 h reperfusion; EPO 1,000 U/kg, administered right before reperfusion, reduced infarct size assessed by TTC staining. Echocardiography examination suggested that EPO administration significantly improved cardiac function following ischemia–reperfusion. TUNEL assay indicated that EPO treatment decreased apoptosis. EPO administration also significantly increased the level of nuclear GATA-4 phosphorylation in the myocardium which was positively correlated with the reduction of MI. Activation of GATA-4 may be one of the mechanisms by which EPO induced protection against myocardial ischemia–reperfusion injury.
Doue et al. (38) studied rats with left coronary artery occlusion randomized to receive either an intravenous injection of 200 U/kg of EPO or saline immediately after release of the occlusion. This study demonstrated that a single treatment with EPO immediately after release of coronary ligation suppressed cardiac remodeling and functional deterioration. (99m)Tc-annexin V autoradiographs and TUNEL staining confirmed that this change was due to a decrease in the extent of myocardial apoptosis in the ischemic–reperfused region.
A model of murine cardiac arrest was employed by Huang et al. (39). Cardiopulmonary resuscitation was started after 6.5 or 9.5 min of asphyxia-induced cardiac arrest. The resuscitated animals received either erythropoietin (1,000, 3,000, or 5,000 U/kg) or placebo intravenously 3 min after return of spontaneous circulation. Erythropoietin 5,000 U/kg improved post-resuscitation myocardial dysfunction and short-term survival only when cardiac arrest lasted 6.5 min.
Burger et al. (40) investigated the role of heme-oxygenase (HO)-l in the anti-apoptotic effects of EPO in isolated fetal murine cardiac myocytes and in vivo. Pretreatment with 2,500 U/kg i.v. of EPO 24 h and 30 min before coronary ligation decreased infarct size as well as ischemia–reperfusion-induced apoptosis in wild-type mice. However, these effects were significantly diminished in HO-l(−/−) mice. Furthermore, EPO given during ischemia reduced infarct size in mice subjected to I/R, and this effect was blocked by chromium mesoporphyrin, a selective inhibitor of HO-1, in wild-type mice. Moreover, inhibition of p38 diminished the cardioprotective effects of EPO. The authors conclude that upregulation of HO-1 expression via p38 signaling contributes to EPO-mediated cardioprotection during myocardial ischemia-reperfusion.
Burger et al. (41) studied in vivo the antiarrhythmic effects of EPO: wild-type (WT) and nNOS null mice were anesthetized and, after a baseline measurement, subjected to myocardial I/R to provoke ventricular arrhythmias. Pretreatment with 2,500 U/kg of i.v. EPO 24 h and 30 min before ischemia increased nNOS expression and significantly reduced the number of premature ventricular contractions and the incidence of ventricular tachycardia in WT mice. In contrast, treatment with EPO had no effect on premature ventricular contractions or the incidence of ventricular tachycardia in nNOS null mice.
French et al. (42) induced myocardial ischemia for 3 h in 10-week-old C57BL6 mice followed by 72 h of reperfusion. EPO (10,000 U/kg) or an equivalent amount of saline vehicle alone was injected i.p. before ligation or immediately after the onset of reperfusion. Assays of residual left ventricular creatine kinase (CK) activity and calculation of left ventricular CK depletion were performed on left ventricular homogenates harvested 72 h after onset of reperfusion for measurement of infarct size, and echocardiography was performed immediately before harvest of tissue for measurement of function. Both mice administered EPO before ligation or after reperfusion had similar infarct size and echo scores compared with those in control mice administered saline.
Schlecht-Bauer et al. (43) subjected rats to 40 min of coronary artery ligation followed by 72 h or 4 weeks reperfusion and received either darbepoetin (DA) (3–30 mg/kg) or vehicle prior to ischemia. In the DA groups reperfused for 72 h, left ventricular shortening fraction and left ventricular ejection fraction were higher than that in the control rats, in agreement with a smaller left ventricular infarct size. The cardioprotective effect of DA was dose-dependent. DA treatment activated the JAK2/Akt signaling pathway, lowered cleaved caspase-3, and increased both phosphorylated-Bad and phosphorylated-GSK-3beta proteins. This was consistent with the decrease of reactive oxygen species production and the lowered binding of Bad to Bcl-xL and Bcl-2 in a DA30 group of rats. Accordingly, at 4 weeks, in the DA group, left ventricular function was better compared to controls.
Tamareille et al. (44) studied rats after 45 min ischemia and 24 h of reperfusion. The control group was compared with ischemic post-conditioning (three cycles of 10 s reperfusion/10 s ischemia) and EPO (1,000 IU/kg i.v.) at the onset of reperfusion. EPO decreased significantly more infarct size and transmurality than ischemic post-conditioning.
Miki et al. (45) tested the hypothesis that endoplasmic reticulum stress in diabetic hearts impairs phospho-glycogen synthase kinase (GSK)-3beta-mediated suppression of mitochondrial permeability transition pore (mPTP) opening, compromising myocardial response to cytoprotective signaling. Infarction was induced by 20-min coronary occlusion and 2-h reperfusion. Administration of 5,000 U/kg of EPO 15 min before ischemia induced phosphorylation of Akt and GSK-3beta and reduced infarct size (percent risk area) from in control hearts. However, neither phosphorylation of Akt and GSK-3beta nor infarct size limitation was induced by EPO in rats with type 2 diabetes. The authors suggest that disruption of protective signals leading to GSK-3beta phosphorylation and increase in mitochondrial GSK-3beta are dual mechanisms by which increased ER stress inhibits EPO-induced suppression of mPTP opening and cardioprotection in diabetic hearts.
Infarct size after 20-min ischemia followed by 2-h reperfusion was larger in a rat model of type 2 diabetes (Otsuka-Long-Evans-Tokushima fatty (OLETF) rat) than in its control (46). Activation of Jak2-mediated signaling by erythropoietin, given 15 min before ischemia (dose and route of administration not reported), limited infarct size in control rats but not in OLETF rats. The results suggest that the diabetic heart is refractory to protection by Jak2-activating ligands such as EPO because of AT1 receptor-mediated upregulation of calcineurin activity.
Treguer et al. (47) used speckle tracking imaging in a rat model of ischemia–reperfusion to accurately detect the reduction of infarct size and transmural extension of MI induced by 1,000 U/kg of EPO i.v. as early as 24 h after reperfusion. A single bolus of EPO was administered at the onset of reperfusion after 45 min ischemia. EPO significantly decreased infarct size and transmural extension of MI. None of the conventional echocardiography parameters was significantly different between the MI-EPO and MI-control groups. These results support the usefulness of speckle tracking imaging in the early evaluation of a cardioprotective strategy in a rat model of ischemia–reperfusion.
Shen et al. (48) studied the cardioprotective and anti-inflammatory effects of EPO in a rat model of 30-min ischemia followed by 3 h reperfusion. Rats receiving single i.v. boluses of rhEPO of 100–5,000 IU/kg 30 min before ischemia exhibited dose-dependent significant reduction in the incidence of ventricular arrhythmias caused by reperfusion, and markedly decreased serum concentrations of IL-6, IL-8, and tumor necrosis factor-alpha compared with the untreated group.
