

Abstract
Timely reperfusion after a myocardial infarction is necessary to salvage the ischemic region; however, reperfusion itself is also a major contributor to the final tissue damage. Currently, there is no clinically relevant therapy available to reduce ischemia-reperfusion injury (IRI). While many drugs have shown promise in reducing IRI in preclinical studies, none of these drugs have demonstrated benefit in large clinical trials. Part of this failure to translate therapies can be attributed to the reliance on small animal models for preclinical studies. While animal models encapsulate the complexity of the systemic in vivo environment, they do not fully recapitulate human cardiac physiology. Furthermore, it is difficult to uncouple the various interacting pathways in vivo. In contrast, in vitro models using isolated cardiomyocytes allow studies of the direct effect of therapeutics on cardiomyocytes. External factors can be controlled in simulated ischemia-reperfusion to allow for better understanding of the mechanisms that drive IRI. In addition, the availability of cardiomyocytes derived from human induced pluripotent stem cells (hIPS-CMs) offers the opportunity to recapitulate human physiology in vitro. Unfortunately, hIPS-CMs are relatively fetal in phenotype, and are more resistant to hypoxia than the mature cells. Tissue engineering platforms can promote cardiomyocyte maturation for a more predictive physiologic response. These platforms can further be improved upon to account for the heterogenous patient populations seen in the clinical settings and facilitate the translation of therapies. Thereby, the current preclinical studies can be further developed using currently available tools to achieve better predictive drug testing and understanding of IRI. In this article, we discuss the state of the art of in vitro modeling of IRI, propose the roles for tissue engineering in studying IRI and testing the new therapeutic modalities, and how the human tissue models can facilitate translation into the clinic.
Introduction
Cardiovascular disease is a major cause of mortality in the world [1]. Myocardial infarction (MI) occurs when coronary blood flow is restricted by occlusion, thus preventing oxygen and nutrient delivery to the downstream ischemic region. The only available therapy to limit damage to the ischemic region following MI is reperfusion of the occluded artery, and reestablishment of nutrient delivery. The time lapse between occlusion and reperfusion is critical for preventing irreversible damage of the myocardium [2]. Paradoxically, reperfusion of the ischemic region can lead to an increase in myocardial damage and in the final infarct size. Ischemia-reperfusion injury (IRI) involves the death of cells upon reperfusion that were still viable at the end of ischemic insult. IRI is recognized as a major contributor to the final damage after an MI, but is also a potentially preventable source of damage (Figure 1). It was first demonstrated by Murry and colleagues in 1986 that ischemic preconditioning of the heart could protect it against IRI [3]. However, preconditioning is not a clinically relevant therapy in the setting of an acute MI. Thus, attention has been turned to the discovery of medicines that can be introduced at the time of reperfusion. Despite promising results in animal models, all of the drugs that made it to phase III clinical trials failed to demonstrate clinical benefits [4–6]. Some criticisms have been levied on the specifics of these clinical trials [7], and the drug development process in general [8]. It is clear that more predictive testing needs to be done during translation of preclinical data into clinical trials. Currently, most of the preclinical testing is done in small animal models, which provide a cost-effective in vivo background to work in, but introduce multiple confounding factors that may obfuscate the effects of drugs [9]. While animal models remain critical for drug development, it may be worth considering human-cell-based in vitro and reductionist models of IRI. The in vitro models are important in that the direct effect of reperfusion and the drug on the cardiomyocytes can be observed, and any external factors can be isolated for and individually controlled.
Figure 1. Timeline of ischemia-reperfusion injury.
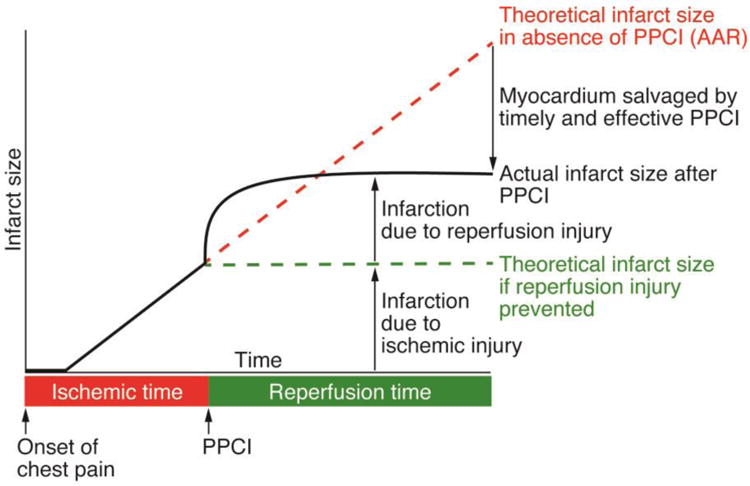
Cell death due to ischemic injury increases with ischemic time (red line). Reperfusion by primary percutaneous coronary intervention (PPCI) halts the progression of ischemic injury (green line), but causes additional damage that leads to a larger final infarct size than predicted by ischemic injury alone (black line). This additional reperfusion injury is potentially preventable, and limiting it through therapeutics would lead to better clinical outcomes. Adapted from Hausenloy D, et al. [10].
There is a growing notion that current preclinical models of IRI fail to translate to the clinical setting [11]. An entirely new approach to preclinical studies of IRI may be needed that would utilize controllable models with sufficient power to predict the human pathophysiology of ischemia and reperfusion. In this article, we discuss the state of the art of in vitro modeling of IRI, and propose the roles for tissue engineering in studying IRI and testing the new therapeutic modalities, and how the human tissue models can facilitate translation into the clinic.
Pathophysiology of Ischemia-Reperfusion Injury
Ischemia
Any improvements in the identification of therapeutic targets and development of effective drugs for myocardial infarction patients can only be based on understanding the progression and basic mechanisms that drive ischemia-reperfusion injury (Figure 1).
MI occurs when a dislodged plaque clogs an artery and occludes blood flow to the downstream ischemic region. This occlusion prevents nutrient and oxygen delivery, and removal of metabolic by-products. During ischemia, the lack of oxygen causes cardiomyocytes to switch from aerobic respiration to anaerobic glycolysis for their primary ATP production. Because glycolysis is far less efficient at producing ATP than mitochondrial oxidative phosphorylation, this transition disrupts ATP turnover. The subsequent decrease in ATP production with an increase in ATP hydrolysis results in increased proton production and intracellular acidification [12]. Glycolysis also results in the production of lactate, which helps restore cytosolic NAD+ stores to support continued ATP generation from glycolysis and helps buffer intracellular pH by extruding protons through the lactate-proton cotransporter [13]. Importantly, the lack of blood flow during ischemia results in extracellular accumulation of lactate that prevents further efflux of lactate. The failure to remove lactate inhibits glycolytic ATP production and contributes to continued intracellular acidosis [14].
The ischemic cardiomyocytes attempt to reestablish a normal intracellular pH by utilizing the sodium-hydrogen exchanger to excrete protons and accumulate sodium [15]. The lack of ATP prevents the sodium-potassium ATPase from excreting sodium. Instead, the high intracellular sodium concentration drives the sodium-calcium exchanger in reverse in an attempt to normalize intracellular sodium concentrations, resulting in intracellular accumulation of calcium (Figure 2a) [16].
