

Abstract
The impact of molecular dynamics (MD) simulations in molecular biology and drug discovery has expanded dramatically in recent years. These simulations capture the behavior of proteins and other biomolecules in full atomic detail and at very fine temporal resolution. Major improvements in simulation speed, accuracy, and accessibility, together with the proliferation of experimental structural data, have increased the appeal of biomolecular simulation to experimentalists—a trend particularly noticeable in , though certainly not limited to, neuroscience. Simulations have proven valuable in deciphering functional mechanisms of proteins and other biomolecules, in uncovering the structural basis for disease, and in the design and optimization of small molecules, peptides, and proteins. Here we describe in practical terms the types of information MD simulations can provide and the ways in which they typically motivate further experimental work.
Introduction
Imagine that an alien lands on Earth, hears about something called a “bicycle,” and wants to understand how it works, how to ride it, and how to fix it when it breaks. Figuring this out given just a picture of a bicycle would be challenging. Watching a movie of someone riding a bicycle would help. Even better, the alien would experiment with an actual bicycle—for example, by turning a pedal and seeing how the wheels respond.
A molecular biologist trying to understand how a protein or other biomolecule works faces a similar challenge. An atomic-level structure is tremendously helpful and typically generates substantial insight about how the biomolecule functions. The atoms in a biomolecule are in constant motion, however, and both molecular function and intermolecular interactions depend on the dynamics of the molecules involved. One would like not just a static snapshot but the ability to watch these biomolecules in action, to perturb them at the atomic level, and to see how they respond. Unfortunately, watching the motions of individual atoms and perturbing them in a desired fashion is difficult. An attractive alternative is to work with an atomic-level computer simulation of the relevant biomolecules.
Molecular dynamics (MD) simulations predict how every atom in a protein or other molecular system will move over time, based on a general model of the physics governing interatomic interactions (Karplus and Mc Cammon, 2002). These simulations can capture a wide variety of important biomolecular processes, including conformational change, ligand binding, and protein folding, revealing the positions of all the atoms at femtosecond temporal resolution. Importantly, such simulations can also predict how biomolecules will respond—at an atomic level—to perturbations such as mutation, phosphorylation, protonation, or the addition or removal of a ligand. MD simulations are often used in combination with a wide variety of experimental structural biology techniques, including x-ray crystallography, cryo-electron microscopy (cryo-EM), nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR), and Förster resonance energy transfer (FRET)
MD simulations are not new. The first MD simulations of simple gasses were performed in the late 1950s (Alder and Wainwright, 1957). The first MD simulation of a protein was performed in the late 1970s (McCammon et al., 1977), and the groundwork that enabled these simulations was among the achievements recognized by the 2013 Nobel Prize in Chemistry (Levitt and Lifson, 1969; Lifson and Warshel, 1968). MD simulations have, however, become substantially more popular and visible in recent years, particularly from the perspective of experimental molecular biologists (Figure 1). Simulations have begun to appear frequently in experimental structural biology papers, where they are used both to interpret experimental results and to guide experimental work. This trend is particularly noticeable in neuroscience: simulations have been used to study proteins critical to neuronal signaling (Dawe et al., 2016; Delemotte et al., 2011; Dror et al., 2013; Jensen et al., 2012; Shi et al., 2008), to assist in the development of drugs targeting the nervous system (Manglik et al., 2016; McCorvy et al., 2018; Spahn et al., 2017), to reveal mechanisms of protein aggregation associated with neurodegenerative disorders (Khandogin and Brooks, 2007; Wu and Shea, 2013), and to provide a foundation for the design of improved optogenetics tools (Takemoto et al., 2015; Kato et al., 2018).
Figure 1. Growth of molecular dynamics simulations in structural biology.
