

Abstract
The developmental lineage of the PHOX2B-expressing neurons in the retrotrapezoid nucleus (RTN) has been extensively studied. These cells are thought to function as central respiratory chemoreceptors, i.e., the mechanism by which brain Pco2 regulates breathing. The molecular and cellular basis of central respiratory chemoreception is based on the detection of CO2 via intrinsic proton receptors (TASK-2, GPR4) as well as synaptic input from peripheral chemoreceptors and other brain regions. Murine models of congenital central hypoventilation syndrome designed with PHOX2B mutations have suggested RTN neuron agenesis. In this review, we examine, through human and experimental animal models, how a restricted number of neurons that express the transcription factor PHOX2B play a crucial role in the control of breathing and autonomic regulation.
Keywords: chemoreflex, CCHS, ventrolateral medulla, breathing
cerebral autonomic neuron dysfunction would be expected to lead to neurological disease. Such is the case with dysfunction of cells in the retrotrapezoid nucleus (RTN). Seminal experiments underscore the importance that RTN neurons play in neurophysiological homeostasis. In 2003, Amiel and colleagues published the first evidence that polyalanine expansions of the paired-like homeobox gene PHOX2B cause central congenital hypoventilation syndrome (CCHS) (Amiel et al. 2003; Weese-Mayer et al. 2005). CCHS patients suffer lower ventilatory responses to hypercapnia and hypoxia compared with normal controls (Weese-Mayer et al. 2005). The proposed mechanism centers on a selective lack of central chemosensory integration rather than respiratory central pattern generator (rCPG) dysfunction (Ikeda et al. 2015; Ruffault et al. 2015; Stornetta et al. 2006). In these patients, breathing may be adequate during wakeful states with ventilation increasing normally during volitional stimulation or exercise (Diedrich et al. 2007). However, when asleep, CCHS patients experience life-threatening hypoventilation or complete apnea. In short, the symptoms of CCHS support the existence of neurons that selectively mediate the chemical drive to breathe and suggest that these neurons are especially important during sleep.
In rodents, PHOX2B expression is required for the development of the nucleus of the solitary tract (NTS), brain stem catecholaminergic neurons, carotid bodies, enteric nervous system, sympathetic ganglionic (but not preganglionic) neurons, and the cranial parasympathetic system but is not required for the development of the serotonergic system (Fig. 1) (Ikeda et al. 2015; Ruffault et al. 2015). PHOX2B expression persists in most brain stem structures after birth (Dauger et al. 2003), including the hypercapnia-sensitive neurons of the RTN and the NTS neurons that mediate the peripheral chemoreflex (Stornetta et al. 2006; Takakura et al. 2014). In short, an uninterrupted neuronal chain involved in chemosensory integration express PHOX2B, whereas the rCPG itself lacks PHOX2B expression. This expression pattern clearly fits with the symptoms of CCHS (normal day-time breathing but greatly reduced peripheral and central chemoreflexes). Furthermore, from a basic science standpoint, the presence of PHOX2B in the RTN and its absence from the rCPG provides an important clue that the respiratory network's pH sensitivity relies heavily on specialized neurons that drive the rCPG, not on the rCPG itself (Guyenet and Bayliss 2015). Indeed, the presence of PHOX2B-positive RTN neurons underscores a compelling disease-based rationale to elucidate their roles in breathing.
Fig. 1.

Schematic view of the control of CO2 homeostasis and the role of PHOX2B-expressing neurons. Retrotrapezoid nucleus (RTN) chemoreceptor neurons are activated by CO2 via their intrinsic pH sensitivity and via inputs from the carotid bodies. RTN neurons target various components of the respiratory central pattern generator (rCPG) and are presumed to play a key role in breathing automaticity during anesthesia, sleep, and quiet waking. The carotid body may also influence the activity of the rCPG neurons through connections (not shown) that bypass the RTN (Stornetta et al. 2006; Takakura et al. 2006). RTN neurons are also inhibited by lung inflation via slowly adapting receptor (SAR) activation and by the fact that the effect of SARs is mediated by GABAergic pump cells located within the nucleus of the solitary tract (NTS) at area postrema level (Moreira et al. 2007). In mammals, PHOX2B expression is required for the development of the carotid bodies (1) (Dauger et al. 2003), NTS (2) (Stornetta et al. 2006), and RTN chemoreceptors neurons (3 and 4) (Moreira et al. 2007; Stornetta et al. 2006), as well as brain stem catecholaminergic neurons, enteric nervous system, sympathetic ganglionic (but not preganglionic) neurons, and the cranial parasympathetic system but is not required for the serotonergic system or the rCPG neurons (5) (Stornetta et al. 2006). 1, Illustration showing the expression of PHOX2B immunoreactive in the carotid body between E13.5 and E16.5 in mice. [Modified from Dauger et al. (2003).]. 2, Awake rats subjected to hypoxia and hypoxia-activated NTS neurons were identified by the presence of Fos-immunoreactive nuclei. The brain tissue was processed for simultaneous detection of FluorGold (FG; native blue fluorescence), PHOX2B immunoreactivity (Alexa 488 fluorescence; green), Fos immunoreactivity (Cy3; red). The photomicrograph taken within the commissural portion of the NTS shows multiple aqua-colored neurons (arrows) with RTN projections, activated by carotid body stimulation and expressing PHOX2B. The color coding for other combinations of markers is indicated. Scale bar, 50 μm. [Modified from Stornetta et al. (2006).] 3, Example of one RTN chemoreceptor neuron (arrows) labeled in vivo with biotinamide [PHOX2B immunoreactivity revealed with Alexa 488 (green) and biotinamide with Cy-3 (red); colocalization shown in yellow]. Scale bar, 50 μm. [Modified from Stornetta et al. (2006).] 4, Example of one RTN chemoreceptor neuron (arrows) labeled in vivo with biotinamide [PHOX2B immunoreactivity revealed with Cy-3 (red) and biotinamide with Alexa 488 (green); colocalization shown in yellow]. [Modified from Moreira et al. (2007).] 5, Biotinamide labeling of the Bötzinger complex. This sample was also reacted for PHOX2B immunoreactivity (Cy3; red) revealing that the recorded neuron (Alexa 488; green) was PHOX2B negative. Scale bar, 50 μm. [Modified from Stornetta et al. (2006).]
Central respiratory chemoreception mechanisms.
Recent literature describes at least four prevalent theories on the cell types responsible for mediating central respiratory chemoreception. These theories attribute chemosensation to 1) specialized respiratory chemoreceptors, 2) broad-spectrum chemoreceptors, 3) ubiquitous chemoreceptors, and 4) pH-insensitive neurons (Guyenet 2008; Guyenet et al. 2010; Guyenet and Bayliss 2015; Nattie 2011). In the present review, we focus on the role of specialized respiratory chemoreceptors. The chemosensitivity process refers to the intrinsic pH sensitivity of brain cells including neurons, glia, and blood vessels, whereas respiratory chemoreception, commonly called central chemoreception, is the process by which central nervous system (CNS) Pco2 drives respiration (Feldman et al. 2003; Guyenet et al. 2010; Guyenet and Bayliss 2015; Nattie 2011). Thus respiratory chemoreception refers both to a subset of chemosensitive cells, the respiratory chemoreceptors, and to their relationship to a specific neuronal network, the breathing network. Intracellular pH changes mediate neuronal pH sensitivity in vitro (Kumar et al. 2015; Mulkey et al. 2004; Ritucci et al. 2005; Wang et al. 2013), presumably attributed to the simultaneous effects of pH on multiple ion channels (Kumar et al. 2015; Putnam et al. 2004). The correct identification of relevant central chemoreceptors is complicated by the fact that brain stem neurons very commonly respond to extracellular fluid acidification in vitro and that, equally common, some pH responses persist after blockade of action potential-dependent transmitter release with tetrodotoxin (Bayliss et al. 2001; Kumar et al. 2015; Mulkey et al. 2004; Wang et al. 2013). Such results have led others to theorize that all rCPG neurons could be equally sensitive to pH and to conclude that the entire respiratory network possesses central respiratory chemosensitivity. That CCHS patients show a virtual loss of central respiratory chemoreception without impairment of daytime breathing challenges the view that the entire rCPG shows chemosensitive properties. An alternative view to which we subscribe is that the widespread pH sensitivity of rCPG neurons observed in vitro does not explain central respiratory chemoreception. Rather, central chemoreception relies on specialized neurons (Loeschcke 1982; Mitchell et al. 1963) that are responsible for the highly sensitive respiratory output resulting from brain pH changes (Eldridge et al. 1984). We propose that RTN neurons exemplify such cells and that others (locus coeruleus, medullary raphe, and the NTS) are yet to be found, presumably within brain stem regions where focal acidification in vivo activates breathing (Biancardi et al. 2008; Dean et al. 1989; Feldman et al. 2003; Guyenet et al. 2010; Guyenet and Bayliss 2015; Nattie 2011; Richerson 2004).
