

Abstract
The primary hurdle in the path to curing multiple myeloma (MM) is defining a validated minimal residual disease (MRD) and its utility in the therapeutic decision making. A better definition of MRD will aid in tailoring MM therapy further to address the clonal heterogeneity and genomic instability and overcome patient’s ineffective immune surveillance. MRD analysis can define the logical endpoint for maintenance therapy, in addition also aids in providing a better clinical end point for studies comparing novel agents in myeloma. MRD is a surrogate for the survival in MM. Guidelines for global incorporation of MRD in myeloma is fraught with lack of standardization, universal availability and abridged physicians’ understanding of MRD modalities. We aimed at addressing some of the frequently asked questions in the MRD assessment and will also place in perspective some arguments in favor of MRD assessment in routine practice and clinical trial scenario.
Keywords: Multiple myeloma, survival, response, residual disease, flow cytometry, sequencing. imaging
INTRODUCTION
One of the important ongoing debates in multiple myeloma (MM) is the possibility of cure in this disease. [1] There are various obstacles in the path to defining cure in MM, one of which is the lack of validated minimal residual disease (MRD) assessment tool and its incorporation in the therapeutic decision making. The term “minimal residual disease” (MRD) in the context of hematological malignancies refers to the persistence of disease undetectable by standard morphology based diagnostic tests requiring additional sensitive techniques. Defining a cure in MM would first entail demonstration of MRD negativity that is sustained for prolonged periods. Obtaining a sustained MRD negativity will likely entail tailor-made regimens to address the clonal heterogeneity and genomic instability, and overcome patient’s ineffective immune surveillance (e.g., tumor escape, inappropriate dendritic cell function and inhibition of T/Natural Killer(NK) cells). We aim at addressing the first of these two aspects, i.e., the role of MRD assessment in the path to the MM cure. We will address some of the frequently asked questions (FAQs) in the MRD assessment and will also place in perspective some arguments in favor of MRD assessment in routine practice and clinical trial scenario.
Why do we need MRD analysis in myeloma:
The beneficial role of depth of response was first evident in the early 2000s from better long-term outcomes in patients achieving optimal overall response (defined as a partial response (PR) or better).[2, 3] The outcomes in patients achieving complete response (CR) were superior to those with near complete response (nCR) and very good partial response (VGPR), further supporting the relevance of depth of response.[2, 3, 4] But achievement of CR does not mean cure of these patients, as most patients relapse despite achieving CR. The definition of CR was thus suboptimal and required further refinement in the form of additional criteria. Modifications to the existing criteria were first done in 2006 by international myeloma working group (IMWG) with the addition of stringent complete response (sCR: defined as conventional CR plus normal free light chain (FLC) ratio and absence of clonal plasma cells in the bone marrow by immunohistochemistry (IHC)). [5] But implementation of these additional criteria did not uniformly translate into improved relapse rates as was anticipated.[6, 7, 8] To further stratify these patients (those with CR), we required complementary tests to evaluate the presence of MRD within the bone marrow (medullary) with higher sensitivity (>10−4) and outside bone marrow (extramedullary). The proof of concept for implementation of MRD testing comes from studies which compared CR and MRD by multi-parameter flowcytometry (MFC), showing around 30% (range: 15–50%) patients being MRD positive despite being in CR.[9, 10, 11, 12, 13, 14, 15] Thus it was proposed that MRD assessment should be made routinely implemented when assessing therapeutic responses.
Importantly, the therapeutic armamentarium of MM is rapidly evolving with newer drugs leading to prolonged progression-free survival (PFS).[16, 17] Post autologous stem cell transplant (ASCT) consolidation and regimens including novel agent have led to CR rates approaching near 100% thus requiring newer sensitive modalities to differentiate their efficacy. [18]
In addition, assessment of MRD status in early phases of therapy and MRD kinetics may help in identification of chemo-sensitivity providing the best prognostic indication among those currently available. Also, MRD testing has the potential for integration into the therapeutic decision making in the standard of care setting (in newly diagnosed multiple myeloma - NDMM) and to tailor the therapy during maintenance to define the optimal duration of maintenance (avoiding overtreatment). Given these, it is important that MRD analysis be incorporated into various study designs assessing newer drugs and regimens. A proposed study design incorporating MRD and therapeutic decision making has been illustrated in Fig. 1. Finally, MM can still be considered an incurable disease; despite newer agents with improved remission rates, patients still relapse but at different timeframes. Many ongoing clinical trials of newer agents or novel regimens have already incorporated MRD testing for assessing the depth of response as a primary or secondary outcome measure (Suppl Table 1). However, one needs to keep in mind that even the most sensitive assessment may fail to detect residual myeloma cells in a given patient and may not reflect true eradication of the tumor clone. Despite this, MRD negativity is an essential step in the quest for of cure, which we can hopefully achieve at some time point.
Fig. 1: Suggested trial design for the assessing newer drugs/ regimens in the future incorporating MRD analysis.
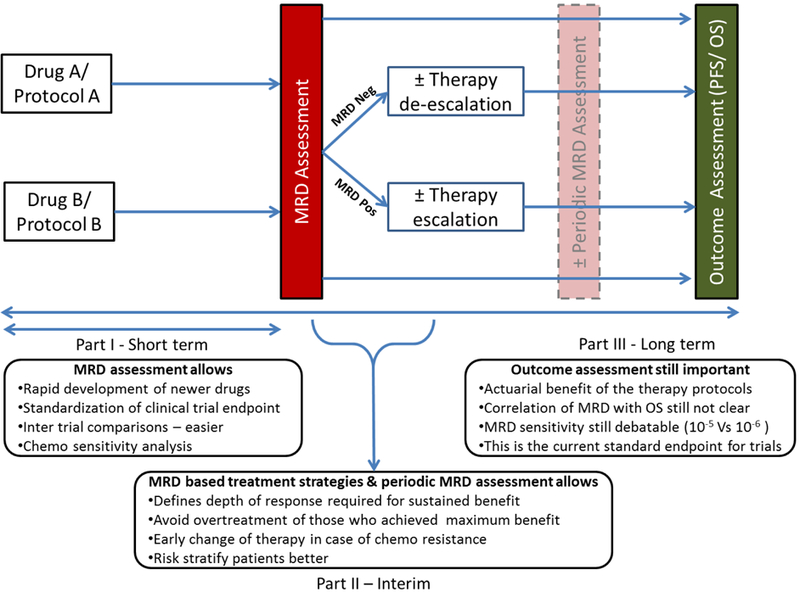
We divided the study protocol into three parts (a) Part I denotes the short-term benefits of the MRD assessment, (b) Part II describes benefits of MRD based treatment strategies and periodic MRD assessment, (c) Part III – Long term evaluation of PFS/ OS and benefits thereof. Investigators can incorporate any of these parts in their study protocol based on the specific end points.
Legend: OS – Overall survival; PFS – Progression free survival; MRD – Minimal residual disease; Neg – Negative; Pos - Positive
WHAT IS AN IDEAL MRD ASSESSMENT TECHNIQUE?
Considering the plausible benefits of the MRD assessment as described above and available literature on improved PFS in patients with MRD negativity, it is important to identify the ideal MRD technique. There are different facets to an ideal MRD marker, and is influenced by disease/ patient characteristics as well as the socio-economic background where MRD is analyzed. Generalizability or ability to use in most patients is important and it has two components. Generalizability is an important attribute, requiring the testing facilities to be available universally considering the different socioeconomic backgrounds, availability of resources and standardization issues of the testing facilities and be applicable to all myeloma patients (considering the heterogeneity in the disease characteristics e.g., oligo-secretory, non-secretory, light chain disease). Further, MRD tests should be highly sensitive (tumor cell detection rate of at least 1 in 105) with high reproducibility, precision, and accuracy. The tests should be validated in multicenter trials. Other hallmarks of ideal assay include excellent feasibility (results obtained in most of the patients), rapid turnaround time and sample requirements which facilitates easy transportation. Currently, none of the MRD techniques used for MM fulfill all of these criteria. With the available techniques, next generation flow (NGF) and next generation sequencing (NGS) are the closest to the ideal MRD techniques.[19]
DEPTH AND DURATION OF MRD: WHAT LEVELS ARE SIGNIFICANT?
Depth of response is associated with better clinical outcomes in patients with MM as discussed earlier.[2, 3, 4] Depth of MRD is more important than the number and type of therapeutic regimens in achieving MRD for predicting disease outcomes, as also reported by the UK Myeloma group.[20, 21, 22] Latest IMWG guidelines suggest the minimal sensitivity of 10−5 or higher for the bone marrow (BM) based MRD testing [19] The real question is the optimal sensitivity for MRD analysis which would be of clinical relevance. Increasing the sensitivity of the MRD assay beyond a certain level can be associated with plausible controversies as illustrated in Fig. 2.
Fig. 2: Depth of MRD and plausible controversies.