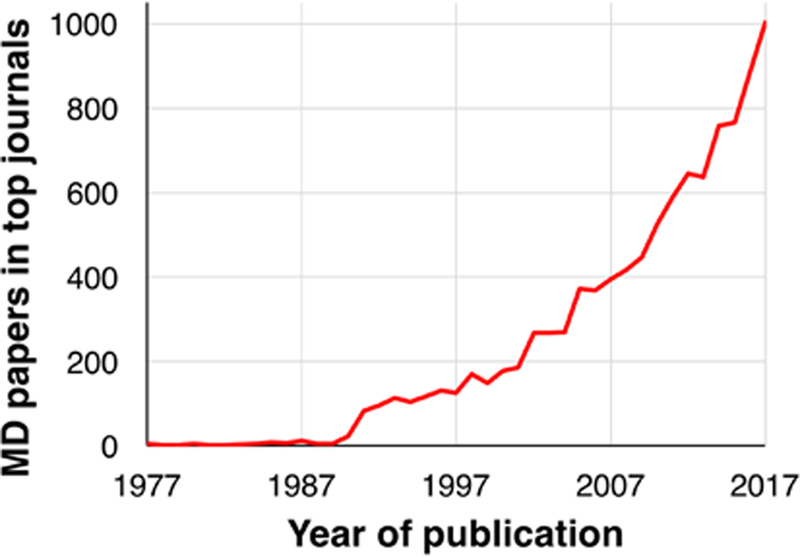
For the top 250 journals by impact factor, we plot the number of publications per year that include the term “molecular dynamics” in either the title, abstract or keywords. Analysis was performed via Web of Science (https://webofknowledge.com/) in February 2018.
The increasing attention to MD simulations has at least two underlying drivers. First, the last few years have seen an explosion in experimental structures of certain classes of molecules that are critical in neuroscience, including molecular families that represent the targets of most neuroscience medications (Coleman et al., 2016; Hilger et al., 2018; Minor, 2007). Many of these—for example, ion channels, neurotransmitter t ransporters, and G protein–coupled receptors (GPCRs)—are membrane proteins. Crystallog raphic structure determination for membrane proteins has historically been difficult, but recent breakthroughs in crystallography have delivered dozens of such structures (recognized by Nobel Prizes in 2003 and 2012), and breakthroughs in cryo-EM (recognized by a Nobel Prize in 2017) are now further accelerating the solution of such structures (Fernandez-Leiro and Scheres, 2016). These experimental structures provide a starting point for MD simulations and have also focused more attention on structural questions that simulation can help address: how key neuronal proteins function, why proteins aggregate pathologically under certain conditions, how one can best carry out structure-based drug design, and how one can best engineer proteins that serve as tools for studying neuronal function (e.g., by optogenetics and imaging).
Second, MD simulations themselves have become much more powerful and accessible over the past few years. Until recently, most high-impact work performed using MD simulations required a supercomputer. Recently introduced computer hardware, particularly graphics processing units (GPUs), allows powerful simulations to be run locally at a modest cost (Salomon-Ferrer et al., 2013; Stone et al., 2016). Software packages for performing MD simulations have also become easier to use, with better support for non-experts. Finally, although the physical models underlying MD simulations are inherently approximations, they have become substantially more accurate.
Our goal in this review is to explain how MD may be useful from the perspective of an experimental structural or molecular biologist. We explain the types of studies one can undertake by simulation, and the types of information they are likely to yield. We also discuss how simulations can generate new experimentally testable hypotheses and thus influence further experimental work. Finally, we provide a basic primer on MD simulations, explain some practical details of using them, and discuss their limitations.
What is an MD simulation: the basics
The basic idea behind an MD simulation is straightforward. Given the positions of all the atoms in a biomolecular system (e.g., a protein surrounded by water and perhaps a lipid bilayer), one can calculate the force exerted on each atom by all the other atoms. One can thus use Newton’s laws of motion to predict the spatial position of each atom as a function of time. In particular, one steps through time, repeatedly calculating the forces on each atom and then using those forces to update the position and velocity of each atom. The resulting trajectory is, in essence, a three-dimensional movie that describes the atomic-level configuration of the system at every point during the simulated time interval.
