

Abstract
Purpose:
The aim of the present study was to investigate the effect of etanercept (ETA) on histopathological and biochemical changes after traumatic brain injury (TBI) in rats.
Materials and Methods:
Thirty-six male Wistar albino rats were distributed into three groups (n = 12 each). Control group rats were not subjected to trauma. Trauma group rats were subjected to TBI only. ETA group rats were subjected to TBI plus ETA (5 mg/kg intraperitoneal [i.p.]). The groups were further subdivided into those sacrificed in the hyperacute stage (1 h after TBI) (control-1, trauma-1, and ETA-1 groups) and the acute stage (6 h after TBI) (control-6, trauma-6, and ETA-6 groups). Tissue levels of tumour necrosis factor-alpha, interleukin-1 beta, malondialdehyde, catalase, glutathione peroxidase, and superoxide dismutase were analyzed. Histopathological and ultrastructural evaluations were also performed.
Results:
i.p. administration of ETA at 1 and 6 h significantly reduced inflammatory cytokine expression, attenuated oxidative stress and lipid peroxidation, prevented apoptosis, and increased antioxidant defense mechanism activity in comparison to trauma group. Histopathological and ultrastructural abnormalities were significantly reduced in ETA-treated rats compared to closed head injury trauma groups.
Conclusions:
ETA significantly improves neural function and prevents post-TBI histopathological damage in rats.
Keywords: Etanercept, rat, traumatic brain injury, tumor necrosis factor-α antagonist
Introduction
Traumatic brain injury (TBI) is a major global public health problem as the leading cause of morbidity and mortality in children and young adults.[1] Head trauma can be fatal or disabling which requires long-term treatment and care and can cause heavy financial burden. Brain injury secondary to trauma consists of the primary damage due to the direct effect of impacting forces and the subsequent secondary damage due to the sequential reactions of various metabolic events.[2,3] TBI is associated with microglial and astrocytic activation and the release of pro-inflammatory cytokines.[4] The most studied pro-inflammatory cytokines related to the development of cerebral edema, breakdown of the blood–brain barrier, and secondary neuronal injury are interleukin-1 (IL-1), IL-6, and tumor necrosis factor-alpha (TNF-α).[5] Post-TBI treatment with anti-inflammatory agents is a promising therapeutic strategy associated with reductions in blood–brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death, which result in improved neurological outcomes.[6] Enbrel (etanercept [ETA]) is a dimeric fusion protein consisting of the extracellular ligand-binding portion of the human 75-kilodalton (p75) TNF receptor linked to the Fc portion of human IgG1. ETA administered systemically at the dosage regimen approved for its licensed indications (approximately 50 mg/week in humans) would not be expected to achieve therapeutic levels in the cerebrospinal fluid (CSF) because of its high molecular weight.[7] Recent studies showed that ETA can inhibit inflammatory responses and reduce neuronal and glial apoptosis in the brain secondary to traumatic injury.[8,9,10] These results suggest that experimental TBI could be affected by ETA therapy. This study was designed to investigate ETA's effect on tissue damage, cytokine expression (TNF-α), lipid peroxidation, and oxidative stress (glutathione peroxidase [GSH-Px], malondialdehyde [MDA], and superoxide dismutase [SOD]), to evaluate whether post-TBI ETA administration in rats has protective effects.
Materials and Methods
Animal care and all experiments were conducted according to the European Communities’ Council Directive of November 24, 1986 (86/609/EEC), on the protection of animals for experimental use and approved by the Ethical Committee of the Ministry of Health Ankara Education and Research Hospital (Date: May 31, 2012; number: 0009).
Animal preparation
Thirty-six adult male Wistar albino rats (weight, approximately 300–350 g) were used. The temperature and relative humidity in the animal facility were maintained at 20°C ± 2°C and 50% ±10%, respectively. Food and water were provided ad libitum.
Surgical procedure and head trauma
Rats were anesthetized by intramuscular administration of 40 mg/kg ketamine HCl (Ketalar®; Pfizer Inc., New York, USA) and 5 mg/kg xylazine HCl (Rompun® 2%; Bayer HealthCare AG, Leverkusen, Germany). A median line scalp incision was made, and the periosteum was opened. A 10 mm diameter metal disc was glued to the cranium at the midline at the intersection of the coronal and lambdoid sutures. Diffuse closed head injury was induced by the method of Marmarou et al. using a 450 g/2 m weight–height impact onto the metal disc on the intact skull of the rats.[11] Thereafter, the trauma and ETA group rats were allowed to wake from anesthesia and examined by a neurosurgeon for neurological deficits.
After sedation, the animals were sacrificed by exsanguination through the aorta. The brain was removed and immediately extracted on ice and divided into two equal parts; half of the tissues were immediately snap-frozen in liquid nitrogen without additives and then transferred to tissue archiving freezer (−80°C) and stored at for biochemical analysis. The other half of the brain tissue was divided into two and used for light and electron microscopic evaluation.