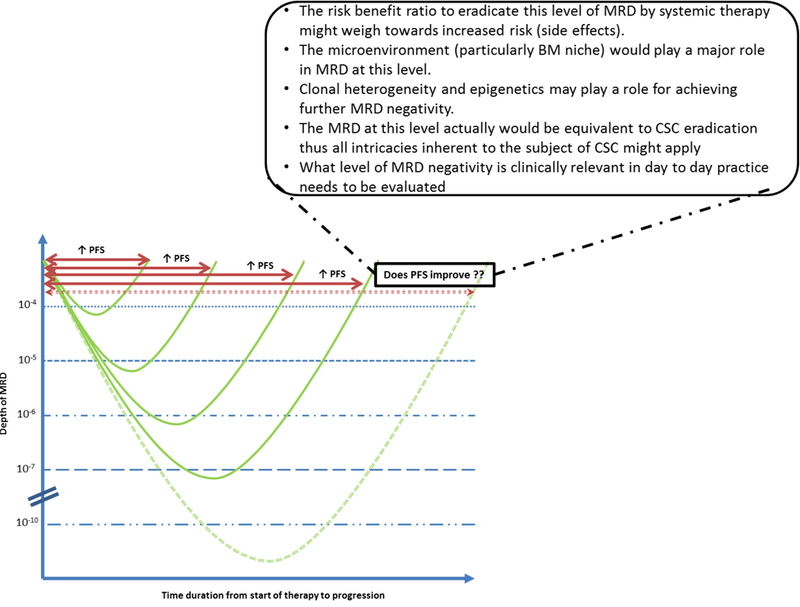
Legend: PFS – Progression free survival; MRD – Minimal residual disease; BM – Bone marrow; CSC – Cancer Stem Cell
The latest IMWG guidelines have also introduced the term sustained MRD negativity for individuals with MRD negativity for more than one year. [19] This is a step towards curing of myeloma, as patients with sustained MRD for prolonged periods can be labeled cured. The ideal duration of MRD negativity to tag it as “sustained” corroborating with near cure of MM needs to be identified in well-designed prospective studies.
MRD ANALYSIS TECHNIQUES
MRD analysis can be done using a multitude of tests which can be classified based on the type of disease detected (medullary or extramedullary disease) or based on the character of tests (bone marrow based testing or imaging). Bone marrow (BM) based testing can be further classified based on whether phenotypic or genotypic features are used for MRD analysis. As reiterated earlier, none of these currently available MRD tests are ideal when performed in isolation, and may have to be performed in combination to complement each other. BM based testing is based on identifying abnormal residual cells on a blind BM biopsy. Heterogeneous/ patchy marrow involvement could potentially give us false negative results. Irrespective of the MRD analysis technique (either cell-based or molecular-based tests) while using BM sample, we are relying on blind biopsies, so we don’t know to what degree that potentially impacts the results. The quality of BM sample is impacted by the peripheral blood dilution and can decrease the sensitivity of the tests. These tests don’t provide any data about the disease at the sanctuary and extramedullary sites.
Imaging by anatomical methods does not help in accurately measuring the tumor load. WBCT is poorest of all modalities in detecting MRD, its use limited to detecting new bone disease, often as a trigger for initiating therapy in patients with suspected myeloma. It can detect extramedullary disease (EMD) as space occupying lesions/ tumorous growths. The lytic bone lesions are a poor marker for follow-up evaluation as the healing lags behind the MRD negativity. MRI is a sensitive method to detect marrow alterations and identify soft tissue masses, but recent chemotherapy, use of G-CSF, presence of inflammation secondary to healing or infections, pathological fractures and use of bone cement can all lead to misinterpretation of MRI in MM. Positron emission tomography (PET) is a functional imaging with good correlation to MRD negativity by BM based testing and PFS.[23] It is the ideal choice for MRD testing in MM patients with evidence of EMD. The absence of standardized methods to report PET in MM is a major drawback. New modalities such as PET-MRI may combine the advantages of both modalities, but data is currently lacking.
MRD by MFC
This technique rests on the concept of detecting phenotypic cell surface markers in differentiating normal from the abnormal plasma cells. The sensitivity of this technique rests on the quality of the BM specimen (can be ensured by the viability of cells above 85% and presence of normal BM constituents i.e., plasma cells, mast cells and B-cell/ myeloid precursors), number of cells analyzed and the antibody panel (it determines the capability of differentiating the normal from the abnormal plasma cells). The literature has variably defined the sensitivity and specificity of this technique due to lack of standardization for the above three components.
Prognostic impact of Flow-MRD:
The prognostic impact of the MFC was studied extensively, with more than 100 patients evaluated for MRD status in GEM2000, GEM2005MENOS65, and MRC IX trials individually.[9, 10, 11, 12, 13, 15, 24] In GEM2000 study on analysis of 125 patients for MRD status, PFS and OS were significantly better in the MRD-negative patients as compared to MRD-positive patients at day 100 after ASCT (71m Vs 37m, p<0.001; median not reached vs 89m; p-0.002 respectively). [9] In MRC IX trial 246 patients were evaluated for the MRD status after intensive chemotherapy with CTD Vs CVAD followed by ASCT. The Median PFS and OS were significantly better in MRD-negative patients than MRD positive irrespective of the induction chemotherapy regimen (28.6m Vs 15.5m, p<0.001; 80.6m Vs 59m; p-0.002 respectively). This trial reiterated the importance of depth of MRD more than the agents required to achieve MRD negativity.[12] In a prospective analysis of 154 patients from GEM2000 and GEM2005MENOS65 trials, the presence of baseline high-risk cytogenetics and persistent MRD positivity at day 100 after ASCT were the only independent predictive markers for unsustained CR. [11, 25]
IMWG 2016 guidelines for Flow MRD:
IMWG suggests the use of MFC techniques with Euroflow standards utilizing eight color combinations in two tubes, identifying clonality by cytoplasmic κ/ λ expression and phenotypically aberrant plasma cell expression. Using individual antibodies can be made further efficient by using lyophilized antibody mixture as it reduces the costs involved, turnaround time and errors. Simultaneous evaluation of the discriminatory markers (CD19, 27, 56, 81, 117) and plasma cell markers (CD38, 45, 138) is necessary. Minimal sensitivity for any of the MFC technique used should be ≥ 1 in 105 nucleated cells. Bone marrow aspirates are the ideal sample for MFC with at least 5 million cells analyzed. Use of bone biopsy washings, or clot reconstitution by dissolving the clot in anticoagulant solutions should be avoided. [19, 26]
Advantages of standard MFC:
The major advantages of flow MRD are the high applicability (>85% of the patients), the rapid turnaround time (<2h) and a relatively simple test with high generalizability. The sensitivity of universally available four or six colors MFC is 10−3 to 10−4. The sensitivity is higher with next generation flow cytometry (NGF), and using two tubes, eight or more color MFC. Also, the normalization and recovery of the normal cells in the compartment can be assessed by MFC which is not possible by the other BM based tests. The greater sensitivity of NGF is primarily due to the use of an optimized combination of fluorochromes and antibody reagents for increased specificity at very low MRD levels, and the 10-fold increase in the number of cells evaluated.
The most important advantage of the flow MRD is the lack of requirement for the patient’s baseline sample, so we can resort to MRD at any stage of therapy and need not be preplanned as in ASO-PCR/NGS. Discrimination between the clonal and normal PCs is possible despite the phenotypic shifts; this attribute allows the MRD assessment in virtually 100% of the patients.
Disadvantages of standard MFC:
All disadvantages of BM based testing as enumerated earlier are applicable to MFC testing as well. Aberrant phenotypes can be missed from detection as no tumor specific antigens are evaluated. The performance of MFC requires fresh samples (preferably <24h). The first and second generation MFCs were less sensitive than the sequencing-based methods, but this has been overcome by NGF.
Lack of standardization is a major problem with MFC. [20, 27, 28] In a survey of 11/26 responders from 30 different institutions specialized in myeloma care, there was huge variability in four studied parameters (number of events, the number of mPCs for MRD positivity and the number of markers studied) with sensitivity varying between 0.01% to 0.0005%.[27] Various efforts have been made to harmonize the results across the world and the best way to overcome this problem is by the implementation of EuroFlow standards.[19, 29, 30, 31, 32] Studies have shown that the standard MFC is inferior to ASO-PCR and NGS particularly at sensitivity levels higher than 10−4.[24, 33] The role of MFC with more than 8 Color (10 or 14 colors) has to be validated in larger studies. Other investigators have recently proposed two tubes 10 color MFC with an aim of standardizing the protocols.[34]
Contemporary issues in MFC:
In 2013, Euro flow consortium had set up a project for increasing the sensitivity and standardization of MFC by next generation flow cytometry (NGF). They use 8–12 color MFC which is highly standardized with automated gating, accurate quantitation, efficient data storage and management with easy data comparison and review. Other advantages are, it is fast (can be completed in 3–4h), with external and internal quality control on a per sample basis and most importantly increased sensitivity in the range of 10−4 to 10−5. But even these techniques are fraught with limitations such as the requirement of fresh samples (<24–48 h) and many cells (e.g., ≥ 5 × 106 for achieving a sensitivity of 10−5). Also, the education and training in NGF are limited across institutions. [35] The NGF should not be confused with standard second generation 8 color MFC, as there are multiple technical differences between them and the sensitivity of the former is much higher than the later.