These simulations are powerful for several reasons. First, they capture the position and motion of every atom at every point in time, which is very difficult with any experimental technique. Second, the simulation conditions are precisely known and can be carefully controlled: the initial conformation of a protein, which ligands are bound to it, whether it has any mutations or post-translational modifications, which other molecules are present in its environment, its protonation state, the temperature, the voltage across a membrane, and so on. By comparing simulations performed under different conditions, one can identify the effects of a wide variety of molecular perturbations.
The forces in an MD simulation are calculated using a model known as a molecular mechanics force field, which is fit to the results of quantum mechanical calculations and, typically, to certain experimental measurements. For example, a typical force field incorporates terms that capture electrostatic (Coulombic) interactions between atoms, spring-like terms that model the preferred length of each covalent bond, and terms capturing several other types of interatomic interactions. Such force fields are inherently approximate. Comparison of simulations to a variety of experimental data indicates that force fields have improved substantially over the past decade (Lindorff-Larsen et al., 2012), but they remain imperfect, and the uncertainty introduced by these approximations should be considered when analyzing simulation results. Moreover, in a classical MD simulation, no covalent bonds form or break. Quantum mechanics/molecular mechanics (QM/MM) simulations, in which a small part of the system is modeled using quantum mechanical calculations and the remainder by MD simulation, are frequently employed to study reactions that involve changes to covalent bonds or are driven by the absorption of light (Senn and Thiel, 2009).
To ensure numerical stability, the time steps in an MD simulation must be short, typically only a few femtoseconds (10–15 s) each. Most of the events of biochemical interest—for example, functionally important structural changes in proteins—take place on timescales of nanoseconds, microseconds, or longer. A typical simulation thus involves millions or billions of time steps. This fact, combined with the millions of interatomic interactions typically evaluated during a single time step, causes simulations to be very computationally demanding.
Over the past several decades, improvements in computing hardware and in the algorithms and software used for MD have allowed longer and cheaper simulations. Recent improvements have been particularly remarkable. Highly specialized hardware (Shaw et al., 2008; Shaw et al., 2014) has led to a major increase in maximum achievable speed, allowing certain simulations to reach millisecond timescales. Perhaps more importantly, GPUs have allowed simulations running on one or two inexpensive computer chips to outperform those previously performed on most supercomputers (Salomon-Ferrer et al., 2013). These GPUs have made simulations on biologically meaningful timescales accessible to far more researchers than ever before.
Indeed, performing simulations is now relatively straightforward (see the practical considerations section of this review), and the computational resources to perform useful amounts of simulation are increasingly widely accessible. What requires expertise is figuring out what questions can be addressed by simulations, designing simulations to address these questions, and interpreting the simulation results. Interpreting simulation results—gaining biological insight from a large amount of trajectory data describing a mass of jiggling atoms— can be particularly challenging. In addition, a wide variety of advanced simulation techniques are available to address questions that are intractable by simple “brute force” simulation.
What information can MD simulations provide?
Molecular dynamics simulations can be used to answer many types of questions (Figure 2). Here we survey some of the most common, with an emphasis on how simulations typically complement experimental molecular biology investigations. Figures 3, 4, and 5 illustrate several of our recent simulation-based studies.
Figure 2. Applications of molecular dynamics simulations.
