

Abstract
Background
Drug repurposing has been an interesting and cost-effective approach, especially for neglected diseases, such as Chagas disease.
Methods
In this work, we studied the activity of the antidepressant drug sertraline against Trypanosoma cruzi trypomastigotes and intracellular amastigotes of the Y and Tulahuen strains, and investigated its action mode using cell biology and in silico approaches.
Results
Sertraline demonstrated in vitro efficacy against intracellular amastigotes of both T. cruzi strains inside different host cells, including cardiomyocytes, with IC50 values between 1 to 10 μM, and activity against bloodstream trypomastigotes, with IC50 of 14 μM. Considering the mammalian cytotoxicity, the drug resulted in a selectivity index of 17.8. Sertraline induced a change in the mitochondrial integrity of T. cruzi, resulting in a decrease in ATP levels, but not affecting reactive oxygen levels or plasma membrane permeability. In silico approaches using chemogenomic target fishing, homology modeling and molecular docking suggested the enzyme isocitrate dehydrogenase 2 of T. cruzi (TcIDH2) as a potential target for sertraline.
Conclusions
The present study demonstrated that sertraline had a lethal effect on different forms and strains of T. cruzi, by affecting the bioenergetic metabolism of the parasite. These findings provide a starting point for future experimental assays and may contribute to the development of new compounds.
Electronic supplementary material
The online version of this article (10.1186/s40409-018-0165-8) contains supplementary material, which is available to authorized users.
Keywords: Trypanosoma cruzi, Drug, Treatment, Sertraline, Drug repurposing, Drug repositioning
Background
Chagas disease is a neglected tropical protozoan disease that affects 8 million people, mainly in South and Central America [1]; however, cases have also been reported in North America, Europe and some other Western countries [2]. The current treatment of Chagas disease is an alarming issue. A recent clinical study re-evaluated the efficacy of benznidazole (BZ), the only available treatment in Brazil; despite a reduction in parasitemia, the study revealed that BZ induced no significant improvements in cardiac clinical outcomes during chronic-phase Chagas disease [3]. According to the non-profit organization Drugs for Neglected Diseases initiative (DNDi), in the next 5 years, 200,000 people living with Chagas disease will die from heart disease and related complications. DNDi also highlighted the urgent need to find better treatments [4]. Additionally, the drug safety of the available treatment is inadequate; BZ is implicated in serious systemic side effects, including anorexia, vomiting, peripheral polyneuropathy, depression of bone marrow, and allergic dermopathy. As a consequence of these adverse reactions, treatment has to be discontinued [5].
The discovery of new therapeutic agents is expensive and may take many years. Several strategies have been implemented in order to reduce the costs and time of the drug discovery process. Drug repositioning has emerged as a promising strategy for drug discovery for Neglected Tropical Diseases (NTDs). Also referred to as drug repurposing, this strategy consists of finding novel indications for approved drugs [6]. Sertraline (SERT), a serotonin reuptake inhibitor, is widely used in the treatment of patients with depression and severe anxiety disorders [7], and has exhibited antifungal [8], antimicrobial [9], and anticancer activities [10]. The drug has shown promising activity against the protozoan Leishmania donovani; it reduced the parasite burden of animals by 72% at doses of 10 mg/kg for 30 days [11].
In the present work, we evaluated the activity of SERT against T. cruzi trypomastigotes and intracellular amastigotes of the Y and Tulahuen strains and investigated its mode of action using cell biology and in silico chemogenomic approaches.
Methods
Additional file 1 shows a Flowchart of the global methodology.
Animals
BALB/c mice were supplied by the animal breeding facility at the Adolfo Lutz Institute of São Paulo whereas Swiss mice were obtained from the Fundação Oswaldo Cruz (FIOCRUZ) Rio de Janeiro. The mice were maintained in sterilized cages under a controlled environment and received water and food ad libitum. Animal procedures were performed with the approval of the Research Ethics Commission, in agreement with the Guidelines for the Care and Use of Laboratory Animals from the National Academy of Sciences. All procedures carried out at Institute Adolfo Lutz were approved by the Committee for Ethics in the Use of Animals (CEUA 04/2016). All procedures performed at FIOCRUZ were in accordance with the guidelines established by the FIOCRUZ Committee for Ethics in the Use of Animals (CEUA LW16/14).
Drugs and chemicals
Resazurin, Roswell Park Memorial Institute medium (RPMI 1640) without phenol red, and Hanks’ Balanced Salt Solution (HBSS), were purchased from Sigma-Aldrich. Sytox Green® and H2CDFDA (2 ‘, 7′-dichlorodihydrofluorescein diacetate) were purchased from Molecular Probes ® (Invitrogen ™). Fetal Bovine Serum (FBS) was obtained from Gibco and gentamicin sulfate from Hipolabor Pharmaceuticals. Benznidazole (BZ) and sertraline (SERT) were purchased from Sigma-Aldrich. All other reagents not mentioned were purchased from Sigma-Aldrich.
Parasites and mammalian host cells
T. cruzi (Y strain - culture trypomastigotes)
Trypomastigotes were maintained in LLC-MK2 cells with RPMI-1640 medium supplemented with 2% fetal bovine serum (FBS) at 37 °C and 5% CO2 in a humidified incubator.
T. cruzi (Y strain - bloodstream trypomastigotes - BT)
Trypomastigotes were obtained from the blood samples of infected albino Swiss mice at the peak of parasitemia. The purified parasites were resuspended in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS as reported previously [12].
Macrophages
Macrophages used in the intracellular amastigote assays were collected from the peritoneal cavity of BALB/c mice by washing with RPMI-1640 medium supplemented with 10% FBS and maintained at 37 °C in an atmosphere of 5% CO2 in a humidified incubator.
Cardiac cell cultures (CC)
Cardiac cells were used in the cytotoxicity and intracellular amastigote assays. Primary cultures of embryonic cardiac cells were obtained from Swiss mice as previously reported [12]. Briefly, after purification, the CC were seeded at a density of (0.2 × 106 cell/well) into 24-well microplates containing gelatin-coated cover slips as previously described. The cardiac cultures were then sustained at 37 °C in DMEM supplemented with 10% horse serum, 5% fetal bovine serum, 2.5 mM CaCl2, 1 mM L-glutamine and 2% chicken embryo extract.
NCTC cells-clone L929
NCTC cells were maintained in medium M-199 supplemented with 10% FBS and were maintained at 37 °C under 5% CO2 in a humidified incubator.
Determination of the Anti-T. cruzi activity
Culture trypomastigotes
To determine the 50% inhibitory concentration (IC50) against T. cruzi, trypomastigotes were counted in a Neubauer hemocytometer and seeded at (1 × 106 cells/well) in 96-well microplates. The drug was dissolved in dimethyl sulfoxide (DMSO), diluted with RPMI-1640 medium at different concentrations for 24 h at 37 °C and placed in a 5% CO2 humidified incubator. The parasite viability was determined using the colorimetric resazurin assay [13]. Benznidazole was used as the standard drug. The optical density was read at 570 nm (FilterMax F5 Multi-Mode Microplate Reader, Molecular Devices). DMSO was used at a maximal concentration of 0.5% in all assays and was incubated with cells as an internal control.