Figure 2. Pathophysiology of ischemia-reperfusion injury after an acute MI.
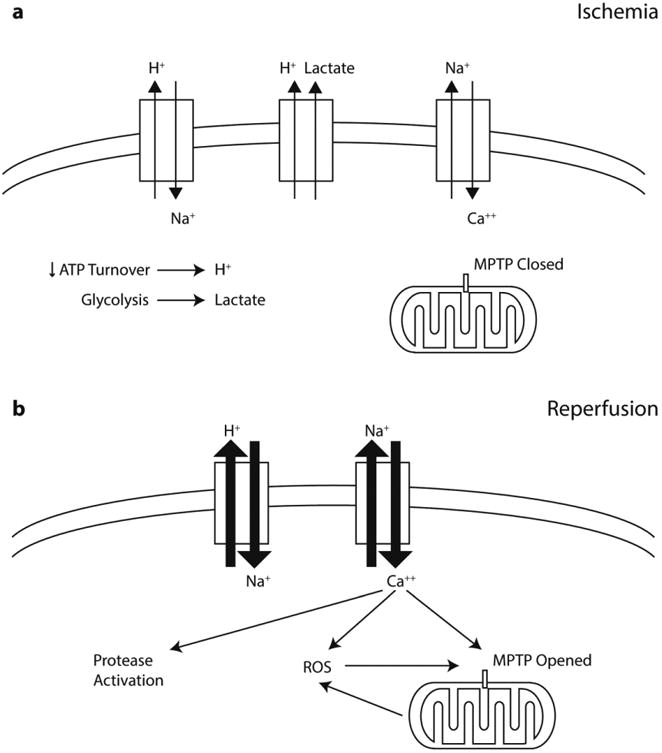
(a) During ischemia, the lack of oxygen drives cells from aerobic respiration to anaerobic glycolysis for ATP production. The subsequent decrease in ATP turnover leads to intracellular acidosis. Lactate from glycolysis helps to buffer the accumulated protons, but the stagnation of blood flow prevents the removal of lactate. Cardiomyocytes attempt to reestablish normal intracellular pH through the sodium-hydrogen exchanger. The subsequent accumulation of intracellular sodium drives the sodium-calcium exchanger in reverse, and causes intracellular accumulation of calcium. (b) During reperfusion, the rapid normalization of extracellular pH exacerbates the conditions seen in ischemia. The large proton gradient increases proton efflux and results in intracellular calcium overload. Reoxygenation also leads to increased reactive oxygen species (ROS) generation. Intracellular calcium overload and rapid ROS generation contribute to opening of the mitochondrial permeability transition pore (MPTP), decoupling of oxidative phosphorylation, and eventually cell death.
Reperfusion
Reperfusion is achieved by unblocking the artery through percutaneous coronary intervention (PCI) or thrombolytic therapy. The restoration of blood flow salvages the ischemic region by reestablishing oxygen and nutrient delivery and removing metabolic by-products; however, reperfusion also leads to an increase in cell death. Lethal reperfusion injury is the death of cardiomyocytes upon reperfusion that were otherwise viable at the end of the ischemic period.
Reperfusion restores extracellular ionic balance and normalizes the extracellular pH. The rapid normalization of the extracellular pH creates a large proton gradient that exacerbates the conditions seen in ischemia. The sodium-hydrogen exchanger accelerates excretion of hydrogen to restore physiological levels of intracellular pH. The rapid normalization of intracellular pH leads in turn to an even greater increase in the intracellular accumulation of calcium through the sodium-calcium exchanger [17]. The high intracellular calcium concentration helps to activate calpains, proteases that target intracellular proteins [18]. Reoxygenation also leads to an increase in the generation of reactive oxygen species (ROS) that contribute to cell injury [19].
Intracellular calcium overload and the generation of reactive oxygen species (ROS) lead to opening of the mitochondria permeability transition pores (MPTP). The opening of the MPTP is thought to be the critical step in IRI, because it causes a loss of mitochondrial membrane potential, decoupling of the electron transport chain, and eventually the failure of ATP production. It also allows water to enter, causing swelling of the mitochondrial membrane. If the mitochondrial membrane is ruptured, cell death signaling proteins are released into the cytosol, resulting in the loss of viability (Figure 2b) [20, 21].
Despite the identification of this major mechanism of reperfusion injury, the exact progression and mechanisms of IRI are still not completely defined. Inhibition of MPTP opening by targeting its regulator, cyclophilin D, has proven promising in animal studies [22]. However, the use of cyclosporine A, an inhibitor of cyclophilin D, has not been successful in clinical trials [4, 23]. It is unclear if the failure of cyclosporine A in clinical trials is due to overemphasizing the importance of the MPTP and some other factors related to the clinical setting [7]. Researchers have also found success in limiting IRI by targeting other pathways, such as the unfolded protein response or histone deacetylation [24, 25]. It is possible that there are more unknown therapeutic targets. It is clear that the field needs to further develop our understanding of the mechanisms driving IRI and develop effective therapeutic modalities.
Animal Models of IRI
Currently, researchers primarily rely on animal models when testing the new therapeutics for IRI. Ischemia-reperfusion in animals is typically simulated by using a suture to occlude the left descending coronary artery for the designated ischemic time, and then releasing the suture to allow for reperfusion [26]. This approach best captures the process of hypoxia-reoxygenation, but does not fully simulate the clinical setting where an atherosclerotic artery is clogged by a dislodged plaque and then reperfused by PCI. Animal models are useful in that they allow for examination of the interactions between various cell types during reperfusion. For example, it has been found that the adhesion of neutrophils to the endothelium can be modified and decreased IRI [27]. These interactions involve paracrine signaling through a multitude of molecules, such as TNF-α and nitric oxide [28], and are difficult to accurately simulate in vitro. These other cell types play important roles, and must be considered when studying IRI.
While animal models are very helpful, they fail to fully emulate human physiology. Pigs are the closest analogues because they have a comparable heart size and heart rate to humans, lack the protective coronary collateral blood flow found in dogs [29], and do not have the innate resistance to MI found in primates [30]. However, large animals are difficult and expensive to work with. Thus, most researchers rely on rodent hearts for their in vivo work. Compared to humans, rodent hearts have a much higher intrinsic beating rate [31], different myosin isoform predominance, higher cardiac basal metabolism [32], and different electrophysiology of the heart [33]. While the relative importance of these differences is undefined, they influence the response to IRI. The fact that the therapeutic demonstrating success in animal studies have not made the transition to clinical utility indicates that these animal models are not adequate.
Researchers have also explored the use of isolated perfused hearts. These models maintain the physiological simulation of ischemia-reperfusion, but allow for more control over the conditions. Researchers have demonstrated their effectiveness by reducing infarct size using various cardioprotective agents [34]. However, ex vivo models – similar to animal models – do not recapitulate human physiology. New preclinical platforms are needed that better emulate human physiology.