Experimental groups
The animals were randomly allocated into three main groups. Control group (n = 12) was not subjected to trauma; trauma group (n = 12) was subjected to TBI, but no treatment was administered; and ETA group (n = 12) was subjected to TBI and received intraperitoneal ETA (5 mg/kg). The control, trauma, and ETA groups were each further subdivided into two subgroups (n = 6 each) according to the time of sacrifice. Rats sacrificed 1 h after TBI (hyperacute stage) were placed into the trauma-1 and ETA-1 groups, whereas those sacrificed 6 h after TBI (acute stage) were placed into the trauma-6 and ETA-6 groups.
Histopathological analysis
Tissue processing for light microscopy
For histological examination, brain tissue samples were fixed in 10% neutral-buffered formalin, dehydrated in a graded series of ethanol concentrations, and embedded in paraffin. Paraffin-embedded tissue samples were cut into 5-μm thick sections, stained with hematoxylin and eosin, and examined by light microscopy (Olympus CX21FS1, Olympus Co., Tokyo, Japan). Histopathological changes during the acute postinjury phase (1–2 days) such as eosin staining of cytoplasm (red neurons), pyknosis and hyperchromasia, satellitosis (perineural oligodendroglia), neutrophil degeneration (spongiosis), gliosis, polymorphonuclear leukocyte infiltration, and vascular congestion were graded on a scale of 0–3 by experienced histologists who were blind to the treatment groups: 0 was nonexistent; 1, mild; 2, moderate; and 3, severe.[12]
Tissue processing for electron microscopy
For electron microscopy, brain specimens were fixed by immersion in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 4–6 h at 4°C, postfixed in 1% osmium tetroxide for 2 h, dehydrated in an ascending alcohol concentration series, and embedded in Araldite. Semi-thin sections (1 μm) were stained with toluidine blue and observed under a light microscope. Ultra-thin sections stained with uranyl acetate and lead citrate were observed under an LEO 906E transmission electron microscope (Carl Zeiss AG, Oberkochen, Germany).
Tissue biochemical analysis
Tumor necrosis factor-alpha and interleukin-1 beta analysis
Tissue TNF-α and IL-1 β concentrations were determined using the double antibody sandwich enzyme-linked immunosorbent assay (R and D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Malondialdehyde analysis
MDA is formed from the breakdown of polyunsaturated fatty acids and serves as an important and reliable index for determining the extent of peroxidation reactions (Tator CH 1991). Tissue MDA levels were determined by thiobarbituric acid (TBA) reaction method. Briefly, samples were mixed with two volumes of cold saline solution containing 0.001% butylated hydroxytoluene (BHT) (200 μL of 0.01% BHT solution in methanol) and 0.07% sodium dodecyl sulfate (SDS) (20 μL of 7% SDS). Next, 1 mL of sample was added to 500 μL of 0.01 NH2SO4 and 500 μL of the TBA reagent (0.67% TBA in 50% acetic acid) to precipitate protein. Samples were heated in boiling water for 60 min, and after cooling, an equal volume (2 mL) of n-butanol was added to each test tube and the solution was mixed. The mixture was centrifuged at 4000 rpm for 10 min at room temperature. The absorbance of the organic layer in a 1 mL cell was read at 535 nm (Molecular Devices Corporation, Sunnyvale, CA, USA). MDA concentrations were expressed as nanomoles per milligram wet tissue weight.
Catalase analysis
Catalase (CAT) activity was determined by the method described by Aebi (Aebi H 1974). The assessment of CAT activity is based on the determination of the rate constant (k, sec-1) or of the hydrogen peroxide decomposition rate at 240 nm. Results were expressed as kU/g of protein.
Glutathione peroxidase analysis
GSH-Px activity was determined by measuring changes in nicotinamide adenine dinucleotide phosphate (NADPH) absorbance at 340 nm.[13] In the activity calculations (international unit [IU]), extinction coefficients of NADPH were used for GSH-Px. Results were expressed as IU/mg protein.
Superoxide dismutase analysis
Total SOD (Cu-Zn and Mn, EC 1.15.1.1) activity was determined according to the method of Sun et al.,[14] which is based on the inhibition of nitro blue tetrazolium (NBT) reduction by the xanthine-xanthine oxidase system, which acts as a superoxide generator. Activity was assessed in the ethanol phase of the supernatant after 1 mL ethanol/chloroform mixture (5:3, v/v) was added to the same volume of sample and centrifuged. One unit of SOD was defined as the enzyme amount causing 50% inhibition in the NBT reduction rate. SOD activity was expressed as U/mg protein.