Chemoreceptors of the RTN.
The respiratory stimulant effects of ventral medullary surface (VMS) acidification and the depressant effects caused by VMS cooling or topical application of inhibitory neurotransmitters lead to the hypothesis that respiratory chemoreception occurs near the VMS (Loeschcke 1982; Millhorn 1986; Millhorn and Eldridge 1986; Mitchell et al. 1963). The most notable chemosensitive region of the VMS is located roughly ventral to the facial motor nucleus and overlaps the region that was defined in 1989 as the RTN (Smith et al. 1989). Smith and colleagues defined the RTN as extending from the caudal end of the trapezoid body to the Bötzinger complex level of the ventral respiratory column (VRC) and from the lateral edge of the pyramids to the trigeminal tract (Smith et al. 2013). Classical lesion/stimulation experiments support the notion that an excitatory drive to the breathing pattern generators derives from RTN neurons (Nattie 2011; Nattie and Li 2000; Takakura et al. 2013). Furthermore, RTN acidification in vivo increases breathing, whereas inhibition of the same area with drugs or neuronal lesions reduces breathing and central chemosensitivity (Nattie 2011; Nattie and Li 2000; Takakura et al. 2008, 2013, 2014). Finally, Fos induction in the RTN occurs when animals or brain stem preparations are exposed to hypercapnia (Fortuna et al. 2009; Sato et al. 1992); these data suggest hypercapnia directly activates RTN cells. In addition, pathological evidence obtained from polyalanine repeat mutation (PARM) CCHS animal models supports the notion that central respiratory chemosensitivity relies on relatively few specialized neurons, such as the chemoreceptor neurons of the RTN (Dubreuil et al. 2008; Ramanantsoa et al. 2011). In this genetic disease, central chemosensitivity is essentially absent; severe hypoventilation occurs during sleep but relatively adequate breathing persists during waking, and some degree of exercise-induced hyperventilation remains, at least in the milder cases (Amiel et al. 2003, 2009; Carroll et al. 2010; Gozal, 1998; Shea et al. 1993). The severity of the symptoms increases according to the number of extra alanine residues in the mutated polyalanine track of PHOX2B (Carroll et al. 2010). The respiratory symptoms of the disease suggest that the central respiratory controller located in the pre-Bötzinger complex of CCHS patients is largely intact but that the brain of these individuals lacks neurons that are either uniquely specialized in detecting CO2 or are specialized in funneling the excitatory influence of CO2 to the respiratory controller. The mouse models of the disease suggest that these missing neurons could be the RTN neurons (Dubreuil et al. 2008; Goridis et al. 2010; Nobuta et al. 2015; Ramanantsoa et al. 2011).
Chemoreceptor neurons in the RTN during development and in adult mammals.
Lineage-tracing experiments suggest that the RTN originates from an Egr-2-dependent embryonic domain (rhombomeres 3/5) and coexpresses Atoh-1 and PHOX2B during late embryogenesis (Dubreuil et al. 2009; Ramanantsoa et al. 2011). Proper RTN development depends on the aforementioned transcription factors (Huang et al. 2012; Ruffault et al. 2015). Perinatally, RTN neurons seem to retain many of their embryonic characteristics, i.e., their pacemaker properties, pH modulation, and ability to activate the pre-Bötzinger complex (Fortin and Thoby-Brisson 2009; Onimaru et al. 2008, 2014; Onimaru and Homma 2003; Thoby-Brisson et al. 2009). In adult rats, RTN neurons function as central chemoreceptors by controlling multiple aspects of breathing including frequency, depth of inspiration, active expiration, and airway patency (Abbott et al. 2013; Guyenet and Bayliss 2015; Silva et al. 2016; Takakura et al. 2014). Several key pieces of evidence support the role of RTN neurons as central chemoreceptors. First, these neurons respond with a massive increase in activity to hypercapnia in vivo (Mulkey et al. 2004; Takakura et al. 2006). The discharge activity rate of RTN chemoreceptors neurons varies by an average of ∼3.0 Hz for a 1% change in arterial Pco2 (PaCO2) (Guyenet et al. 2005). These cells are silent at low levels of CO2, and most of the RTN cells have a threshold (CO2 level above which the cells are active) that is below that of the respiratory output (Guyenet et al. 2005; Takakura et al. 2006). In addition to their extreme pH sensitivity, it is also important to point out that RTN neurons in vivo presumably rely on a combination of synaptic drives (peripheral chemoreceptors, raphe, hypothalamic neurons, and astrocytes) and intrinsic pH sensitivity (Barna et al. 2014; Guyenet and Bayliss 2015; Nattie and Li 2002; Takakura et al. 2014). We believe that such integrative characteristics are expected from neurons (central chemoreceptors) that drive the respiratory system.
In summary, multiple studies performed on RTN neurons support a role in central chemosensation and include 1) their intrinsic acid sensitivity (Kumar et al. 2015; Wang et al. 2013) and 2) their integration of excitatory inputs from the peripheral chemoreceptors, raphe, and hypothalamus and from neighboring astrocytes that modulate respiratory drive (Barna et al. 2014; Fortuna et al. 2009; Moreira et al. 2015; Nattie 2011; Richerson 2004; Takakura et al. 2006; Wenker et al. 2010).
CCHS: insights into pathophysiology from clinical, molecular, and genetic studies.
CCHS was first described by modern medicine in 1970 (Mellins et al. 1970) and is classically characterized as a genetically inherited disorder with alveolar hypoventilation and monotonous respiratory rates despite abnormal PaCO2 and pH concentrations, especially during non-rapid eye movement (NREM) sleep (Patwari et al. 2010). CCHS patients fail to arouse from sleep despite progressive hypercapnia or hypoxemia. Amiel et al. (2003) first described that mutations in the homeobox transcription factor PHOX2B cause CCHS. The inheritance is considered overall most consistent with autosomal dominant inheritance pattern. However, cases of parental mosaic PHOX2B status or incomplete penetrance have characterized some families (Trochet et al. 2008). The mechanisms of variable penetrance are unknown but have been postulated to be due to other modifier mutations that may occur concurrently in CCHS patients. For example, mutations in HASH1 lead to worsening noradrenergic phenotypes in experimental animals with CCHS mutations (Amiel et al. 2003; de Pontual et al. 2003). Furthermore, in addition to HASH1, other pertinent co-mutations found in some CCHS patients include RET, GFRA1, and PHOX2A (Sasaki et al. 2003). Family members of CCHS patients have a higher incidence of autonomic nervous system dysfunction, Hirschprung's disease, neuroblastoma, and sudden infant death syndrome (Berry-Kravis et al. 2006; Devriendt et al. 2000; Weese-Mayer et al. 1993).