In Vitro Models of IRI
An alternative to animal models is to use the in vitro models based on isolated cardiomyocytes. While these models do not recapitulate the complexity of the heart, they allow examining the direct effect of drugs on cardiomyocytes, with better manipulation and control of the various confounding factors found in animal models. In vitro models could provide a platform to rigorously study the important pathways of IRI, and test the candidate therapeutic options in settings that are more predictive of the pathophysiology of ischemia-reperfusion than animal models.
Modeling of ischemic conditions in vitro
In vitro models examine IRI in a non-physiological setting, and simulate ischemia and reperfusion by manipulating the cellular environment. In addition to providing a hypoxic environment, simulation of the ischemic state must also encompass other factors, including hyperkalemia, acidosis, nutrient deprivation, and waste accumulation [35].
In vitro studies have attempted to simulate ischemic conditions by culturing cells in physiological salt solutions that are free of metabolic substrates, supplemented with additional potassium to achieve hyperkalemia, and titrated to an acidic pH [36]. Additionally, lactate is added to simulate its accumulation due to anaerobic glycolysis. Some studies have also added 2-deoxy-d-glucose, a nonmetabolizable glucose analogue, into the ischemic solution to inhibit glycolysis and further shutdown cellular metabolism [37]. It is unknown if the non-metabolized 2-deoxy-d-glucose phosphate interferes with the reperfusion process, and its effect needs to be further studied before being included in a simulated IRI model. The precise concentrations and compositions of the salt solutions vary across the different studies reviewed; however, a potassium concentration of 12 mM, lactate concentration of 20 mM, and pH of 6.5 are most commonly used [36, 38]. The cells are placed in a gas-tight chamber, which is then flushed with anoxic gas (95% N2, 5% CO2) to achieve a hypoxic environment.
Another method to achieve a hypoxic state has depended on the pelleting of cells with a minimal amount of media under a layer of mineral oil that hindered oxygen diffusion to create a hypoxic environment [39]. The key idea behind this model is to achieve ischemia through a more physiological means, with gradual reduction of nutrients and accumulation of wastes. However, this method is more variable and the pelleting through centrifugation can lead to mechanical damage to the cells.
Metabolic inhibition through compounds, such as cyanide, to decrease oxygen consumption has also been explored [40]. These chemicals may be hard to wash out or have irreversible effects that can disrupt a physiologically accurate reperfusion process. It is recommended to utilize a salt solution simulating ischemic conditions and a hypoxic chamber to maximize control over the in vitro model.
The necessary ischemic time in vitro has also not been precisely determined. In animal experiments, ischemic times are relatively short and well defined. Thirty minutes is all that is needed to achieve an ischemic state within the tissue [26, 41]. However, isolated cardiomyocytes cultured in vitro are at different levels of maturity and oxygen dependency than those in intact mature tissue. The embryonic heart is more resistant to hypoxia than the adult heart, and has improved recovery after an ischemic insult [42]. As a result, much longer ischemic times, ranging from 90 minutes [43] to 9 hours [26], are needed to demonstrate a decrease in cell viability. The necessary duration of ischemia is dependent on the cell source used, but can also vary within the cell types used. One study had to adjust the ischemic time to two to five hours [25].
Further complicating the matter, it has been found in vivo that after a certain length of ischemia, preconditioning no longer has cardioprotective effects with respect to the final infarct size [3]. This time dependence of the effectiveness of ischemic conditioning must be accounted for when assessing the potential of a therapeutic. The European Society of Cardiology Working Group Cellular Biology of the Heart has recommended that the combined ischemic and reperfusion times should be selected to result in 50% cell death [9]. Each in vitro model must therefore independently establish an ischemic time that causes enough cell death, but is not too long to affect possible preconditioning or drug benefits.
Simulating reperfusion in vitro
Reperfusion must also be simulated through non-physiological means in vitro. In clinical settings, the aim of reperfusion therapy is to restore nutrient and oxygen delivery and to remove the accumulated metabolic by-products. Reperfusion is typically simulated in vitro by replacing the ischemic solution to remove metabolic waste, adding normal culture media for nutrient delivery, and removing the system from the hypoxic chamber to return the environmental levels of oxygen to normoxia [44, 45].
The composition of culture media added to simulate reperfusion must be carefully selected. Most in vitro studies utilize commercially available basal media supplemented with serum. However, it is unclear if normal cell culture media best simulates physiological conditions. The reperfusion solution needs to provide nutrients to the cells and normalize extracellular pH and ion balance in order to cause intracellular calcium overload and allow for the generation of ROS. Cell culture media provide nutrients in the forms of glucose in the basal media, and lipids in the serum. Cardiomyocytes are more dependent on lipids for metabolism [46]; however, serum supplementation may be problematic. The exact composition of serum is unknown and has lot-to-lot variations, which introduces many variables that may obfuscate the effect of the therapeutic of interest. Serum also contains antioxidants that may ameliorate the oxidative stress seen in IRI. Serum-free media are utilized in cardiomyocytes derived from human iPS cells [47] and a chemically defined reperfusion solution can help establish a consistent response to IRI.
It is important to also re-establish the physiological extracellular calcium concentration (1-2 mM) [48] because intracellular calcium overload is one of the important effectors of reperfusion damage [35]. Certain types of basal media contain less calcium, and it has been recognized that physiological calcium levels are required for normal function of cardiomyocytes [49].
The time after reperfusion before measuring cell viability is also important. Using LDH release as a measure for cell injury it was determined that there were two separate peaks of LDH release. Cardioprotective treatments affected one or both of these peaks [50]. The assessment of a drug thus needs to take into consideration all these factors in order to properly determine therapeutic benefit.
Isolated cardiomyocytes as models of ischemia and reperfusion
In vitro models of IRI primarily rely on isolated cardiomyocytes derived from animals, immortalized cell lines, or human pluripotent stem cells (iPS cells). The cardiomyocyte source affects its response to IRI, and thus, its predictive power for pharmacologic testing.
Most of the in vitro work has utilized cardiomyocytes derived from animals. Primary cardiomyocytes have been isolated for study from a variety of animal types, including rats, pigs, and rabbits, and from animals of different levels of maturity. Cardiomyocytes derived from adult animals are more relevant for the studies of IRI, because the immature cells derived from neonatal animals are more resistant to hypoxia than adult human cardiomyocytes [42]. This resilience causes neonatal hearts to respond very differently to IRI. For example, ischemic preconditioning does not protect neonatal hearts [51], and neonatal cardiomyocyte mitochondria are not as sensitive to IRI-induced membrane permeabilization [52]. These physiological differences are significant, and need to be accounted for when comparing preclinical data. Therefore, therapeutics should be tested in adult-derived cardiomyocytes.
Researchers have also explored the use of cell lines, such as immortalized mouse atrial HL-1 cells or rat H9c2 cardiomyoblasts. They are more cost effective than primary cardiomyocytes, and have demonstrated similar drug responses to IRI as primary cardiomyocytes [26, 53]. However, these cell lines do not recapitulate the cardiac physiology. The cells are dividing, unlike mature cardiomyocytes, and display differences in their cell death pathways and bioenergetics [54, 55]. Because mitochondria are important effectors of IRI, caution must be exercised when extrapolating results obtained with cell lines.