Statistical analyses
Data analyses were performed by SPSS for Windows version 11.5 (SPSS Inc., Chicago, IL, USA). To determine if the distributions of continuous variables were normally distributed or not, we used Shapiro–Wilk test. Levene test was used to evaluate the homogeneity of variances. The data are shown as mean ± standard error of the mean (SEM). While the differences in normally distributed variables among groups were analyzed using one-way ANOVA, Kruskal–Wallis test was applied in not normally distributed data. When the P value from one-way ANOVA or Kruskal–Wallis test, statistics were statistically significant; the post hoc Tukey's honest significant difference or Conover's nonparametric multiple comparison test were used to determine which group differed from which other groups, and P < 0.05 was considered statistically significant. The data are shown as mean ± SEM.
Results
Histopathological analysis
Light microscopic examination of cerebral cortex demonstrated that white and gray matter had normal cellular composition, with typical neurons and neuroglial cells, in the control groups. The brain parenchyma neuropil in the cortex, which consists of dendrites, unmyelinated axons, and glial cell processes, was homogenous in appearance. There were no vascular changes [Figure 1]. Light microscopic examination of the brain tissue of the trauma groups showed signs of acute neuronal damage such as edema in the neuropil (spongiosis) and local vascular congestion. Neuronal cells showed hyperchromasia and pyknotic nuclei, as well as eosin staining of the cytoplasm (red neuron), which indicated dysfunctional mitochondrial metabolism that led to neuronal degeneration. Some of the damaged neurons were surrounded by an elevated number of glial cells (oligodendrocytes), which is known as satellitosis. Some focal gliosis was also observed. Perivascular edema caused by astrocytic foot processes swelling around blood vessels was evident. The white matter also showed axonal edema and oligodendrogliosis [Figure 2]. In the ETA-1 group, vascular dilatation and perivascular edema were observed in the cerebral cortex. Neuropil edema was less severe than that observed in the trauma-1 and -6 groups. In the cortex, there were many damaged red neurons, in addition to neurons with normal euchromatic nuclei. In the white matter, an abundance of glial cells was observed around axon extensions [Figure 3]. In the ETA-6 group, histopathological findings were similar to those found in the ETA-1 group but were less severe. Capillary dilatation and stasis were found in the cortex. In addition to normal neurons, red neurons with eosin staining in the cytoplasm were observed although fewer of these cells were found in the ETA-6 group compared to the ETA-1 group [Figure 3]. Electron microscope observations revealed that neurons in the control group had intact cellular organelles and normal vascular structures [Figure 4]. However, the trauma groups showed perivascular edema and structural disorganization in the neuropil. Watery cytoplasm was observed in neuronal cells, and microglial cells (MC) were found. The pathological alterations were more severe in the 6 h trauma groups [Figure 5]. One hour after treatment, ultrastructural analysis of the cerebral cortex in ETA-treated groups showed mild perivascular edema and neurons surrounded by MC. Six hours after treatment, the neuropil, blood vessels, and neurons exhibited good structural appearance [Figure 6].
Figure 1.
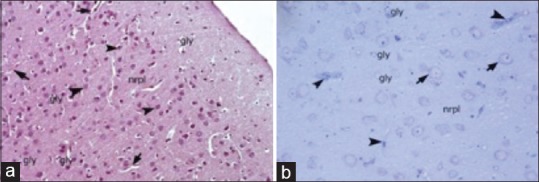
Light microscopic examination of the cerebral cortex of the control group. Gly: Glia cells; arrowheads: Neuron soma; arrows: Capillaries; nrpl: Neuropil; (H and E, ×20). Semi-thin section arrowhead: Capillaries; arrows: Neuron soma with clear nucleus and nucleolus, gly: Neuroglia; ×40, Toluidine blue staining
Figure 2.
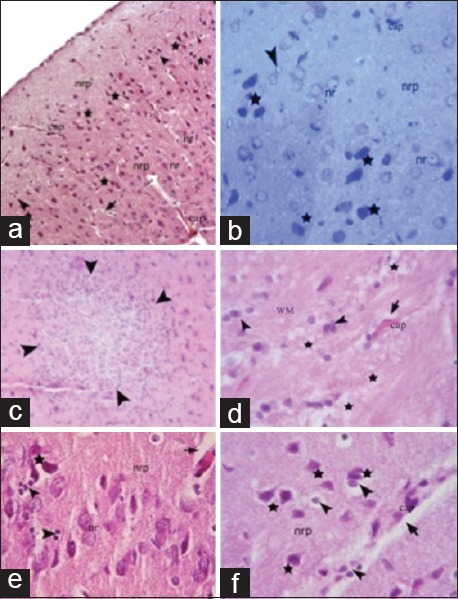
Light microscopic examination of the sham trauma groups 1 and 6 h after injury. One-hour trauma group: (a and b); 6-h trauma group; (c-f). nr: Normal neuron; nrp: Neuropil edema; arrowheads, satellitosis; arrows, perivascular edema; stars, degenerated neurons; cap, brain capillary
Figure 3.