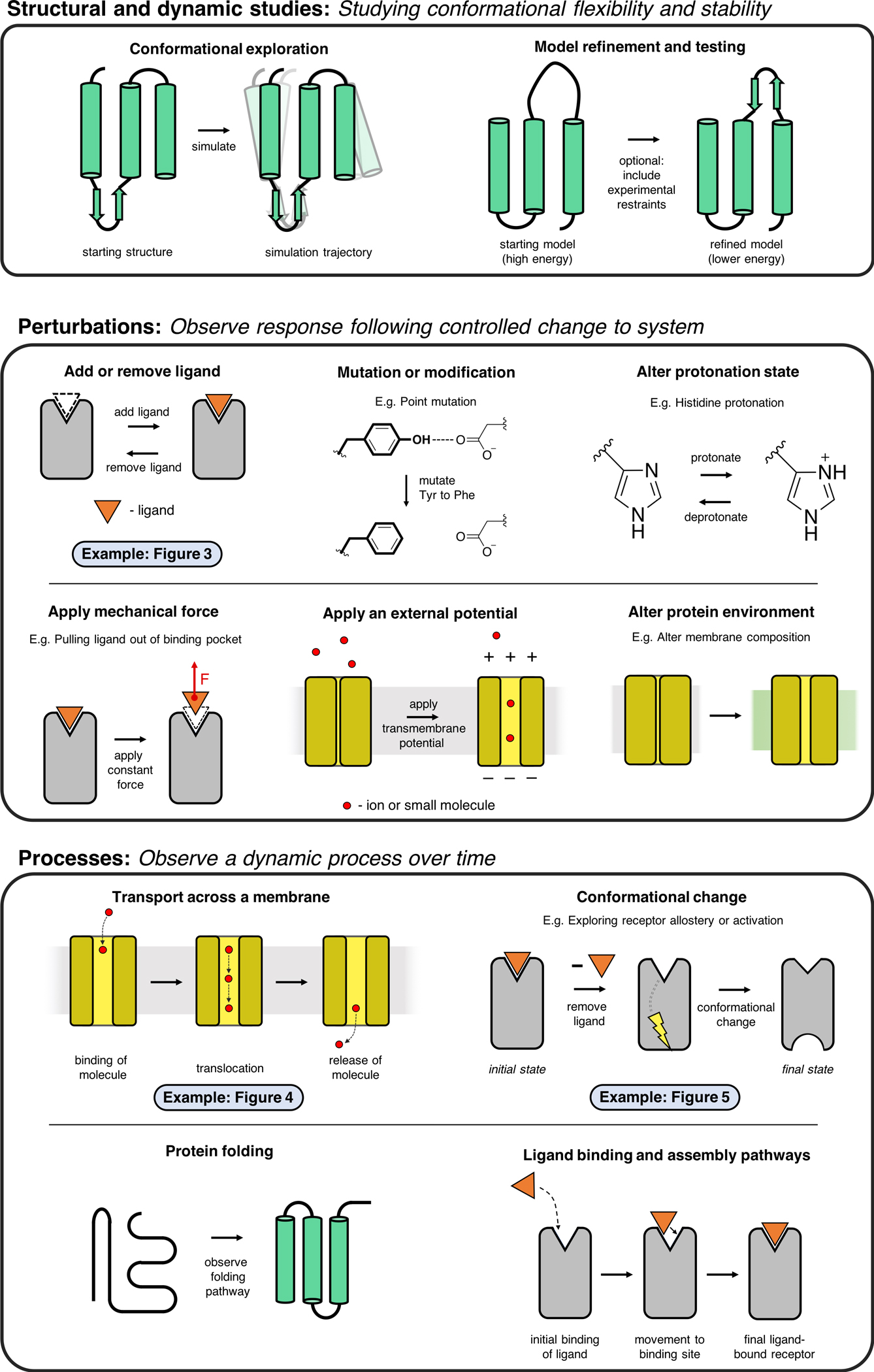
Here we illustrate some of the most common applications of MD simulations.
Figure 3. Case study: structural basis of allosteric modulation in GPCRs.