In the United States, CCHS clinical diagnostic and management guidelines are rendered by the American Thoracic Society (ATS). Specifically, in patients with alveolar hypoventilation, a documented PHOX2B mutation represents the sine qua non for CCHS diagnosis (Weese-Mayer et al. 2010). PHOX2B (human chromosome 4p12) has a 20-alanine stretch in exon 3. CCHS-causing mutations include 1) polyalanine repeat mutations (PARM) in the 20-alanine repeat of exon 3 resulting in polyalanine tracts ranging from 25–35 alanines, or 2) non-polyalanine repeat mutations (NPARM) resulting in exon 3 deletions that cause frameshift mutations, missense mutations, or nonsense mutations (Weese-Mayer et al. 2003). Of note, PHOX2B mutations resulting in loss of alanines manifest as PHOX2B20/15 genotypes; these mutations exist in 1–1.5% of the general population and are not diagnostic of CCHS even if the patient suffers hypoventilation (Weese-Mayer et al. 2003). ATS guidelines recommend biannual and then annual in-hospital comprehensive evaluations during awake and asleep states so that ventilatory needs during all stages of sleep and a wide range of activity can be determined. These comprehensive evaluations include 1) 72-h Holter monitoring, 2) echocardiogram, 3) autonomic nervous system (ANS) evaluation across other organs, and 4) neurocognitive evaluation. These guidelines represent an attempt to decrease acute episodes of hypoventilation. However, the extent to which long-term autonomic dysfunction affects other outcomes, including neurocognitive state or other quality of life measures, is unclear.
CCHS is a rare genetic disorder, yet its exact incidence and prevalence is unknown. Using the definition originally proposed by Trang et al. (2005), the French CCHS working group estimated an incidence of 1 in 200,000 births. Furthermore, incidence may vary with ethnicity, with some studies demonstrating up to 90% of their affected CCHS population being Caucasian (Weese-Mayer et al. 2010). To date, no study has suggested a gender bias in CCHS. Accurate epidemiological CCHS data utilizing ATS diagnostic criteria have not been generated throughout the world. Further confounding accurate statistics, surveys have shown loose adherence to ATS guidelines in North America (Amin et al. 2013). In summary, CCHS represents a rare genetic disorder without gender bias, with most experts believing that the incidence will increase if the ATS diagnostic criteria become widely implemented.
CCHS patients suffer a spectrum of severity. Continuous ventilatory support is required in 35% of PARM patients and 67% of NPARM patients (Weese-Mayer et al. 2010). For example, patients with a PHOX2B20/25 phenotype never need 24-h ventilatory support. In contrast, patients with PHOX2B20/28-20/33 typically require continuous ventilatory support and patients with PHOX2B20/26 show variable awake needs depending on activity (Berry-Kravis et al. 2006; Matera et al. 2004; Weese-Mayer et al. 2003). Patients with PHOX2B20/25 have the mildest problems, may present only after exposure to respiratory depressants or severe respiratory infection, and are typically managed with nocturnal ventilatory support (Antic et al. 2006; Repetto et al. 2009; Weese-Mayer et al. 2005). Other ANS manifestations correlate with PHOX2B genetic aberrations. For example, studies measuring cardiac asystole in CCHS patients demonstrate no sinus pauses of >3 s in patients with PHOX2B20/25, whereas this is found in 19% and 83% of patients with PHOX2B20/26 and PHOX2B20/27, respectively. Hirschprung's disease occurs in 19% of PARM patients but in 80% of NPARM patients; neural crest tumors are found in 1% of PARM patients and 41% of NPARM patients. In summary, when PARM clinical phenotypes are compared, polyalanine expansion positively correlates with disease severity. Furthermore, although alveolar hypoventilation and neural crest tumors are clinically critical events, other sequelae of long-term autonomic dysfunction are unknown and are not addressed in current clinical management guidelines.
The molecular mechanisms by which PHOX2B mutations cause dysfunction are not entirely known. Molecular studies do not indicate that these PHOX2B mutations yield simple “dominant negative” proteins. Bachetti et al. (2005) demonstrated that polyalanine expansions cause aggregate formation and reduced transcriptional activities. These findings are widely reproducible and are illustrated in Fig. 2A. The PHOX2A promoter, a direct target of PHOX2B, can be tested in a luciferase transcriptional assay. A PHOX2B frameshift mutation (PHOX2BΔ8) characterized by an eight-nucleotide deletion found in a CCHS case (Otero et al. 2011) shows robust activation of the PHOX2A promoter, whereas a polyalanine expansion mutation shows no such activation. The dopamine β-hydroxylase promoter, a direct target of both PHOX2B and PHOX2A proteins, can be activated by PHOX2B and PHOX2A. Of note, synergistic activation can be achieved by cotransfection of PHOX2A + PHOX2B or PHOX2A + PHOX2BΔ8. Overexpression of PHOX2B-polyalanine (PHOX2B-PA) in 293FT cells results in localization of this transcription factor in the cytoplasm, at times as a discoid aggregate (Fig. 2, B3 and B4); PHOX2B and PHOX2BΔ8 show only nuclear localization in such assays. As determined by proximity ligation assay (Fig. 2C), both PHOX2B-PA and PHOX2BΔ8 are capable of binding to PHOX2A and PHOX2B; of note, PHOX2B-PA interaction can occur in nucleus and cytoplasm. In summary, although both NPARM and PARM mutations cause alveolar hypoventilation, these proteins likely induce the defects through distinct molecular mechanisms as has been postulated by Wu et al. (2009). These distinct molecular mechanisms may mediate the varying clinical phenotypes observed between PARM and NPARM patients.
Fig. 2.

Summary of molecular studies of PHOX2B mutation function. A: luciferase assay of PHOX2A promoter (top) and dopamine β-hydroxylase (DBH; bottom). Y-axis shows relative fluorescence units normalized to Renilla luciferase. Both PHOX2B and PHOX2BΔ8 show activation of this promoter. For the DBH studies, both PHOX2B and PHOX2A cDNA constructs can transactivate the DBH promoter. Note that cotransfection of PHOX2A+PHOX2B as well as PHOX2A+PHOX2BΔ8 shows synergistic activity relative to PHOX2A or PHOX2B alone. This synergy is not noted with PHOX2B-polyalanine (PA). B: immunofluorescent analyses of PHOX2B constructs expressed in 293FT cells. The transfected construct is delineated at top right and a color code for immunofluorescent staining at bottom left of each panel. Cells were transfected with PHOX2B, PHOX2B-PA, or PHOX2BΔ8 cDNA and immunostained using NH2-terminal (N-TERM) or COOH-terminal (C-TERM; which cannot detect PHOX2BΔ8) PHOX2B antibodies (Santa Cruz Biotechnology), c-Myc epitope tag antibody (MYC; in frame with PHOX2B and PHOX2B-PA reading frame but not in frame with PHOX2BΔ8 reading frame). Cells are counterstained with DAPI in blue, and colocalization of both antigens is indicated in yellow. Note that PHOX2B-PA shows significant cytoplasmic localization (B3 and B4), whereas PHOX2B and PHOX2BΔ8 show nuclear localization. These findings support the notion that PARM PHOX2B mutations lead to accumulation in the cytoplasm. C: proximity ligation assay [PLA; see Supplemental Material (available for this article online at the Journal of Neurophysiology website) for epitopes and antibodies used] determines the proximity of 2 epitopes. Epitopes need to lie within 70 nm for a signal to be elicited and thus represent a way to identify histologically whether 2 proteins interact. PLA signal is red, and cells are counterstained with DAPI in blue. The transfected constructs in which PLA reaction is performed are shown at top right of each panel. PLA demonstrates nuclear localization of interactions between PHOX2A and PHOX2B (C1), PHOX2A and PHOX2BΔ8 (C3), and PHOX2B and PHOX2BΔ8 (C5). In contrast, significant cytoplasmic localization is noted between PHOX2A and PHOX2B-PA (C2) and PHOX2B-PHOX2B-PA (C4) interactions. C6 shows interactions of PHOX2BΔ8 with a separate PHOX2BΔ8 protein, indicating that NPARM mutations can homodimerize.
Experimental models of CCHS and comparison to human pathology.