Human cardiomyocytes would be ideal for in vitro models of IRI, because they can provide a more biomimetic platform to conduct studies in by avoiding the problem of interspecies comparisons. Animal and adult human cardiomyocytes differ in many aspects, such as the beating rate, myosin isoform predominance, ATP utilization, and electrophysiological properties [56, 57]. These differences can affect the cell response to IRI and would make it difficult to extrapolate results from animals to humans. The challenge is that human primary cardiomyocytes are impossible to work with because they are terminally differentiated and have limited proliferative potential, and are also very difficult to obtain.
In an attempt to provide models based on adult human cardiomyocytes, researchers have developed a proliferating human cardiomyocyte cell line (AC16) by fusing primary cells from human ventricular tissue with transformed fibroblasts [58]. These cells are differentiated following transfer into mitogen-depleted media that stops their proliferation, and they express cardiac-specific proteins and maintain cardiac nuclear and mitochondrial DNA. AC16 cardiomyocytes have been used and validated in an in vitro study that examined the effect of asiatic acid of IRI [59]. The results were consistent with other studies in a rat model and an in vitro H9c2 cardiomyoblast model that also demonstrated asiatic acid attenuation of IRI [60, 61]. Still, there are concerns about the physiologic relevance of transformed and fused cell lines.
The most attractive cell source for biomimetic studies of IRI are cardiomyocytes derived from human induced pluripotent stem cells (hiPS-CMs) or embryonic stem cells (hESC-CMs) (Table 1). It has been demonstrated that human stem cell derived cardiomyocytes can be cultured and differentiated in a defined and reproducible manner [47], and they have been used in IRI studies with some success.
Table 1. Cardiomyocyte sources for in vitro models of Ischemia-Reperfusion Injury.
| Cardiomyocyte Source | Ease of use | Predictive Utility | Cell Source Issue |
|---|---|---|---|
| Animal cell source | |||
| Isolated primary cardiomyocytes | |||
| Adult cardiomyocytes | Low | Medium | Interspecies physiolocal differences |
| Neonatal cardiomyocytes | Low | Low-Medium | Immature phenotype, Interspecies physiological differences |
| Immortalized cell lines (e.g. HL-1, H9c2) | High | Low | Proliferating cell line, Interspecies physiolocal differences |
| Human cell source | |||
| Transformed cell line (e.g. AC16) | High | Low | Proliferating cell line |
| Cardiomyocytes derived from Pluripotent Stem Cells | |||
| hESC-CMs | Medium | Low-Medium | Immature phenotype |
| hIPS-CMs | Medium | Low-Medium | Immature phenotype |
Hsieh et al found that the simulation of IRI in hESC-CMs caused an increase in the qPCR ratio of anti-apopototic BCL-2 to the apoptotic BAX gene following a pretreatment with sodium nitrite [62]. Paloczi et al demonstrated the cardioprotective effect of exogenous nitric oxide supplementation in simulated IRI experiments on hESC-CMs derived from embryoid bodies (EB) [45]. However, these studies failed to specifically separate the cardiomyocytes from the other cells in the EBs, and instead relied on a GFP expression to identify cardiac regions and quantify cell death. The narrow range of methods that these studies employed makes it unclear if hESC-CMs are a suitable cell source to study IRI.
The establishment of hIPSCs as a source of human cardiomyocytes has allowed researchers to avoid the ethical concerns of using hESCs and to also derive patient-specific cell lines. Specifically, hiPS-CMs allow for studies of patient-specific disease backgrounds [63]. There is a great interest and some success in utilizing hIPS-CMs in pharmacological screening [64, 65], but very little work has been done in the setting of reperfusion injury. In one of the few IRI studies done using hIPS-CMs, Wei et al. demonstrated their utility by confirming the cardioprotective effects of Danshen in vitro [66]. These results were consistent with previous studies in animal models demonstrating the therapeutic benefit of Danshen [41]. However, hIPS-CMs are relatively immature and more closely resemble fetal cardiomyocytes than adult cardiomyocytes [67]. As known, the fetal phenotype and bioenergetics are associated with non-physiologic responses to reperfusion injury, and can pose challenges to predictive testing of drug efficacy and safety.
Organ-On-Chip Models
Because the fetal nature of hIPS-CMs limits their use in IRI studies, tissue engineering provides an intriguing avenue for maturing these cells into a more adult phenotype. Previous studies have demonstrated that incorporating hIPS-CMs into tissue engineered heart constructs has helped mature their gene expression profile, ultrastructure, and electrophysiological properties [49, 68–72]. However, these cells still fall short of adult cardiomyocyte function, but much progress is being made.
One study utilized an engineered heart tissue model of IRI [73], but unfortunately, the tissue constructs were based on neonatal rat cardiomyocytes. Nevertheless, it is instructive to look into how tissue engineering can help advance the IRI research. The study examined how hypoxia affected electrical conduction in the tissue, and used protein expression to characterize reperfusion injury. Cyclosporine A, akin to animal studies [74], showed cardioprotective effects in this system. This study helped establish tissue engineering as a useful tool for studying IRI and testing of therapeutic options.
Tissue engineered heart constructs are of interest because of the potential for real-time measurements of contractile function. The twitch and peak stresses of engineered heart tissues can be determined by calculating the deflection of a cantilever from video recordings [75]. The model was precise enough to capture the changes in force due to genetic mutations in the patient-derived hiPS cell line. The force generated by engineered cardiac tissue could potentially be correlated to the ejection fraction of the heart. Currently, most animal studies use the infarct size as the readout, but physicians are much more interested in left ventricular function. Using force as the measurement in vitro will allow for better correlation with the clinical outcomes.
Overall, in vitro models can capture the effects of treatments or drugs on cardiomyocytes, while avoiding the confounding factors encountered in animal models. As detailed above, the source and maturity of cardiomyocytes can help obtain a more physiological response to IRI. These models can be further improved by taking into consideration the aspects of IRI that affect the clinical setting (Figure 3).
Figure 3. Potential improvements to preclinical studies of IRI.
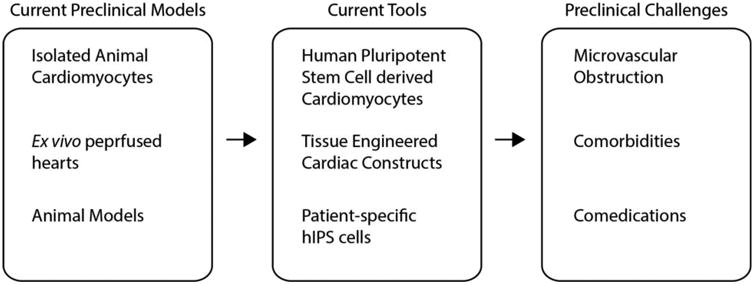
Current preclinical models of IRI can be improved by utilizing human cardiomyocytes that better recapitulate human physiology and response to IRI. In vitro models can also be improved by accounting for the heterogeneous patient population seen in the clinical settings.