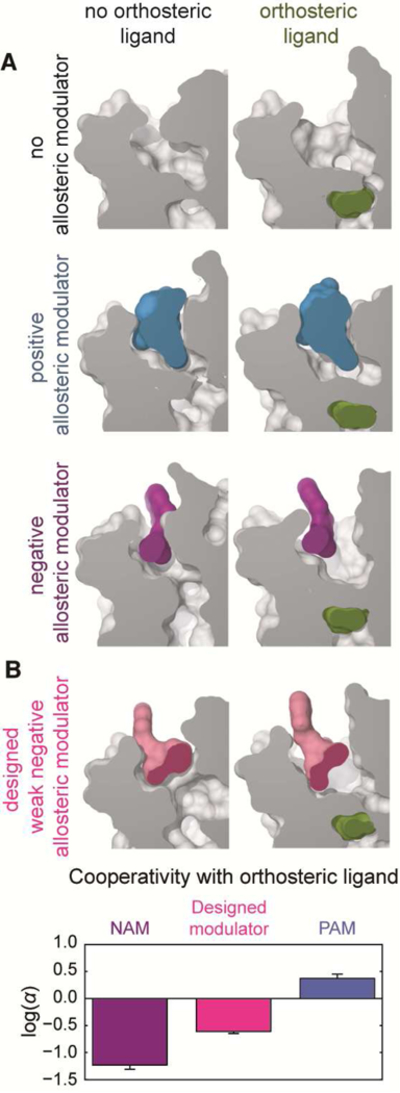
We used MD simulations to determine how allosteric modulators bind to a GPCR, the M2 muscarinic acetylcholine receptor, and how these allosteric modulators increase or decrease binding affinity of orthosteric ligands. A) The conformations of the orthosteric and allosteric binding sites in the presence or absence of different ligands, as determined by MD simulations. The orthosteric ligand N-Methyl Scopolamine (NMS) favors an enlarged allosteric site. Binding of the positive allosteric modulator (PAM) alcuronium requires a larger allosteric site to bind, whereas the negative allosteric modulator (NAM) C7/3-phth does not. B) To validate the proposed mechanism of allostery, we designed a modified version of the NAM that would require a larger allosteric pocket to bind, and is thus predicted to have less negative cooperativity. Indeed, radioligand binding experiments revealed that the cooperativity of the designed modulator is fourfold less negative than that of the original NAM, even though the affinity of the designed modulator is higher. Adapted from (Dror et al., 2013), with permission.
Figure 4. Case study: atomic-level mechanism of an alternating access transporter.
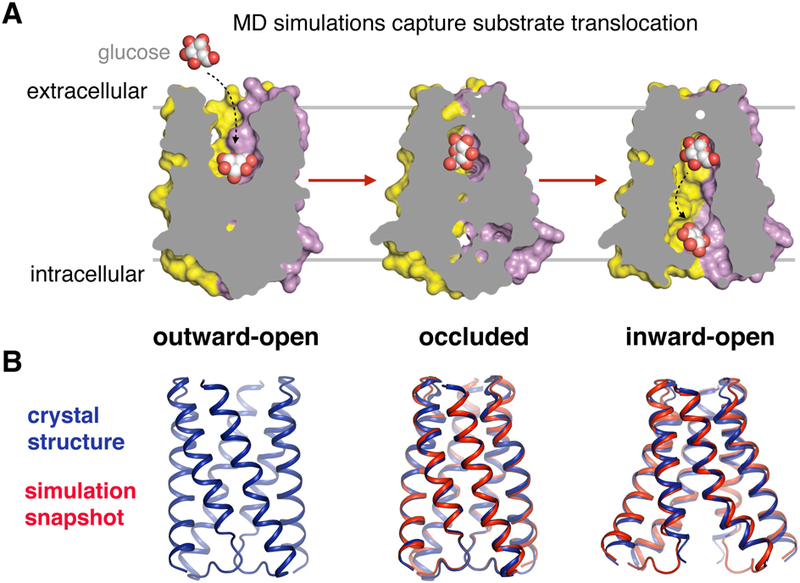
A) MD simulations captured the spontaneous transition of the sugar transporter SemiSWEET from its outward-open state (where the substrate-binding pocket is accessible to the outside of the cell) to its inward-open state, along with the accompanying substrate translocation process. This simulation study addressed several long-standing questions such as what drives the structural changes associated with transport, how the presence of the substrate affects the conformations the transporter adopts, and how the inner and outer gates avoid opening simultaneously. B) Overlays of simulation snapshots and the corresponding crystal structures of the occluded and inward facing states show that conformations visited in simulation are nearly identical to those observed crystallographically. Mutagenesis studies further validated simulation results. Adapted from (Latorraca et al., 2017), with permission.