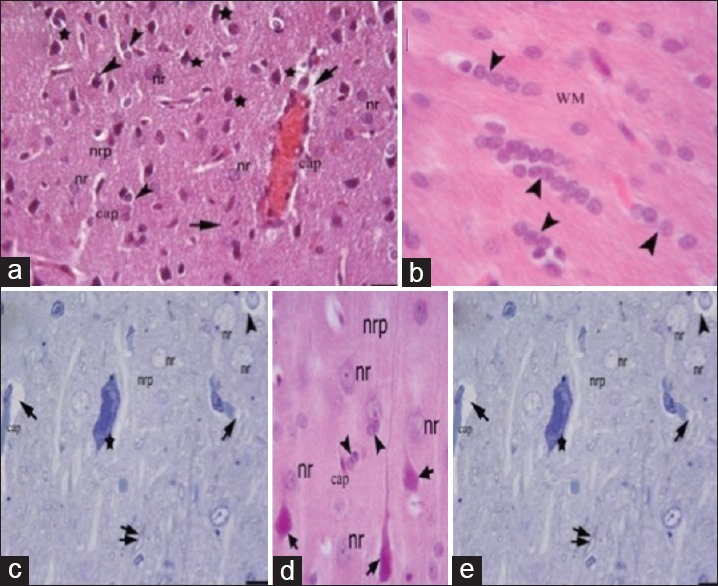
Light microscopic examination of the etanercept-treated trauma groups. Etanercept-1 (1 h after trauma) group: (a and b) Etanercept-6 (6 h after trauma) group: (c-e) nr: Nucleus of a neuron; nrp: Neuropil; cap: Capillaries; star, degenerated neurons; arrowheads, neuroglia; arrows, perivascular edema; WM: White matter; pia: Pia mater; arrows, degenerated red neurons; double arrow, myelinated axons
Figure 4.
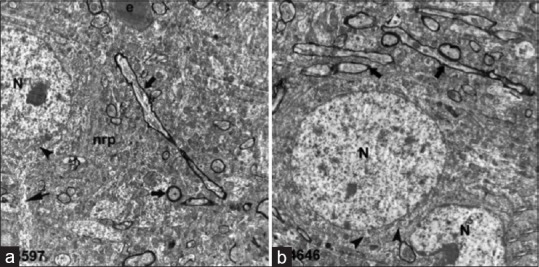
Electron microscopic examination of control group: (a and b). Neuronal cell nucleus (N) and mitochondria (arrowhead) in the cytoplasm. Neural cell process (thin arrow) and myelinated fibers (thick arrows) in the neuropil (nrp)
Figure 5.
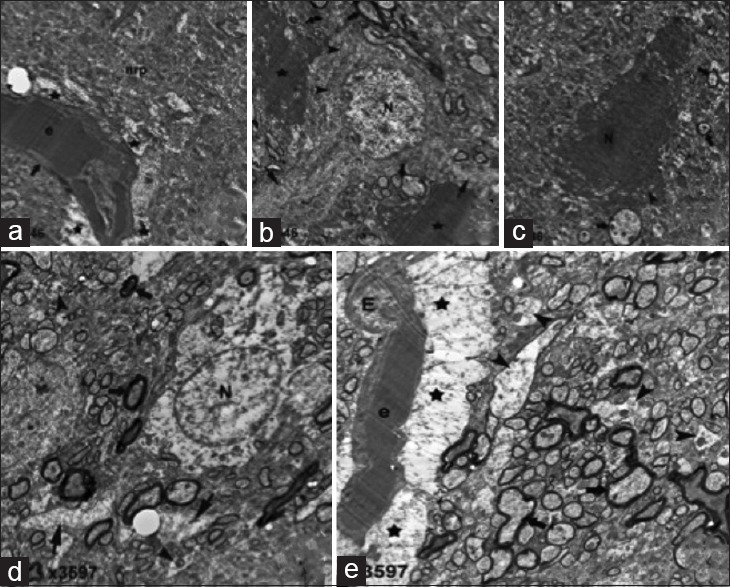
Electron microscopy 1 h after traumatic brain injury (a-c). (a) Brain blood vessel (thick arrow) with erythrocytes (e), perivascular edema (star), neutrophil (nrp). (b) Neuron (N) with processes (thick arrows), mitochondria (arrowheads) surrounded by microglia (stars). (c) Mitochondria (arrowhead), cytoplasm (thick arrow). Six hours after traumatic brain injury (d-e). (d) Neuron (N) with swollen neuronal process (thin arrows), myelinated axons (thick arrows), naked axons (arrowheads). (e) Endothelial cell (E), erythrocytes (e), astrocytic foot process swelling (stars), myelinated (thick arrows), naked axons (arrowhead)
Figure 6.