Modeling Microvascular Obstruction
In addition to the direct effects of reperfusion on the cardiomyocytes, a long-lasting source of damage is microvascular obstruction [76–78]. In the heart, microvascular obstruction is the failure of coronary artery reperfusion to fully reperfuse the underlying myocardial microvasculature. This is an independent risk factor that correlates with worsened clinical outcomes [79]. Reperfusion causes the influx of leukocytes and platelets, endothelial damage, and vasoconstriction, which all contribute to microvascular obstruction [80–82]. These phenomena could be studied in vitro in a heart muscle engineered to contain microvasculature. There have been many advances in engineering vascularized cardiac tissue for implantation [83–85], and the existing platforms could potentially be further developed to examine microvascular obstruction in a tissue model of IRI. With the functional microvasculature and surrounding pericytes, it would be possible to study in vitro the endothelial damage and vasoconstriction associated with microvascular obstruction. Tissue engineered constructs with a functional microvasculature will help better recapitulate the clinical setting of IRI.
Accounting for Comorbidities
A common criticism levied on preclinical IRI studies (and in fact most preclinical studies, both in cell culture and in animal models) is that therapies are tested on a homogenous, young, and healthy population. In contrast, myocardial infarction primarily affects a diverse older population with many different comorbidities, such as diabetes and cardiac hypertrophy. These additional factors affect the cardiomyocyte response in different ways, usually by increasing their susceptibility to reperfusion injury and decreasing the effectiveness of treatments [86–88].
In recent years, the researchers have begun demonstrating that hiPSCs can recapitulate the key features of chronic diseases. Drawnel et al. established a diabetic hiPS-CMs screening platform using cells exposed to a prodiabetic environment and hiPS cell lines derived from diabetic patients [89]. The diabetic patient-specific hIPS-CMs were not exposed to any diabetes-specific environmental signals, and yet they demonstrated a diabetic phenotype and the corresponding responses to drugs. These findings indicate that the hIPS-CMs recapitulating the disease phenotype should be a part of patient-specific diabetic models. A diabetic phenotype was also demonstrated in an engineered heart tissue model, utilizing neonatal rat cardiomyocytes [90]. In addition, diabetic susceptibility to IRI has been demonstrated in an animal model [91], but not in vitro using human cells or tissues.
Tissue engineering could be instrumental for modeling comorbidities in vitro. In some models, engineered heart tissue was suspended between two attachment sites to allow for mechanical stress and auxotonic contractions [92]. Hirt et al. was able to expand upon this concept by inserting metal braces to increase the afterload sensed by the cardiac tissue. Consequently, the cardiomyocytes developed a pathologic cardiac hypertrophic phenotype. The results were validated against a hypertrophy-promoting pharmacologic conditioning protocol [93]. In summary, there is increasing evidence that tissue engineering enables various approaches to model comorbidities of interest, and could thus provide a more biomimetic model of IRI.
Concomitant Drugs
It has been found that many of the medications taken to treat the comorbid conditions found in patients can alter the effects of IRI and could be cardioprotective themselves. For example, statins are cardioprotective, but also interfere with the cardioprotective effects of ischemic preconditioning [94]. Some of the anesthetics used during PCI to reperfuse the artery are also cardioprotective, while others are not [95, 96]. Further complicating the matter, it has been found that comorbid conditions, such as diabetes, can interfere with the cardioprotective effect of anesthetics and statins [97, 98]. These interactions are not being systematically tested and identified in preclinical animal models, and may play a role in clinical trials of promising drugs. In fact, the concomitant drug may be providing cardioprotection through the same pathway as the therapeutic of interest, or may be actively hindering the therapy.
The cardioprotective features of anesthetics have been demonstrated on isolated human atrial trabeculae ex vivo [99]. In vitro models would be ideal for performing these studies in a high-throughput manner. In particular, tissue engineered cardiac constructs derived from patient-specific hIPS-CMs will be of value in identifying the drug-drug interactions. As the patient population grows older and requires more drugs for treating multiple comorbidities, such screenings will become even more important. Reductionist models could be very helpful for understanding how different drugs and conditions interact, in order to avoid false negative results.
Conclusion
IRI has been extensively studied ever since it was discovered that ischemic preconditioning could provide cardioprotection. However, the field has failed to produce therapeutic modalities to treat IRI in the clinical setting. There have been many issues in the drug development process that have contributed to the lack of success, emphasizing the need to develop better in vitro models. These reductionist models would allow for better control and study of various contributing factors. Much of the previous work in vitro has been performed using animal-derived cells, but the recent uptick of research into hiPS-CMs provides an avenue to model IRI using human cardiomyocytes. Tissue engineering techniques further these recent developments by helping to mature hIPS-CMs into a more adult phenotype. The engineered platforms can be further modified to include clinical comorbidities and thereby provide a more accurate setting for drug screening and testing for possible interactions between the drugs and comorbidities. There is a great need to develop therapeutics to treat IRI, and the current tools hold promise to further our understanding of the progression of IRI and support testing of drugs under controllable conditions.
Future Work
Future directions should first establish the presence of IRI in a tissue engineered cardiac construct utilizing hIPS-CMs. Next, the use of patient-specific hIPS-CMs and tissue engineering techniques to form disease models will allow us to better understand the effect different comorbidities have on IRI and its potential treatments.
Lay Summary.
Heart attacks are treated by unblocking the occluded blood vessel; however, the restoration of blood flow to the damaged area causes additional injury, termed ischemia-reperfusion injury (IRI). Many treatments have successfully decreased IRI in animals, but none have shown the same success in humans. This failure may be because animal hearts have important functional differences compared to human hearts. Human engineered tissue models are important tools to overcome this hurdle because they utilize human cells in a highly controllable environment. In this article, we discuss models of IRI and how they can facilitate the development of treatments for patient use.
Acknowledgments
We gratefully acknowledge the NIH funding of the work related to this article (grants HL076485, EB002520, EB17103 and EB025765).