Stein et al. (49) comparatively assessed the beneficial effects of intra-myocardial application of umbilical cord blood expanded endothelial progenitor cells (eEPCs) and EPO as compared to eEPCs or EPO alone in experimental MI. Nude rats underwent ligation of the left anterior descending coronary artery for 30 min followed by reperfusion. After intra-myocardial transplantation of eEPC and 200 U of EPO, CD68+ leukocyte count and vessel density were enhanced in the border zone of the infarct area. Moreover, apoptosis of transplanted CD31 +TUNEL+eEPC was decreased as compared to transplantation of eEPCs alone. Regional wall motion of the left ventricle was measured using Magnetic Resonance Imaging 4 weeks after infarction. After injection of eEPC in the presence of EPO regional wall motion significantly improved as compared to injection of eEPCs or EPO alone, while global ejection fraction was not affected. In conclusion, intra-myocardial transplantation of eEPC in the presence of EPO during experimental MI improves regional wall motion.
Yano et al. (50) induced MI by 20-min coronary occlusion followed by 2 h reperfusion in spontaneously hypertensive stroke-prone rats (SHR-SPs) and their controls (Wistar-Kyoto rats (WKYs)). Infarct size expressed as a percentage of area-at-risk was larger by 29% in SHR-SPs than in WKYs. Pretreatment with 5,000 U/kg of EPO i.v. 15 min before ischemia significantly limited infarct size in WKYs but not in SHR-SPs. The results suggest that enhanced opening of mitochondrial permeability transition pores by reactive oxygen species and modification of the signal downstream of phospho-glycogen synthase kinase-3b in the mitochondria underlie the increased vulnerability to infarction and the lack of anti-infarct tolerance by EPO, respectively, in hypertensive hypertrophied hearts.
The straightforward clinical transferability of results obtained in models of cardiac ischemia–reperfusion requires some final comments.
In vivo animal studies with EPO in cardiac ischemia–reperfusion model suggest the following:
Practically all studies on small rodents show that doses of EPO as low as 100 U/kg reduce infarct size and improve left ventricular function
Most dose regimens included a dose given before ischemia (clinically unfeasible), other studies showed that EPO was protective even when administered right before or even after the onset of reperfusion (21, 38, 42); in one study the darbepoetin was effective even administered 24 h after the onset of reperfusion (31).
Two out of four studies on larger animals (dog, sheep and two studies with pig) reported the lack of effect of EPO, even if EPO was administered before ischemia and/or right before reperfusion (23, 26,29, 33); one study in the pig showed better left ventricular function than controls, in the presence of unchanged size of infarct.
1.2. Clinical trials
Pharmacokinetics and pharmacodynamics of erythropoietin in healthy volunteers have been well established (51). A single subcutaneous dose of erythropoietin as high as 2,400 IU/kg is well tolerated by healthy people (higher dose was not tested), and while with respect to erythropoiesis the effect of EPO is dose-dependent, the dose higher than 1,800 IU/kg not followed by further increase of reticulocytes in blood. However, the safety and tolerance of erythropoietin in patients, especially patients with developing MI needed to be tested. 33,000 IU of rhEPO (about 450 IU/kg) had been well tolerated by patients when injected during the first 3 days after stroke (52). 200 U/kg had been also injected intravenously to 29 patients during the first 3 days of acute MI with ST-segment elevation additionally to PCI and standard therapy (53). EPO treatment did not result in any complications. The bleeding time, platelet function assay closure time and plasma levels of von Willebrand factor were not altered by the EPO treatment. Safety of EPO in patients in acute stage of MI was also evaluated in the safety arms of clinical trials described below and it was found mostly safe.
Published to date results of completed phase I and II clinical trials are summarized in the Table 3. The first small study (20 patients with first acute ST-segment elevation MI and indications for PCI) was concluded in 2006 in the Netherlands (54). 60,000 IU of Darbepoetin-alfa (long-acting EPO with half-life of 21 h), which is approximately equivalent to 800 IU/kg in the average patient, or placebo were injected intravenously immediately before PCI. Time between PCI and symptom onset was not reported. The serum EPO level was significantly increased 24 h and the progenitor endothelial cells were significantly increased 72 h after treatment in EPO treated vs. placebo cohorts. EPO treatment was well tolerated and no adverse effect of EPO treatment had been noted during 30 days after treatment. However, the LV function measured 4 months after intervention by radionuclide ventriculography showed no differences in LVEF between two groups.
Table 3.
Clinical trials
| Average time to drug |
Primary end-point |
Secondary end-point |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Name/year/ references |
Country | Number of pt |
EPO, type and dose |
From onset |
From PCI | Measurements | Outcome | Measurements | Outcome |
| Lipsic 2006 (21) | The Netherlands |
20 | Darbepoetin alia 60,000 IU |
NA | 0 min | LVEF at 4 m | NS | NA | NA |
| Liem, 2009 (22) | The Netherlands |
51 | Epoetin alfa 40,000 IU |
8 h from elevated tro– ponin |
NA | MI size (enzymes) |
NS | NA | NA |
| Binbrec, 2009 (23, 24) |
Dubai | 236 | Epoetin beta 30,000 IU |
6 h | At the time of thrombolysis |
MI size (enzymes) |
NS | Echo at discharge |
NS |
| EPO/AMI-1, 2010 (25, 26) |
Japan | 36 | Epoetin beta 12,000 IU |
Within 48 h |
Within 24 h | LVEF 4 day and 6 month |
NS | NA | NA |
| HEBE III 2010 (27, 28) |
The Netherlands, Germany |
529 | Epoetin alfa 60,000 IU |
12–24 h | Within 3 h | LVEF at 6 weeks |
NS | MI size (enzymes) |
NS |
| REVIVAL-3, 2010(29) |
Germany | 138 | Epoetin beta 33,300 IU |
Within 24 h | 0 min (add. doses at 24 and 48 h) |
LVEF at 6 month |
NS | ΔLVEF and MI | NS |
| EPOC-AMI, 2010(30) |
Japan | 35 | Epoetin beta 6,000 IU |
Within 24 h | 0 min (add. doses at 24 and 48 h) |
MI size 4 days and 6 month |
NS | NA | NA |
| REVEAL, 2011(31, 32) |
USA | 131 | Epoetin alfa 60,000 IU |
Within 8 h | Within 4 h | MI size 2 to 6 days and 12 weeks |
NS | LV remodeling | NS |
| Ferrario et al., 2011(33) |
Italy | 30 | Epo 33,000 IU |
Within 6 h | 0 min (add. doses at 24 and 48 h) |
MI size (enzymes) LVEF at admission and at 6 months |
MI ↓ p< 0.02 EF ↑ p<0.05 |
Progen. cells at 72 h |
↑ p<0.002 |
A pilot study involving 51 patients with non-STEMI was also conducted in the Netherlands (55). Single intravenous dose of 40,000 IU of Epoetin-alfa was given within 8 h from elevated troponin level to 26 patients. Enzymatic MI size measured at non-STEMI was not different between experimental and placebo groups. Elevation of BP was noted among patients treated with EPO.
In the study conducted in Dubai (56, 57) 236 patients with STEMI, admitted less than 6 h after onset of chest pain were subjected to thrombolytic treatment. 115 of them were additionally treated with intravenous injection of 30,000 of epoetin-beta prior to thrombolytic therapy. The primary end-point was an enzymatically estimated MI size, secondary end-point—echocardiographic measurements at the time of discharge. There were no differences between groups at any of end-point parameters.
Results of a small (16 control, 20 EPO) prospective randomized, single-blind, multicentered trial (EPO/AMI-1) conducted in Japan were reported in 2010 (58, 59). AMI patients with ST-segment elevation were admitted within 24 h after onset of symptoms. They undergo successful PCI and thrombolysis. EPO, 12,000 IU (epoetin-beta) was given within 24 h after PCI. LVEF measured via photon emission CT 4 days after PCI was not different between groups, but at 6-month follow-up LVEF was significantly higher in EPO treated group. Nevertheless, the LVEF increments between 4 days and 6 months measurements were not different between groups.