Bloodstream trypomastigotes (BT)
The trypomastigotes (5 × 106/mL) were incubated for 24 h at 37 °C in RPMI medium in the presence or absence of serial dilutions of the drug (0 to 50 μM). After 24 h of incubation, the parasite death rate was determined by light microscopy through direct quantification of the number of living parasites using a Neubauer chamber, and the IC50 was then calculated.
Determination of cytotoxicity
Cytotoxicity against mammalian cells
NCTC cells 929 (6 × 104 cells/well) in 96-well microplates were incubated with the drug for 48 h at 37 °C in a 5% CO2 incubator. The selectivity index (SI) was determined using the following formula: CC50 against mammalian cells/ IC50 against parasites. The cell viability was determined using the colorimetric resazurin assay [13]. The obtained data represent the mean of two independent experiments performed in duplicate.
Cytotoxicity against CC
Cardiac cells were incubated at 37 °C for different periods of time (24–48 h) with increasing concentrations of the drug and were diluted in DMEM (without phenol red). The mammalian cell morphology and spontaneous contractibility were evaluated by light microscopy, whereas the cellular viability was determined by the colorimetric resazurin assay. After incubation for 24 h, the absorbance was determined at 570 nm [14]. The obtained data represent the mean of two independent experiments performed in duplicate.
Intracellular amastigotes of T. cruzi in peritoneal macrophages
After the mammalian cytotoxicity studies, the effect of sertraline was investigated against intracellular amastigotes. Peritoneal macrophages (1 × 105 cells/well) were dispensed in 16-well chamber slides (NUNC, Thermo, USA) and maintained for 24 h in the same medium at 37 °C in a 5% CO2 humidified incubator for attachment. Non-adherent cells were removed by two-step washings with medium. After 24 h, these cells were infected with (1 × 106 culture trypomastigote) forms for 4 h (parasite-to-macrophage ratio 10:1). Subsequently, infected cells were incubated with the drug for 48 h. Finally, the slides were fixed with methanol, stained with Giemsa, and observed via light microscopy. The parasite load was defined by counting 400 macrophages/well by evaluating the number of infected macrophages. Benznidazole was used as the standard drug. The obtained data represent the mean of two independent experiments performed in duplicate.
Intracellular amastigotes of cardiac cell cultures
For analysis of the effect against intracellular amastigotes from the Y strain, after 24 h of parasite-host cell interaction, the infected cardiac cell cultures were washed to remove free parasites and were then incubated for another 48 h with increasing concentrations of the drug. Cardiac cell cultures were maintained at 37 °C in an atmosphere of 5% CO2 and air, and the medium was replaced every 24 h. Then, untreated and treated infected cardiac cell cultures were fixed and stained with Giemsa solution, and the mean number of infected host cells and mean number of parasites per infected cell were scored. Only characteristic T. cruzi nuclei and kinetoplasts were counted as living parasites since irregular structures may indicate parasites undergoing death. The compound activity was estimated by calculating the infection index (II - percentage of infected cells times the average number of intracellular amastigotes per infected host cell) [15]. The obtained data represent the mean of two independent experiments performed in duplicate.
Intracellular amastigotes inside L929 cell lines
The effect against intracellular forms was also investigated in L929 cell lineages infected with tissue culture-derived trypomastigotes (Tulahuen strain expressing the Escherichia coli β-galactosidase gene), employing a parasite-to-host-cell ratio of 10:1. After incubation with the drug for 96 h, the viability of parasites was determined colorimetrically as previously reported [14]. The obtained data represent the mean of two independent experiments performed in duplicate.
Mode of action studies of SERT in T. cruzi
Spectrofluorimetric detection of the permeability of the cell membrane
Culture trypomastigotes were washed with PBS (phosphate buffered saline), deposited on a microplate (2 × 106 cells/well) and incubated with SYTOX Green® (1 μM) for 15 min at 24 °C [16]. Sertraline was added at the IC50 value (2 μM), and the fluorescence was measured after 20, 40 and 60 min. The maximum permeability was observed with 0.1% Triton X-100 (positive control). The fluorescence intensity was determined using a plate spectrofluorimeter (FilterMax F5 Multi-Mode MicroplateReader-Molecular Devices) with excitation and emission wavelengths of 485 and 520 nm, respectively. Untreated trypomastigotes and 0.5% (v/v) DMSO-treated parasites were used in all assays as negative controls. The obtained data represent the mean of two independent experiments performed in triplicate.
Effect of sertraline on the mitochondrial integrity
Culture trypomastigotes were washed with PBS, deposited on a microplate (2 × 106 cells/well) and incubated with sertraline at the IC50 value (2 μM) for 60 min at 37 °C. MitoTracker Red CM-H2XROS (500 nM) was added and the incubation continued for 40 min in the dark. Parasites were washed twice with HBSS (Hanks’ Balanced Salt Solution), and the fluorescence was measured using a plate spectrofluorimeter (FilterMax F5 Multi-Mode Microplate Reader-Molecular Devices) with excitation and emission wavelengths of 540 and 595 nm, respectively [17]. Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 10 μM) was used as a positive control [18]. The obtained data represent the mean of two independent experiments performed in triplicate.
Analysis of reactive oxygen species (ROS)
Culture trypomastigotes (2 × 106 cells/well) were washed in HBSS (Hanks’ Balanced Salt Solution) and incubated with sertraline at the IC50 value (2 μM) for 60 min at 37 °C. H2DCF-DA (5 μM) was added, and the cells were incubated for 15 min. The fluorescence intensity was detected using a plate spectrofluorimeter (FilterMax F5 Multi-ModeMicroplate Reader-Molecular Devices) at 485 and 520 nm for excitation and emission, respectively [19]. The obtained data represent the mean of two independent experiments performed in triplicate.
Measurement of cellular ATP content
The intracellular adenosine triphosphate (ATP) content was measured by a luciferin–luciferase bioluminescence assay using a specific kit (Life Technologies, USA) according to the manufacturer’s instructions. ATP concentrations were calculated using the ATP standard curve kit. Culture trypomastigotes were washed twice with PBS, seeded at (2 × 106 cells/well) and incubated at the IC50 value (2 μM) of sertraline in the presence or absence (control) of 0.1% Triton X-100 for 1 h. The ATP level was rapidly measured by a coupled luciferin-luciferase reaction [20]. The obtained data represent the mean of two independent experiments performed in triplicate.
In silico studies
Prediction of sertraline targets using publicly available databases
We carried out a literature search using PubMed, PubChem Bioassay, ChEMBL and BindingDB in order to identify all possible SERT targets for all organisms. Our definition of a “sertraline target” embraces in vitro assays of SERT against any enzyme, receptor or channel with inhibition ≤40 μM. Then, individual information for each SERT target (primary amino acid sequence in FASTA format, target name and organism) was obtained from the UNIPROT database and was subsequently allocated into a single Excel file (Additional file 2).
Pairwise protein alignment
Superimposed structures allow a comparison of functionally relevant features, conserved residues necessary for catalysis, and residues critical for ligand binding. Therefore, SERT targets were aligned with all T. cruzi proteins using pairwise BLAST. We considered the T. cruzi target to be druggable if it has ≥80% overlap of the corresponding SERT target and an expected value (E-value) ≤ 10− 20. The E-value represents the number of hits with an alignment score “Z” equal to or greater than the “Z” that would be expected by chance when searching a database, which is the expected number of times a homology will occur at random from a given set of trials.