References
- 1.Sanchis-Gomar F, Perez-Quilis C, Leischik R, Lucia A. Epidemiology of coronary heart disease and acute coronary syndrome. Ann Transl Med. 2016;4:256–256. doi: 10.21037/atm.2016.06.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McNamara RL, Wang Y, Herrin J, et al. Effect of door-to-balloon time on mortality in patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2006;47:2180–6. doi: 10.1016/j.jacc.2005.12.072. [DOI] [PubMed] [Google Scholar]
- 3.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 4.Cung TT, Morel O, Cayla G, et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N Engl J Med. 2015;373:1021–1031. doi: 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
- 5.Dominguez-Rodriguez A, Abreu-Gonzalez P, de la Torre-Hernandez JM, et al. Effect of intravenous and intracoronary melatonin as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: Results of the Melatonin Adjunct in the acute myocaRdial Infarction treated with Angioplasty trial. J Pineal Res. 2017;62:e12374. doi: 10.1111/jpi.12374. [DOI] [PubMed] [Google Scholar]
- 6.Hausenloy DJ, Garcia-Dorado D, Bøtker HE, et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc Res. 2017;113:564–585. doi: 10.1093/cvr/cvx049. [DOI] [PubMed] [Google Scholar]
- 7.Hausenloy DJ, Yellon DM. Targeting Myocardial Reperfusion Injury--The Search Continues. N Engl J Med. 2015;373:1073–5. doi: 10.1056/NEJMe1509718. [DOI] [PubMed] [Google Scholar]
- 8.Rossello X, Yellon DM. A critical review on the translational journey of cardioprotective therapies! Int J Cardiol. 2016;220:176–184. doi: 10.1016/j.ijcard.2016.06.131. [DOI] [PubMed] [Google Scholar]
- 9.Lecour S, Botker HE, Condorelli G, et al. ESC Working Group Cellular Biology of the Heart: Position Paper: improving the preclinical assessment of novel cardioprotective therapies. Cardiovasc Res. 2014;104:399–411. doi: 10.1093/cvr/cvu225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossello X, Yellon DM. Cardioprotection: The Disconnect Between Bench and Bedside. Circulation. 2016;134:574–5. doi: 10.1161/CIRCULATIONAHA.116.022829. [DOI] [PubMed] [Google Scholar]
- 12.Vághy PL. Role of mitochondrial oxidative phosphorylation in the maintenance of intracellular pH. J Mol Cell Cardiol. 1979;11:933–40. doi: 10.1016/0022-2828(79)90385-7. [DOI] [PubMed] [Google Scholar]
- 13.Robergs RA. Biochemistry of exercise-induced metabolic acidosis. AJP Regul Integr Comp Physiol. 2004;287:R502–R516. doi: 10.1152/ajpregu.00114.2004. [DOI] [PubMed] [Google Scholar]
- 14.Halestrap AP, Wang X, Poole RC, et al. Lactate transport in heart in relation to myocardial ischemia. Am J Cardiol. 1997;80:17A–25A. doi: 10.1016/S0002-9149(97)00454-2. [DOI] [PubMed] [Google Scholar]
- 15.Theroux P, Chaitman BR, Danchin N, et al. Inhibition of the Sodium-Hydrogen Exchanger With Cariporide to Prevent Myocardial Infarction in High-Risk Ischemic Situations : Main Results of the GUARDIAN Trial. Circulation. 2000;102:3032–3038. doi: 10.1161/01.CIR.102.25.3032. [DOI] [PubMed] [Google Scholar]
- 16.Marban E, Kitakaze M, Kusuoka H, et al. Intracellular free calcium concentration measured with 19F NMR spectroscopy in intact ferret hearts. Proc Natl Acad Sci U S A. 1987;84:6005–9. doi: 10.1073/pnas.84.16.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am J Physiol. 1997;273:C1783–92. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
- 18.Hernando V, Inserte J, Sartório CL, et al. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J Mol Cell Cardiol. 2010;49:271–279. doi: 10.1016/j.yjmcc.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 19.Raedschelders K, Ansley DM, Chen DDY. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol Ther. 2012;133:230–255. doi: 10.1016/j.pharmthera.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol. 2009;104:181–188. doi: 10.1007/s00395-009-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307(Pt 1):93–8. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 23.Ottani F, Latini R, Staszewsky L, et al. Cyclosporine A in Reperfused Myocardial Infarction the Multicenter, Controlled, Open-Label CYCLE Trial. J Am Coll Cardiol. 2016;67:365–374. doi: 10.1016/j.jacc.2015.10.081. [DOI] [PubMed] [Google Scholar]
- 24.Zhang C, Tang Y, Li Y, et al. Unfolded protein response plays a critical role in heart damage after myocardial ischemia/reperfusion in rats. PLoS One. 2017;12:e0179042. doi: 10.1371/journal.pone.0179042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie M, Kong Y, Tan W, et al. Histone Deacetylase Inhibition Blunts Ischemia/Reperfusion Injury by Inducing Cardiomyocyte Autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He X, Li S, Liu B, et al. Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc Natl Acad Sci U S A. 2017;114:E4582–E4591. doi: 10.1073/pnas.1621384114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pabla R, Buda AJ, Flynn DM, et al. Nitric oxide attenuates neutrophil-mediated myocardial contractile dysfunction after ischemia and reperfusion. Circ Res. 1996;78:65–72. doi: 10.1161/01.RES.78.1.65. [DOI] [PubMed] [Google Scholar]
- 28.Thielmann M, Dörge H, Martin C, et al. Myocardial dysfunction with coronary microembolization: signal transduction through a sequence of nitric oxide, tumor necrosis factor-alpha, and sphingosine. Circ Res. 2002;90:807–13. doi: 10.1161/01.RES.0000014451.75415.36. [DOI] [PubMed] [Google Scholar]
- 29.Heusch G, Skyschally A, Schulz R. The in-situ pig heart with regional ischemia/reperfusion -Ready for translation. J Mol Cell Cardiol. 2011;50:951–963. doi: 10.1016/j.yjmcc.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 30.Shen YT, Fallon JT, Iwase M, Vatner SF. Innate protection of baboon myocardium: effects of coronary artery occlusion and reperfusion. Am J Physiol. 1996;270:H1812–8. doi: 10.1152/ajpheart.1996.270.5.H1812. [DOI] [PubMed] [Google Scholar]
- 31.Hamlin RL, Altschuld RA. Extrapolation from mouse to man. Circ Cardiovasc Imaging. 2011;4:2–4. doi: 10.1161/CIRCIMAGING.110.961979. [DOI] [PubMed] [Google Scholar]
- 32.Gibbs CL. Cardiac energetics: Sense and nonsense. Clin Exp Pharmacol Physiol. 2003;30:598–603. doi: 10.1046/j.1440-1681.2003.03878.x. [DOI] [PubMed] [Google Scholar]
- 33.O'Hara T, Rudy Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. AJP Hear Circ Physiol. 2012;302:H1023–H1030. doi: 10.1152/ajpheart.00785.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: A form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res. 2004;95:230–232. doi: 10.1161/01.RES.0000138303.76488.fe. [DOI] [PubMed] [Google Scholar]
- 35.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DIAZ R, WILSON G. Studying ischemic preconditioning in isolated cardiomyocyte models. Cardiovasc Res. 2006;70:286–296. doi: 10.1016/j.cardiores.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Diaz RJ, Harvey K, Boloorchi A, et al. Enhanced cell volume regulation: a key mechanism in local and remote ischemic preconditioning. Am J Physiol Cell Physiol. 2014;306:C1191–9. doi: 10.1152/ajpcell.00259.2013. [DOI] [PubMed] [Google Scholar]