Hollingsworth and Dror review modern molecular dynamics (MD) simulations, with an emphasis on how such simulations complement wet-lab experiments. MD simulations capture biomolecular motion in atomic detail and have come into widespread use thanks to recent technological and scientific advances.
Figure 5. Case study: how GPCRs cause arrestin activation.
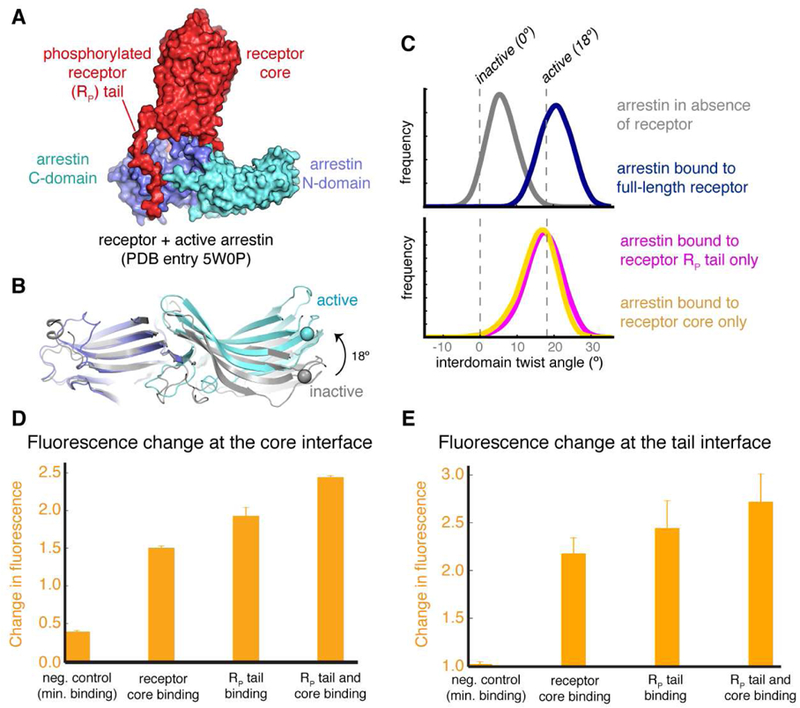
A) A crystal structure of GPCR-bound, active-state arrestin. The receptor’s core and phosphorylated tail (RP tail) bind to distinct surfaces of arrestin, and their respective influences on arrestin conformation have been unclear. Upon activation, the C-domain of arrestin twists 18º relative to the N-domain. C) Distributions of the interdomain twist angles under different simulation conditions are shown. Simulations indicate that binding of either the receptor core or the RP tail is sufficient to activate arrestin, with binding of both the core and RP tail leading to an even larger activation effect. D, E) Site-directed fluorescence spectroscopy experiments support these computational results. These experiments probe conformational change in arrest in at either the core interface or the RP tail interface (E) and show that receptor constructs that bind only at the core interface or only at the RP tail cause conformational changes at both interfaces. Adapted from (Latorraca et al., 2018), with permission.
Perhaps the most basic and intuitive application of simulation is to assess the mobility or flexibility of various regions of a biomolecule. Experimental structure determination methods such as x-ray crystallography and cryo-EM generally yield an average structure. By simply examining a simulation of such a structure, one can quantify how much various regions of the molecule move at equilibrium, and what types of structural fluctuations they undergo. Such simulations can also reveal the dynamic behavior of water molecules and salt ions, which are often critical for protein function and ligand binding (Berneche and Roux, 2001; Khafizov et al., 2012; Li et al., 2013).
Simulation can also be used to test the accuracy of a modeled structure or even to refine it. For example, a crystal structure may suffer from artifacts due to crystal lattice packing or, for a membrane protein, to the absence of a lipid bilayer. One can often correct such artifacts by performing a simulation starting from the crystal structure but in an appropriate solvated environment and allowing the structure to relax to a more favorable conformation, if one exists (Burg et al., 2015). A similar approach is often used to test modeled binding poses of ligands: a pose that is stable in simulation is more likely to be accurate than one that is unstable (Clark et al., 2016). Such efforts have proven effective in determining ligand poses in cryo-EM structures with ambiguous ligand density (Koehl et al., 2018). MD simulations are sometimes useful in refining protein homology models, but many attempts to do this have not been successful (Mirjalili and Feig, 2013; Raval et al., 2012).