Combined efforts of clinical groups in the Netherlands and Germany resulted in prospective, randomized, blinded end-points study, HEBE III (60, 61). Patients with first ST-segment elevation MI were hospitalized within 12–24 h of symptom onset and randomized after successful PCI. A single bolus of 60,000 IU of epoetin-alfa or placebo was injected intravenously within 3 h after PCI. MI size measured enzymatically was slightly (but statistically not significant) reduced. However, LVEF 6 weeks after MI was not different between groups.
In the German clinical trial REVIVAL-3 (62) patients diagnosed with ST-segment elevation MI were undergoing primary PCI within 24 h of symptom onset. Epoetin-beta (33,300 IU/kg) was given immediately after PCI. Additional doses were given 24 and 48 h later. Cardiac MRI was performed 5 days and 6 months after MI. There were no differences between EPO treated and control groups in any measured parameter at 5 days or 6 months. The changes of MI size and EF between two groups were also similar.
Japanese clinical trial, EPOC-AMI (63) was similar in design with REVIVAL-3, but EPO dose was lower. Patients with SR-segment elevated MI were subjected to a primary PCI within 12 h of symptom onset and treated with 6,000 IU of epoetin-beta immediately after that. This dose was repeated on day 2 and 4. MI size was measured via Tc99m-tetrofosmin SPECT 4 days and 6 months after PCI. There were no differences between treated and untreated groups at either measurement. However, comparison of these two measurements showed the reduction of MI and some increase of EF with time in EPO-treated group, while changes in untreated group were not statistically significant.
The only clinical trial in the USA was recently completed (64,65). It was randomized, double-blind, placebo-controlled, multicenter phase I–II trial evaluating the effect of epoietin-alpha in the patients with large MI. In the dose escalating safety phase the dose of EPO was tested in the cohorts receiving 15,000, 30,000, and 60,000 IU. In the phase II, 65 patients received EPO within 4 h of PCI (within 8 h of symptom onset). Equal number of patients received placebo at that time. Main outcome was an MI size estimated as a percent of LV mass measured by cardiac magnetic resonance imaging 2–6 days after treatment and 12 weeks later. MI size was not reduced by EPO measured either during the first CMR, or 12 weeks later. There was a tendency of increased MI size and the number of adverse effect in older patients.
A small clinical trial, which authors considered a pilot, completed recently in Italy (66) was the only one so far with encouraging outcome. It was a single-center, randomized, placebo controlled, double-blind study. Thirty patients with a first ST-segment elevation AMI underwent PCI within 6 h of symptoms onset. EPO, 33,000 IU (type is not reported) or placebo were delivered intravenously in 50 mL of saline over 30 min, starting before and continued during PCI. This dose was repeated 24 and 48 h after PCI. MI size was estimated enzymatically. Echocardiography and cardiac magnetic resonance studies were performed shortly after admission and repeated after 6 months. Seventy-two hours after initiation of treatment the level of CD34+ cells was reported four times higher in the EPO group compared with placebo group (p=0.002). Enzymatically measured MI was smaller (p<0.03) in the EPO group. LVEF measured by CMR improved after 6 months in the EPO group vs. placebo. EF measured by echocardiography was not different at 6 months between groups, but in the EPO group EDV did not change and ESV was smaller, while in placebo group these changes were reversed.
Finally, a clinical trial, EPAMINONDAS (Exogenous erythropoietin in Acute Myocardial Infarction New Outlook aNd Dose), is underway in Italy (67). The results are not available at this time. It is a multicenter, randomized controlled trial that enrolls >100 patients with ST-segment elevation MI. Two doses of epoetin-alfa will be tested (100 and 200 IU/kg). EPO treatment (or placebo) will be repeated in the first 3 days after primary PCI with the first dose given within 12 h of procedure. Primary end-points will be an enzymatically measured MI size and results of cardiac MRI. Secondary end-points will be 12-month cardiac remodeling and safety events.
Therefore, all completed clinical trials except one (66) failed to demonstrate any or almost any beneficial effects of EPO treatment in the setting of acute stage of MI. All these trials have one common trait—timing of application of experimental drug is different from that used in the experiment on animals. Experimental data obtained in animal research clearly indicated that effectiveness of EPO treatment reversely proportional to a time elapsed since a coronary occlusion. In experiments involving the model of permanent coronary occlusion the best result were achieved when EPO was applied at the time of occlusion. In ischemia-reperfusion model the most effective results were observed when treatment was applied no later than at the time of reperfusion, i.e., 30–90 min from coronary occlusion. In reviewed clinical trials, even if EPO was injected at the time of PCI, the time from the symptom onset was sometime as long as 24 h. In the only trial with promising outcome (66) the EPO was injected at the time of PCI and the PCI was done within 6 h of the beginning of symptoms.
We propose that critical therapeutic window for using EPO in MI patients should be counted from the time of symptom onset, not from the time of reperfusion. With this proposal in mind we conducted an experiment to test a hypothesis that design of above clinical trials made them impossible to succeed (68). Experiment was designed to closely replicate one of the clinical trials, REVEAL (64, 65). In five groups of 2-month old male Wistar rats, the left descending coronary artery was occluded either by a permanent (two groups) or a temporary ligature (three groups). In the three temporary ligature groups the occluding ligature was released 2 h after occlusion. All animals were sacrificed 24 h after beginning of occlusion and MI size was measured histologically. Animals from one of the permanent occlusion groups and one of the temporary occlusion groups remained untreated. Animals from one of the temporary occlusion groups received intraperitoneal injection of 3,000 IU/kg of epoetin-alfa immediately after reperfusion. Another group received the same dose of EPO 4 h after reperfusion, i.e., 6 h after coronary occlusion. The last group (permanent occlusion) also received the same dose of EPO 6 h after occlusion. Results of MI size 24 h after occlusion are presented in Fig. 1. Our hypothesis had been confirmed: MI size 24 h after coronary occlusion was 42 ± 2% of the area at risk in untreated animals; it was reduced by ~50% (19±2% of the AAR, p<0.0001) only in the group that was treated at the time of reperfusion; MI size in groups in which treatment was delayed by 4 h after reperfusion or by 6 h after permanent occlusion was not different from untreated animals.
Fig. 1.