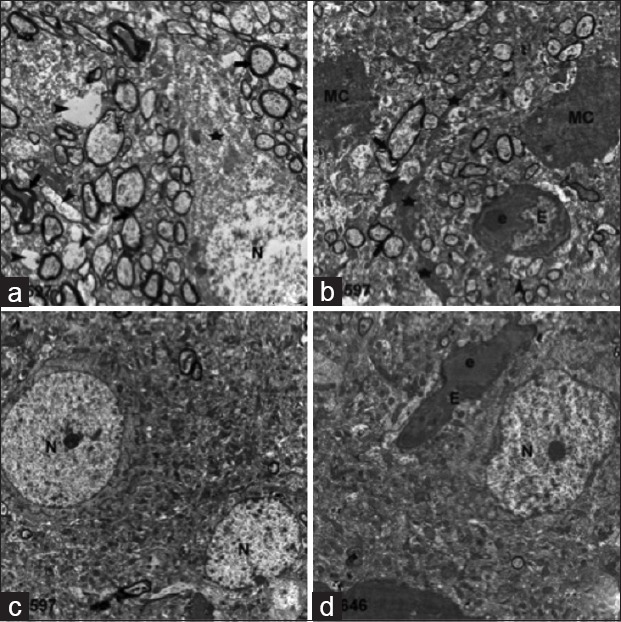
Electron microscopic examination of 1 h after injury in the etanercept-treated trauma group: (a and b). (a) Moderate damage (star) of a neuron (N), myelinated (thick arrows) and naked (arrowheads) axons. (b) Microglial cells and their extensions (stars), endothelial cell (E), erythrocyte (e). Six hours after injury in the etanercept-treated trauma group: (c and d). neural cells (N), endothelial cell (E), and erythrocyte (e)
Tissue biochemical analysis
Tissue tumor necrosis factor-alpha levels
Tissue TNF-α levels were significantly increased in rats of the trauma group 1 h after head injury, compared to control group (P < 0.001). The ETA-treated group exhibited significantly lower MDA levels compared to trauma group (P < 0.001). There was a statistically significant difference in MDA levels between the control- and ETA-treated groups (P < 0.001). Similarly, the ETA-treated group also exhibited significantly lower levels of MDA compared to the trauma group 6 h after head injury (P < 0.001).
Tissue interleukin 1 levels
IL-1 levels after 1 h were significantly elevated in the brain tissue of diffuse closed head injury-exposed rats, in comparison to the control group (P < 0.001). Treatment with ETA significantly inhibited this increase in IL-1 level relative to trauma group (P < 0.001) although IL-1 levels in ETA-treated rats were still significantly higher than in control group animals (P < 0.001). In addition, IL-1 levels after 6 h trauma were significantly decreased in ETA-treated rats compared to trauma group (P < 0.001).
Tissue malondialdehyde levels
Significantly higher MDA levels were observed in the head injury group relative to control group animals after 1 h (P < 0.001). ETA significantly decreased MDA levels compared to the trauma group (P < 0.001). MDA levels in the trauma group were also significantly higher than in the ETA-treated group (P < 0.001) 6 h after trauma. Treatment with ETA did not reduce MDA to baseline levels, as MDA levels in both treatment groups at 1 and 6 h were significantly greater than in the control group (P < 0.001 and P = 0.001, respectively).
Tissue catalase, glutathione peroxidase levels, and superoxide dismutase levels
CAT enzyme activity reduced significantly in rats 1 and 6 h after head injury in comparison with the control group (P < 0.001 for both). ETA treatment caused a statistically significant increase in CAT activity when compared with the trauma group (P = 0.002 for 1 h and P = 0.005 for 6 h). The ETA-treated group also showed a statistically significant increase in GSH-Px enzyme activity at 1 and 6 h after diffuse closed head injury (P < 0.001 for both). Similarly, levels of SOD activity in trauma group were significantly decreased compared to levels in the control group (P < 0.001 for 1 and 6 h). ETA treatments at 1 and 6 h significantly increased SOD activity compared to the trauma group (P < 0.001 for both). However, treatment with ETA did not increase the antioxidant enzyme activity to the levels of the control groups, as CAT, GSH-Px, and SOD levels in both treatment groups at 1 and 6 h were significantly lower than in the control groups. All biochemical results are shown in Table 1.
Table 1.
Biochemical results relevant to the study groups

Discussion
The present study provides evidence for the protective effect of intraperitoneally administered ETA against TBI in a rat model. The beneficial effects of ETA were observed both histologically and neurologically. The method described by Marmarou et al.[11] was used as the trauma model in our study; in this method, the area between the coronal and lambdoid sutures was targeted, and a diffuse brain injury was caused by allowing an object weighing 450 g to fall freely from an altitude of 2 m through a Plexiglas tube. A possible fracture of the calvarium was prevented by covering it with a stainless steel plate. Although this model has high mortality and a high risk of posttraumatic seizure, none of the rats in the present study died of trauma. Several groups[6,15,16,17,18,19] have reported increased TNF-α and other cytokine levels 1 h post-TBI and peak levels 4 h post-TBI, after which levels returned to baseline so that we assessed the effect of ETA on histopathological and biochemical changes 1 and 6 h after TBI in rats.