- 38.Si J, Wang N, Wang H, et al. HIF-1α signaling activation by post-ischemia treatment with astragaloside iv attenuates myocardial ischemia-reperfusion injury. PLoS One. 2014;9:1–10. doi: 10.1371/journal.pone.0107832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strijdom H, Genade S, Lochner A. Nitric Oxide synthase (NOS) does not contribute to simulated ischaemic preconditioning in an isolated rat cardiomyocyte model. Cardiovasc drugs Ther. 2004;18:99–112. doi: 10.1023/B:CARD.0000029027.50796.84. [DOI] [PubMed] [Google Scholar]
- 40.Cavalheiro RA, Marin RM, Rocco SA, et al. Potent cardioprotective effect of the 4-anilinoquinazoline derivative PD153035: involvement of mitochondrial K(ATP) channel activation. PLoS One. 2010;5:e10666. doi: 10.1371/journal.pone.0010666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song M, Huang L, Zhao G, Song Y. Beneficial effects of a polysaccharide from Salvia miltiorrhiza on myocardial ischemia-reperfusion injury in rats. Carbohydr Polym. 2013;98:1631–6. doi: 10.1016/j.carbpol.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 42.Sedmera D, Kucera P, Raddatz E. Developmental changes in cardiac recovery from anoxia-reoxygenation. Am J Physiol Regul Integr Comp Physiol. 2002;283:R379–R388. doi: 10.1152/ajpregu.00534.2001. [DOI] [PubMed] [Google Scholar]
- 43.Portal L, Martin V, Assaly R, et al. A Model of Hypoxia-Reoxygenation on Isolated Adult Mouse Cardiomyocytes: Characterization, Comparison With Ischemia-Reperfusion, and Application to the Cardioprotective Effect of Regular Treadmill Exercise. J Cardiovasc Pharmacol Ther. 2013;18:367–375. doi: 10.1177/1074248412475158. [DOI] [PubMed] [Google Scholar]
- 44.Chang G, Zhang D, Liu J, et al. Exenatide protects against hypoxia/reoxygenation-induced apoptosis by improving mitochondrial function in H9c2 cells. Exp Biol Med (Maywood) 2014;239:414–22. doi: 10.1177/1535370214522177. [DOI] [PubMed] [Google Scholar]
- 45.Pálóczi J, Varga ZV, Apáti Á, et al. Exogenous Nitric Oxide Protects Human Embryonic Stem Cell-Derived Cardiomyocytes against Ischemia/Reperfusion Injury. Oxid Med Cell Longev. 2016;2016:1–9. doi: 10.1155/2016/4298945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopaschuk GD, Ussher JR, Folmes CDL, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009.. [DOI] [PubMed] [Google Scholar]
- 47.Burridge PW, Matsa E, Shukla P, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–60. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larsson L, Ohman S. Serum ionized calcium and corrected total calcium in borderline hyperparathyroidism. Clin Chem. 1978;24:1962–5. [PubMed] [Google Scholar]
- 49.Tiburcy M, Hudson JE, Balfanz P, et al. Defined Engineered Human Myocardium With Advanced Maturation for Applications in Heart Failure Modeling and Repair. Circulation. 2017;135:1832–1847. doi: 10.1161/CIRCULATIONAHA.116.024145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Povlsen JA, Løfgren B, Dalgas C, et al. Frequent biomarker analysis in the isolated perfused heart reveals two distinct phases of reperfusion injury. Int J Cardiol. 2014;171:9–14. doi: 10.1016/j.ijcard.2013.11.035. [DOI] [PubMed] [Google Scholar]
- 51.Ostadalova I, Ostadal B, Kolár F, et al. Tolerance to ischaemia and ischaemic preconditioning in neonatal rat heart. J Mol Cell Cardiol. 1998;30:857–65. doi: 10.1006/jmcc.1998.0653. [DOI] [PubMed] [Google Scholar]
- 52.Milerova M, Charvatova Z, Skarka L, et al. Neonatal cardiac mitochondria and ischemia/reperfusion injury. Mol Cell Biochem. 2010;335:147–153. doi: 10.1007/s11010-009-0251-x. [DOI] [PubMed] [Google Scholar]
- 53.Teixeira G, Abrial M, Portier K, et al. Synergistic protective effect of cyclosporin A and rotenone against hypoxia-reoxygenation in cardiomyocytes. J Mol Cell Cardiol. 2013;56:55–62. doi: 10.1016/j.yjmcc.2012.11.023. [DOI] [PubMed] [Google Scholar]
- 54.Cao X, Wang X, Ling Y, et al. Comparison of the degree of autophagy in neonatal rat cardiomyocytes and H9c2 cells exposed to hypoxia/reoxygenation. Clin Lab. 2014;60:809–814. doi: 10.7754/Clin.Lab.2013.130521. [DOI] [PubMed] [Google Scholar]
- 55.Kuznetsov AV, Javadov S, Sickinger S, et al. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim Biophys Acta - Mol Cell Res. 2015;1853:276–284. doi: 10.1016/j.bbamcr.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu HR, Mariën R, Saels a, De Clerck F. Species plays an important role in drug-induced prolongation of action potential duration and early afterdepolarizations in isolated Purkinje fibers. J Cardiovasc Electrophysiol. 2001;12:93–102. doi: 10.1046/j.1540-8167.2001.00093.x. doi:lu01. [DOI] [PubMed] [Google Scholar]
- 57.Rundell VLM, Manaves V, Martin AF, de Tombe PP. Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Hear Circ Physiol. 2005;288:H896–903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- 58.Davidson MM, Nesti C, Palenzuela L, et al. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 59.Wu K, Hu M, Chen Z, et al. Asiatic acid enhances survival of human AC16 cardiomyocytes under hypoxia by upregulating miR-1290. IUBMB Life. 2017:660–667. doi: 10.1002/iub.1648. [DOI] [PubMed] [Google Scholar]
- 60.Huo L, Shi W, Chong L, et al. Asiatic acid inhibits left ventricular remodeling and improves cardiac function in a rat model of myocardial infarction. Exp Ther Med. 2016;11:57–64. doi: 10.3892/etm.2015.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang X, Zuo L, Lv Y, et al. Asiatic acid attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3β/HIF-1α signaling in rat H9c2 cardiomyocytes. Molecules. 2016;21:1–14. doi: 10.3390/molecules21091248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hsieh A, Feric NT, Radisic M. Combined hypoxia and sodium nitrite pretreatment for cardiomyocyte protection in vitro. Biotechnol Prog. 2015;31:482–92. doi: 10.1002/btpr.2039. [DOI] [PubMed] [Google Scholar]
- 63.Sun N, Yazawa M, Liu J, et al. Patient-Specific Induced Pluripotent Stem Cells as a Model for Familial Dilated Cardiomyopathy. Sci Transl Med. 2012;4:130ra47–130ra47. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khan JM, Lyon AR, Harding SE. The case for induced pluripotent stem cell-derived cardiomyocytes in pharmacological screening. Br J Pharmacol. 2013;169:304–317. doi: 10.1111/j.1476-5381.2012.02118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paşca SP, Portmann T, Voineagu I, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei W, Liu Y, Zhang Q, et al. Danshen-Enhanced Cardioprotective Effect of Cardioplegia on Ischemia Reperfusion Injury in a Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Model. Artif Organs. 2016 doi: 10.1111/aor.12801. [DOI] [PubMed] [Google Scholar]
- 67.Guo L, Abrams RMC, Babiarz JE, et al. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci. 2011;123:281–289. doi: 10.1093/toxsci/kfr158. [DOI] [PubMed] [Google Scholar]