Use of the single tube versus two tube techniques is another contemporary issue. The need to rapidly process and run two eight-color tubes with the acquisition of 5×106 cells per tube places heavy demands on limited resources, including instrument time, analysis time, sample quality control, and data storage. Single tube MFC aids in rapid turnaround time, the requirement for the smaller sample (fewer acquisitions) and lesser reagents in busy labs with no need for inferential reasoning between tubes. [36] Use of monoclonal antibody therapies in MM can cause difficulties with MFC/ NGF-based MRD analysis (discussed in later part of this article).
MRD BY ASO-PCR
Allele-specific oligonucleotide polymerase chain reaction (ASO-PCR) for clonal PC specific IgH gene rearrangement (particularly VDJ rearrangement) is a form of genotypic/ molecular MRD assay with the capability of detecting up to 1 in 105 plasma cells. It is superior to the previous PCR techniques in providing accurate quantification. This technique requires preparation of patient-specific primers to the VDJ portion of the IgH gene necessitating baseline BM samples.
Korthals et al studied the role of pre-transplant MRD levels by ASO-PCR as an independent prognostic marker for survival outcomes (both EFS and OS).[37] Putkonen et al studied the relevance of post-transplant MRD levels by ASO-PCR at the time of achievement of CR or nCR. A cutoff of 0.01% for ASO-PCR assay 3–6 months after ASCT the difference in the PFS was statistically significant (70m vs. 19m; p-0.003) between the two groups. [38] ASO-PCR was also used to study the significance of post-transplant consolidation with novel therapy for achieving molecular remissions.[39] Martinelli et al studied the molecular remission rate using ASO-PCR in patients undergoing allogeneic stem cell transplant (Allo-HSCT) and ASCT. The patients with molecular remission had significantly lower relapse rate (41% vs. 16%; p < 0.05). [40]
Advantages of ASO-PCR:
It is more sensitive and specific than traditional MFC.[24, 41] It requires a smaller sample (<1 × 106 cells) for testing. Sample processing can be delayed, because testing can be done both on fresh and stored samples, unlike MFC.
Disadvantages of MRD by ASO-PCR:
The requirement of the baseline BM samples is another major disadvantage using this technique. This limits the applicability to only patients with preplanned MRD analysis. The presence of multiple somatic mutations in Ig genes can cause suboptimal performance due to the requirement of patient-specific primers/ probes. Patients with minimal plasma cell infiltration at baseline can have technical difficulties in obtaining these extra probes/ primers. It is applicable to only 60–70% patients for the above-mentioned difficulties. It is more time consuming than the MFC testing as it is labor intensive and can take several days in preparing patient-specific probes, thus also increasing the turnaround time. Internal quality control of the sample is not possible and requires additional tests. EuroMRD is yet to standardize ASO-PCR for MM yet. [14]
Use of CD138 selection can overcome the suboptimal performance of the ASO-PCR by increasing identification of the target VDJ rearrangements in this group. Use of droplet digital PCR for assessing rearrangements can increase the applicability and decrease the labor.[42]
MRD BY NGS
Sequencing-based MRD testing is a genotypic BM based testing technique. It can further be of two varieties, high throughput VDJ sequencing and high throughput exome sequencing. The difference between the two is that VDJ sequencing is only focusing on VDJ, whereas in exome sequencing there is no particular restriction on focus, so it includes the whole exome. Only VDJ sequencing technique has been studied in clinical trials for the benefit associated with MRD negativity. So our discussion primarily would focus on VDJ sequencing when referring to NGS. LymphoSIGHT platform (Sequenta Inc, USA) was used for NGS (VDJ sequencing) in most clinical trials as per the published literature.[33]
Martinez et al studied the role of NGS in MRD detection in both transplant ineligible and post ASCT settings. MRD positivity at a sensitivity level of 1 in 106 was seen in 73% of the individuals. This study also elucidated a good agreement between MFC, ASO-PCR, and NGS.[33] In IFM 2009 trial, patients were evaluated for MRD using both MFC and NGS. Patients with MRD negativity had a significantly higher PFS as compared to the patients with MRD positivity (87% vs. 42% pre-maintenance, 83% vs. 30% post maintenance).[21]
IMWG 2016 guidelines for MRD by NGS:
IMWG criteria suggests NGS on BM aspirates using 3 primers (IgH for VDJH, IgH for DJH, Ig kappa) and use of LymphoSIGHT (or any other equivalent validated) platform for interpreting results by NGS with a minimum sensitivity of 10−5. The MRD negativity is defined as less than two identical sequencing reads.[19]
Advantages of MRD by NGS:
NGS has universal applicability as with MFC and with sensitivity up to 10−6. It can also be done from peripheral blood, though with low sensitivity, likely related to dilutional effects and concentration of myeloma cells in the marrow. One major advantage of this technique over any other MRD assay is the ability to detect clonal evolution (best with genome/ exome sequencing and limited with VDJ sequencing). There is no requirement for the fresh sample; stored samples can be used for the assays.
Disadvantages of MRD by NGS:
The major disadvantage is the large turnaround time (around one week with VDJ sequencing and many weeks with exome/ genome sequencing). Baseline samples are still required for identification of dominant clonotype (alternatively a stored sample when the disease was detectable can be used as reference standard).
MRD by PET/CT
Positron emission tomography (PET) is a functional imaging modality based on the increased fluoro-deoxy-glucose (FDG) uptake by the malignant cells enabling tumor metabolic activity assessment. An addition of low-dose CT aids in screening associated bone disease and also in supplementing anatomical details to the functional imaging. Patients with documented remission on PET/CT after ASCT were associated with superior outcomes compared to patients with persisting PET positivity (PFS: 47% vs 32%, p-0.017; OS: 79% Vs 66%, p-0.018). Stratification for survival outcomes by PET/CT was superior to conventional serological based CR.[43] Caldarella C et al showed usefulness of PET/CT in monitoring response to treatment and evaluation of possible sites of recurrent or progressive disease.[44, 45] Prospective IMAJEM study (part of IFM/DFCI 2009 trial) has proven the superiority of PET/CT to MRI in the prognosticating and monitoring of MM patients.[23, 46] PET/CT had good correlation with MFC in the prospective PIPET trial.[47]
IMWG 2016 guidelines for MRD by PET/CT:
The imaging positive, MRD-negative group has been added to the MM response criteria for the first time by IMWG in 2016.[19] It is imperative to have MRD negativity by NGF or NGS before applying these imaging criteria. MRD negativity by imaging requires disappearance of increased metabolic activity found at baseline or a prior PET/CT or decrease to SUV less than mediastinal blood pool or decrease to less than that of surrounding normal tissue. A modified Deauville’s criteria or those used by Zamagni et al can be used for reporting PET [48, 49, 50] Sum of perpendicular diameters (SPD) should be used while monitoring tumor size on PET (except for those with skin lesions, where physical measurement of the lesion by a ruler can substitute for SPD). Positive lesions were recognized by the presence of focal areas of increased SUV within bones on at least two consecutive slices, without a compulsory requirement of the underlying lesion on CT. SUV cut-offs have been defined for osteolytic CT lesions.
Advantages of PET/CT:
PET/CT is crucial for detection of the EMD in patients with MM as none of the current MRD techniques fail to detect the same. EMD can be present in 10% of the patients at diagnosis and in a higher proportion of patients at relapse. Presence of EMD at baseline is associated with poor prognosis (both PFS and OS).[43] PET can be of great help in non-secretory, oligo-secretory and occult MM as these patients can be falsely labelled to be in CR.[51, 52, 53] Simultaneous evaluation of the marrow avidity can provide limited information on the medullary disease status as demonstrated by Sager S et al.[54] Thus, patients can be spared from repeated invasive BM aspiration procedures if PET negative as in Hodgkin’s lymphoma, but this approach needs to be validated. There are contemplations on its benefits in concurrently ruling out any infections and secondary malignancies.
Disadvantages of PET/CT:
Radiation exposure, cost, and availability are the major issues with the routine use of PET/CT in monitoring MM. The exact frequency of PET for MRD in MM is not defined. Lack of standardization in reporting is another drawback in the routine use of PET/CT. The Italian group has recently proposed a Deauville based scoring system, though it has not been clinically validated.[50] A similar scoring system as part of PIPET trial was validated in patients undergoing pre-transplant PET/CT in 64 patients which was presented recently.[55] Though there is no compulsory requirement for the PET scan at diagnosis but it is necessary to have a previous PET at some stage of disease for comparison.