Cytokines seem to play an important pathophysiological role in inflammatory diseases of the central nervous system (CNS).[20] Accumulating evidence has demonstrated that local synthesis of cytokines by resident cells in the CNS is initiated in response to TBI. Local induction of TNF-α has been demonstrated in various head trauma models such as experimental cortical contusion, surgical brain injury, fluid percussion trauma, and experimental axonal injury.[21] In accordance with data derived from animal experiments, clinical studies have demonstrated elevated levels of TNF-α in CSF and serum from head trauma patients.[3,4] Inhibition of TNF-α with ETA effectively attenuates the formation of TBI-induced cerebral contusions, motor and cognitive dysfunction, astrocytic and microglial activation, and inflammation. Although the central effects of ETA administered systemically have been reported in the literature, ETA may have exerted the effect through its ability to block peripheral TNF-α, which is produced by the liver as part of the peripheral response to acute CNS inflammation or TBI.[5,22] Inhibition of TNF-α activity reduces hepatic chemokine expression, thus decreasing the mobilization of neutrophils from the bone marrow into the circulation and reducing the number available for recruitment to the brain. In addition, TNF-α causes a neuroinflammatory response in the injury penumbra and an increase in blood–brain barrier permeability which may result in cerebral edema.[23,24,25]
In the present study, tissue biochemical analysis showed that TNF-α and IL-1 β levels in the ETA group were significantly lower than those of the trauma group in the hyperacute and acute stages. Reactive oxygen species are key mediators of secondary injury induced by trauma.[26] The brain is particularly vulnerable to oxidative injury because of its high rate of oxygen consumption, intense production of reactive radicals, and high levels of transition ETA, such as iron, which catalyze the production of reactive radicals.[27,28] When tissues are exposed to oxidative stress, they increase the activity and expression of antioxidant enzymes as a compensatory mechanism against free radical-mediated damage. Nevertheless, increased antioxidant enzyme activity may be insufficient to counteract the damage produced by oxidative stress during many pathological conditions. In the present study, we demonstrated that tissue CAT, GSH-Px, and SOD levels decreased significantly after TBI. We hypothesize that this decrease is due to highly elevated oxidative stress. After the administration of ETA, expression of these antioxidant enzymes increased significantly at 1 and 6 h after TBI. It has been shown that TNF-α inhibited neurite outgrowth of cultured dorsal root ganglia and hippocampal neurons[20] which immediate ETA therapy enhanced the rate of axonal regeneration after nerve injury in rats.[29] In addition, endotoxin-induced inflammation impaired neurogenesis, whereas blocking inflammation restored neurogenesis in the adult brain following ischemic insult.[30] Silver and Miller proposed that overcoming the inhibitory environment of the glial scar might allow for long-distance functional regeneration after TBI.[31] Indeed, as demonstrated in the current results, neuropil edema in the ETA-1 group was less severe than that of the sham groups. In the ETA-6 group, histopathological findings were similar to those found in the ETA-1 group but less severe. ETA-1 group scores were similar to those of the sham-1 group; in contrast, ETA-6 group scores were half the magnitude of those of the sham-6 group. Therefore, ETA might enhance functional neuronal regeneration by inhibiting glial scar formation during TBI.
The dose of ETA used in this study, 5 mg/kg, is more than five times the normal human weekly ETA dose. High-dose treatment might have been resulted in penetration of ETA into the CSF. In addition, broken down blood–brain barrier after TBI as an adjunct to further ameliorate the delivery of ETA into the CSF might also have proved ETA to have a potential therapeutic role in TBI. Similarly, in a recent Alzheimer model, high doses of ETA given peripherally were necessary to achieve a therapeutic effect.[32] Perispinal or other forms of administration designed to selectively improve CNS delivery may enhance the therapeutic effect of ETA for treatment of brain disorders.[8,33,34,35,36,37,38] Recent clinical data provide evidence of the effectiveness of perispinal ETA for the treatment of chronic brain dysfunction following stroke and TBI.[36] However, systemically administration of ETA penetrates directly into the contused brain parenchyma and improves the outcomes by reducing TNF-α and also by stimulating newly formed neurogenesis.[9]