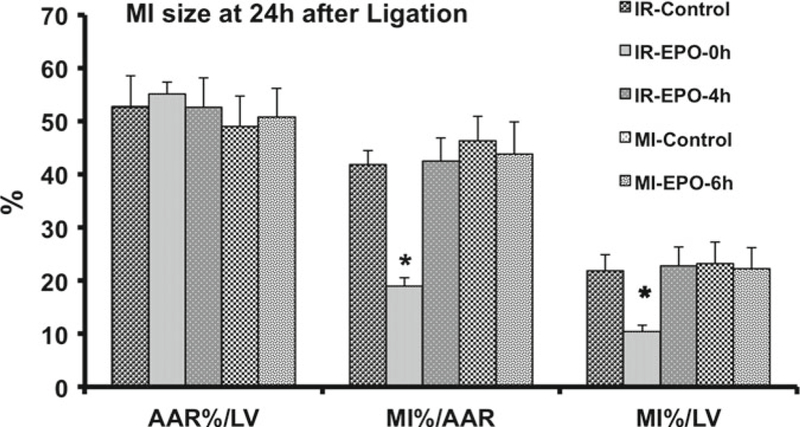
Area of myocardium at risk (AAR) and size of myocardial infarction (MI) 24 h after permanent occlusion of the left descending coronary artery or after 2 h of occlusion followed by 22 h of reperfusion. rhEPO is administered at the time of reperfusion (IR-EPO-0 h) or 4 h after beginning of reperfusion (IR-EPO-4 h) or 6 h after permanent coronary occlusion (MI-EFO-6 h). Data presented as means± SEMI. AAR is presented as percent of left ventricle (LV). MI is presented as percent of AAR or percent of LV *p < 0.05 vs. all other group (Bonferroni post hoc comparison)
1.3. Conclusion
Comparison of experimental data with design of several completed to date clinical trials clearly suggests that failure of these trials to improve clinical outcome by using erythropoietin in the acute phase of MI might be related to design of clinical trials. Experimental animal data definitely indicated that therapeutic window for erythropoietin is narrow and dose-dependent. In the doses that substantially exceed customary therapeutic doses for erythropoietin, the window between coronary occlusion and EPO injection could not exceed 12 h. In doses approaching acceptable therapeutic doses the efficacy of EPO was lost even with small delay from the time of coronary occlusion. These considerations drawn on the basis of animal experiments were disregarded in the clinical trials design. The issue whether this chasm stemmed from regulatory difficulties in conducting clinical trial involving a patient in clinical emergency that creates a pressure on clinicians to shorten the door-to-balloon time and makes it difficult to obtain informed consent early enough, or the problem is much deeper and have its roots in a growing culture among clinical scientist not to accept data obtained from animal research for anything more than proof of concept that can serve only as a trigger to start independent clinical evaluation deserves special discussion elsewhere.
1.4. Experimental Myocardial Models of Infarction
Two, mostly used experimental MI models are permanent occlusion of a coronary artery or temporary coronary occlusion followed by a reperfusion (I/R model). Permanent occlusion is the most “pure” experimental model, which allows studying and intervening into the effects of ischemic damage of the myocardium per se. Nevertheless, reperfusion following acute MI is the most effective intervention to reduce the extent of injury and to improve clinical outcomes, and contemporary treatment of MI always includes reperfusion either by thrombolysis or by percutaneous coronary intervention (PCI). In the light of this clinical mandate I/R experimental model is more clinically relevant model with respect to translation of experimental finding into clinical practice, However, in the past 30 years, it has become apparent that reperfusion may bring additional damage to a part of the viable myocardium via ROS; inflammatory response to reperfusion following ischemia has been identified as a major player in the so called “reperfusion injury”. In large animals (dogs, minipigs) MI can be induced through balloon occlusion of a coronary artery; this technique is similar to clinical percutaneous angioplasty (PCI) and is not discussed here.
The most common small rodents used for this kind of experiments are the mouse and the rat.
The instruments, reagents, and apparatuses used for the two most widely used models of MI in small rodents, permanent ischemia and ischemia–reperfusion, are the same. Therefore, materials are fisted once with specifications when requested by differences between the two models.
2. Materials
Operating table with safe aspiration of anesthetic gas.
Perspex chamber for induction of anesthesia, before tracheal intubation.
Inhalation anesthesia: isoflurane, oxygen.
Vaporizer.
Ventilator for rodents.
23-gauge intravenous cannula (for mouse intubation).
Drugs: ampicilfin; polyvinylpyrrolidone; Evans blue 5% w/v; tetrazolium chloride (TTC).
3. Methods
3.1. Surgery
All procedures are conducted in conformity with the institutional guidelines that are in compliance with national and international laws and policies.
Anesthesia is induced in a small chamber at 5% isoflurane in oxygen and animals are quickly moved from the chamber to the operating table for tracheal intubation.
Anesthesia is maintained with isoflurane 1.5% in oxygen under mechanical ventilation with a ventilator (tidal volume 8–10 mL/kg; 120 strokes/min for the mouse and 70 strokes/ min for the rat) through the endotracheal cannula.
The left anterior descending coronary artery is ligated with a 7-O silk (Ethicon) suture after exteriorization of the heart through a small opening at the fourth-intercostal space.
An overhand knot is tied with two pieces of suture to arrest blood flow for 20–90 min before release of the knot (reperfusion); a permanent knot is tied in case of permanent ischemia. Body temperature of the mouse/rat must be strictly controlled and maintained at 37°C to obtain reproducible infarcts with ischemia-reperfusion. In case of permanent ischemia, temperature is not so critical since the procedure is much shorter: chest is closed under negative pressure right after coronary ligation.
Proper placement of the ligature is confirmed by the appearance of ventricular ectopy and blanching of the myocardium. The chest is the closed under negative pressure and mice are weaned from mechanical ventilation. Postsurgical analgesia is achieved by buprenorphine (0.1 mg/kg s.c. q12h for 1 day).
3.2. In vivo imaging for serial assessment of progression of cardiac remodeling, function and Ml size
The two most widely used noninvasive imaging techniques in studies in small rodents are echocardiography and magnetic resonance imaging.
Echocardiography, which can easily be performed on awake or lightly sedated mice, has become the procedure of choice for most studies of the mouse heart for its flexibility. While echocardiography in small rodents, first in the rat then in the mouse, has been performed with apparatuses used for clinical activity, choosing mostly vascular probes of 7–15 MHz, in the last decade, new instruments designed for small animal exams have been made commercially available. The high-frequency probes (30 MHz) allow a higher spatial resolution (30 mn) and, consequently, a more accurate assessment of heart dimensions and function. However, the major difficulties of this technique are the geometric assumptions, image positioning errors, and use of subjective visual methods thus making necessary a skilled sonographer. Other limitations are LV wall motion assessment because of postsurgical adhesions and chest deformations, and a not well visualization of the right ventricle.
Magnetic resonance imaging (MRT) represents the gold standard for measurement of cardiac morphology and function either in humans or in small rodents. It is noninvasive, accurate, free from geometric assumptions compared to echocardiography. The contiguous stack of short-axis images of the heart covers the entire length of the LV and allows for an accurate estimation of both left and right ventricular mass, volumes, and ejection fraction. Even though MRI is a more reliable method than echocardiography for the characterization of the pathophysiological consequences of experimental MI and of treatments response in rodents, MRI exams usually last more than 1 h, require deep anesthesia, and is much more expensive. For these reasons it is not routinely used in large series of animals.
Transthoracic echocardiography can be performed with an instrument for clinical use on conscious, previously trained mice with the use of a 12–15 MHz linear transducer at high frame rate imaging (102 Hz) and a 7.5 MHz phased array probe for pulsed- wave Doppler measurements. An echocardiograph, especially design for mice, is used to examine anesthetized (0.5–1.5% isoflurane) animals. High frequency of its probes, 30 MHz, allows higher spatial resolution. The rat cannot be easily trained, therefore, needs to be evaluated under sedation, with ketamine and imidazolam for example, or under anesthesia with 0.5–1.5% isoflurane.
Short and long-axis two dimensional (2D) views and M-mode at the level of infarction is analyzed in real time and recorded on magneto-optic disk for off-line analysis by a sonographer blind to study groups. Left atrial diameter, end-diastolic and end-systolic wall thicknesses and left ventricular internal dimensions are measured, as recommended by the American Society of Echocardiography (see Note 1).
As for echocardiography, more than one model and set up for MRI exist on the market. Since the detailed description of the instrumental settings goes far beyond the aims of the present chapter, the setting used in the laboratory of one of the authors (RL) is described (see Note 2).
All animals are anesthetized by facemask with isoflurane (induction in chamber at 3–5%, maintenance 0.5–1.5%) and 0.3 L/min O2.