- 68.Nunes SS, Miklas JW, Liu J, et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods. 2013;10:781–7. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shadrin IY, Allen BW, Qian Y, et al. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat Commun. 2017;8:1825. doi: 10.1038/s41467-017-01946-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ronaldson K, Ma S, Yeager K, et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature. doi: 10.1038/s41586-018-0016-3. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li RA, Keung W, Cashman TJ, et al. Bioengineering an electro-mechanically functional miniature ventricular heart chamber from human pluripotent stem cells. Biomaterials. 2018;163:116–127. doi: 10.1016/j.biomaterials.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Agarwal A, Goss JA, Cho A, et al. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip. 2013;13:3599–608. doi: 10.1039/c3lc50350j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Katare RG, Ando M, Kakinuma Y, Sato T. Engineered Heart Tissue: A Novel Tool to Study the Ischemic Changes of the Heart In Vitro. PLoS One. 2010;5:e9275. doi: 10.1371/journal.pone.0009275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Argaud L, Gateau-Roesch O, Muntean D, et al. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–374. doi: 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 75.Wang G, McCain ML, Yang L, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616–23. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kloner RA, Ganote CE, Jennings RB. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest. 1974;54:1496–508. doi: 10.1172/JCI107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skyschally A, Walter B, Heusch G. Coronary microembolization during early reperfusion: Infarct extension, but protection by ischaemic postconditioning. Eur Heart J. 2013;34:3314–3321. doi: 10.1093/eurheartj/ehs434. [DOI] [PubMed] [Google Scholar]
- 78.Hamirani YS, Wong A, Kramer CM, Salerno M. Effect of microvascular obstruction and intramyocardial hemorrhage by CMR on LV remodeling and outcomes after myocardial infarction: A systematic review and meta-analysis. JACC Cardiovasc Imaging. 2014;7:940–952. doi: 10.1016/j.jcmg.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brosh D, Assali AR, Mager A, et al. Effect of no-reflow during primary percutaneous coronary intervention for acute myocardial infarction on six-month mortality. Am J Cardiol. 2007;99:442–5. doi: 10.1016/j.amjcard.2006.08.054. [DOI] [PubMed] [Google Scholar]
- 80.Engler RL, Schmid-Schönbein GW, Pavelec RS. Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am J Pathol. 1983;111:98–111. [PMC free article] [PubMed] [Google Scholar]
- 81.Ito BR, Schmid-Schönbein G, Engler RL. Effects of leukocyte activation on myocardial vascular resistance. Blood Cells. 1990;16:145-63–6. [PubMed] [Google Scholar]
- 82.Galaup A, Gomez E, Souktani R, et al. Protection against myocardial infarction and no-reflow through preservation of vascular integrity by angiopoietin-like 4. Circulation. 2012;125:140–149. doi: 10.1161/CIRCULATIONAHA.111.049072. [DOI] [PubMed] [Google Scholar]
- 83.Zhang B, Montgomery M, Chamberlain MD, et al. Biodegradable scaffold with built-in vasculature for organ-on-a-chip engineering and direct surgical anastomosis. Nat Mater. 2016 doi: 10.1038/nmat4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiu LLY, Montgomery M, Liang Y, et al. Perfusable branching microvessel bed for vascularization of engineered tissues. Proc Natl Acad Sci U S A. 2012;109:E3414–23. doi: 10.1073/pnas.1210580109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sekine H, Shimizu T, Sakaguchi K, et al. In vitro fabrication of functional three-dimensional tissues with perfusable blood vessels. Nat Commun. 2013;4:1399. doi: 10.1038/ncomms2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferdinandy P, Hausenloy DJ, Heusch G, et al. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev. 2014;66:1142–74. doi: 10.1124/pr.113.008300. [DOI] [PubMed] [Google Scholar]
- 87.Sivaraman V, Hausenloy DJ, Wynne AM, Yellon DM. Preconditioning the diabetic human myocardium. J Cell Mol Med. 2010;14:1740–1746. doi: 10.1111/j.1582-4934.2009.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Szilvassy Z, Ferdinandy P, Szilvassy J, et al. The loss of pacing-induced preconditioning in atherosclerotic rabbits: role of hypercholesterolemia. J Mol Cell Cardiol. 1995;27:2559–69. doi: 10.1006/jmcc.1995.0043. [DOI] [PubMed] [Google Scholar]
- 89.Drawnel FM, Boccardo S, Prummer M, et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014;9:810–820. doi: 10.1016/j.celrep.2014.09.055. [DOI] [PubMed] [Google Scholar]
- 90.Song H, Zandstra PW, Radisic M. Engineered heart tissue model of diabetic myocardium. Tissue Eng Part A. 2011;17:1869–78. doi: 10.1089/ten.TEA.2010.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li H, Liu Z, Wang J, et al. Susceptibility to myocardial ischemia reperfusion injury at early stage of type 1 diabetes in rats. Cardiovasc Diabetol. 2013;12:133. doi: 10.1186/1475-2840-12-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hansen A, Eder A, Bönstrup M, et al. Development of a drug screening platform based on engineered heart tissue. Circ Res. 2010;107:35–44. doi: 10.1161/CIRCRESAHA.109.211458. [DOI] [PubMed] [Google Scholar]
- 93.Hirt MN, Sörensen Na, Bartholdt LM, et al. Increased afterload induces pathological cardiac hypertrophy: a new in vitro model. Basic Res Cardiol. 2012;107:307. doi: 10.1007/s00395-012-0307-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kocsis GF, Pipis J, Fekete V, et al. Lovastatin interferes with the infarct size-limiting effect of ischemic preconditioning and postconditioning in rat hearts. Am J Physiol Heart Circ Physiol. 2008;294:H2406–H2409. doi: 10.1152/ajpheart.00862.2007. [DOI] [PubMed] [Google Scholar]
- 95.Cope DK, Impastato WK, Cohen MV, Downey JM. Volatile anesthetics protect the ischemic rabbit myocardium from infarction. Anesthesiology. 1997;86:699–709. doi: 10.1167/8.5.1.. [DOI] [PubMed] [Google Scholar]
- 96.Kottenberg E, Thielmann M, Bergmann L, et al. Protection by remote ischemic preconditioning during coronary artery bypass graft surgery with isoflurane but not propofol - a clinical trial. Acta Anaesthesiol Scand. 2012;56:30–8. doi: 10.1111/j.1399-6576.2011.02585.x. [DOI] [PubMed] [Google Scholar]
- 97.Tai W, Shi E, Yan L, et al. Diabetes abolishes the cardioprotection induced by sevoflurane postconditioning in the rat heart in vivo: Roles of glycogen synthase kinase-3β and its upstream pathways. J Surg Res. 2012;178:96–104. doi: 10.1016/j.jss.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 98.Fan Y, Yang S, Zhang X, et al. Comparison of cardioprotective efficacy resulting from a combination of atorvastatin and ischaemic post-conditioning in diabetic and non-diabetic rats. Clin Exp Pharmacol Physiol. 2012;39:938–943. doi: 10.1111/1440-1681.12014. [DOI] [PubMed] [Google Scholar]
- 99.Lemoine S, Durand C, Zhu L, et al. Desflurane-induced postconditioning of diabetic human right atrial myocardium in vitro. Diabetes Metab. 2010;36:21–28. doi: 10.1016/j.diabet.2009.06.006. [DOI] [PubMed] [Google Scholar]