Comparison of functional regions
The ConSurf server [21] is a bioinformatics tool for estimating the evolutionary conservation of amino-acid positions in a protein based on the phylogenetic relations between homologous sequences. We used ConSurf for an additional characterization of the functional regions (active site conservation) in T. cruzi targets. Therefore, the degree of conservation of the amino acids from the active site was estimated using 150 homologue proteins with similar sequences retrieved from the UNIPROT database and was identified by the PSI–BLAST method (E-value cutoff ≤1− 10) [22]. The sequences were clustered, and highly similar (> 95%) sequences were removed using CD-HIT [23]. A multiple sequence alignment (MSA) of the homologous sequences was constructed using MAFFT-L-INS-I [23]. Subsequently, the MSA was employed to construct a phylogenetic tree using the neighbor-joining algorithm [24]. Position-specific conservation scores were analyzed using the empirical Bayesian method. Subsequently, functional regions were visually compared with corresponding SERT targets and were classified as conserved (≥ 70%) or non-conserved (< 70%). The obtained results are described in Additional files 3, 4, 5 and 6.
Homology modeling and molecular docking
In the absence of available experimental data, a homology model of T. cruzi isocitrate dehydrogenase 2 (TcIDH2) was constructed using homology modeling by comparing the sequence of this target protein with sequences of other proteins (template) for which experimental structures are available. The TcIDH2 sequence was obtained from the TriTrypDB database (Accession: Tc00.1047053506925.319). A BLAST search was carried out with the Protein Data Bank for the identification of a template structure. Three IDH crystal structures were found: human (HsIDH2), Sus scrofa and Mycobacterium tuberculosis (PDB IDs: 4JA8, 1LWD, 4HCX, respectively). Based on the resolution, the crystal structure of HsIDH2 bound to the allosteric inhibitor AGI-6780 (PDB ID 4JA8) [25], was chosen as a template for homology modeling, which presented 66.5% of sequence identity in relation to TcIDH2. SWISS-MODEL program [26] was used for generation of the homology model of TcIDH2. The quality of the model was assessed using the programs PROCHECK [27], VERIFY 3D [28] and ERRAT.
Molecular docking studies were performed to investigate the intermolecular interactions between SERT and the amino-acid residues of TcIDH2 as well as to predict the binding affinity. The generated homology model of TcIDH2 was imported into Maestro v. 10.0 [29] and was prepared using the Protein Preparation Wizard workflow as follows: hydrogen atoms were added according to Epik v. 2.7 (pH 7.4 ± 1.0) and minimized using the OPLS-2005 force field. Next, the structure of SERT was imported from the ChemSpider database and 300 conformations were generated using OMEGA v. 2.5.1.4 [30]. Subsequently, the conformers had their most favorable ionization state calculated at pH 7.4 using the ‘fixpka’ function, and AM1-BCC charges were added using QUACPAC v.1.6.3.1 [29]. Before the docking studies, two different grids were defined to include the catalytic site and one allosteric site of TcIDH2. The catalytic site grid was built with the dimensions of 26.3 Å × 15.8 Å × 27.9 Å (x, y and z) and a volume of 11,645 Å3. The allosteric site grid had the dimensions of 16.3 Å × 20.3 Å × 19.7 Å and a volume of 6531 Å3. Finally, molecular docking of SERT with TcIDH2 was investigated using the software FRED, which is available in OEDocking suite v. 3.0.1 [31] using the high-resolution precision and ChemGauss 4 scoring function.
Statistical analysis
The obtained data represent the mean of three independent experiments performed in duplicate. The IC50 and CC50 values were calculated using sigmoid dose-response curves generated by the software GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA). The ANOVA test was performed to evaluate the significance (p < 0.05) of data.
Results
Antiparasitic activity and mammalian cytotoxicity of sertraline
The anti-trypanosomal activity of SERT in cell culture-derived trypomastigotes (Y strain) was determined colorimetrically by resazurin. After 24 h, 100% of the parasites were eliminated, resulting in an IC50 value of 1.8 μM (± 0.8). Using blood-derived trypomastigotes, SERT showed an IC50 value of 14.2 μM (± 5.5) (Table 1).
Table 1.
Antiparasitic activity and mammalian cytotoxicity of sertraline
| T. cruzi IC50 μM (±SD) | CC50 μM (±SD) | ||||||
|---|---|---|---|---|---|---|---|
| Drugs | Trypomastigote (Y) | Trypomastigote BT (Y) | Amastigote L929 (Tulahuen) | Amastigote CC (Y) | Amastigote MØ (Y) | L929 | CC |
| SERT | 1.8 ± 0.8 | 14.2 ± 5.5 | 10.0 ± 1.7 | 6.6 ± 1.4 | 1.4 ± 0.6 | 11.5 ± 2.4 | 25.0 ± 13.2 |
| BZ | 17.7 ± 1.9 | 11.5 ± 1.0 | 2.5 ± 0.9 | 3.6 ± 1.0 | 5.0 ± 1.5 | 190.6 ± 13.4 | > 200 |
IC50 50% inhibitory concentration, CC50 50% cytotoxic concentration, CC cardiac cells, SERT sertraline, BZ benznidazole, SD standard deviation, BT bloodstream, MØ peritoneal macrophage
The drug was also effective against the intracellular amastigotes in peritoneal macrophages, resulting in an IC50 value of 1.4 (± 0.6) μM; using intracellular amastigotes in cardiac cell cultures, the IC50 value was 6.6 μM (± 1.4). The intracellular amastigotes of the / a Tulahuen strain inside fibroblasts (L929 cells) were also susceptible, resulting in an IC50 value of 10.0 μM (± 1.7) (Table 1). The mammalian cytotoxicity was determined in L929 cells and cardiac cell cultures, and resulted in respective CC50 values of 11.5 μM (± 2.48) and 25.0 μM (± 13.2). Benznidazole was used as the standard and resulted in IC50 values of 17.7 μM (± 1.9) against trypomastigotes (Y strain) and 5 μM (± 1.5) against intracellular amastigotes (Y strain) (Table 1).
Action mode studies of SERT in T. cruzi
Permeability of plasma membrane
To evaluate the possible effect of SERT on the plasma membrane permeability of culture trypomastigotes of T. cruzi, SERT was incubated for 60 min with trypomastigotes at the corresponding IC50 value, after which the permeability of the membrane was evaluated fluorimetrically using the vital dye SYTOX Green®. The data demonstrated no increase in the fluorescence levels, suggesting that sertraline did not interfere with the plasma membrane permeability of the parasite. Triton X-100 was used as a positive control (data not shown).
Effects on mitochondrial integrity and ATP production
The mitochondrial integrity of trypomastigotes was investigated in the presence of SERT at the IC50 value using the fluorescent probe Mitotracker Red®. After 60 min of incubation, SERT induced an intense and significant depolarization (p < 0.05), with the fluorescence intensity decreasing by 100% when compared to untreated trypomastigotes (control) (Fig. 1a). FCCP was used as a positive control and reduced the fluorescence levels by approximately 54% compared to untreated parasites. The production of ATP by trypomastigotes was investigated in the presence of SERT using a luminescent assay with luciferase (ATP kit - Thermo). At 60 min of incubation, an intense decrease in ATP levels of approximately 70% was observed compared to untreated parasites (p < 0.05) (Fig. 1b). Sodium azide was used as a positive control and resulted in a 100% decrease in the ATP levels of trypomastigotes.