Animals are positioned in a purpose-built cradle and ECG electrodes are attached to the front paws. NOTE: ad hoc properly shielded ECG leads and cables must be purchased in order to avoid the instrument-derived noise that would completely mask the ECG signal.
A pressure-transducer for respiratory gating is positioned above the abdomen.
A fiber optic probe is used for monitoring the rectal temperature.
ECG and respiratory signals are processed and displayed using a gating device and temperature is maintained at 37±0.5°C during the whole exam (60–90 min).
After the image plane orientation from coronal, axial and oblique LV long-axis, a 2 chamber long-axis view is obtained and orthogonal to it a 4 chamber long-axis is acquired followed by 1 mm serial short-axis slices covering the entire LV length. Seven to ten short-axis slices are acquired from base to apex. Sixteen frames per slice for one cine sequence are saved. Images are exported in DICOM format and analyzed off-line.
End-diastolic and end-systolic variables are measured in selected frames according to the visual estimation of the maximal and minimal ventricular cavity respectively.
LV anterior and posterior diameters and wall thicknesses are measured at the mid-papillary level. End-diastolic and systolic endocardial areas of each slice are traced manually. LV volumes are calculated by the modified formula for Simpson’s rule.
As for echocardiography, it is recommended that two investigators blind to the experimental conditions read MRI recordings even though the better definition of the images reduce the inter and intra-reader variability.
3.3. Hemodynamics
Prior to euthanasia LV pressure or pressure–volume loop analyses can be performed. Anesthesia can be provided by different routes and with different agents. The authors routinely use (a) isoflurane (2% in oxygen), rats are intubated and mechanically ventilated, or (b) pentobarbital 40–60 mg/kg i.p.
Two approaches are possible:
The less invasive approach is by pushing a Millar pressure catheter (indicatively SPC-320 for the rat, SPR-671 for the mouse) or a Millar pressure–conductance catheter through the right carotid artery to the left ventricle.
The open chest approach requires a bilateral thoracotomy in fourth and fifth intercostal. After opening the pericardium, a catheter (Millar Instruments Inc., Houston, TX) is inserted into the LV from the apex.
Naturally, pressure catheters measures are limited to a LV pressures and their derivatives. Pressure-conductance catheter allow for a full array of pressure–volume loop analyses: LV end-diastolic pressure (EDP), end-diastolic (EDV) and end-systolic(ESV) volumes, stroke volume (SV), +dP/dt, −dP/dt, isovolumic relaxation time (tau), and arterial elastance (Ea) are determined in 10–20 digitally averaged cardiac cycles while the ventilator is stopped. LV end-systolic elastance (Ees), preload recruitable stroke work (PRSW) and end-diastolic stiffness (Eed) are measured using a graded preload reduction technique. Arterioventricular (AV) coupling is calculated as Ea/Ees. The test is concluded by advancing the catheter into thoracic aorta to measure arterial blood pressure.
3.4. Gross Pathology and Histological Assessment
The hearts and lungs are removed and weighed (wet weight). Hearts are processed and the histological analyses are performed at different time depending on experiment design. Three approaches are used by the authors:
Hearts are cut into two pieces through the short axis. The basal half is fast frozen and stored for different assays, and the apical half is used for histological analysis.
Hearts are slightly frozen to reach a consistency which favors thin and precise cutting, are cut with a chopper into 1 mm-thick sections which are then incubated in TTC for determination of infarct size and the area at risk (AAR is stained in the red color).
Hearts are cut into two to three transverse sections (perpendicular to the long axis) starting from slightly above the coronary ligature (in case of permanent ischemia the thread is in place and visible), or above the visible area of injury (in case of ischemia-reperfusion the thread is preferably not left in place to decrease the occurrence of adhesions between epicardium and chest wall). The sections are put in 10% buffered formalin to be processed within 1 month for paraffin embedding.
Methods 1 and 2 are usually employed in case of long-term experiments (>7 days follow-up), while method 2 is for short-term experiments (2–24 h).
Myocardial tissue sections are subjected to Masson’s trichrome and hematoxylin-eosin staining. Myocyte cell size and density are measured in H&E-stained sections (see Note 3).
To avoid likely biases, it is crucial that the person assessing all histological slides is blinded to a grouping.
Footnotes
Notes
Fractional shortening (FS) is calculated from the composite LV internal, diastolic (LVIDd) and systolic (LVIDs) dimensions as: FS = ((LVIDd−LVIDs)/LVIDd)× 100 from M-mode short axis view. Left ventricular (LV) volumes and LVEF are calculated by the modified simple plane Simpson’s rule from the parasternal long-axis view. The MI size at the mid-papillary muscle level is estimated from 2D short axis LV images and expressed as a percent of the LV endocardial circumference. Infarct area is identified as a sharply demarcated section of the LV free wall that fails to thicken during systole. The length of the akinetic part of the LV endocardial circumference is measured from freeze-frame images at end-diastole. Aortic outflow and diastolic transmitral LV inflow velocities are measured from 4 to 5 apical chamber views respectively by pulsed-wave Doppler with a sample volume length of 3.5–7.5 mm; the ultrasound beam is aligned as parallel as possible to Color Doppler flow and to record the highest velocities.
Usually the anesthetized rat/mouse is placed in a horizontal bore 7 T Biospec preclinical scanner with a shielded gradient insert (BGA 12, 40 mT/m; rise time, 110 ms). A quadrature or linear volume coil and rat/mouse surface coil are used to transmit/receive the magnetic resonance signals (Quadrature or Linear volume coil inner diameter, 72 mm; rat/mouse surface coil inner diameter, 20 mm/10 mm). After localization of the heart, scans (segmented double-gated FLASH imaging) are run to ensure correct positioning; slice-selective shimming and flip angle calibration are performed manually before each experiment. The parameters of Cine-MRI pulse-sequence are: matrix, 256×128; FOV, 40.0×20.0 mm; echo time, 2.98 ms; flip angle, 15°; slice thickness, 1 mm; number of averages, 6; repetition time, 12 ms.
Only myocytes which nuclei are clearly identified are counted. Myocyte diameter is measured as the shortest distance across the nucleus in transverse cell sections. Diameters of 100 myocytes from five randomly selected microscope fields (×200 magnification) from the LV posterior wall are averaged to represent the myocyte diameter of a given specimen. Myocyte density is calculated from the same area in the same fashion. Myocardial tissue fibrosis is measured in Masson’s trichrome-stained sections and is expressed as a fraction of a microscopic field (×l00 magnification) of the LV posterior wall. An average of five randomly selected fields represented results of a given specimen. Collagen content in the thoracic aorta wall is measured on 7 mm fresh frozen sections stained with Masson’s trichrome. Digital images of stained sections are obtained from light microscopy and analyzed using a digital imaging analysis system (MCID, InterFocus Imaging Ltd, Cambridge, UK). The collagen content in aortic wall is calculated as a percentage of the total wall thickness or tunica media.
Contributor Information
Mark I. Talan, Laboratory of Cardiovascular Sciences, National Institute on Aging, NIH, Baltimore, MD, USA
Roberto Latini, Department of Cardiovascular Research, Istituto Mario Negri, Milan, Italy.