CCHS pathobiology research is unique in that murine experimental models of the disease were generated before the extent of human anatomic pathology was fully understood. This was because, in addition to CCHS being a highly rare disease, patients with CCHS usually die in childhood or late adulthood. It is therefore difficult for health care providers to discuss consents to medical autopsies in this disease because of the sensitivity required for a normal grieving process. Nevertheless, animal models have yielded significant insights. One of the first clues that the embryonic loss of RTN neurons leads to breathing abnormalities came from studies of ATOH1 knockout mice. Such ATOH1−/− mice show ablation of the ATOH1 locus and die due to breathing problems shortly after birth (Ben-Arie et al. 1997). Indeed, localization of RTN neurons in the ventrolateral medulla and the RTN's ventral respiratory column connections requires ATOH1 (Huang et al. 2012). The age of CCHS transgenic mouse biology emerged with the publication of a seminal manuscript by Dubreuil et al. (2008) whereby they achieved a knockin to the PHOX2B locus of a modified exon 3 harboring a 7-alanine expansion (Fig. 3B). This results in embryonic development with all cells showing the PHOX2B20/KI-27 genotype, which is the most common PHOX2B mutation in CCHS patients. These animals had unique breathing abnormalities. Specifically, 3 of 18 animals showed intermittent gasping at room air, with the remainder showing a continuum of rhythmic breathing abnormalities ranging from normal (albeit slower) rhythmic breathing rates to intermittent interruption by apneic episodes. However, fully penetrant in all of the PHOX2B20/KI-27 animals is a lack of responsiveness to hypercapnia. These animals show loss of RTN neurons and precursor populations by embryonic day 15.5 (E15.5), with other areas, including the locus coeruleus, A5, and carotid bodies, showing no significant developmental pathology.
Fig. 3.

Genetic design of CCHS-type mutations in mouse experimental models. A: the gene structure of PHOX2B, located on mouse chromosome 5, is composed of 3 exons. B: knockin mice were generated with an extra 7 alanines to generate the PHOX2B20/Ki-27 genotype. The neomycin resistance cassette was removed by interbreeding chimeric mice with PGK-cre mice. Experiments were performed with neomycin cassette-intact and neomycin cassette-ablated mice to ensure that the neomycin cassette did not affect the phenotype. C: humanized PHOX2B20/27 conditional knockout mouse design. mE3 is flanked by loxP sites. The neomycin cassette was inserted between mE3 and the loxP site 5′ to mE3 and was removed by interbreeding founders. Located 3′ to the mE3 site is a humanized E3 with the additional 7 alanines and a human 3′-UTR. NPARM knockin models for 5 nucleotide deletions (D) and 8 nucleotide deletions (E) were done on mE3. Note that in D and E, the neomycin sites are located 3′ to the mutated E3 and were removed by in vitro Cre expression. F: a humanized conditional knockin strategy for NPARM analysis. mE3 is flanked by loxP sites. The neomycin cassette flanked by Frt sites was located between mE3 and the 3′ loxP site; this neomycin cassette was removed by interbreeding with flp-expressing mice. The humanized PHOX2BΔ8 construct contains the human 3′-UTR and GFP after an internal ribosome entry site (IRES) sequence. After Cre-mediated recombination, proteins are generated from this locus: PHOX2BΔ8 and green fluorescent protein (GFP). ALA, alanine; mE3, mouse exon 3.
Although abnormal breathing was not a primary outcome of their work, additional insights in CCHS pathology were obtained from transgenic mice generated by Nagashimada et al. (2012), who focused their analyses on NPARM of PHOX2B. These mice were constructed to have deletions of 5 nucleotides and 8 nucleotides in PHOX2B exon 3 (Fig. 3D for PHOX2B20/Ki-Δ5 and Fig. 3E for PHOX2B20/Ki-Δ8). Both genotypes show significant defects in enteric nervous system development and other peripheral nervous system structures. Interestingly, both genotypes also show loss of PHOX2B-positive neurons in the ventrolateral medulla, implying RTN neuron hypoplasia. However, these mice differed in their effects on the 7th cranial nerve nucleus, which was completely ablated in the PHOX2B20/Ki-Δ8 animal yet intact in the PHOX2B20/Ki-Δ5. Delivery by caesarean section at E18.5 showed that pups from both genotypes suffer from persistent cyanosis, which suggests that these mice had trouble initiating breathing at room air.
The above knockin models of PARM and NPARM CCHS suffer the caveats that the experimental mice show significant defects at birth, which precludes the ability to propagate these mice. Several idiosyncrasies about the PHOX2B locus require special consideration for the generation of conditional experimental models of PARM CCHS (Ramanantsoa et al. 2011) and NPARM CCHS (Nobuta et al. 2015). The primary structures of mouse and human PHOX2B protein are identical, yet significant differences exist at the genomic level. This is important to consider, because frameshift mutations, such as the PHOX2B20/Ki-Δ8, would generate murine C-terminal fragments completely different from the human disease if the deletion were engineered in mouse exon 3. Additional differences include differences in the 3′-untranslated region (UTR) between mice and humans. Luckily, the 3′ splice acceptor site for exon 3 is 100% identical between mouse and human genes, which facilitated the genetic design of conditional, humanized PHOX2B PARM (Fig. 3C, hereafter referred to as PHOX2B20/E3-cKi-27) and NPARM (Fig. 3F, hereafter referred to as PHOX2B20/E3-cKi-Δ8-ires-GFP) experimental models. These studies were then able to express the mouse mutants either in germline with PGK-cre (Lallemand et al. 1998) or HPRT-cre mice (Tang et al. 2002), in the nervous system with Brn4-cre (Ahn et al. 2001) and BLBP1-cre mice (Hegedus et al. 2007), or in rhombomeres 3 and 5, which give rise to RTN neurons (Ramanantsoa et al. 2011), with Egr2-cre mice (Voiculescu et al. 2000).
The most instructive information on respiratory physiology can be obtained from the PARM PHOX2B20/E3-cKi-27 studies. Both PGK-cre, PHOX2B20/E3-cKi-27 and Brn4-cre, PHOX2B20/E3-cKi-27 showed findings similar to the PHOX2B20/KI-27 genotype, including perinatal lethality. To focus the pathological misexpression in RTN neurons, PHOX2B20/E3-cKi-27 mice were then bred to Egr2-cre, which induces Cre-mediated recombination in rhombomeres 3/5. These mice suffer a massive loss of RTN neurons with normal phrenic nerve root activity, indicating that downstream components of the RTN neural network are intact. Marked lack of chemosensation occurs up until postnatal day 9; these mice also show reduced mean ventilation in normoxia and a mild metabolic acidosis. Switching the animals to 100% oxygen leads to deep breaths interrupted by prolonged apneas, underscoring that the main defect in respiratory drive is in the CO2 detection cells.
The above-described findings in PHOX2B20/E3-cKi-27 mice demonstrate a clear link between chemosensation and PHOX2B mutations at a functional and neuroanatomical level. However, complicating the translation of these findings into humans is the fact that the anatomical location of the human RTN remains elusive. One study, based on PHOX2B immunohistochemistry, suggested that the human RTN is located between the 7th cranial nerve and the superior olivary nuclear complex (Rudzinski and Kapur 2010); however, this interpretation is fraught with caveats. Specifically, the RTN is principally defined functionally, and PHOX2B immunohistochemistry shows diffuse immunoreactivity in the brain stem. Therefore, immunohistochemistry-based analyses are insufficient to determine with certainty RTN-like phenotypes in human brain stem sections. In addition, PHOX2B immunohistochemistry is notoriously inconsistent in human postmortem brain stem applications. Thus the hypothesis that the human RTN is located between the 7th cranial nerve and superior olivary nuclear complex cannot be accepted or rejected using immunohistochemical approaches. Further understanding of the human neuropathology of CCHS is thus required determine the extent to which human clinical phenotypes are caused by RTN dysfunction.