Fig. 1.
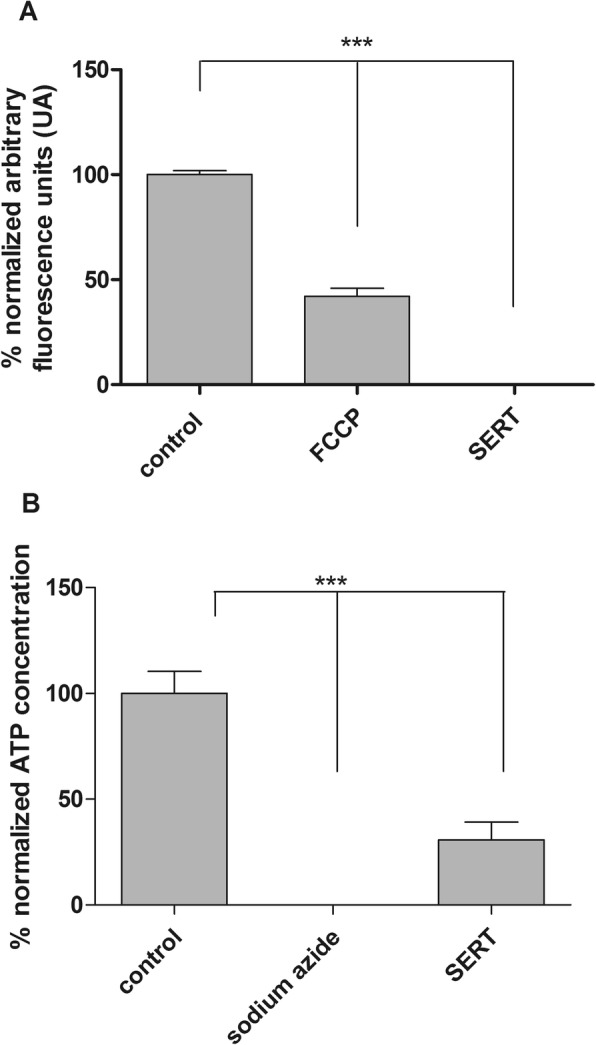
Evaluation of the bioenergetic parameters of T. cruzi. a Mitochondrial membrane potential of T. cruzi trypomastigotes treated with sertraline for 1 h at 2 μM, positive control (FCCP) and negative control (untreated cells). The fluorescence was determined using a fluorimetric microplate reader (FilterMax F5 Multi-Mode Microplate Reader) at 540 and 595 nm for excitation and emission, respectively. *** (p < 0.05); b Evaluation of the ATP levels of T. cruzi incubated for 1 h with sertraline at 2 μM. The levels were measured in a spectroluminometer plate reader (FilterMax F5 Multi-Mode Microplate Reader) using the ATP determination Kit (Life technologies). *** p < 0.05
Reactive oxygen species (ROS) production
Considering the interference of SERT in mitochondrial metabolism, the ROS production by trypomastigotes was evaluated using the fluorescent probe H2DCf-DA in the presence of the drug. The results indicated no alterations in ROS levels after 60 or 120 min compared to untreated parasites. Oligomycin was used as a positive control, and it upregulated ROS levels in trypomastigotes, as indicated by increased fluorescence intensity (data not shown).
In silico studies
To identify SERT targets that were experimentally determined in other organisms, we performed a literature search in PubMed, PubChem Bioassay, BindingDB and ChEMBL. Using a chemogenomic target fishing strategy, we identified 15 similar targets in T. cruzi (Additional file 2). Following these conditions, we identified three potential targets of SERT in T. cruzi (Table 2).
Table 2.
List of potential sertraline targets in T. cruzi
| SERT target | T. cruzi target | Biological process | E-value | Overlap | Active Site Conservation (%) |
|---|---|---|---|---|---|
| Isocitrate dehydrogenase 1 | Isocitrate dehydrogenase 2 | isocitrate metabolism | 0 | 100% | High conservation (97%) |
| Ubiquitin-conjugating enzyme E2 N | Ubiquitin-conjugating enzyme E2, purative | ubiquitin cycle | 1−68 | 96% | High conservation (76%) |
| Cyclin-dependent kinase 1 | CDC2-related protein kinase 1, putative | protein phosphorylation | 2− 113 | 95% | High conservation (78%) |
Homology modeling and molecular docking
To build the homology model of TcIDH2, the following criteria were used for template selection: the template should have high coverage, good sequence identity and good X-ray crystallography resolution (< 2.0 Å). Therefore, the selected template, HsIDH2 (PDB ID: 4JA8), presented 66.5% sequence identity with TcIDH2, coverage of 0.99 and a resolution of 1.5 Å. The stereochemical quality of the TcIDH2 model was evaluated using PROCHECK. This analysis revealed that 91.7% of residues were within most favored regions, 8.2% of residues were in additional allowed regions, and only 0.1% of residues were in disallowed regions of the Ramachandran plot, demonstrating the good quality of the generated model (Fig. 2).
Fig. 2.
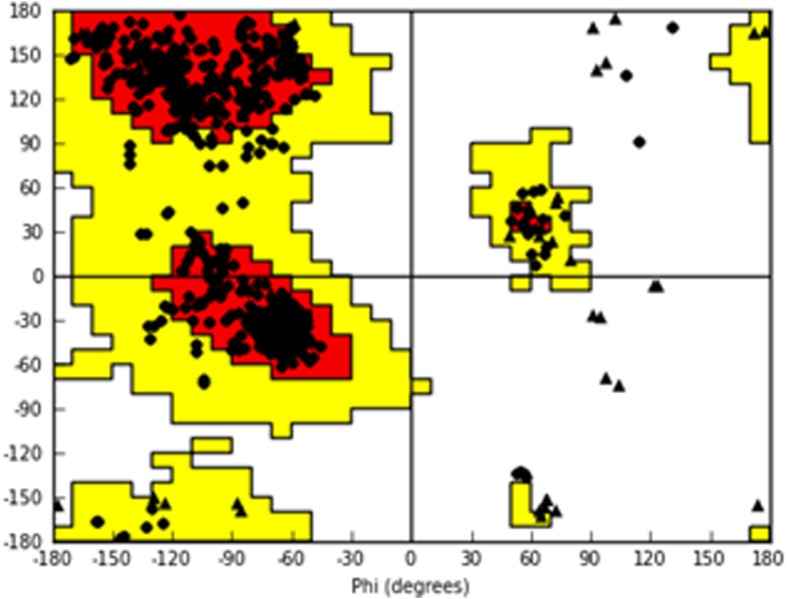
Ramachandran plot for the TcIDH2 homology model. The amino acids are represented as circles, except for glycine, represented as triangles. The white area shows sterically disallowed regions for all amino acids, except glycine. The yellow areas show the sterically allowed regions. The red areas indicate the most sterically favorable regions, i.e. regions where amino acids conformations have no steric clashes
Additionally, the quality of the homology model was evaluated using VERIFY 3D, through which the TcIDH2 model presented a score of 92.6%. An overall quality factor of 86.6% was obtained from the analysis on ERRAT. The homology model of TcIDH2 had a satisfactory quality and could be used in molecular docking studies. Figure 3a shows the generated 3D structure of the TcIDH2 homology model. Docking studies were performed at two different binding sites of TcIDH2, the catalytic site, i.e., the binding site of the substrate isocitrate, and in the allosteric site, which is located in the interface between the two chains of the homodimeric protein. The predicted binding affinities of SERT in the catalytic and allosteric sites were - 5.5 kcal/mol and - 12.2 kcal/mol, respectively. The remarkable difference in binding affinities indicates that SERT may be an allosteric inhibitor of TcIDH2.