Contemporary issues in PET/CT:
Although the prognostic value of 18FDG-PET has been well documented, more specific tracers addressing hallmarks of myeloma biology, e.g. paraprotein biosynthesis, are needed. A study by Lückerath K et al evaluated the amino acid tracers L-methyl-[11C]-methionine (11C-MET) and [18F]-fluoroethyl-L-tyrosine ((18F-FET) for their potential to image myeloma and to characterize tumor heterogeneity.[56] Using myeloma cell lines and patient-derived CD138⁺ PCs, the relative uptake of 18C-MET and 18F-FET exceeded that of 18F-FDG. Importantly, 11C-MET uptake significantly differed between indolent PCs and those with worse prognosis (e.g. t(4;14)). These preclinical data led to the use of 18C-MET in a prospective cohort of 43 patients and compared to routine functional imaging with 18FDG. MET scanning appeared to be superior to 18FDG for evaluating both intramedullary and extramedullary lesions post therapy (re-staging).[57]
MRI
The role of MRI in detecting bone lesions for diagnosing MM was endorsed by the IMWG consensus criteria in 2014.[58] This was of particular interest in relabeling high-risk patients with SMM as MM.[59] With the increasing interest in MRI (both WB-MRI and limited MRI of the spine), studies were conducted to study its relevance for MRD in MM. [46, 60] MRI was found to be of clinical value at diagnosis, but its relevance in patients on therapy for monitoring MRD was not statistically significant.[46, 60] In IMAJEM study (a sub study of IFM2009), MRI was compared to PET/CT with primary endpoint being comparison of number of bone lesions at diagnosis and secondary endpoint being determination of the prognostic impact after 3 cycles of induction therapy and before maintenance (correlation of PFS/OS and PET or MRI negativity). MRI of spine and pelvis and whole-body PET/CT were equally effective in determining bone involvement at diagnosis (McNemar test = 0.94; P = 0.33). MRI had no impact on PFS or OS during or end of therapy. PET/CT negativity after 3 cycles of chemotherapy and before maintenance was prognostic for PFS, whereas PET/CT negativity before maintenance is prognostic for OS.44
Comparison of MRD techniques:
As there is no gold standard MRD analysis technique, the comparisons to assess the sensitivity and specificity of the techniques are very difficult. These comparisons are not meant to judge but for providing insight for the investigators into these MRD techniques. Suppl Table 2 illustrates a comparison of some these available techniques.
TIMING OF MRD ASSESMENT
It is important for bedside practice to define the ideal frequency of MRD testing, i.e., after how many cycles of chemotherapy, role in pre-and post-transplant settings and frequency during maintenance therapy. Though we don’t have literature to dictate the exact frequency of MRD monitoring, but there are few time points which can be deduced based on the previous literature for remission/ response assessment. These time points are at the first documentation of CR (to assess the accuracy of remission and to evaluate the depth of response), after fixed number of chemotherapy cycles (can be investigator-driven in the clinical trial), prior to ASCT (based on the data on the prognostic role of MRD negativity prior to transplant), post ASCT at day 100 (this the time point where disease evaluation is done for the response to HDT and ASCT and most of the literature on prognostic impact of MRD is based on this time point) and at periodic intervals among patients on maintenance therapy post ASCT to detect emerging MRD negativity or to assess duration of response. It is strongly recommended that more reliance is placed on the MRD kinetics than MRD analysis at any single time point, as MRD kinetics may discriminate chemosensitive vs. chemoresistant patients.[12, 41, 61]
Relevance of MRD in prognosticating patients with MM was most studied in the setting of the autologous transplantation particularly at day 100 post-ASCT.[4, 7, 9, 13, 43] IFM-DFCI has studied the MRD in the setting of early and delayed transplant. In the IFM arm of the study, patients receiving upfront ASCT had a higher number of patients with MRD negativity. [21, 46] Galimberti et al studied compared the MRD status post non-myeloablative Allo-HSCT and post ASCT among MM patients by PCR-based testing. A higher percentage of patients achieved MRD negativity in the Allo-HSCT arm (60% Vs 15%). MRD negative status was also associated 2y OS.[62, 63] A study by Martinelli et al also suggested similar results of higher molecular remissions using ASO-PCR in patients undergoing Allo-HSCT vis-à-vis ASCT (p<0.01).[40] Many trials are currently underway on the role of MRD status in setting of Allo-HSCT (Suppl Table 1).
NOVEL AGENTS AND THE MRD ANALYSIS
In patients receiving immunotherapy, there can be persistence of minimal quantities of monoclonal protein even after effective therapy, causing difficulty in labelling remission status based on the current criteria. The use of MRD analysis can clearly delineate patients with response to therapy.
Immunotherapy and MRD by MFC Analysis:
The use of monoclonal antibodies such as anti CD38 (e.g., Daratumumab) and anti CD138 (e.g., Indatuximab Ravtansine) in MM can cause interference with the protein electrophoresis (when monoclonal antibody shares the same isotype with the monoclonal protein) and MFC analysis (as MFC panel uses these cell surface markers for the categorizing clonal PCs). Use of mass spectrometry based testing and anti-idiotype antibodies that bind the offending drug can mitigate the difficulties with electrophoresis. Use of other markers such as CD54, 229, 319 and VS38c can help in clonal plasma cell recognition in such patients. These monoclonal antibodies do not interfere with the NGS, thus making it the ideal test in these scenarios. [64, 65, 66]
Identifying ideal induction remission protocol in the era of novel agents:
Previously studies have used endpoints such as PFS and OS in identifying better regimens/ protocols employing newer agents. There are inherent disadvantages with the use of these end points including (a) The study has to be carried for a prolonged period before identifying the efficacy of the agents. (b) The lack of standardization in identifying these end points. (c) Difficult to compare across trials. The use of MRD-based endpoints in these remission induction trials can partly overcome the above-mentioned disadvantages and also have the advantages of better objectivity, early detection of insensitivity and relapse allowing prompt changes and avoiding patients from getting suboptimal therapy.
APPLICABILITY TO REAL WORLD SETTING
Most of the techniques in the current IMWG recommendation are either not available or unaffordable even if present in the real-world settings, especially in the developing countries. It is imperative for the scientific community to come up with alternative recommendations delineating what’s optional, optimal and absolutely necessary in MRD analysis in resource constraints. The industry should try to make accessible to the developing countries with equipment for MRD analysis at economically viable prices. The scientists should be encouraged for innovations and studies which can satisfy the local needs (e.g., combination of cheaper first generation MRD techniques whose results can equate to a recommended higher generation procedure).
RECENT ADVANCES
Peripheral blood-based testing:
One of the major disadvantages of BM based MRD analysis is the requirement of invasive BM aspiration procedure. Clonotypic target tryptic peptides are a serum based analysis of Ig light chains by mass spectrometry which is patient specific (peptides prepared at baseline/diagnosis by digestion of abundant M protein from individual patients) and used thereafter to monitor MRD. [67, 68, 69] This method has higher sensitivity than BM-based methodologies, but the clinical relevance and validity across institutions needs evaluation. The potential benefits of this assay are being less invasive; a serum-based test capable of detecting very low concentrations of disease burden and potential to detect patchy or focal disease that BM based tests might miss. The disadvantages are the necessity of the baseline sample as in genotypic BM methods. [69, 70] Korthals et al studied the role of MRD in peripheral blood by quantitative real-time IgH-PCR (IgH-qPCR). The MRD levels of the peripheral blood PCR was 40 fold lower than the mean MRD by BM based PCR. They found no correlation with serological remissions, but the patients with MRD positivity had significantly lower PFS. [71] Ongoing studies are evaluating the role of “liquid biopsies”, based on circulating tumor DNA in the peripheral blood as sensitive MRD markers.[72] MicroRNA (miRNA) has been studied in the setting of myeloma, particularly from disease biology standpoint, with its role in detecting MRD unclear at this point.[73]
Hevylite assays:
Hevylite assay enables quantitation of the specific pairs of the heavy/ light chains by developing antibodies directed to the conjunction epitopes between light and heavy chain. This enables detection of both involved and the polyclonal non-involved pair. It is of great relevance in oligo-secretory MM and MRD monitoring. They have a distinct additional advantage of assessing functional reconstitution of the normal B-cells and plasma cells during immune recovery other than the quantitation of MRD. There are limited studies on the prognostic impact of the Hevylite assay as a MRD marker in MM.[74, 75]
Genomic instability and phenotypic/ genotypic shifts of MRD cells:
The question of “whether the MRD plasma cells (PCs) are unique or are they identical to the PCs at diagnosis” needs to be answered. Selection of the clonal PCs occurs at the MRD levels. The differences in the PCs at these time points can be assessed by the phenotype, genotype, and gene expression profiling.[76] The impact of therapy and relapse in selecting a specific clone from the clonally heterogeneous population can explain these differences.[77] Phenotypically the MRD cells are different from the clonal PCs at diagnosis, but on analysis of independent surface CD markers, there can be overlapping features. This can be explained by selection of a different major molecular clone which would further be responsible for the relapses.[78] Paino T et al demonstrated that these different phenotypic clones have distinct cytogenetic profiles.[41] The immunophenotypic protein expression profiles of the MRD cells have a shift from the diagnostic clonal PCs.[79, 80] The phenotypic shifts in diagnostic-PCs to MRD-PCs are not merely immaturity shifts (more immaturity markers) as seen in BM-PCs to peripheral-PCs.[41] These shifts are related to a different genetic background in MRD-PCs compared to diagnostic-PCs by the acquisition of new chromosomal abnormalities secondary to linear evolution or branching evolution leading to genomic instability. These genetic shifts are clinically relevant as proven by Paiva et al, that MRD-positive patients with high-risk cytogenetic profile have inferior survival or outcomes.[11]
CONCLUSION AND FUTURE DIRECTIONS
The MRD analysis in patients with MM is a step forward towards the cure of this disease. A better definition of MRD, standardization of the available techniques and implementation of these in the routine clinical practice through universal availability can aid in tailoring the MM therapy further. Most characteristics of the MRD analysis discussed till now are based on limited literature, with the lack of larger studies. There are many questions requiring further exactitude or remain unanswered. These are potential areas of research needing to be addressed in large prospective trials designed specifically to answer these.