The first human neuropathological reports of CCHS began in 1997, before the discovery of PHOX2B mutations; these patients were classified as CCHS due to clinical criteria of the time (Cutz et al. 1997). These patients showed severe loss of the carotid bodies and a decrease in dense core vesicles of glomus cells, with an increase in the number of sustentacular cells estimated at up to a twofold increase above normal. In the lung, these patients showed increased frequency and size of neuroepithelial bodies. These findings shed light on the finding that CCHS patients suffer reduced respiratory drive in the setting of hypoxia, which was not identified in murine PARM experimental models. Radiographic studies have shown concurrence of other brain stem malformations, including Chiari type I malformations, in CCHS patients (Bachetti et al. 2006). Kumar et al. (2008) reported diffuse neuropathology in CCHS using in vivo imaging modalities. Specifically, they measured radial diffusivity and axial diffusivity, measures of myelin alterations and axonal injury, respectively. These patients showed increased axial diffusivity in the lateral medulla, and clusters of injured areas were noted from the dorsal midbrain through the basal pons. Tomcyz et al. (2010) evaluated histopathologically a case suspicious for CCHS (without a confirmatory PHOX2B mutation) and found significantly decreased tyrosine hydroxylase staining in the locus coeruleus and cortical malformations. These cortical malformations included a developmentally inappropriate operculum, small superior temporal gyrus, an abnormally split posterior temporal gyrus, hyperconvoluted gyri, fused sulci, and focal abnormal laminations in cortical layers I–IV.
The only histopathological findings of CCHS with confirmed PHOX2B mutations was reported by Nobuta et al. (2015). This data set included two CCHS patients, one PARM and one NPARM (Fig. 4 for simplified pedigrees). The NPARM patient suffered an 8-nucleotide deletion in PHOX2B exon 3, whereas the PARM patient showed a PHOX2B20/27 phenotype. The NPARM patient's pathology was significant for markedly diffuse abnormalities. These included significantly atrophic locus coeruleus, decreases in the dorsal motor nucleus of the vagus nerve, absent mesencephalic trigeminal nucleus, decreased serotonergic median raphe neurons, decreased dopamine β-hydroxylase-positive adrenal medullary neurons, and marked colonic aganglionosis characterized by loss of enteric ganglia from 10 cm distal to the ligament of Treitz to anus. Of note, the intestinal aganglionosis showed pathological features distinct from classical Hirschprung's disease. Specifically, the NPARM proband was remarkable for loss of both chromogranin-positive neurons and S100 protein-positive Schwann cells. In contrast, classical Hirschprung's pathology manifests histologically as loss of neurons with normal to increased S100 protein-positive peripheral nerves. Unfortunately, the NPARM patient's carotid body was not sampled. The PARM patient's histopathology was also characterized by marked reduction in dopamine β-hydroxylase-positive neurons of the locus coeruleus. Other comorbidities found in the PARM patient included marked hypoxic-ischemic encephalopathy, likely due to the patient's premature birth. The PARM patient showed an intact dorsal motor nucleus of the vagus nerve and a median raphe. However, diffuse brain stem astrogliosis was noted in the PARM patient with significant accentuation in the median raphe. The PARM patient's adrenal gland, intestine, and the carotid body were not sampled. In both patients, an intact facial nerve nucleus was identified.
Fig. 4.
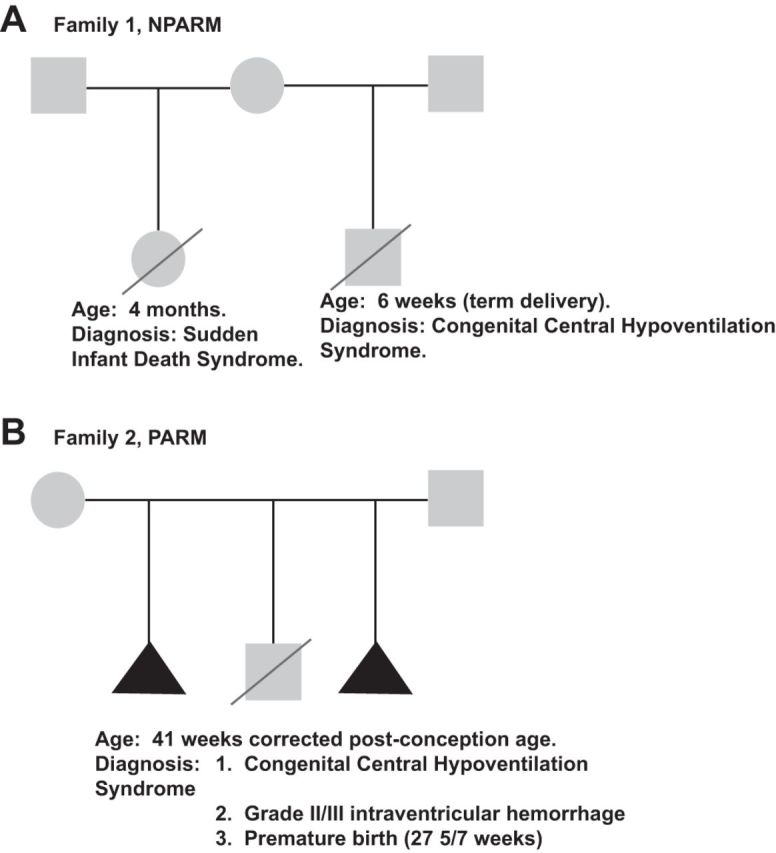
Pedigrees of 2 PHOX2B mutation confirmed CCHS probands reported in literature with histopathological findings. A: family 1, NPARM. Note that the NPARM proband has a half-sibling deceased at 4 mo of age due to sudden infant death syndrome (with co-sleeping). B: family 2, PARM. The clinical history is complicated by a complex maternal obstetrical history, including 2 pregnancies not taken to term for unknown reasons. The PARM proband suffered preterm birth and was initially diagnosed with apnea of prematurity. This child could not become ventilator independent at ∼32–33 wk corrected postgestational age, an age at which central nervous system respiratory center maturation occurs. Squares represent males, circles represent females, triangles represent pregnancies not taken to term, and diagonal lines indicate deceased.
Although these in vivo and postmortem analyses of CCHS patients provide insight into the pathological processes afflicted by CCHS patients, the nature of these does not allow one to incisively test hypotheses regarding which pathological processes represent developmental syndrome versus a developmental sequence. For these reasons, we developed a humanized NPARM murine experimental model based on the NPARM patients described by Nobuta et al. (2015). In this experiment, we used Hprt-cre interbreeding to evaluate the neuropathology of mice showing early recombination of the PHOX2B20/E3-cKi-Δ8-ires-GFP locus and compared these findings with recombination in later stages of CNS development with BLBP1-cre mice. An added benefit of this design is that cells that express the PHOX2BΔ8 protein also show concurrent cytoplasmic GFP expression. We found that HPRT-cre, PHOX2B20/E3-cKi-Δ8-ires-GFP mice were not born at expected Mendelian ratios, and those that were died soon after birth. These mice showed intestinal aganglionosis and loss of the RTN and facial nerve nucleus. Of particular relevance was that in HPRT-cre, PHOX2B20/E3-cKi-Δ8-ires-GFP mice, several NeuN-positive neurons colabeled for GFP and PHOX2B in an area anatomically appropriate for the locus coeruleus, but these cells did not show significant tyrosine hydroxylase staining. Recombining the PHOX2B20/E3-cKi-Δ8-ires-GFP in later neural development by interbreeding with Blbp-cre mice showed preserved tyrosine hydroxylase-positive locus coeruleus neurons. However, the RTN and the facial nerve nucleus of Blbp-cre, PHOX2B20/E3-cKi-Δ8-ires-GFP mice were both essentially absent. Blbp-cre, PHOX2B20/E3-cKi-Δ8-ires-GFP mice were viable at birth and maintained spontaneous breathing. Although respiratory physiology was not a primary endpoint of the NPARM PHOX2B20/E3-cKi-Δ8-ires-GFP studies, these studies underscore the requirement of noradrenergic neuron function for appropriate respiration at birth. In summary, locus coeruleus pathology has been reported in two CCHS patients with confirmed PHOX2B mutations, in one case suspicious for CCHS, and in a humanized NPARM model.
Conclusion.