Fig. 3.

3D structure of TcIDH2 homology model. a Regions in blue represent Chain A and regions in yellow represent Chain B. The Ca2+ ions are indicated by green spheres. The two ligands presented in the stick model represent the co-factor NADP. b Interactions of sertraline with the allosteric binding site of TcIDH2 were predicted by docking. The residues of chain A are in blue and chain B is colored in yellow, indicating the interface that constitutes the allosteric binding site. In the center, sertraline carbon atoms are colored gray, nitrogen in blue, chlorine atoms are green and hydrogens are white. The hydrogen bond between sertraline and Gln276A is indicated by a green dashed line
Moreover, docking results showed that the predicted binding mode of SERT at the TcIDH2 allosteric site is in agreement with the experimental X-ray binding mode of the co-crystalized ligand on HsIDH2. In particular, SERT can establish a hydrogen bond between its secondary amine hydrogen and the amino acid residue Gln276A. Additional hydrophobic interactions were observed between the SERT aromatic ring and residues Trp266B, Trp124B and Leu120B (Fig. 3b). The presence of a hydrophobic pocket represented by Leu120A, Leu120B, Trp124B, Ile128B, Ile130B and Trp266B can provide hints for the structural optimization of SERT, thus enabling the design of new inhibitors of TcIDH2.
Discussion
Drug repurposing is a successful approach with numerous advantages, including reduced time and costs in the drug discovery process. Sertraline, an FDA-approved drug, belongs to the class of antidepressant agents known as selective serotonin-reuptake inhibitors (SSRIs). The literature reports several attempts to repurpose the drug SERT towards a range of microorganisms [32, 33].
Considering the broad range of biological activities of SERT, our work investigated the in vitro anti-T. cruzi efficacy against trypomastigotes and intracellular amastigotes. Based on the 50% Inhibitory Concentration (IC50), SERT demonstrated similar in vitro efficacy to the standard drug (BZ) against trypomastigotes and intracellular amastigotes (Y strain). The drug effectively eliminated the intracellular amastigotes of different strains and DTUs of T. crui [34]. It was active against the Y strain (DTU II) in macrophages and cardiac cells at micromolar concentrations. Sertraline was also effective against intracellular amastigotes of the Tulahuen strain (DTU VI), an important feature for a hit compound. Also, it is important to note that the difference between these DTUs may vary in activity and therefore should be taken into account in the testing of new drugs [35]. The Y strain was approximately 7-fold more susceptible to SERT than the Tulahuen strain. In addition to its intracellular activity against amastigotes, SERT has been valued as a hit candidate for presenting activity against the trypomastigote forms. Katsuno and co-workers [36] stated that drugs that only target the replicating stages of the parasite may leave non-replicating forms, such as trypomastigotes, capable of maintaining infections long after the end of the treatment; i.e., they are a clinical form responsible for relapses.
One of the advantages of repositioning FDA-approved drugs is the existing medical data on the drug, including dosing, side-effects, tolerance profiles and the pharmacokinetic and pharmacodynamics parameters (PKPD). Although sertraline demonstrated in vitro activity and selectivity against T. cruzi in our study, one should consider that for in vivo efficacy, adequate tissue distribution and serum levels must be achieved to eliminate the parasite. In the literature, SERT administered in humans resulted in serum levels of approximately 58 nM [37], a considerably smaller value than those needed to eliminate T. cruzi intracellular amastigotes (or bloodstream trypomastigotes) in our in vitro assays. Future experimental association studies should be performed to assess sertraline and benznidazole; if synergic combinations are found, reduced doses may be required to treat T. cruzi infected animals. Our data also suggest that sertraline may be a useful candidate for both acute and chronic phases of the disease, as the drug is effective against bloodstream trypomastigotes and intracellular amastigotes. However, considering that sertraline is metabolized to desmethylsertraline in the liver, the in vivo efficacy of the drug may be evaluated in the future using both mouse models (acute and chronic).
Considering the promising in vitro efficacy of SERT in T. cruzi parasites, we investigated the possible mode of action using different approaches, including cell biology techniques and in silico approaches. Using short-time incubations to evaluate initial cellular damage in T. cruzi, SERT induced a change in the mitochondrial integrity of trypomastigotes, resulting in a significant decrease in ATP levels within 1 h without affecting plasma membrane permeability. The depletion of ATP is an early event in mitochondrial dysfunction, which generates irreversible damage and cell death [38].
Trypanosomatids display a single mitochondrion with several peculiar features, such as the presence of different energetic and antioxidant enzymes and a specific arrangement of mitochondrial DNA (kinetoplast DNA) [39]. Due to mitochondrial differences between mammals and trypanosomatids, this organelle is a favorable candidate for drug intervention. Multiple mechanisms and targets are often involved in drug- or xenobiotic-associated mitochondrial impairment [40].
In protozoan parasites such as L. donovani, the action mode of sertraline was attributed to the decreased cytoplasmic ATP levels and oxygen consumption rate in promastigotes, suggesting an apoptosis-like death in treated parasites [11]. Previous studies have suggested that the decrease in oxygen consumption and reduction in cytoplasmic ATP levels, resulting from the inhibition of the respiratory chain, are essential events at the beginning of apoptosis in L. donovani [41–44]. The effect of sertraline in mitochondria was also demonstrated in hepatocytes. According to Li and co-workers [45], SERT began to decrease ATP levels in as early as 30 min in rat primary hepatocytes, targeting complexes I and V in the mitochondria. These data also corroborate a previous study of SERT that resulted in concentration-dependent mitochondrial swelling in hepatocytes [45]. In our study, SERT also seems to affect the mitochondria of T. cruzi. Whereas SERT disturbs the bioenergetic system of eukaryotic cells, including Leishmania, the standard drug benznidazole demonstrates a different action mode in T. cruzi. It is important to note that the action mode of BZ is apparently related to the formation of free radicals and electrophilic metabolites that are generated when its nitro-group is reduced to an amino group by the action of nitroreductases [46, 47]. Thus, it is hypothesized that the trypanocidal effect of BZ is caused by the covalent attachment of its reduced metabolites to macromolecules of the parasite [48].
Currently, several in silico chemogenomic strategies have been applied to drug repositioning against parasitic diseases. The main goal of the chemogenomic approach is to identify new therapeutic targets and drugs. To achieve that goal, various public drug databases that integrate information about gene/protein–drug–disease interactions, such as the Therapeutic Target Database (TTD), DrugBank and STITCH, are valuable resources to develop these strategies. Based on the concept that “similar targets have similar ligands”, homology-based inverse virtual screening allowed the prediction of potential targets of SERT in T. cruzi [49].