We also performed electron microscopy analyses to support the biochemical and histopathological results of the present study. Results of the electron microscopic evaluation were in accordance with elevated microglial activation subsequent to TBI which was attenuated in ETA-1 and -2 treatment groups. Several lines of study have demonstrated the evidence of elevated microglial activity in head injury,[39,40,41,42] and ETA showed greater tissue-protective effects as shown by a significant attenuation of microglial activation. The findings were consistent with the previously reported effects of ETA in rat model of spinal cord injury, hepatic encephalopathy, and glaucoma.[43,44,45]
Conclusions
A better understanding of secondary damage mechanisms will allow researchers to develop treatments that target particular biochemical signaling pathways, which will reduce the severity of head trauma-related injury and the rate of head trauma-related mortality. The results of the present study indicate that ETA possesses promising protective effects against TBI, which it may produce by depressing inflammation. These findings suggest that ETA could become a new therapeutic agent for the treatment of TBI.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Masters SJ. Evaluation of head trauma: Efficacy of skull films. AJR Am J Roentgenol. 1980;135:539–47. doi: 10.2214/ajr.135.3.539. [DOI] [PubMed] [Google Scholar]
- 2.Ates O, Cayli S, Altinoz E, Gurses I, Yucel N, Sener M, et al. Neuroprotection by resveratrol against traumatic brain injury in rats. Mol Cell Biochem. 2007;294:137–44. doi: 10.1007/s11010-006-9253-0. [DOI] [PubMed] [Google Scholar]
- 3.Nishio S, Yunoki M, Noguchi Y, Kawauchi M, Asari S, Ohmoto T. Detection of lipid peroxidation and hydroxyl radicals in brain contusion of rats. Acta Neurochir Suppl. 1997;70:84–6. doi: 10.1007/978-3-7091-6837-0_26. [DOI] [PubMed] [Google Scholar]
- 4.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–40. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hailer NP, Vogt C, Korf HW, Dehghani F. Interleukin-1beta exacerbates and interleukin-1 receptor antagonist attenuates neuronal injury and microglial activation after excitotoxic damage in organotypic hippocampal slice cultures. Eur J Neurosci. 2005;21:2347–60. doi: 10.1111/j.1460-9568.2005.04067.x. [DOI] [PubMed] [Google Scholar]
- 6.Stahel PF, Shohami E, Younis FM, Kariya K, Otto VI, Lenzlinger PM, et al. Experimental closed head injury: analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J Cereb Blood Flow Metab. 2000;20:369–80. doi: 10.1097/00004647-200002000-00019. [DOI] [PubMed] [Google Scholar]
- 7.Francis J, Chu Y, Johnson AK, Weiss RM, Felder RB. Acute myocardial infarction induces hypothalamic cytokine synthesis. Am J Physiol Heart Circ Physiol. 2004;286:H2264–71. doi: 10.1152/ajpheart.01072.2003. [DOI] [PubMed] [Google Scholar]
- 8.Chio CC, Lin JW, Chang MW, Wang CC, Kuo JR, Yang CZ, et al. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J Neurochem. 2010;115:921–9. doi: 10.1111/j.1471-4159.2010.06969.x. [DOI] [PubMed] [Google Scholar]
- 9.Cheong CU, Chang CP, Chao CM, Cheng BC, Yang CZ, Chio CC. Etanercept attenuates traumatic brain injury in rats by reducing brain TNF-α contents and by stimulating newly formed neurogenesis. Mediators Inflamm 2013. 2013 doi: 10.1155/2013/620837. 620837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chio CC, Chang CH, Wang CC, Cheong CU, Chao CM, Cheng BC, et al. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-α. BMC Neurosci. 2013;14:33. doi: 10.1186/1471-2202-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- 12.Mena H, Cadavid D, Rushing EJ. Human cerebral infarct: a proposed histopathologic classification based on 137 cases. Acta Neuropathol. 2004;108:524–30. doi: 10.1007/s00401-004-0918-z. [DOI] [PubMed] [Google Scholar]
- 13.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70:158–69. [PubMed] [Google Scholar]
- 14.Sun Y, Oberley LW, Li Y. A simple method for clinical assay of superoxide dismutase. Clin Chem. 1988;34:497–500. [PubMed] [Google Scholar]
- 15.Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol. 1993;42:177–85. doi: 10.1016/0165-5728(93)90008-m. [DOI] [PubMed] [Google Scholar]
- 16.Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72:169–77. doi: 10.1016/s0165-5728(96)00181-6. [DOI] [PubMed] [Google Scholar]
- 17.Holmin S, Schalling M, Höjeberg B, Nordqvist AC, Skeftruna AK, Mathiesen T. Delayed cytokine expression in rat brain following experimental contusion. J Neurosurg. 1997;86:493–504. doi: 10.3171/jns.1997.86.3.0493. [DOI] [PubMed] [Google Scholar]
- 18.Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95:115–25. doi: 10.1016/s0165-5728(98)00273-2. [DOI] [PubMed] [Google Scholar]
- 19.Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–9. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- 20.Larsson K, Rydevik B, Olmarker K. Disc related cytokines inhibit axonal outgrowth from dorsal root ganglion cells in vitro . Spine (Phila Pa 1976) 2005;30:621–4. doi: 10.1097/01.brs.0000155410.48700.9e. [DOI] [PubMed] [Google Scholar]
- 21.Bethea JR, Nagashima H, Acosta MC, Briceno C, Gomez F, Marcillo AE, et al. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma. 1999;16:851–63. doi: 10.1089/neu.1999.16.851. [DOI] [PubMed] [Google Scholar]