References
- 1.Bogoyevitch MA (2004) An update on the cardiac effects of erythropoietin cardioprotection by erythropoietin and the lessons learnt from studies in neuroprotection. Cardiovasc Res 63(2):208–216, Review [DOI] [PubMed] [Google Scholar]
- 2.Koul D, Dhar S, Chen-Scarabelli C, Guglin M, Scarabelli TM (2007) Erythropoietin: new horizon in cardiovascular medicine. Recent Pat Cardiovasc Drug Discov 2(1):5–12, Review [DOI] [PubMed] [Google Scholar]
- 3.Riksen NP, Hausenloy DJ, Yellon DM (2008) Erythropoietin: ready for prime-time cardioprotection. Trends Pharmacol Sei 29(5):258– 267, Review [DOI] [PubMed] [Google Scholar]
- 4.Latini R, Brines M, Fiordaliso F (2008) Do non-hemopoietic effects of erythropoietin play a beneficial role in heart failure? Heart Fail Rev 13(4):415–423, Review [DOI] [PubMed] [Google Scholar]
- 5.Vogiatzi G, Briasoulis A, Tousoulis D, Papageorgiou N, Stefanadis C (2010) Is there a role for erythropoietin in cardiovascular disease? Expert Opin Biol Ther 10(2):251–264, Review [DOI] [PubMed] [Google Scholar]
- 6.Lipsic E, Schoemaker RG, van der Meer P, Voors AA, van Veldhuisen DJ, van Gilst WH (2006) Protective effects of erythropoietin in cardiac ischemia: from bench to bedside. J Am Coll Cardiol 48(11):2161–2167, Epub 2006 Nov 9. Review [DOI] [PubMed] [Google Scholar]
- 7.Tramontano AF, Muniyappa R, Black AD, Blendea MC, Cohen I, Deng L, Sowers JR, CutaiaMV El-SherifN (2003) Erythropoietin protects cardiac myocytes from hypoxia- induced apoptosis through an Akt-dependent pathway. Biochem Biophys Res Commun 308(4):990–994 [DOI] [PubMed] [Google Scholar]
- 8.Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG, Talan MI (2003) Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc Natl Acad Sei USA 100(20):11612–11617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon C, Krawczyk M, Paik D, Lakatta EG, Talan MI (2005) Cardioprotection by recombinant human erythropoietin following acute experimental myocardial infarction: dose response and therapeutic window. Cardiovasc Drugs Ther 19(4):243–250 [DOI] [PubMed] [Google Scholar]
- 10.Moon C, Krawczyk M, Lakatta EG, Talan MI (2006) Therapeutic effectiveness of a single vs. multiple doses of erythropoietin after experimental myocardial infarction in rats. Cardiovasc Drugs Ther 20(4):245–251 [DOI] [PubMed] [Google Scholar]
- 11.Moon C, Krawczyk M, Paik D, Coleman T, Brines M, Juhaszova M, Sollott SJ, Lakatta EG, Talan MI (2006) Erythropoietin, modified to not stimulate red blood cell production, retains its cardioprotective properties. J Pharmacol Exp Ther 316(3):999–1005 [DOI] [PubMed] [Google Scholar]
- 12.Ahmet I, Tae HJ, Juhaszova M, Riordon DR, Boheler KR, Sollott SJ, Brines M, Cerami A, Lakatta EG, Talan MI (2011) A small non- erythropoietic helix B surface peptide based upon erythropoietin structure is cardioprotective against ischemic myocardial damage. Mol Med 17(3—4):194–200. doi: 10.2119/molmed.2010.00235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB, Thompson RB, Pctrofski JA, Annex BH, Stamler JS, Koch WJ (2003) A novel protective effect of erythropoietin in the infkrcted heart. J Clin Invest 112(7):999–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Meer P, Lipsic E, Henning RH, Boddeus K, van der Velden J, Voors AA, van Veldhuisen DJ, van Gilst WH, Schoemaker RG (2005) Erythropoietin induces neovascularization and improves cardiac function in rats with heart failure after myocardial infarction. J Am Coll Cardiol 46(1):125–133 [DOI] [PubMed] [Google Scholar]
- 15.Hale SL, Sesti C, Kloner RA (2005) Administration of erythropoietin fails to improve long-term healing or cardiac function after myocardial infarction in the rat. J Cardiovasc Pharmacol 46(2):211–215 [DOI] [PubMed] [Google Scholar]
- 16.Hirata A, Minamino T, Asanuma H, Fujita M, Wakeno M, Myoishi M, Tsukamoto O, Okada K, Koyama H, Komamura K, Takashima S, Shinozaki Y, Mori H, Shiraga M, Kitakaze M, Hori M (2006) Erythropoietin enhances neovascularization of ischemic myocardium and improves left ventricular dysfunction after myocardial infarction in dogs. J Am Coll Cardiol 48(1):176–184, Epub 2006 Jun 2 [DOI] [PubMed] [Google Scholar]
- 17.Chua S, Leu S, Lin YC, Sheu JJ, Sun CK, Chung SY, Chai HT, Lee FY, Kao YH, Wu CJ, Chang LT, Ko SF, Yip HK (2011) Early erythropoietin therapy attenuates remodeling and preserves function of left ventricle in porcine myocardial infarction. J Investig Med 59(3):574–586 [DOI] [PubMed] [Google Scholar]
- 18.Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, Salio M, Cerami A, Brines M (2003) Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sei USA 100:4802–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsa CJ, Kim J, Riel RU, Pascal LS, Thompson RB, Petrofski JA, Matsumoto A, Stamler JS, Koch WJ (2004) Cardioprotective effects of erythropoietin in the reperfused ischemic heart: a potential role for cardiac fibroblasts. J Biol Chem 279:20655–20662 [DOI] [PubMed] [Google Scholar]
- 20.Abdelrahman M, Sharpies EJ, McDonald MC, Collin M, Patel NS, Yaqoob MM, Thiemermann C (2004) Erythropoietin attenuates the tissue injury associated with hemorrhagic shock and myocardial ischemia. Shock 22:63–69 [DOI] [PubMed] [Google Scholar]
- 21.Lipsic E, van der Meer P, Henning RH, Suurmeijer AJ, Boddeus KM, van Veldhuisen DJ, van Gilst WH, Schoemaker RG (2004) Timing of erythropoietin treatment for cardioprotection in ischemia/reperfusion. J Cardiovasc Pharmacol 44:473–479 [DOI] [PubMed] [Google Scholar]
- 22.Rui T, Feng Q, Lei M, Peng T, Zhang J, Xu M, Abel ED, Xenocostas A, Kvietys PR (2005) Erythropoietin prevents the acute myocardial inflammatory response induced by ischemia/ reperfusion via induction of AP-1. Cardiovasc Res 65:719–727 [DOI] [PubMed] [Google Scholar]
- 23.Hirata A, Minamino T, Asanuma H, Sanada S, Fujita M, Tsukamoto O, Wakeno M, Myoishi M, Okada K, Koyama H, Komamura K, Takashima S, Shinozaki Y, Mori H, Tomoike H, Hori M, Kitakaze M (2005) Erythropoietin just before reperfusion reduces both lethal arrhythmias and infarct size via the phosphatidylinositol-3 kinase-dependent pathway in canine hearts. Cardiovasc Drugs Ther 19:33–40 [DOI] [PubMed] [Google Scholar]
- 24.Xu B, Dong GH, Liu H, Wang YQ, Wu HW, Jing H (2005) Recombinant human erythropoietin pretreatment attenuates myocardial infarct size: a possible mechanism involves heat shock Protein 70 and attenuation of nuclear factor-kappa B. Ann Clin Lab Sei 35:161–168 [PubMed] [Google Scholar]