To identify SERT targets that were experimentally determined in other organisms, we performed a target fishing approach using a literature search in PubMed, PubChem Bioassay, BindingDB and ChEMBL. Using this chemogenomic strategy, we identified 15 similar targets in T. cruzi (Additional file 2). Most target databases are only starting to emerge, and the predicted T. cruzi targets are not yet scored for druggability. The druggability concept adds a structural dimension and evaluates the likelihood that small drug-like molecules can bind a given target with sufficient potency to alter its activity [50]. Therefore, the predicted T. cruzi targets were considered druggable if they presented an overlap ≥80% of the SERT target, an E-value ≤10− 20 and conservation of the functional regions. Thus, overlap sequences and conserved functional regions analysis of positions among SERT targets and T. cruzi targets revealed the importance of each position for protein function and also the possible preservation of affinity for SERT.
Among the predicted targets, isocitrate dehydrogenase 2 (IDH2; Accession: Tc00.1047053506925.319), an enzyme that catalyzes the oxidative decarboxylation of isocitrate to produce 2-oxoglutarate, CO2 and NADPH [51], was suggested. A search of the T. cruzi genome database (http://www.genedb.org/Homepage) showed the presence of two IDHs, namely IDH1 and IDH2. Both IDHs exhibited remarkable relatedness (> 65% identity) and revealed an equivalent degree of similarity compared to the NADP-linked mammalian counterparts, such as human IDH1. In T. cruzi, IDH1 is restricted to the tricarboxylic acid cycle (Krebs cycle) and provides a portion of the NADH utilized for ATP production by oxidative phosphorylation [52]. In contrast, NADP-linked IDH2 localizes in the peroxisomes, mitochondria and cytosol, where their biological roles are associated with multiple functions, such as intracellular redox homeostasis, β-oxidation of fatty acids, and lipogenesis. Furthermore, the expression levels of IDH2 are significantly increased in amastigotes and trypomastigotes compared to epimastigotes. Conversely, IDH1 appears to be more abundant in the insect stage of T. cruzi [51]. Therefore, our in silico study suggests that SERT is able to inhibit T. cruzi IDH2, a homologue of human IDH1, with higher overlap and conservation of the active site than the other predicted targets.
In addition to the targets highlighted above, 15 other T. cruzi targets were similar to the SERT targets in other organisms. In all cases, we considered the numerical parameters (overlap, conserved functional regions, and E-value) for target homology to be sufficiently significant to infer the predicted target with a high degree of confidence. They were not discussed in detail due to their low druggability profile. For example, T. cruzi mitogen-activated protein kinase (E-value = 5− 91; overlap = 95%) has low conservation of functional regions (61% of predicted residues are conserved), which is insufficient to infer the predicted target with a satisfactory degree of confidence.
Conclusions
Our studies demonstrated that SERT has a rapid and lethal effect on different forms and strains of T. cruzi, affecting the bioenergetic metabolism of the parasite. The depletion of ATP levels in trypomastigotes and the change in mitochondrial integrity may be initial effects of the drug, contributing to the death of T. cruzi. However, considering the multi-target characteristic of SERT, our in silico studies also suggested that sertraline affects the parasite TcIDH2, a parasitic enzyme possibly involved in the mechanisms of resistance to oxidative stress. Due to the high homology between the TcIDH1 and TcIDH2, SERT may affect both enzymes. Therefore, further in vitro enzymatic studies involving the predicted enzymes are required to confirm the potential inhibitory effect of SERT. Considering our in vitro experimental studies and the homology-based inverse virtual approach, sertraline should be considered a new hit compound for T. cruzi. These findings provide a starting point for future experimental assays and may contribute to the development of new compounds.
Additional files
{kind=link}
Flowchart of the global methodology (PNG 147 kb)
List of compiled and predicted T. cruzi targets (XLSX 166 kb)
{kind=link}
Alignment and functional region analysis of the T. cruzi IDH2, purative (Sbjct) and human IDH1 (Query) (PNG 312 kb)
{kind=link}
Alignment and functional region analysis of the T. cruzi ubiquitin-conjugating enzyme E2, putative (Sbjct) and human ubiquitin-conjugating enzyme E2 N (Query) (PNG 143 kb)
{kind=link}
Alignment and functional region analysis of the T. cruzi CRK1 (Sbjct) and human cyclin-dependent kinase 1(Query) (PNG 238 kb)
{kind=link}
Alignment and functional region analysis of the T. cruzi mitogen-activated protein kinase (Sbjct) and human mitogen-activated protein kinase 1 (Query) (PNG 277 kb)
Acknowledgements
We thank Ms. Matilia Ana do Nascimento and Mr. Vicente Duarte for laboratory assistance. We are also grateful to OpenEye Scientific Software, Inc., for providing academic licenses for their software to us.
Funding
The authors thank São Paulo Research Foundation (FAPESP) for financial support (grant 2015/23403–9 for AGT); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for scientific awards to AGT, CHA, MNCS; and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação de Amparo à Pesquisa do Estado de Goiás(FAPEG) for financial support and fellowships. The authors are also grateful to FAPERJ and Fiocruz. MNCS is Cientista do Nosso Estado (CNE –FAPERJ). This publication was supported by the Coordination for the Improvement of Higher Education Personnel (CAPES) through Programa Editoração CAPES – Edital No. 13/2016, No. do Auxílio 0722/2017, No. do Processo 88881.142062/2017–01 and from the National Council for Scientific and Technological Development (CNPq) Programa Editorial CNPq/CAPES process No. 26/2017, Proc. No. 440954/2017–7.
Availability of data and materials
Available by request to the corresponding author.
Abbreviations
- ATP
Adenosine triphosphate
- BT
Bloodstream trypomastigotes
- BZ
Benznidazole
- CC
Cardiac cell
- CC50
50% cytotoxic concentration
- DMSO
Dimethyl sulfoxide
- DNDi
Drugs for Neglected Diseases Initiative
- FBS
Fetal bovine serum
- FCCP
Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- FIOCRUZ
Fundação Oswaldo Cruz
- HBSS
Hanks’ Balanced Salt Solution
- IC50
50% inhibitory concentration
- NTDs
Neglected Tropical Diseases
- PBS
Phosphate-buffered saline
- ROS
Reactive oxygen species
- RPMI
Roswell Park Memorial Institute Medium
- SERT
Sertraline
- TTD
Therapeutic Target Database
Authors’ contributions
DDF a PhD student, developed the all experimental work; MMR developed part of the in vitro action mechanism studies; JTM developed part of the in vitro action mechanism studies, TADC-S developed part of the in vitro action mechanism studies; DDGJB developed part of the in vitro studies, CFDS; ANSDG developed part of the in vitro studies; BJN developed the in silico studies, CCM-F developed the in silico studies, MDNCS developed and analyzed the in vitro studies, CHA developed and analyzed the in silico studies and AGT is responsible for the design and general ideation of the work and also analyzed the in vitro studies. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Animal procedures were performed with the approval of the Research Ethics Commission, in agreement with the Guidelines for the Care and Use of Laboratory Animals from the National Academy of Sciences. All procedures carried out at Institute Adolfo Lutz were approved by the Committee for Ethics in the Use of Animals (CEUA 04/2016). All procedures performed at Fiocruz were in accordance with the guidelines established by the Fiocruz Committee for Ethics in the Use of Animals (CEUA LW16/14).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Daiane Dias Ferreira, Email: daiane_bio09@hotmail.com.