- 22.Campbell SJ, Jiang Y, Davis AE, Farrands R, Holbrook J, Leppert D, et al. Immunomodulatory effects of etanercept in a model of brain injury act through attenuation of the acute-phase response. J Neurochem. 2007;103:2245–55. doi: 10.1111/j.1471-4159.2007.04928.x. [DOI] [PubMed] [Google Scholar]
- 23.Kim KS, Wass CA, Cross AS, Opal SM. Modulation of blood-brain barrier permeability by tumor necrosis factor and antibody to tumor necrosis factor in the rat. Lymphokine Cytokine Res. 1992;11:293–8. [PubMed] [Google Scholar]
- 24.Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. 2004;190:446–55. doi: 10.1016/j.expneurol.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Lv S, Song HL, Zhou Y, Li LX, Cui W, Wang W, et al. Tumour necrosis factor-alpha affects blood-brain barrier permeability and tight junction-associated occludin in acute liver failure. Liver Int. 2010;30:1198–210. doi: 10.1111/j.1478-3231.2010.02211.x. [DOI] [PubMed] [Google Scholar]
- 26.Tyurin VA, Tyurina YY, Borisenko GG, Sokolova TV, Ritov VB, Quinn PJ, et al. Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J Neurochem. 2000;75:2178–89. doi: 10.1046/j.1471-4159.2000.0752178.x. [DOI] [PubMed] [Google Scholar]
- 27.Evans PH. Free radicals in brain metabolism and pathology. Br Med Bull. 1993;49:577–87. doi: 10.1093/oxfordjournals.bmb.a072632. [DOI] [PubMed] [Google Scholar]
- 28.Ozdemir D, Uysal N, Gonenc S, Acikgoz O, Sonmez A, Topcu A, et al. Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiol Res. 2005;54:631–7. [PubMed] [Google Scholar]
- 29.Kato K, Liu H, Kikuchi S, Myers RR, Shubayev VI. Immediate anti-tumor necrosis factor-alpha (etanercept) therapy enhances axonal regeneration after sciatic nerve crush. J Neurosci Res. 2010;88:360–8. doi: 10.1002/jnr.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718–24. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- 31.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–56. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 32.Detrait ER, Danis B, Lamberty Y, Foerch P. Peripheral administration of an anti-TNF-α receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-α levels and memory deficits in mice. Neurochem Int. 2014;72:10–3. doi: 10.1016/j.neuint.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Tobinick E. Perispinal etanercept: a new therapeutic paradigm in neurology. Expert Rev Neurother. 2010;10:985–1002. doi: 10.1586/ern.10.52. [DOI] [PubMed] [Google Scholar]
- 34.Clark IA, Alleva LM, Vissel B. The roles of TNF in brain dysfunction and disease. Pharmacol Ther. 2010;128:519–48. doi: 10.1016/j.pharmthera.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Clark I. New hope for survivors of stroke and traumatic brain injury. CNS Drugs. 2012;26:1071–2. doi: 10.1007/s40263-012-0014-1. [DOI] [PubMed] [Google Scholar]
- 36.Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs. 2012;26:1051–70. doi: 10.1007/s40263-012-0013-2. [DOI] [PubMed] [Google Scholar]
- 37.Camara ML, Corrigan F, Jaehne EJ, Jawahar MC, Anscomb H, Baune BT. Effects of centrally administered etanercept on behavior, microglia, and astrocytes in mice following a peripheral immune challenge. Neuropsychopharmacology. 2015;40:502–12. doi: 10.1038/npp.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sedger LM, McDermott MF. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants – Past, present and future. Cytokine Growth Factor Rev. 2014;25:453–72. doi: 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 39.Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WS, et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 40.Folkersma H, Boellaard R, Yaqub M, Kloet RW, Windhorst AD, Lammertsma AA, et al. Widespread and prolonged increase in (R)-(11) C-PK11195 binding after traumatic brain injury. J Nucl Med. 2011;52:1235–9. doi: 10.2967/jnumed.110.084061. [DOI] [PubMed] [Google Scholar]
- 41.Folkersma H, Foster Dingley JC, van Berckel BN, Rozemuller A, Boellaard R, Huisman MC, et al. Increased cerebral (R)-[(11) C] PK11195 uptake and glutamate release in a rat model of traumatic brain injury: a longitudinal pilot study. J Neuroinflammation. 2011;8:67. doi: 10.1186/1742-2094-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 43.Marchand F, Tsantoulas C, Singh D, Grist J, Clark AK, Bradbury EJ, et al. Effects of etanercept and minocycline in a rat model of spinal cord injury. Eur J Pain. 2009;13:673–81. doi: 10.1016/j.ejpain.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Chastre A, Bélanger M, Beauchesne E, Nguyen BN, Desjardins P, Butterworth RF. Inflammatory cascades driven by tumor necrosis factor-alpha play a major role in the progression of acute liver failure and its neurological complications. PLoS One. 2012;7:e49670. doi: 10.1371/journal.pone.0049670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roh M, Zhang Y, Murakami Y, Thanos A, Lee SC, Vavvas DG, et al. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF-α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One. 2012;7:e40065. doi: 10.1371/journal.pone.0040065. [DOI] [PMC free article] [PubMed] [Google Scholar]