In the brain, we have a well-organized network to maintain one of the most important systems to life, the respiratory system. One important task of the neural control of breathing is to maintain within normal ranges, most of the time, the levels of CO2 and pH in the body (Dempsey and Smith 2014; Guyenet and Bayliss 2015; Smith et al. 2015). Central respiratory chemoreception is a mechanism by which very small changes in tissue CO2/H+ produce very large changes in breathing. One region that is involved in central respiratory chemoreception is the retrotrapezoid nucleus (RTN) (Guyenet et al. 2010; Guyenet and Bayliss 2015; Nattie 2011). This region is located in the ventrolateral surface of the medulla with neurons that have a genetic lineage (derived from neurons that express PHOX2B, Atoh-1, and Egr-2) and express different phenotypes (PHOX2B, NK1 receptors, VGlut2, TASK-2, GPR4, and galanin but not GABA, glycine, acetylcholine, or catecholamines) (Kumar et al. 2015; Lazarenko et al. 2009; Ruffault et al. 2015; Takakura et al. 2008, 2014; Wang et al. 2013). In this concise review, we discuss, on the basis of clinical and genetic studies in human respiratory disease and evidence from neurophysiology and experimental animal models, how a restricted number of neurons, located in the VMS, expressing and depending on the PHOX2B transcription factor could be essential in the control of breathing and autonomic regulation. The neurons in the VMS named RTN chemoreceptors neurons are considered the principal central respiratory chemoreceptors (Guyenet and Bayliss 2015). Such knowledge will certainly enable new approaches (genetics, physiology, pharmacology, and biochemistry) to elucidate how the breathing networks are assembled and configured in normal and pathological conditions. Elucidating the mechanisms by which these neural networks regulate respiratory control represents a fertile area for future investigation. Furthermore, current unanswered questions regarding clinical presentation of CCHS include 1) determining the genetic mechanisms to CCHS's known variable penetrance, 2) developing uniform diagnostic and patient management guidelines to improve prognosis and quality of life for patients with CCHS, and 3) determining the extent to which long-term suboptimal management of ventilator drive contributes to other neurocognitive/neuropsychiatric manifestations of the disease.
GRANTS
This work was supported by São Paulo Research Foundation (FAPESP) Grants 2014/22406-1 (to A. C. Takakura), 2009/54888-7 (to T. S. Moreira), and 2013/10573-8 (to T. S. Moreira), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) Grants 471744/2011-5 (to A. C. Takakura), 471263/2013-3 (to A. C. Takakura), and 471283/2012-6 (to T. S. Moreira), and CNPq Fellowships 305533/2012-6 (to T. S. Moreira) and 301651/2013-2 (to A. C. Takakura). J. J. Otero acknowledges support from National Institutes of Health (NIH) Grant 1R21EB017539-01A1. J. J. Otero and C. Czeisler were supported by NIH Grant R011HL132355-01A1.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.S.M., A.C.T., C.C., and J.J.O. conception and design of research; T.S.M., A.C.T., C.C., and J.J.O. performed experiments; T.S.M., A.C.T., C.C., and J.J.O. analyzed data; T.S.M., A.C.T., C.C., and J.J.O. interpreted results of experiments; T.S.M., A.C.T., C.C., and J.J.O. prepared figures; T.S.M., A.C.T., C.C., and J.J.O. drafted manuscript; T.S.M., A.C.T., C.C., and J.J.O. edited and revised manuscript; T.S.M., A.C.T., and J.J.O. approved final version of manuscript.
Supplemental Data
REFERENCES
- Abbott SB, DePuy SD, Nguyen T, Coates MB, Stornetta RL, Guyenet PG. Selective optogenetic activation of rostral ventrolateral medullary catecholaminergic neurons produces cardiorespiratory stimulation in conscious mice. J Neurosci : 3164–3177, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott SB, Stornetta RL, Coates MB, Guyenet PG. PHOX2B-expressing neurons of the parafacial region regulate breathing rate, inspiration, and expiration in conscious rats. J Neurosci : 16410–16422, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Mishina Y, Hanks MC, Behringer RR, Crenshaw EB 3rd. BMPR-IA signaling is required for the formation of the apical ectodermal ridge and dorsal-ventral patterning of the limb. Development : 4449–4461, 2001. [DOI] [PubMed] [Google Scholar]
- Amiel J, Dubreuil V, Ramanantsoa N, Fortin G, Gallego J, Brunet JF, Goridis C. PHOX2B in respiratory control: lessons from congenital central hypoventilation syndrome and its mouse models. Respir Physiol Neurobiol : 125–132, 2009. [DOI] [PubMed] [Google Scholar]
- Amiel J, Laudier B, Attie-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, Vekemans M, Munnich A, Gaultier C, Lyonnet S. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet : 459–461, 2003. [DOI] [PubMed] [Google Scholar]
- Amin R, Moraes TJ, Skitch A, Irwin MS, Meyn S, Witmans M. Diagnostic practices and disease surveillance in Canadian children with congenital central hypoventilation syndrome. Can Respir J : 165–170, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antic NA, Malow BA, Lange N, McEvoy RD, Olson AL, Turkington P, Windisch W, Samuels M, Stevens CA, Berry-Kravis EM, Weese-Mayer DE. PHOX2B mutation-confirmed congenital central hypoventilation syndrome: presentation in adulthood. Am J Respir Crit Care Med : 923–927, 2006. [DOI] [PubMed] [Google Scholar]
- Bachetti T, Matera I, Borghini S, Di Duca M, Ravazzolo R, Ceccherini I. Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Hum Mol Genet : 1815–1824, 2005. [DOI] [PubMed] [Google Scholar]
- Bachetti T, Robbiano A, Parodi S, Matera I, Merello E, Capra V, Baglietto MP, Rossi A, Ceccherini I, Ottonello G. Brainstem anomalies in two patients affected by congenital central hypoventilation syndrome. Am J Respir Crit Care Med : 706–709, 2006. [DOI] [PubMed] [Google Scholar]
- Barna BF, Takakura AC, Moreira TS. Acute exercise-induced activation of PHOX2B-expressing neurons of the retrotrapezoid nucleus in rats may involve the hypothalamus. Neuroscience : 355–363, 2014. [DOI] [PubMed] [Google Scholar]
- Bayliss DA, Talley EM, Sirois JE, Lei Q. TASK-1 is a highly modulated pH-sensitive ‘leak’ K+ channel expressed in brainstem respiratory neurons. Respir Physiol : 159–174, 2001. [DOI] [PubMed] [Google Scholar]
- Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, Matzuk MM, Zoghbi HY. Math1 is essential for genesis of cerebellar granule neurons. Nature : 169–172, 1997. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med : 1139–1144, 2006. [DOI] [PubMed] [Google Scholar]
- Biancardi V, Bícego KC, Almeida MC, Gargaglioni LH. Locus coeruleus noradrenergic neurons and CO2 drive to breathing. Pflügers Arch : 1119–1128, 2008. [DOI] [PubMed] [Google Scholar]
- Carroll JL, Agarwal A. Development of ventilatory control in infants. Paediatr Respir Rev : 199–207, 2010. [DOI] [PubMed] [Google Scholar]
- Cutz E, Ma TK, Perrin DG, Moore AM, Becker LE. Peripheral chemoreceptors in congenital central hypoventilation syndrome. Am J Respir Crit Care Med : 358–363, 1997. [DOI] [PubMed] [Google Scholar]
- Dauger S, Pattyn A, Lofaso F, Gaultier C, Goridis C, Gallego J, Brunet JF. PHOX2B controls the development of peripheral chemoreceptors and afferent visceral pathways. Development : 6635–6642, 2003. [DOI] [PubMed] [Google Scholar]
- Dean JB, Lawing WL, Millhorn DE. CO2 decreases membrane conductance and depolarizes neurons in the nucleus tractus solitarii. Exp Brain Res : 656–661, 1989. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Smith CA. Pathophysiology of human ventilatory control. Eur Respir J : 495–512, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pontual L, Nepote V, Attie-Bitach T, Al Halabiah H, Trang H, Elghouzzi V, Levacher B, Benihoud K, Auge J, Faure C, Laudier B, Vekemans M, Munnich A, Perricaudet M, Guillemot F, Gaultier C, Lyonnet S, Simonneau M, Amiel J. Noradrenergic neuronal development is impaired by mutation of the proneural HASH-1 gene in congenital central hypoventilation syndrome (Ondine's curse). Hum Mol Genet : 3173–3180, 2003. [DOI] [PubMed] [Google Scholar]