Supplementary Material
Footnotes
Disclosure of conflicts of Interest: Uday Yanamandra declares no conflicts. Shaji Kumar: Celgene (Consultancy and Research Funding), Millennium (Consultancy and Research Funding), Novartis (Research Funding), Onyx (Consultancy and Research Funding), AbbVie (Research Funding), Janssen (Consultancy and Research Funding), BMS (Consultancy and Research Funding).
BIBLIOGRAPHY
- 1.Barlogie B, Mitchell A, van Rhee F, Epstein J, Morgan GJ, Crowley J. Curing myeloma at last: defining criteria and providing the evidence. Blood. 2014;124:3043–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gay F, Larocca A, Wijermans P, Cavallo F, Rossi D, Schaafsma R, Genuardi M, Romano A, Liberati AM, Siniscalchi A, Petrucci MT, Nozzoli C, Patriarca F, Offidani M, Ria R, Omede P, Bruno B, Passera R, Musto P, Boccadoro M, Sonneveld P, Palumbo A. Complete response correlates with long-term progression-free and overall survival in elderly myeloma treated with novel agents: analysis of 1175 patients. Blood. 2011;117:3025–31. Epub 2011/01/14. [DOI] [PubMed] [Google Scholar]
- 3.van de Velde HJ, Liu X, Chen G, Cakana A, Deraedt W, Bayssas M. Complete response correlates with long-term survival and progression-free survival in high-dose therapy in multiple myeloma. Haematologica. 2007;92:1399–406. Epub 2007/11/21. [DOI] [PubMed] [Google Scholar]
- 4.Lahuerta JJ, Mateos MV, Martinez-Lopez J, Rosinol L, Sureda A, de la Rubia J, Garcia-Larana J, Martinez-Martinez R, Hernandez-Garcia MT, Carrera D, Besalduch J, de Arriba F, Ribera JM, Escoda L, Hernandez-Ruiz B, Garcia-Frade J, Rivas-Gonzalez C, Alegre A, Blade J, San Miguel JF. Influence of pre- and post-transplantation responses on outcome of patients with multiple myeloma: sequential improvement of response and achievement of complete response are associated with longer survival. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:5775–82. Epub 2008/11/13. [DOI] [PubMed] [Google Scholar]
- 5.Durie BG, Harousseau JL, Miguel JS, Blade J, Barlogie B, Anderson K, Gertz M, Dimopoulos M, Westin J, Sonneveld P, Ludwig H, Gahrton G, Beksac M, Crowley J, Belch A, Boccadaro M, Cavo M, Turesson I, Joshua D, Vesole D, Kyle R, Alexanian R, Tricot G, Attal M, Merlini G, Powles R, Richardson P, Shimizu K, Tosi P, Morgan G, Rajkumar SV. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467–73. Epub 2006/07/21. [DOI] [PubMed] [Google Scholar]
- 6.Martinez-Lopez J, Paiva B, Lopez-Anglada L, Mateos MV, Cedena T, Vidriales MB, Saez-Gomez MA, Contreras T, Oriol A, Rapado I, Teruel AI, Cordon L, Blanchard MJ, Bengoechea E, Palomera L, de Arriba F, Cueto-Felgueroso C, Orfao A, Blade J, San Miguel JF, Lahuerta JJ. Critical analysis of the stringent complete response in multiple myeloma: contribution of sFLC and bone marrow clonality. Blood. 2015;126:858–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapoor P, Kumar SK, Dispenzieri A, Lacy MQ, Buadi F, Dingli D, Russell SJ, Hayman SR, Witzig TE, Lust JA, Leung N, Lin Y, Zeldenrust SR, McCurdy A, Greipp PR, Kyle RA, Rajkumar SV, Gertz MA. Importance of achieving stringent complete response after autologous stem-cell transplantation in multiple myeloma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:4529–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radocha J, Pour L, Pika T, Maisnar V, Spicka I, Gregora E, Krejci M, Minarik J, Machalkova K, Straub J, Pavlicek P, Hajek R, Zak P. Multicentered patient-based evidence of the role of free light chain ratio normalization in multiple myeloma disease relapse. Eur J Haematol. 2016;96:119–27. [DOI] [PubMed] [Google Scholar]
- 9.Paiva B, Vidriales MB, Cervero J, Mateo G, Perez JJ, Montalban MA, Sureda A, Montejano L, Gutierrez NC, Garcia de Coca A, de Las Heras N, Mateos MV, Lopez-Berges MC, Garcia-Boyero R, Galende J, Hernandez J, Palomera L, Carrera D, Martinez R, de la Rubia J, Martin A, Blade J, Lahuerta JJ, Orfao A, San Miguel JF, Groups GPCS. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood. 2008;112:4017–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paiva B, Martinez-Lopez J, Vidriales MB, Mateos MV, Montalban MA, Fernandez-Redondo E, Alonso L, Oriol A, Teruel AI, de Paz R, Larana JG, Bengoechea E, Martin A, Mediavilla JD, Palomera L, de Arriba F, Blade J, Orfao A, Lahuerta JJ, San Miguel JF. Comparison of immunofixation, serum free light chain, and immunophenotyping for response evaluation and prognostication in multiple myeloma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1627–33. Epub 2011/03/16. [DOI] [PubMed] [Google Scholar]
- 11.Paiva B, Gutierrez NC, Rosinol L, Vidriales MB, Montalban MA, Martinez-Lopez J, Mateos MV, Cibeira MT, Cordon L, Oriol A, Terol MJ, Echeveste MA, de Paz R, de Arriba F, Palomera L, de la Rubia J, Diaz-Mediavilla J, Sureda A, Gorosquieta A, Alegre A, Martin A, Hernandez MT, Lahuerta JJ, Blade J, San Miguel JF, Groups PGCS. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood. 2012;119:687–91. Epub 2011/12/01. [DOI] [PubMed] [Google Scholar]
- 12.Rawstron AC, Child JA, de Tute RM, Davies FE, Gregory WM, Bell SE, Szubert AJ, Navarro-Coy N, Drayson MT, Feyler S, Ross FM, Cook G, Jackson GH, Morgan GJ, Owen RG. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:2540–7. Epub 2013/06/05. [DOI] [PubMed] [Google Scholar]
- 13.Roussel M, Lauwers-Cances V, Robillard N, Hulin C, Leleu X, Benboubker L, Marit G, Moreau P, Pegourie B, Caillot D, Fruchart C, Stoppa AM, Gentil C, Wuilleme S, Huynh A, Hebraud B, Corre J, Chretien ML, Facon T, Avet-Loiseau H, Attal M. Front-line transplantation program with lenalidomide, bortezomib, and dexamethasone combination as induction and consolidation followed by lenalidomide maintenance in patients with multiple myeloma: a phase II study by the Intergroupe Francophone du Myelome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:2712–7. Epub 2014/07/16. [DOI] [PubMed] [Google Scholar]
- 14.Rawstron AC, Paiva B, Stetler-Stevenson M. Assessment of minimal residual disease in myeloma and the need for a consensus approach. Cytometry B Clin Cytom. 2016;90:21–5. Epub 2015/07/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarasquete ME, Garcia-Sanz R, Gonzalez D, Martinez J, Mateo G, Martinez P, Ribera JM, Hernandez JM, Lahuerta JJ, Orfao A, Gonzalez M, San Miguel JF. Minimal residual disease monitoring in multiple myeloma: a comparison between allelic-specific oligonucleotide real-time quantitative polymerase chain reaction and flow cytometry. Haematologica. 2005;90:1365–72. Epub 2005/10/13. [PubMed] [Google Scholar]
- 16.Dhakal B, Girnius S, Hari P. Recent advances in understanding multiple myeloma. F1000Res. 1000;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonsalves WI, Milani P, Derudas D, Buadi FK. The next generation of novel therapies for the management of relapsed multiple myeloma. Future Oncol. 2016;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paiva B, Puig N, García-Sanz R, San Miguel JF. Is this the time to introduce minimal residual disease in multiple myeloma clinical practice? Clinical Cancer Research. 2015;21:2001–8. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, Munshi N, Lonial S, Blade J, Mateos MV, Dimopoulos M, Kastritis E, Boccadoro M, Orlowski R, Goldschmidt H, Spencer A, Hou J, Chng WJ, Usmani SZ, Zamagni E, Shimizu K, Jagannath S, Johnsen HE, Terpos E, Reiman A, Kyle RA, Sonneveld P, Richardson PG, McCarthy P, Ludwig H, Chen W, Cavo M, Harousseau JL, Lentzsch S, Hillengass J, Palumbo A, Orfao A, Rajkumar SV, San Miguel J, Avet-Loiseau H. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. [DOI] [PubMed] [Google Scholar]