- 25.Bullard AJ, Govewalla P, Yellon DM (2005) Erythropoietin protects the myocardium against reperfusion injury in vitro and in vivo. Basic Res Cardiol 100:397–403 [DOI] [PubMed] [Google Scholar]
- 26.Kristensen J, Maeng M, Rehling M, Berg JS, Mortensen UM, Nielsen SS, Nielsen TT (2005) Lack of acute cardioprotective effect from pre-ischaemic erythropoietin administration in a porcine coronary occlusion model. Clin Physiol Funct Imaging 25:305–310 [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Xie W, Liu P, Duan M, Jia Z, Li W, Xu J (2006) Mechanism of the cardioprotection of rhEPO pretreatment on suppressing the inflammatory response in ischemia-reperfusion. Life Sei 78:2255–2264 [DOI] [PubMed] [Google Scholar]
- 28.Nishihara M, Miura T, Miki T, Sakamoto J, Tanno M, Kobayashi H, Ikeda Y, Ohori K, Takahashi A, Shimamoto K (2006) Erythropoietin affords additional cardioprotection to preconditioned hearts by enhanced phosphorylation of glycogen synthase kinase-3 beta. Am J Physiol Heart Circ Physiol 29LH748–H755 [DOI] [PubMed]
- 29.Olea FD, Vera Janavel G, De Lorenzi A, Cuniberti L, Yannarelli G, Cabeza Meckert P, Cearras M, Laguens R, Crottogini A (2006) High-dose erythropoietin has no long-term protective effects in sheep with reperfused myocardial infarction. J Cardiovasc Pharmacol 47:736–741 [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Zhou Z, Feng X, Jia Z, Jin Y, Xu J (2006) Cyclooxygenase-2 plays an essential part in cardioprotection of delayed phase of recombinant human erythropoietin preconditioning in rats. Postgrad Med J 82:588–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao E, Boucher M, Chuprun JK, Zhou RH, Eckhart AD, Koch WJ (2007) Darbepoetin alfa, a long-acting erythropoietin analog, offers novel and delayed cardioprotection for the ischemic heart. Am J Physiol Heart Circ Physiol 293:H60–H68 [DOI] [PubMed] [Google Scholar]
- 32.Baker JE, Kozik D, Hsu AK, Fu X, Tweddell JS, Gross GJ (2007) Darbepoetin-alfa protects the rat heart against infarction: dose-response, phase of action, and mechanisms. J Cardiovasc Pharmacol 49:337–345 [DOI] [PubMed] [Google Scholar]
- 33.Toma C, Letts DP, Tanabe M, Gorcsan J III, Counihan PJ (2007) Positive effect of darbepoetin on peri-infarction remodeling in a porcine model of myocardial ischemia-reperfusion. J Mol Cell Cardiol 43:130–136 [DOI] [PubMed] [Google Scholar]
- 34.Singh D, Kolarova JD, Wang S, Ayoub IM, Gazmuri RJ (2007) Myocardial protection by erythropoietin during resuscitation from ventricular fibrillation. Am J Ther 14:361–368 [DOI] [PubMed] [Google Scholar]
- 35.Boucher M, Pesant S, Lei YH, Nanton N, Most P, Eckhart AD, Koch WJ, Gao E (2008) Simultaneous administration of insulin-like growth factor-1 and darbepoetin-alfa protects the rat myocardium against myocardial infarction and enhances angiogenesis. Clin Transi Sei 1:13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prunier F, Pottier P, Clairand R, Mercier A, Hajjar RJ, Planchon B, Furber A (2009) Chronic erythropoietin treatment decreases post-infarct myocardial damage in rats without venous thrombogenic effect. Cardiology 112:129–134 [DOI] [PubMed] [Google Scholar]
- 37.Shan X, Xu X, Cao B, Wang Y, Guo L, Zhu Q, Li J, Que L, Chen Q, Ha T, Li C, Li Y (2009) Transcription factor GATA-4 is involved in erythropoietin-induced cardioprotection against myocardial ischemia/reperfusion injury. Int J Cardiol 134:384–392 [DOI] [PubMed] [Google Scholar]
- 38.Doue T, Ohtsuki K, Ogawa K, Ueda M, Azuma A, Saji H, Strauss HW, Matsubara H (2008) Cardioprotective effects of erythropoietin in rats subjected to ischemia-reperfusion injury: assessment of infarct size with 99mTc-annexin V. J Nucl Med 49:1694–1700 [DOI] [PubMed] [Google Scholar]
- 39.Huang CH, Hsu CY, Tsai MS, Wang TD, Chang WT ChenWJ (2008. ) Cardioprotective effects of erythropoietin on post-resuscitation myocardial dysfunction in appropriate therapeutic windows. Crit Care Med 36: S467–S473 [DOI] [PubMed] [Google Scholar]
- 40.Burger D, Xiang F, Hammoud L, Lu X, Feng Q (2009) Role of heme oxygenase-1 in the cardioprotective effects of erythropoietin during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol 296:H84–H93 [DOI] [PubMed] [Google Scholar]
- 41.Burger DE, Xiang FL, Hammoud L, Jones DL, Feng Q (2009) Erythropoietin protects the heart from ventricular arrhythmia during ischemia and reperfusion via neuronal nitric- oxide synthase. J Pharmacol Exp Ther 329:900–907 [DOI] [PubMed] [Google Scholar]
- 42.French CJ, Zaman AK, Sobel BE (2009) Failure of erythropoietin to render jeopardized ischemic myocardium amenable to incremental salvage by early reperfusion. Coron Artery Dis 20:295–299 [DOI] [PubMed] [Google Scholar]
- 43.Schlecht-Bauer D, Antier D, Machet MC, Hyvelin JM (2009) Short- and long-term cardioprotective effect of darbepoetin-alpha: role of Bcl-2 family proteins. J Cardiovasc Pharmacol 54:223–231 [DOI] [PubMed] [Google Scholar]
- 44.Tamareille S, Ghaboura N, Treguer F, Khachman D, Croué A, Henrion D, Furber A, Prunier F (2009) Myocardial reperfusion injury management: erythropoietin compared with postconditioning. Am J Physiol Heart Circ Physiol 297:H2035–H2043 [DOI] [PubMed] [Google Scholar]
- 45.Miki T, Miura T, Hotta H, Tanno M, Yano T, Sato T, Terashima Y, Takada A, Ishikawa S, Shimamoto K (2009) Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes 58:2863–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hotta H, Miura T, Miki T, Togashi N, Maeda T, Kim SJ, Tanno M, Yano T, Kuno A, Itoh T, Satoh T, Terashima Y, Ishikawa S, Shimamoto K (2010) Angiotensin II type 1 receptor- mediated upregulation of caldneurin activity underlies impairment of cardioprotective signaling in diabetic hearts. Circ Res 106:129–132 [DOI] [PubMed] [Google Scholar]
- 47.Treguer F, Donal E, Tamareille S, Ghaboura N, Derumeaux G, Furber A, Prunier F (2010) Speckle tracking imaging improves in vivo assessment of EPO-induced myocardial salvage early after ischemia-reperfusion in rats. Am J Physiol Heart Circ Physiol 298:H1679–H1686 [DOI] [PubMed] [Google Scholar]
- 48.Shen Y, Wang Y, Li D, Wang C, Xu B, Dong G, Huang H, Jing H (2010) Recombinant human erythropoietin pretreatment attenuates heart ischemia-reperfusion injury in rats by suppressing the systemic inflammatory response. Transplant Proc 42:1595–1597 [DOI] [PubMed] [Google Scholar]