- Devriendt K, Fryns JP, Naulaers G, Devlieger H, Alliet P. Neuroblastoma in a mother and congenital central hypoventilation in her daughter: variable expression of the same genetic disorder? Am J Med Genet : 430–431, 2000. [PubMed] [Google Scholar]
- Diedrich A, Malow BA, Antic NA, Sato K, McEvoy RD, Mathias CJ, Robertson D, Berry-Kravis EM, Weese-Mayer DE. Vagal and sympathetic heart rate and blood pressure control in adult onset PHOX2B mutation-confirmed congenital central hypoventilation syndrome. Clin Auton Res : 177–185, 2007. [DOI] [PubMed] [Google Scholar]
- Dubreuil V, Ramanantsoa N, Trochet D, Vaubourg V, Amiel J, Gallego J, Brunet JF, Goridis C. A human mutation in PHOX2B causes lack of CO2 chemosensitivity, fatal central apnoea and specific loss of parafacial neurons. Proc Natl Acad Sci USA : 1067–1072, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil V, Thoby-Brisson M, Rallu M, Persson K, Pattyn A, Birchmeier C, Brunet JF, Fortin G, Goridis C. Defective respiratory rhythmogenesis and loss of central chemosensitivity in PHOX2B mutants targeting retrotrapezoid nucleus neurons. J Neurosci : 14836–14846, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge FL, Kiley JP, Millhorn DE. Respiratory effects of carbon dioxide-induced changes of medullary extracellular fluid pH in cats. J Physiol : 177–189, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci : 239–266, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin G, Thoby-Brisson M. Embryonic emergence of the respiratory rhythm generator. Respir Physiol Neurobiol : 86–91, 2009. [DOI] [PubMed] [Google Scholar]
- Fortuna MG, Stornetta RL, West GH, Guyenet PG. Activation of the retrotrapezoid nucleus by posterior hypothalamic stimulation. J Physiol : 5121–5138, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortuna MG, West GH, Stornetta RL, Guyenet PG. Botzinger expiratory-augmenting neurons and the parafacial respiratory group. J Neurosci : 2506–2515, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goridis C, Brunet JF. Central chemoreception: lessons from mouse and human genetics. Respir Physiol Neurobiol : 312–321, 2010. [DOI] [PubMed] [Google Scholar]
- Gozal D. Sleep-disordered breathing and school performance in children. Pediatrics : 616–620, 1998. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The 2008 Carl Ludwig Lecture: retrotrapezoid nucleus, CO2 homeostasis, and breathing automaticity. J Appl Physiol : 404–416, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Bayliss DA. Neural control of breathing and CO2 homeostasis. Neuron : 946–961, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Mulkey DK, Stornetta RL, Bayliss DA. Regulation of ventral surface chemoreceptors by the central respiratory pattern generator. J Neurosci : 8938–8947, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Bayliss DA. Central respiratory chemoreception. J Comp Neurol : 3883–3906, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus B, Dasgupta B, Shin JE, Emnett RJ, Hart-Mahon EK, Elghazi L, Bernal-Mizrachi E, Gutmann DH. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell : 443–457, 2007. [DOI] [PubMed] [Google Scholar]
- Huang WH, Tupal S, Huang TW, Ward CS, Neul JL, Klisch TJ, Gray PA, Zoghbi HY. Atoh1 governs the migration of postmitotic neurons that shape respiratory effectiveness at birth and chemoresponsiveness in adulthood. Neuron : 799–809, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Takahashi M, Sato S, Igarashi H, Ishizuka T, Yawo H, Arata S, Southard-Smith EM, Kawakami K, Onimaru H. A PHOX2B BAC transgenic rat line useful for understanding respiratory rhythm generator neural circuitry. PLoS One : e0132475, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Macey PM, Woo MA, Alger JR, Harper RM. Diffusion tensor imaging demonstrates brainstem and cerebellar abnormalities in congenital central hypoventilation syndrome. Pediatr Res : 275–280, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar NN, Velic A, Soliz J, Shi Y, Li K, Wang S, Weaver JL, Sen J, Abbott SB, Lazarenko RM, Ludwig MG, Perez-Reyes E, Mohebbi N, Bettoni C, Gassmann M, Suply T, Seuwen K, Guyenet PG, Wagner CA, Bayliss DA. Regulation of breathing by CO2 requires the proton-activated receptor GPR4 in retrotrapezoid nucleus neurons. Science : 1255–1260, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand Y, Luria V, Haffner-Krausz R, Lonai P. Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res : 105–112, 1998. [DOI] [PubMed] [Google Scholar]
- Lazarenko RM, Milner TA, Depuy SD, Stornetta RL, West GH, Kievits JA, Bayliss DA, Guyenet PG. Acid sensitivity and ultrastructure of the retrotrapezoid nucleus in PHOX2B-EGFP transgenic mice. J Comp Neurol : 69–86, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeschcke HH. Central chemosensitivity and the reaction theory. J Physiol : 1–24, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera I, Bachetti T, Puppo F, Di Duca M, Morandi F, Casiraghi GM, Cilio MR, Hennekam R, Hofstra R, Schober JG, Ravazzolo R, Ottonello G, Ceccherini I. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset central hypoventilation syndrome. J Med Genet : 373–380, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellins RB, Balfour HH Jr, Turino GM, Winters RW. Failure of automatic control of ventilation (Ondine's curse). Report of an infant born with this syndrome and review of the literature. Medicine (Baltimore) : 487–504, 1970. [PubMed] [Google Scholar]
- Millhorn DE. Neural respiratory and circulatory interaction during chemoreceptor stimulation and cooling of ventral medulla in cats. J Physiol : 217–231, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL. Role of ventrolateral medulla in regulation of respiratory and cardiovascular systems. J Appl Physiol : 1249–1263, 1986. [DOI] [PubMed] [Google Scholar]
- Mitchell RA, Loeschcke HH, Massion WH, Severinghaus JW. Respiratory responses mediated through superficial chemosensitive areas on the medulla. J Appl Physiol : 523–533, 1963. [DOI] [PubMed] [Google Scholar]
- Moreira TS, Takakura AC, Colombari E, West GH, Guyenet PG. Inhibitory input from slowly adapting lung stretch receptors to retrotrapezoid nucleus chemoreceptors. J Physiol : 285–300, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira TS, Wenker IC, Sobrinho CR, Barna BF, Takakura AC, Mulkey DK. Independent purinergic mechanisms of central and peripheral chemoreception in the rostral ventrolateral medulla. J Physiol : 1067–1074, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Stornetta RL, Weston MC, Simmons JR, Parker A, Bayliss DA, Guyenet PG. Respiratory control by ventral surface chemoreceptor neurons in rats. Nat Neurosci : 1360–1369, 2004. [DOI] [PubMed] [Google Scholar]
- Nagashimada M, Ohta H, Li C, Nakao K, Uesaka T, Brunet JF, Amiel J, Trochet D, Wakayama T, Enomoto H. Autonomic neurocristopathy-associated mutations in PHOX2B dysregulate Sox10 expression. J Clin Invest : 3145–3158, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E. Julius H, Comroe Jr., distinguished lecture: central chemoreception: then … and now. J Appl Physiol : 1–8, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E, Li A. Muscimol dialysis in the retrotrapezoid nucleus region inhibits breathing in the awake rat. J Appl Physiol : 153–162, 2000. [DOI] [PubMed] [Google Scholar]
- Nattie EE, Li A. Substance P-saporin lesion of neurons with NK1 receptors in one chemoreceptor site in rats decreases ventilation and chemosensitivity. J Physiol : 603–616, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobuta H, Cilio MR, Danhaive O, Tsai HH, Tupal S, Chang SM, Murnen A, Kreitzer F, Bravo V, Czeisler C, Gokozan HN, Gygli P, Bush S, Weese-Mayer DE, Conklin B, Yee SP, Huang EJ, Gray PA, Rowitch D, Otero JJ. Dysregulation of locus coeruleus development in congenital central hypoventilation syndrome. Acta Neuropathol : 171–183, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Homma I. A novel functional neuron group for respiratory rhythm generation in the ventral medulla. J Neurosci : 1478–1486, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K, Kawakami K. CO2-sensitive preinspiratory neurons of the parafacial respiratory group express PHOX2B in the neonatal rat. J Neurosci : 12845–12850, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K, Mariho T, Kawakami K. Cytoarchitecture and CO2 sensitivity of PHOX2B-positive parafacial neurons in the newborn rat medulla. Prog Brain Res : 57–71, 2014. [DOI] [PubMed] [Google Scholar]