- 20.Mailankody S, Korde N, Lesokhin AM, Lendvai N, Hassoun H, Stetler-Stevenson M, Landgren O. Minimal residual disease in multiple myeloma: bringing the bench to the bedside. Nat Rev Clin Oncol. 2015;12:286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avet-Loiseau H, Corre J, Lauwers-Cances V, Chretien M-L, Robillard N, Leleu X, Hulin C, Gentil C, Arnulf B, Belhadj K, Brechignac S, Garderet L, Karlin L, Marit G, Benboubker L, Orsini-Piocelle F, Royer B, Drenou B, Tiab M, Lamy T, MACRO M, Richardson PG, Anderson KC, Faham M, Facon T, Moreau P, Attal M, Munshi NC. Evaluation of Minimal Residual Disease (MRD) By Next Generation Sequencing (NGS) Is Highly Predictive of Progression Free Survival in the IFM/DFCI 2009 Trial. Blood. 2015;126:191-.26160184 [Google Scholar]
- 22.Landgren O, Owen RG. Better therapy requires better response evaluation: Paving the way for minimal residual disease testing for every myeloma patient. Cytometry B Clin Cytom. 2016;90:14–20. Epub 2015/07/07. [DOI] [PubMed] [Google Scholar]
- 23.Moreau P, Attal M, Caillot D, Macro M, Karlin L, Garderet L, Facon T, Benboubker L, Escoffre-Barbe M, Stoppa AM, Laribi K, Hulin C, Perrot A, Marit G, Eveillard JR, Caillon F, Bodet-Milin C, Pegourie B, Dorvaux V, Chaleteix C, Anderson K, Richardson P, Munshi NC, Avet-Loiseau H, Gaultier A, Nguyen JM, Dupas B, Frampas E, Kraeber-Bodere F. Prospective Evaluation of Magnetic Resonance Imaging and [18F]Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography at Diagnosis and Before Maintenance Therapy in Symptomatic Patients With Multiple Myeloma Included in the IFM/DFCI 2009 Trial: Results of the IMAJEM Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017:Jco2017722975. Epub 2017/07/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puig N, Sarasquete ME, Balanzategui A, Martinez J, Paiva B, Garcia H, Fumero S, Jimenez C, Alcoceba M, Chillon MC, Sebastian E, Marin L, Montalban MA, Mateos MV, Oriol A, Palomera L, de la Rubia J, Vidriales MB, Blade J, Lahuerta JJ, Gonzalez M, Miguel JF, Garcia-Sanz R. Critical evaluation of ASO RQ-PCR for minimal residual disease evaluation in multiple myeloma. A comparative analysis with flow cytometry. Leukemia. 2014;28:391–7. Epub 2013/07/19. [DOI] [PubMed] [Google Scholar]
- 25.Lahuerta J-J, Paiva B, Vidriales M-B, Cordón L, Cedena M- T, Puig N, Martinez-Lopez J, Rosiñol L, Gutierrez NC, Martín-Ramos M- L, Oriol A, Teruel A- I, Echeveste M- A, Paz Rd, Arriba Fd, Hernandez MT, Palomera L, Martinez R, Martin A, Alegre A, Rubia JDl, Orfao A, Mateos M-V, Blade J, San-Miguel JF, Group obotGPCS. Depth of Response in Multiple Myeloma: A Pooled Analysis of Three PETHEMA/GEM Clinical Trials. Journal of Clinical Oncology.0:JCO.201669517. [Google Scholar]
- 26.Flores-Montero J, de Tute R, Paiva B, Perez JJ, Bottcher S, Wind H, Sanoja L, Puig N, Lecrevisse Q, Vidriales MB, van Dongen JJ, Orfao A. Immunophenotype of normal vs. myeloma plasma cells: Toward antibody panel specifications for MRD detection in multiple myeloma. Cytometry B Clin Cytom. 2016;90:61–72. Epub 2015/06/24. [DOI] [PubMed] [Google Scholar]
- 27.Flanders A, Stetler-Stevenson M, Landgren O, Mailankody S, Korde N, Lesokhin AM, Lendvai N, Hassoun H, Keeney M, Halley JG, Rhoads DD, Ansari MQ, Kussick SJ, Karlon WJ, Mehta KU, Dorfman DM, Linden MA, Arroz M, Came N, Lin P, Chen W, Yuan C, Lagoo A, Monreal M, de Tute R, Vergilio JA, Rawstron AC, Paiva B. Minimal residual disease testing in multiple myeloma by flow cytometry: major heterogeneity Minimal residual disease in multiple myeloma: bringing the bench to the bedside Marked Variability in Reported Minimal Residual Disease Lower Level of Detection of 4 Hematolymphoid Neoplasms: A Survey of Participants in the College of American Pathologists Flow Cytometry Proficiency Testing Program Consensus guidelines on plasma cell myeloma minimal residual disease analysis and reporting. [Google Scholar]
- 28.Keeney M, Halley JG, Rhoads DD, Ansari MQ, Kussick SJ, Karlon WJ, Mehta KU, Dorfman DM, Linden MA. Marked Variability in Reported Minimal Residual Disease Lower Level of Detection of 4 Hematolymphoid Neoplasms: A Survey of Participants in the College of American Pathologists Flow Cytometry Proficiency Testing Program. Arch Pathol Lab Med. 2015;139:1276–80. Epub 2015/02/20. [DOI] [PubMed] [Google Scholar]
- 29.Arroz M, Came N, Lin P, Chen W, Yuan C, Lagoo A, Monreal M, de Tute R, Vergilio JA, Rawstron AC, Paiva B. Consensus guidelines on plasma cell myeloma minimal residual disease analysis and reporting. Cytometry B Clin Cytom. 2016;90:31–9. Epub 2015/01/27. [DOI] [PubMed] [Google Scholar]
- 30.Flores-Montero J, de Tute R, Paiva B, Perez JJ, Bottcher S, Wind H, Sanoja L, Puig N, Lecrevisse Q, Vidriales MB, van Dongen JJ, Orfao A. Immunophenotype of normal vs. myeloma plasma cells: Toward antibody panel specifications for MRD detection in multiple myeloma. Cytometry Part B, Clinical cytometry. 2016;90:61–72. Epub 2015/06/24. [DOI] [PubMed] [Google Scholar]
- 31.Pojero F, Flores-Montero J, Sanoja L, Perez JJ, Puig N, Paiva B, Bottcher S, van Dongen JJ, Orfao A. Utility of CD54, CD229, and CD319 for the identification of plasma cells in patients with clonal plasma cell diseases. Cytometry B Clin Cytom. 2016;90:91–100. Epub 2015/07/02. [DOI] [PubMed] [Google Scholar]
- 32.Rawstron AC, Paiva B, Stetler-Stevenson M. Assessment of minimal residual disease in myeloma and the need for a consensus approach. Cytometry Part B, Clinical cytometry. 2016;90:21–5. Epub 2015/07/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Lopez J, Lahuerta JJ, Pepin F, Gonzalez M, Barrio S, Ayala R, Puig N, Montalban MA, Paiva B, Weng L, Jimenez C, Sopena M, Moorhead M, Cedena T, Rapado I, Mateos MV, Rosinol L, Oriol A, Blanchard MJ, Martinez R, Blade J, San Miguel J, Faham M, Garcia-Sanz R. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. 2014;123:3073–9. Epub 2014/03/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landgren O, Gormley N, Turley D, Owen RG, Rawstron A, Paiva B, Barnett D, Arroz M, Wallace P, Durie B, Yuan C, Dogan A, Stetler-Stevenson M, Marti GE. Flow cytometry detection of minimal residual disease in multiple myeloma: Lessons learned at FDA-NCI roundtable symposium. American journal of hematology. 2014;89:1159–60. Epub 2014/08/19. [DOI] [PubMed] [Google Scholar]
- 35.van Dongen JJ, van der Velden VH, Bruggemann M, Orfao A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125:3996–4009. Epub 2015/05/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Royston DJ, Gao Q, Nguyen N, Maslak P, Dogan A, Roshal M. Single-Tube 10-Fluorochrome Analysis for Efficient Flow Cytometric Evaluation of Minimal Residual Disease in Plasma Cell Myeloma. American journal of clinical pathology. 2016;146:41–9. Epub 2016/07/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korthals M, Sehnke N, Kronenwett R, Bruns I, Mau J, Zohren F, Haas R, Kobbe G, Fenk. The level of minimal residual disease in the bone marrow of patients with multiple myeloma before high-dose therapy and autologous blood stem cell transplantation is an independent predictive parameter. [DOI] [PubMed] [Google Scholar]