Juliana Tonini Mesquita, Email: jt_mesquita@hotmail.com.
Thais Alves da Costa Silva, Email: tha_isbio@yahoo.com.br.
Maiara Maria Romanelli, Email: ma.romaneli@gmail.com.
Denise da Gama Jaen Batista, Phone: (21) 2598-4220, Email: denisegama@ioc.fiocruz.br.
Cristiane França da Silva, Phone: (21) 2598-4220, Email: franca@ioc.fiocruz.br.
Aline Nefertiti Silva da Gama, Phone: (21) 2598-4220, Email: alinenefertitigama@gmail.com.
Bruno Junior Neves, Email: bruno.labmol@gmail.com.
Cleber Camilo Melo-Filho, Email: ccmf89@gmail.com.
Maria de Nazare Correia Soeiro, Phone: (21) 2598-4220, Email: soeiro@ioc.fiocruz.br.
Carolina Horta Andrade, Email: andradech@yahoo.com.
Andre Gustavo Tempone, Email: andre.tempone@ial.sp.gov.br, Email: atempone@usp.br.
References
- 1.Pereira PCM, Navarro EC. Challenges and perspectives of Chagas disease: a review. J Venom Anim Toxins incl Trop Dis. 2013;19(1):34. doi: 10.1186/1678-9199-19-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gascon J, Bern C, Pinazo MJ. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010;115(1–2):22–27. doi: 10.1016/j.actatropica.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 3.Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A, Jr, Rosas F, et al. Randomized trial of benznidazole for chronic chagas’ cardiomyopathy. N Engl J Med. 2015;373(14):1295–1306. doi: 10.1056/NEJMoa1507574. [DOI] [PubMed] [Google Scholar]
- 4.Pecoul B, Batista C, Stobbaerts E, Ribeiro I, Vilasanjuan R, Gascon J, et al. The BENEFIT trial: where do we go from here? PLoS Negl Trop Dis. 2016;10(2):e0004343. doi: 10.1371/journal.pntd.0004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tempone AG, Sartorelli P, Mady C, Fernandes F. Natural products to anti-trypanosomal drugs: an overview of new drug prototypes for American trypanosomiasis. Cardiovasc Hematol Agents Med Chem. 2007;5(3):222–235. doi: 10.2174/187152507781058726. [DOI] [PubMed] [Google Scholar]
- 6.Berman J. Miltefosine, an FDA-approved drug for the “orphan disease”, leishmaniasis. Expert Opin Orphan Drugs. 2015;3(6):727–735. doi: 10.1517/21678707.2015.1039510. [DOI] [Google Scholar]
- 7.Duan W, Peng Q, Masuda N, Ford E, Tryggestad E, Ladenheim B, et al. Sertraline slows disease progression and increases neurogenesis in N171-82Q mouse model of Huntington's disease. Neurobiol Dis. 2008;30(3):312–322. doi: 10.1016/j.nbd.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhai B, Wu C, Wang L, Sachs MS, Lin X. The antidepressant sertraline provides a promising therapeutic option for neurotropic cryptococcal infections. Antimicrob Agents Chemother. 2012;56(7):3758–3766. doi: 10.1128/AAC.00212-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munoz-Bellido J, Munoz-Criado S, Garcìa-Rodrìguez JA. Antimicrobial activity of psychotropic drugs: selective serotonin reuptake inhibitors. Int J Antimicrob Agents. 2000;14(3):177–180. doi: 10.1016/S0924-8579(99)00154-5. [DOI] [PubMed] [Google Scholar]
- 10.Tuynder M, Fiucci G, Prieur S, Lespagnol A, Géant A, Beaucourt S, et al. Translationally controlled tumor protein is a target of tumor reversion. Proc Natl Acad Sci U S A. 2004;101(43):15364–15369. doi: 10.1073/pnas.0406776101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palit P, Ali N. Oral therapy with sertraline, a selective serotonin reuptake inhibitor, shows activity against Leishmania donovani. J Antimicrob Chemother. 2008;61(5):1120–1124. doi: 10.1093/jac/dkn046. [DOI] [PubMed] [Google Scholar]
- 12.Meirelles MN, de Araujo-Jorge TC, Miranda CF, de Souza W, Barbosa HS. Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur J Cell Biol. 1986;41(2):198–206. [PubMed] [Google Scholar]
- 13.Martins LF, Mesquita JT, Pinto EG, Costa-Silva TA, Borborema SE, Galisteo Junior AJ, et al. Analogues of marine guanidine alkaloids are in vitro effective against Trypanosoma cruzi and selectively eliminate Leishmania (L.) infantum intracellular amastigotes. J Nat Prod. 2016;79(9):2202–2210. doi: 10.1021/acs.jnatprod.6b00256. [DOI] [PubMed] [Google Scholar]
- 14.Romanha AJ, Castro SL, Soeiro Mde N, Lannes-Vieira J, Ribeiro I, Talvani A, et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem Inst Oswaldo Cruz. 2010;105(2):233–238. doi: 10.1590/S0074-02762010000200022. [DOI] [PubMed] [Google Scholar]
- 15.da Silva CF, Batista Dda G, Oliveira GM, de Souza EM, Hammer ER, da Silva PB, et al. In vitro and in vivo investigation of the efficacy of arylimidamide DB1831 and its mesylated salt form--DB1965--against Trypanosoma cruzi infection. PLoS One. 2012;7(1):e30356. doi: 10.1371/journal.pone.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mesquita JT, da Costa-Silva TA, Borborema SET, Tempone AG. Activity of imidazole compounds on Leishmania (L.) infantum chagasi: reactive oxygen species induced by econazole. Mol Cell Biochem. 2014;389(1–2):293–300. doi: 10.1007/s11010-013-1954-6. [DOI] [PubMed] [Google Scholar]
- 17.Williams RAM, Smith TK, Cull B, Mottram JC, Coombs GH. ATG5 is essential for ATG8-dependent autophagy and mitochondrial homeostasis in Leishmania major. PLoS Pathog. 2012;8(5):e1002695. doi: 10.1371/journal.ppat.1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V, et al. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J Am Heart Assoc. 2012;1(5):e003012. doi: 10.1161/JAHA.112.003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ribeiro GA, Cunha-Júnior EF, Pinheiro RO, da-Silva SA, Canto-Cavalheiro MM, da Silva AJ, et al. LQB-118, an orally active pterocarpanquinone, induces selective oxidative stress and apoptosis in Leishmania amazonensis. J Antimicrob Chemother. 2013;68(4):789–799. doi: 10.1093/jac/dks498. [DOI] [PubMed] [Google Scholar]
- 20.Villa-Bellosta R, Wang X, Millán JL, Dubyak GR, O’Neill WC. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2011;301(1):H61–H68. doi: 10.1152/ajpheart.01020.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biegert A, Söding J. Sequence context-specific profiles for homology searching. Proc Natl Acad Sci U S A. 2009;106(10):3770–3775. doi: 10.1073/pnas.0810767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 24.Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics. 2002;18(Suppl 1):S71–S77. doi: 10.1093/bioinformatics/18.suppl_1.S71. [DOI] [PubMed] [Google Scholar]
- 25.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 26.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 28.Bowie JU, Lüthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional stucture. Science. 1991;253(5016):164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 29.Zhu K, Day T, Warshaviak D, Murrett C, Friesner R, Pearlman D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins. 2014;82(8):1646–1655. doi: 10.1002/prot.24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.OMEGA, v2.5.1.4. https://docs.eyesopen.com/omega/citation.html. Accessed 26 July 2017.