- Otero J, Danhaive O, Cilio M, Huang E, Rowitch D. A six-week old male with Haddad syndrome: clinical, genetic, and pathological evaluation. J Neuropathol Exp Neurol : 544, 2011. [Google Scholar]
- Patwari PP, Carroll MS, Rand CM, Kumar R, Harper R, Weese-Mayer DE. Congenital central hypoventilation syndrome and the PHOX2B gene: a model of respiratory and autonomic dysregulation. Respir Physiol Neurobiol : 322–335, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam RW, Filosa JA, Ritucci NA. Cellular mechanisms involved in CO2 and acid signaling in chemosensitive neurons. Am J Physiol Cell Physiol : C1493–C1526, 2004. [DOI] [PubMed] [Google Scholar]
- Ramanantsoa N, Hirsch MR, Thoby-Brisson M, Dubreuil V, Bouvier J, Ruffault PL, Matrot B, Fortin G, Brunet JF, Gallego J, Goridis C. Breathing without CO2 chemosensitivity in conditional PHOX2B mutants. J Neurosci : 12880–12888, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repetto GM, Corrales RJ, Abara SG, Zhou L, Berry-Kravis EM, Rand CM, Weese-Mayer DE. Later-onset congenital central hypoventilation syndrome due to a heterozygous 24-polyalanine repeat expansion mutation in the PHOX2B gene. Acta Paediatr : 192–195, 2009. [DOI] [PubMed] [Google Scholar]
- Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci : 449–461, 2004. [DOI] [PubMed] [Google Scholar]
- Ritucci NA, Erlichman JS, Leiter JC, Putnam RW. Response of membrane potential and intracellular pH to hypercapnia in neurons and astrocytes from rat retrotrapezoid nucleus. Am J Physiol Regul Integr Comp Physiol : R851–R861, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudzinski E, Kapur RP. PHOX2B Immunolocalization of the candidate human retrotrapezoid nucleus. Pediatr Dev Pathol : 291–299, 2010. [DOI] [PubMed] [Google Scholar]
- Ruffault PL, D'Autréaux F, Hayes JA, Nomaksteinsky M, Autran S, Fujiyama T, Hoshino M, Hägglund M, Kiehn O, Brunet JF, Fortin G, Goridis C. The retrotrapezoid nucleus neurons expressing Atoh1 and PHOX2B are essential for the respiratory response to CO2. Elife : e07051, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Kanai M, Kijima K, Akaba K, Hashimoto M, Hasegawa H, Otaki S, Koizumi T, Kusuda S, Ogawa Y, Tuchiya K, Yamamoto W, Nakamura T, Hayasaka K. Molecular analysis of congenital central hypoventilation syndrome. Hum Genet : 22–26, 2003. [DOI] [PubMed] [Google Scholar]
- Sato M, Severinghaus JW, Basbaum AI. Medullary CO2 chemoreceptor neuron identification by c-fos immunocytochemistry. J Appl Physiol : 96–100, 1992. [DOI] [PubMed] [Google Scholar]
- Shea SA, Andres LP, Shannon DC, Banzett RB. Ventilatory responses to exercise in humans lacking ventilatory chemosensitivity. J Physiol : 623–640, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JN, Tanabe FM, Moreira TS, Takakura AC. Neuroanatomical and physiological evidence that the retrotrapezoid nucleus/parafacial region regulates expiration in adult rats. Respir Physiol Neurobiol : 9–22, 2016. [DOI] [PubMed] [Google Scholar]
- Smith CA, Blain GM, Henderson KS, Dempsey JA. Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO2: role of carotid body CO2. J Physiol : 4225–4243, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Borgmann A, Rybak IA, Paton JF. Brainstem respiratory networks: building blocks and microcircuits. Trends Neurosci : 152–162, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Morrison DE, Ellenberger HH, Otto MR, Feldman JL. Brainstem projections to the major respiratory neuron populations in the medulla of the cat. J Comp Neurol : 69–96, 1989. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Moreira TS, Takakura AC, Kang BJ, Chang DA, West GH, Brunet JF, Mulkey DK, Bayliss DA, Guyenet PG. Expression of PHOX2B by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci : 10305–10314, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Barna BF, Cruz JC, Colombari E, Moreira TS. PHOX2B-expressing retrotrapezoid neurons and the integration of central and peripheral chemosensory control of breathing in conscious rats. Exp Physiol : 571–585, 2014. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL, Guyenet PG. Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2-sensitive neurons in rats. J Physiol : 503–523, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, De Paula PM, Menani JV, Colombari E. Control of breathing and blood pressure by parafacial neurons in conscious rats. Exp Physiol : 304–315, 2013. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Stornetta RL, West GH, Gwilt JM, Guyenet PG. Selective lesion of retrotrapezoid PHOX2B-expressing neurons raises the apnoeic threshold in rats. J Physiol : 2975–2991, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SH, Silva FJ, Tsark WM, Mann JR. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis : 199–202, 2002. [DOI] [PubMed] [Google Scholar]
- Thoby-Brisson M, Karlén M, Wu N, Charnay P, Champagnat J, Fortin G. Genetic identification of an embryonic parafacial oscillator coupling to the preBötzinger complex. Nat Neurosci : 1028–1035, 2009. [DOI] [PubMed] [Google Scholar]
- Tomycz ND, Haynes RL, Schmidt EF, Ackerson K, Kinney HC. Novel neuropathologic findings in the Haddad syndrome. Acta Neuropathol : 261–269, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gaultier C; French C.C.H.S. Working Group. The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest : 72–79, 2005. [DOI] [PubMed] [Google Scholar]
- Trochet D, de Pontual L, Straus C, Gozal D, Trang H, Landrieu P, Munnich A, Lyonnet S, Gaultier C, Amiel J. PHOX2B germline and somatic mutations in late-onset central hypoventilation syndrome. Am J Respir Crit Care Med : 906–911, 2008. [DOI] [PubMed] [Google Scholar]
- Voiculescu O, Charnay P, Schneider-Maunoury S. Expression pattern of a Krox-20/Cre knock-in allele in the developing hindbrain, bones, and peripheral nervous system. Genesis : 123–126, 2000. [DOI] [PubMed] [Google Scholar]
- Wang S, Benamer N, Zanella S, Kumar NN, Shi Y, Bévengut M, Penton D, Guyenet PG, Lesage F, Gestreau C, Barhanin J, Bayliss DA. TASK-2 channels contribute to pH sensitivity of retrotrapezoid nucleus chemoreceptor neurons. J Neurosci : 16033–16044, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H; ATS Congenital Central Hypoventilation Syndrome Subcommittee. An official ATS clinical policy statement: Congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med : 626–644, 2010. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Zhou L. Adult identified with congenital central hypoventilation syndrome-mutation in PHOX2B gene and late-onset CHS. Am J Respir Crit Care Med : 88, 2005. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Silvestri JM, Curran ME, Marazita ML. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2B. Am J Med Genet A : 267–278, 2003. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Silvestri JM, Marazita ML, Hoo JJ. Congenital central hypoventilation syndrome: inheritance and relation to sudden infant death syndrome. Am J Med Genet : 360–367, 1993. [DOI] [PubMed] [Google Scholar]
- Wenker IC, Kréneisz O, Nishiyama A, Mulkey DK. Astrocytes in the retrotrapezoid nucleus sense H+ by inhibition of a Kir4.1-Kir5.1-like current and may contribute to chemoreception by a purinergic mechanism. J Neurophysiol : 3042–3052, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HT, Su YN, Hung CC, Hsieh WS, Wu KJ. Interaction between PHOX2B and CREBBP mediates synergistic activation: mechanistic implications of PHOX2B mutants. Hum Mutat : 655–660, 2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.