- 38.Putkonen M, Kairisto V, Juvonen V, Pelliniemi TT, Rauhala A, Itala-Remes M, Remes K. Depth of response assessed by quantitative ASO-PCR predicts the outcome after stem cell transplantation in multiple myeloma. [DOI] [PubMed] [Google Scholar]
- 39.Ladetto M, Pagliano G, Ferrero S, Cavallo F, Drandi D, Santo L, Crippa C, De Rosa L, Pregno P, Grasso M, Liberati AM, Caravita T, Pisani F, Guglielmelli T, Callea V, Musto P, Cangialosi C, Passera R, Boccadoro M, Palumbo A. Major tumor shrinking and persistent molecular remissions after consolidation with bortezomib, thalidomide, and dexamethasone in patients with autografted myeloma. [DOI] [PubMed] [Google Scholar]
- 40.Martinelli G, Terragna C, Zamagni E, Ronconi S, Tosi P, Lemoli RM, Bandini G, Motta MR, Testoni N, Amabile M, Ottaviani E, Vianelli N, de Vivo A, Gozzetti A, Tura S, Cavo M. Molecular remission after allogeneic or autologous transplantation of hematopoietic stem cells for multiple myeloma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000;18:2273–81. Epub 2000/06/01. [DOI] [PubMed] [Google Scholar]
- 41.Paino T, Paiva B, Sayagues JM, Mota I, Carvalheiro T, Corchete LA, Aires-Mejia I, Perez JJ, Sanchez ML, Barcena P, Ocio EM, San-Segundo L, Sarasquete ME, Garcia-Sanz R, Vidriales MB, Oriol A, Hernandez MT, Echeveste MA, Paiva A, Blade J, Lahuerta JJ, Orfao A, Mateos MV, Gutierrez NC, San-Miguel JF. Phenotypic identification of subclones in multiple myeloma with different chemoresistant, cytogenetic and clonogenic potential. Leukemia. 2015;29:1186–94. Epub 2014/11/13. [DOI] [PubMed] [Google Scholar]
- 42.Drandi D, Kubiczkova-Besse L, Ferrero S, Dani N, Passera R, Mantoan B, Gambella M, Monitillo L, Saraci E, Ghione P, Genuardi E, Barbero D, Omede P, Barberio D, Hajek R, Vitolo U, Palumbo A, Cortelazzo S, Boccadoro M, Inghirami G, Ladetto M. Minimal Residual Disease Detection by Droplet Digital PCR in Multiple Myeloma, Mantle Cell Lymphoma, and Follicular Lymphoma: A Comparison with Real-Time PCR. J Mol Diagn. 2015;17:652–60. [DOI] [PubMed] [Google Scholar]
- 43.Zamagni E, Patriarca F, Nanni C, Zannetti B, Englaro E, Pezzi A, Tacchetti P, Buttignol S, Perrone G, Brioli A, Pantani L, Terragna C, Carobolante F, Baccarani M, Fanin R, Fanti S, Cavo M. Prognostic relevance of 18-F FDG PET/CT in newly diagnosed multiple myeloma patients treated with up-front autologous transplantation. Blood. 2011;118:5989–95. Epub 2011/09/09. [DOI] [PubMed] [Google Scholar]
- 44.Caldarella C, Isgro MA, Treglia I, Treglia G. Is fluorine-18-fluorodeoxyglucose positron emission tomography useful in monitoring the response to treatment in patients with multiple myeloma? Int J Hematol. 2012;96:685–91. [DOI] [PubMed] [Google Scholar]
- 45.Caldarella C, Treglia G, Isgro MA, Treglia I, Giordano A. The role of fluorine-18-fluorodeoxyglucose positron emission tomography in evaluating the response to treatment in patients with multiple myeloma. Int J Mol Imaging. 2012;175803:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreau P, Attal M, Karlin L, Garderet L, Facon T, Macro M, Benboubker L, Caillot D, Escoffre M, Stoppa A-M, Laribi K, Hulin C, Marit G, Eveillard JR, Caillon F, Bodet C, Pégourie B, Dorvaux V, Chaleteix C, Richardson PG, Anderson KC, Avet-Loiseau H, Gaultier A, Nguyen j-M, Dupas B, Bodere F. Prospective Evaluation of MRI and PET-CT at Diagnosis and before Maintenance Therapy in Symptomatic Patients with Multiple Myeloma Included in the IFM/DFCI 2009 Trial. Blood. 2015;126:395–. [Google Scholar]
- 47.Yanamandra U, Mittal BR, Upadesh M, Reddy A, Sharma P, Agarwal K, Khadwal A, Prakash G, Varma N, Varma S, Malhotra P. Is 18F-FDG-PET/CT a good MRD marker in patients with multiple myeloma? Comparison and correlation with biochemical markers/flowcytometry. ASCO Meeting Abstracts. 2016;34:8029. [Google Scholar]
- 48.Zamagni E, Nanni C, Mancuso K, Tacchetti P, Pezzi A, Pantani L, Zannetti B, Rambaldi I, Brioli A, Rocchi S, Terragna C, Martello M, Marzocchi G, Borsi E, Rizzello I, Fanti S, Cavo M. PET/CT Improves the Definition of Complete Response and Allows to Detect Otherwise Unidentifiable Skeletal Progression in Multiple Myeloma. Clin Cancer Res. 2015;21:4384–90. [DOI] [PubMed] [Google Scholar]
- 49.Usmani SZ, Mitchell A, Waheed S, Crowley J, Hoering A, Petty N, Brown T, Bartel T, Anaissie E, van Rhee F, Barlogie B. Prognostic implications of serial 18-fluoro-deoxyglucose emission tomography in multiple myeloma treated with total therapy 3. Blood. 2013;121:1819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nanni C, Zamagni E, Versari A, Chauvie S, Bianchi A, Rensi M, Bello M, Rambaldi I, Gallamini A, Patriarca F, Gay F, Gamberi B, Cavo M, Fanti S. Image interpretation criteria for FDG PET/CT in multiple myeloma: a new proposal from an Italian expert panel. IMPeTUs (Italian Myeloma criteria for PET USe). Eur J Nucl Med Mol Imaging. 2016;43:414–21. [DOI] [PubMed] [Google Scholar]
- 51.Dammacco F, Rubini G, Ferrari C, Vacca A, Racanelli V. (1)(8)F-FDG PET/CT: a review of diagnostic and prognostic features in multiple myeloma and related disorders. Clinical and experimental medicine. 2015;15:1–18. Epub 2014/09/15. [DOI] [PubMed] [Google Scholar]
- 52.Lonial S, Kaufman JL. Non-secretory myeloma: a clinician’s guide. Oncology (Williston Park, NY). 2013;27:924–8, 30. Epub 2013/11/29. [PubMed] [Google Scholar]
- 53.Orchard K, Barrington S, Buscombe J, Hilson A, Prentice HG, Mehta A. Fluoro-deoxyglucose positron emission tomography imaging for the detection of occult disease in multiple myeloma. British journal of haematology. 2002;117:133–5. Epub 2002/03/29. [DOI] [PubMed] [Google Scholar]
- 54.Sager S, Ergul N, Ciftci H, Cetin G, Guner SI, Cermik TF. The value of FDG PET/CT in the initial staging and bone marrow involvement of patients with multiple myeloma. Skeletal Radiol. 2011;40:843–7. [DOI] [PubMed] [Google Scholar]
- 55.Yanamandra U, Mittal BR, Reddy A, Agarwal K, Khadwal A, Prakash G, Varma N, Varma S, Malhotra P. Role of PET/CT in prognosticating post-transplant outcomes based on a new scoring system: Results of PIPET-M TRIAL. ASCO Meeting Abstracts. 2016;34:8028. [Google Scholar]
- 56.Luckerath K, Lapa C, Spahmann A, Jorg G, Samnick S, Rosenwald A, Einsele H, Knop S, Buck AK. Targeting paraprotein biosynthesis for non-invasive characterization of myeloma biology. PLoS One. 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lapa C, Knop S, Schreder M, Rudelius M, Knott M, Jorg G, Samnick S, Herrmann K, Buck AK, Einsele H, Luckerath K. 11C-Methionine-PET in Multiple Myeloma: Correlation with Clinical Parameters and Bone Marrow Involvement. Theranostics. 2016;6:254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P, Landgren O, Paiva B, Dispenzieri A, Weiss B, LeLeu X, Zweegman S, Lonial S, Rosinol L, Zamagni E, Jagannath S, Sezer O, Kristinsson SY, Caers J, Usmani SZ, Lahuerta JJ, Johnsen HE, Beksac M, Cavo M, Goldschmidt H, Terpos E, Kyle RA, Anderson KC, Durie BG, Miguel JF. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:70442–5. [DOI] [PubMed] [Google Scholar]