- 49.Stein A, Knödler M, Makowski M, Kühnei S, Nekolla S, Keithahn A, Weidl E, Groha P, Schürmann M, Saraste A, Botnar R, Oostendorp RA, Ott I (2010) Local erythropoietin and endothelial progenitor cells improve regional cardiac function in acute myocardial infarction. BMC Cardiovasc Disord 10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yano T, Miki T, Tanno M, Kuno A, Itoh T, Takada A, Sato T, Kouzu H, Shimamoto K, Miura T (2011) Hypertensive hypertrophied myocardium is vulnerable to infarction and refractory to erythropoietin-induced protection. Hypertension 57:110–115 [DOI] [PubMed] [Google Scholar]
- 51.Cheung WK, Goon BL, Guilfoyle MC, Wacholtz MC (1998) Pharmacokinetics and pharmacodynamics of recombinant human erythropoietin after single and multiple subcutaneous doses to healthy subjects. Clin Pharmacol Ther 64(4):412–423 [DOI] [PubMed] [Google Scholar]
- 52.Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Rüther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Sirén AL (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 8(8):495–505 [PMC free article] [PubMed] [Google Scholar]
- 53.Tang YD, Hasan F, Giordano FJ, Pfau S, Rinder HM, Katz SD (2009) Effects of recombinant human erythropoietin on platelet activation in acute myocardial infarction: results of a double-blind, placebo-controlled, randomized trial. Am Heart J 158(6):941–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lipsic E, van der Meer P, Voors AA, Westenbrink BD, van den Heuvel AF, de Boer HC, van Zonneveld AJ, Schoemaker RG, van Gilst WH, Zijlstra F, van Veldhuisen DJ (2006) A single bolus of a long-acting erythropoietin analogue darbepoetin-alfa in patients with acute myocardial infarction: a randomized feasibility and safety study. Cardiovasc Drugs Ther 20(2):135–141 [DOI] [PubMed] [Google Scholar]
- 55.Liem A, van de Woestijne AP, Bruijns E, Roeters van Lennep HW, de Boo JA, van Halteren HK, van Es TP, Jukema JW, van der Laarse A, Zwinderman AH, van Veldhuisen DJ (2009) Effect of EPO administration on myocardial infarct size in patients with non-STE acute coronary syndromes; results from a pilot study. Int J Cardiol 131(2):285–287, Epub 2007 Nov 1 [DOI] [PubMed] [Google Scholar]
- 56.Binbrek AS, Mittal B, Rao KN, Sobel BE (2007) The potential of erythropoietin for conferring cardioprotection complementing reperfusion. Coron Artery Dis 18(7):583–585, Review [DOI] [PubMed] [Google Scholar]
- 57.Binbrek AS, Rao NS, Al Khaja N, Assaqqaf J, Sobel BE (2009) Erythropoietin to augment myocardial salvage induced by coronary thrombolysis in patients with ST segment elevation acute myocardial infarction. Am J Cardiol 104(8):1035–1040 [DOI] [PubMed] [Google Scholar]
- 58.Ozawa T, Toba K, Suzuki H, Kato K, Iso Y, Akutsu Y, Kobayashi Y, Takeyama Y, Kobayashi N, Yoshimura N, Akazawa K, Aizawa Y, EPO/ AMI-I Pilot Study Researchers (2010) Singledose intravenous administration of recombinant human erythropoietin is a promising treatment for patients with acute myocardial infarction—randomized controlled pilot trial of EPO/AMI-1 study. Cire J 74(7):1415–1423 [DOI] [PubMed] [Google Scholar]
- 59.Yoshimura N, Toba K, Ozawa T, Aizawa Y, Hosoya T (2010) A novel program to accurately quantify infarction volume by (99 m)Tc MIBI SPECT, and its application for re-analyzing the effect of erythropoietin administration in patients with acute myocardial infarction. Cire J 74(12):2741–2743 [DOI] [PubMed] [Google Scholar]
- 60.Belonje AM, Voors AA, van Gilst WH, Anker SD, Slart RH, Tio RA, Zijlstra F, van Veldhuisen DJ, HEBE III investigators (2008) Effects of erythropoietin after an acute myocardial infarction: rationale and study design of a prospective, randomized, clinical trial (HEBE III). Am Heart J 155(5):817–822 [DOI] [PubMed] [Google Scholar]
- 61.Voors AA, Belonje AM, Zijlstra F, Hillege HL, Anker SD, Slart RH, Tio RA, van’t Hof A, Jukema JW, Peels HO, Henriques JP, Ten Berg JM, Vos J, van Gilst WH, van Veldhuisen DJ, HEBE III Investigators (2010) A single dose of erythropoietin in ST-elevation myocardial infarction. Eur Heart J 31(21):2593–2600 [DOI] [PubMed] [Google Scholar]
- 62.Ott I, Schulz S, Mehilli J, Fichtner S, Hadamitzky M, Hoppe K, Ibrahim T, Martinoff S, Massberg S, Laugwitz KL, Dirschinger J, Schwaiger M, Kastrati A, Schmig A, REVIVAL-3 Study Investigators (2010) Erythropoietin in patients with acute ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention: a randomized, double-blind trial. Circ Cardiovasc Interv 3(5):408–413 [DOI] [PubMed] [Google Scholar]
- 63.Taniguchi N, Nakamura T,Sawada T,Matsubara K, Furukawa K, Hadase M, Nakahara Y, Nakamura T, Matsubara H (2010) Erythropoietin prevention trial of coronary restenosis and cardiac remodeling after ST-elevated acute myocardial infarction (EPOC-AMI): a pilot, randomized, placebo-controlled study. Cire J 74(11 ):2365–2371 [DOI] [PubMed] [Google Scholar]
- 64.Melloni C, Rao SV, Povsic TJ, Melton L, Kim RJ, Kilaru R, Patel MR, Talan M, Ferrucci L, Longo DL, Lakatta EG, Najjar SS, Harrington RA (2010) Design and rationale of the reduction of infarct expansion and ventricular remodeling with erythropoietin after large myocardial infarction (REVEAL) trial. Am Heart J 160(5):795–803, e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Najjar SS, Rao SV, Melloni C, Raman SV, Povsic TJ, Melton L, Bareness GW, Prather K, Heitner JF, Kilaru R, Gruberg L, Hasselblad V, Greenbaum AB, Patel M, Kim RJ, Talan M, Ferrucci L, Longo DL, Lakatta EG, Harrington RA, REVEAL Investigators (2011) Intravenous erythropoietin in patients with ST-segment elevation myocardial infarction: REVEAL: a randomized controlled trial. JAMA 305(18): 1863–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferrario M, Arbustini E, Massa M, Rosti V, Marziliano N, Raineri C, Campanelli R, Bertoletti A, De Ferrari GM, Klersy C, Angoli L, Bramucci E, Marinoni B, Ferlini M, Moretti E, Raisaro A, Repetto A, Schwartz PJ, Tavazzi L (2011) High-dose erythropoietin in patients with acute myocardial infarction: a pilot, randomised, placebo-controlled study. Int J Cardiol 147(1 ):124–131 [DOI] [PubMed] [Google Scholar]
- 67.Andreotti F, Agati L, Conti E, Santucci E, Rio T, Tarantino F, Natale L, Berardi D, Mattatelli A, Musumeci B, Bonomo L, Volpe M, Crea F, Autore C (2009) Update on phase II studies of erythropoietin in acute myocardial infarction. Rationale and design of Exogenous erythroPoietin in Acute Myocardial Infarction: New Outlook aNd Dose Association Study (EPAMINONDAS). J Thromb Thrombolysis 28(4):489–495 [DOI] [PubMed] [Google Scholar]
- 68.Ahmet I, Lakatta EG, Talan MI (2011) Therapeutic window for cardioprotection by recombinant human erythropoietin (rhEPO) in the rat model of ischemia-reperfusion: implications for clinical trial design. AHA