- 31.McGann M. FRED pose prediction and virtual screening accuracy. J Chem Inf Model. 2011;51(3):578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 32.Weeks JC, Roberts WM, Leasure C, Suzuki BM, Robinson KJ, Currey H, et al. Sertraline, paroxetine, and chlorpromazine are rapidly acting anthelmintic drugs capable of clinical repurposing. Sci Rep. 2018;8(1):975. doi: 10.1038/s41598-017-18457-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren J, Zhao Y, Fry EE, Stuart DI. Target identification and mode of action of four chemically divergent drugs against ebolavirus infection. J Med Chem. 2018;61(3):724–733. doi: 10.1021/acs.jmedchem.7b01249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zingales B, Andrade SG, Briones MR, Campbell DA, Chiari E, Fernandes O, et al. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz. 2009;104(7):1051–1054. doi: 10.1590/S0074-02762009000700021. [DOI] [PubMed] [Google Scholar]
- 35.Zingales B, Miles MA, Moraes CB, Luquetti A, Guhl F, Schijman AG, et al. Drug discovery for Chagas disease should consider Trypanosoma cruzi strain diversity. Mem Inst Oswaldo Cruz. 2014;109(6):828–833. doi: 10.1590/0074-0276140156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katsuno K, Burrows JN, Duncan K, Hooft van Huijsduijnen R, Kaneko T, Kita K, et al. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat Rev Drug Discov. 2015;14(11):751–758. doi: 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- 37.Reis M, Aamo T, Ahlner J, Druid H. Reference concentrations of antidepressants. A compilation of postmortem and therapeutic levels. J Anal Toxicol. 2007;31(5):254–264. doi: 10.1093/jat/31.5.254. [DOI] [PubMed] [Google Scholar]
- 38.Vercesi AE, Hoffmann ME, Bernardes CF, Docampo R. ATP and Ca2+ homeostasis in Trypanosoma cruzi. Braz J Med Biol Res. 1993;26(4):355–363. [PubMed] [Google Scholar]
- 39.Menna-Barreto RFS, de Castro SL. The double-edged sword in pathogenic trypanosomatids: the pivotal role of mitochondria in oxidative stress and bioenergetics. Biomed Res Int. 2014;2014:614014. doi: 10.1155/2014/614014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Labbe G, Pessayre D, Fromenty B. Drug-induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol. 2008;22(4):335–353. doi: 10.1111/j.1472-8206.2008.00608.x. [DOI] [PubMed] [Google Scholar]
- 41.Bermúdez R, Dagger F, D'Aquino JA, Benaim G, Dawidowicz K. Characterization of mitochondrial electron-transfer in Leishmania mexicana. Mol Biochem Parasitol. 1997;90(1):43–54. doi: 10.1016/S0166-6851(97)00131-X. [DOI] [PubMed] [Google Scholar]
- 42.Sen N, Das BB, Ganguly A, Mukherjee T, Tripathi G, Bandyopadhyay S, et al. Camptothecin induced mitochondrial dysfunction leading to programmed cell death in unicellular hemoflagellate Leishmania donovani. Cell Death Differ. 2004;11(8):924–936. doi: 10.1038/sj.cdd.4401435. [DOI] [PubMed] [Google Scholar]
- 43.Mehata A, Shaha C. Apoptotic death in Leishmania donovani promastigotes in response to respiratory chain inhibition: complex II inhibition results in increased pentamidine cytotoxicity. J Biol Chem. 2004;279(12):11798–11813. doi: 10.1074/jbc.M309341200. [DOI] [PubMed] [Google Scholar]
- 44.Sen N, Das BB, Ganguly A, Mukherjee T, Bandyopadhyay S, Majumder HK. Camptothecin-induced imbalance in intracellular cation homeostasis regulates programmed cell death in unicellular hemoflagellate Leishmania donovani. J Biol Chem. 2004;279(50):52366–52375. doi: 10.1074/jbc.M406705200. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Couch L, Higuchi M, Fang JL, Guo L. Mitochondrial dysfunction induced by sertraline, an antidepressant agent. Toxicol Sci. 2012;127(2):582–591. doi: 10.1093/toxsci/kfs100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maya JD, Cassels BK, Iturriaga-Vásquez P, Ferreira J, Faúndez M, Galanti N, et al. Mode of action of natural and synthetic drugs against Trypanosoma cruzi and their interaction with the mammalian host. Comp Biochem Physiol A Mol Integr Physiol. 2007;146(4):601–620. doi: 10.1016/j.cbpa.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 47.Wilkinson SR, Taylor MC, Horn D, Kelly JM, Cheeseman I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci U S A. 2008;105(13):5022–5027. doi: 10.1073/pnas.0711014105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maya JD, Rodríguez A, Pino L, Pabón A, Ferreira J, Pavani M, et al. Effects of buthionine sulfoximine nifurtimox and benznidazole upon trypanothione and metallothionein proteins in Trypanosoma cruzi. Biol Res. 2004;37(1):61–69. doi: 10.4067/S0716-97602004000100007. [DOI] [PubMed] [Google Scholar]
- 49.Rognan D. Chemogenomic approaches to rational drug design. Br J Pharmacol. 2007;152(1):38–52. doi: 10.1038/sj.bjp.0707307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neves BJ, Braga RC, Bezerra JCB, Cravo PVL, Andrade CH. In silico repositioning-chemogenomics strategy identifies new drugs with potential activity against multiple life stages of Schistosoma mansoni. PLoS Negl Trop Dis. 2015;9(1):e3435. doi: 10.1371/journal.pntd.0003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leroux AE, Maugeri DA, Cazzulo JJ, Nowicki C. Functional characterization of NADP-dependent isocitrate dehydrogenase isozymes from Trypanosoma cruzi. Mol Biochem Parasitol. 2011;177(1):61–64. doi: 10.1016/j.molbiopara.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 52.Adroher FJ, Osuna A, Lupiañez JA. Differential energetic metabolism during Trypanosoma cruzi differentiation. I. Citrate synthase, NADP-isocitrate dehydrogenase, and succinate dehydrogenase. Arch Biochem Biophys. 1988;267(1):252–261. doi: 10.1016/0003-9861(88)90030-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flowchart of the global methodology (PNG 147 kb)
List of compiled and predicted T. cruzi targets (XLSX 166 kb)
Alignment and functional region analysis of the T. cruzi IDH2, purative (Sbjct) and human IDH1 (Query) (PNG 312 kb)
Alignment and functional region analysis of the T. cruzi ubiquitin-conjugating enzyme E2, putative (Sbjct) and human ubiquitin-conjugating enzyme E2 N (Query) (PNG 143 kb)
Alignment and functional region analysis of the T. cruzi CRK1 (Sbjct) and human cyclin-dependent kinase 1(Query) (PNG 238 kb)
Alignment and functional region analysis of the T. cruzi mitogen-activated protein kinase (Sbjct) and human mitogen-activated protein kinase 1 (Query) (PNG 277 kb)
Data Availability Statement
Available by request to the corresponding author.