- 59.Bhutani M, Landgren O. [Imaging in smoldering (asymptomatic) multiple myeloma. Past, present and future]. Der Radiologe. 2014;54:572, 4–81. Epub 2014/06/15. [DOI] [PubMed] [Google Scholar]
- 60.Cascini GL, Falcone C, Console D, Restuccia A, Rossi M, Parlati A, Tamburrini O. Whole-body MRI and PET/CT in multiple myeloma patients during staging and after treatment: personal experience in a longitudinal study. Radiol Med. 2013;118:930–48. [DOI] [PubMed] [Google Scholar]
- 61.Ferrero S, Ladetto M, Drandi D, Cavallo F, Genuardi E, Urbano M, Caltagirone S, Grasso M, Rossini F, Guglielmelli T, Cangialosi C, Liberati AM, Callea V, Carovita T, Crippa C, De Rosa L, Pisani F, Falcone AP, Pregno P, Oliva S, Terragna C, Musto P, Passera R, Boccadoro M, Palumbo A. Long-term results of the GIMEMA VEL-03–096 trial in MM patients receiving VTD consolidation after ASCT: MRD kinetics’ impact on survival. Leukemia. 2015;29:689–95. Epub 2014/07/17. [DOI] [PubMed] [Google Scholar]
- 62.Galimberti S, Morabito F, Guerrini F, Palumbo GA, Azzara A, Martino M, Benedetti E, Di Raimondo F, Petrini M. Peripheral blood stem cell contamination evaluated by a highly sensitive molecular method fails to predict outcome of autotransplanted multiple myeloma patients. British journal of haematology. 2003;120:405–12. Epub 2003/02/13. [DOI] [PubMed] [Google Scholar]
- 63.Galimberti S, Benedetti E, Morabito F, Papineschi F, Callea V, Fazzi R, Stelitano C, Andreazzoli F, Guerrini F, Ciabatti E, Martino M, Nobile F, Iacopino P, Petrini M. Prognostic role of minimal residual disease in multiple myeloma patients after non-myeloablative allogeneic transplantation. Leukemia research. 2005;29:961–6. Epub 2005/06/28. [DOI] [PubMed] [Google Scholar]
- 64.Chanan-Khan A, Somlo G, Heffner LT, Siegel DS, Zimmerman TM, Jagannath S, Munshi NC, Lonial S, Roy V, Ruehle M, Chavan S, Patel P, Rothenburger M, Wartenberg-Demand A, Haeder T, Anderson KC. Indatuximab Ravtansine (BT062) In Combination With Lenalidomide and Low-Dose Dexamethasone In Patients With Relapsed and/Or Refractory Multiple Myeloma: Clinical Activity In Len/Dex-Refractory Patients. Blood. 2013;122:758–. [Google Scholar]
- 65.McCudden CR, Voorhees PM, Hainsworth SA, Whinna HC, Chapman JF, Hammett-Stabler CA, Willis MS. Interference of monoclonal antibody therapies with serum protein electrophoresis tests. Clinical chemistry. 2010;56:1897–9. [DOI] [PubMed] [Google Scholar]
- 66.Genzen JR, Kawaguchi KR, Furman RR. Detection of a monoclonal antibody therapy (ofatumumab) by serum protein and immunofixation electrophoresis. British journal of haematology. 2011;155:123–5. [DOI] [PubMed] [Google Scholar]
- 67.Dekker LJ, Zeneyedpour L, Brouwer E, van Duijn MM, Sillevis Smitt PA, Luider TM. An antibody-based biomarker discovery method by mass spectrometry sequencing of complementarity determining regions. Anal Bioanal Chem. 2011;399:1081–91. Epub 2010/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Remily-Wood ER, Benson K, Baz RC, Chen YA, Hussein M, Hartley-Brown MA, Sprung RW, Perez B, Liu RZ, Yoder SJ, Teer JK, Eschrich SA, Koomen JM. Quantification of peptides from immunoglobulin constant and variable regions by LC-MRM MS for assessment of multiple myeloma patients. Proteomics Clin Appl. 2014;8:783–95. Epub 2014/04/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barnidge DR, Tschumper RC, Theis JD, Snyder MR, Jelinek DF, Katzmann JA, Dispenzieri A, Murray DL. Monitoring M-proteins in patients with multiple myeloma using heavy-chain variable region clonotypic peptides and LC-MS/MS. J Proteome Res. 2014;13:1905–10. Epub 2014/02/21. [DOI] [PubMed] [Google Scholar]
- 70.Bergen HR, 3rd, Dasari S, Dispenzieri A, Mills JR, Ramirez-Alvarado M, Tschumper RC, Jelinek DF, Barnidge DR, Murray DL. Clonotypic Light Chain Peptides Identified for Monitoring Minimal Residual Disease in Multiple Myeloma without Bone Marrow Aspiration. Clinical chemistry. 2016;62:243–51. Epub 2015/10/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Korthals M, Sehnke N, Kronenwett R, Schroeder T, Strapatsas T, Kobbe G, Haas R, Fenk R. Molecular monitoring of minimal residual disease in the peripheral blood of patients with multiple myeloma. [DOI] [PubMed] [Google Scholar]
- 72.Kaedbey R, Kis O, Danesh A, Dowar M, Li T, Li ZH, Liu J, Mansour M, Sukhai M, Zhang T, Kamel-Reid S, Kukreti V, Reece D, Chen C, Tiedemann R, Prica A, Pugh TJ, Trudel S. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating cell free DNA (cfDNA) in multiple myeloma (MM). Clinical Lymphoma Myeloma and Leukemia.15:e45–e6. [Google Scholar]
- 73.Hocking J, Mithraprabhu S, Kalff A, Spencer A. Liquid biopsies for liquid tumors: emerging potential of circulating free nucleic acid evaluation for the management of hematologic malignancies. Cancer biology & medicine. 2016;13:215–25. Epub 2016/07/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tovar N, Fernandez de Larrea C, Elena M, Cibeira MT, Arostegui JI, Rosinol L, Filella X, Yague J, Blade J. Prognostic impact of serum immunoglobulin heavy/light chain ratio in patients with multiple myeloma in complete remission after autologous stem cell transplantation. Biol Blood Marrow Transplant. 2012;18:1076–9. [DOI] [PubMed] [Google Scholar]
- 75.Ludwig H, Milosavljevic D, Zojer N, Faint JM, Bradwell AR, Hubl W, Harding SJ. Immunoglobulin heavy/light chain ratios improve paraprotein detection and monitoring, identify residual disease and correlate with survival in multiple myeloma patients. Leukemia. 2013;27:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, Middha S, Asmann Y, Schmidt J, Braggio E, Keats JJ, Fonseca R, Bergsagel PL, Craig DW, Carpten JD, Stewart AK. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120:1060–6. Epub 2012/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, Van Wier S, Blackburn PR, Baker AS, Dispenzieri A, Kumar S, Rajkumar SV, Carpten JD, Barrett M, Fonseca R, Stewart AK, Bergsagel PL. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120:1067–76. Epub 2012/04/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nature reviews Cancer. 2012;12:335–48. Epub 2012/04/13. [DOI] [PubMed] [Google Scholar]
- 79.Paiva B, Paino T, Sayagues JM, Garayoa M, San-Segundo L, Martin M, Mota I, Sanchez ML, Barcena P, Aires-Mejia I, Corchete L, Jimenez C, Garcia-Sanz R, Gutierrez NC, Ocio EM, Mateos MV, Vidriales MB, Orfao A, San Miguel JF. Detailed characterization of multiple myeloma circulating tumor cells shows unique phenotypic, cytogenetic, functional, and circadian distribution profile. Blood. 2013;122:3591–8. Epub 2013/09/28. [DOI] [PubMed] [Google Scholar]
- 80.Paiva B, Corchete LA, Vidriales MB, Puig N, Maiso P, Rodriguez I, Alignani D, Burgos L, Sanchez ML, Barcena P, Echeveste MA, Hernandez MT, Garcia-Sanz R, Ocio EM, Oriol A, Gironella M, Palomera L, De Arriba F, Gonzalez Y, Johnson SK, Epstein J, Barlogie B, Lahuerta JJ, Blade J, Orfao A, Mateos MV, San Miguel JF. Phenotypic and genomic analysis of multiple myeloma minimal residual disease tumor cells: a new model to understand chemoresistance. Blood. 2016;127:1896–906. Epub 2016/01/13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.