On the other hand, MD simulations are widely used to build or refine structural models based on experimental structural biology data. X-ray crystal structures, for example, are frequently refined by an MD-based simulated annealing protocol that fits the model to the experimental data while maintaining a physically reasonable structure (Afonine et al., 2012; Brunger and Adams, 2002). This approach has been shown to overcome model errors that least squares regression cannot. An MD-based protocol is often used to build atomic-level molecular models from low-resolution cryo-EM density maps, particularly when high-resolution structures of individual components of a complex are separately available (Trabuco et al., 2008; Zhao et al., 2013). MD simulations have also been used to recover ensembles of conformations—as opposed to a single structure—from NMR data (Lindorff-Larse n et al., 2005). In each of these cases, themolecular mechanics force field is supplemented by terms that depend on the experimental data, and that result in a lower energy for structures (or structural ensembles) that agree better with the experimental data.
A particularly important application of MD simulation is to determine how a biomolecular system will respond to some perturbation. For example, one might do any of the following: (1) remove a bound ligand from an experimentally determined protein structure and then simulate to see how the ligand’s removal affects protein conformation (Dror et al., 2013; Wacker et al., 2017b) (Figure 3); (2) replace a bound ligand by a different ligand, or add a ligand where none was present in the experimental structure (McCorvy et al., 2018; Provasi et al., 2011); (3) mutate one or more amino-acid residues in the protein—for example, to explain or predict the functional effect of a mutation, or to recover the wild-type structure in cases where the experimentally resolved construct differed from the wild-type (Cordero-Morales et al., 2007); (4) phosphorylate an amino acid or add some other post-translational modification (Fields et al., 2017; Groban et al., 2006); (5) change the protonation state of an acidic or basic amino acid (Liu et al., 2015); (6) apply external forces to simulated atoms to capture the effect of transmembrane voltage or of mechanical strain (Delemotte et al., 2011); (7) change the molecular environment of a simulated protein, such as the salt concentration or the composition of lipids in a membrane. In each of these cases, one should generally perform several simulations of both the perturbed and unperturbed systems in order to identify consistent differences in the results.
Many MD simulation studies aim to observe biomolecular processes in action, particularly important functional processes such as ligand binding, ligand- or voltage-induced conformational change, protein folding, or membrane transport. This can allow one to answer questions about the structural basis for those events that are difficult to address experimentally: In what order do substructures form during protein folding (Lindorff-Larsen et al., 2011; Snow et al., 2002)? How does binding of a ligand to a GPCR’s extracellular surface cause changes on the intracellular side, where the G protein binds (Dror et al., 2011a)? More generally, what is the structural basis for allostery in proteins (Hertig et al., 2016; Figure 5)? How do alternating access transporters ensure that their outer and inner gates will not open simultaneously (Gu et al., 2009; Latorraca et al., 2017; Stelzl et al., 2014; Figure 4)? What are the factors controlling ligand binding and dissociation kinetics (Buch et al., 2011; Dror et al., 2011b; González et al., 2011; Wacker et al., 2017b)? What is the structural basis for water and ion transport across a membrane (Liang et al., 2016; Suomivuori et al., 2017; Tajkhorshid et al., 2002; Watanabe et al., 2010)? How do intrinsically disordered proteins assemble to form fibrils (Dedmon et al., 2005; Nguyen and Hall, 2004)?
In some cases, a single, unguided simulation can capture such a process in its entirety. When this is not possible—for example, because the relevant timescales are too long or because reactive chemistry is involved—one can often still reconstruct the process by simulating parts of it separately or by using a variety of enhanced sampling simulation methods (Bernardi et al., 2015; Harpole and Delemotte, 2018; Hertig et al., 2016; Schwantes et al., 2014).
In addition, MD simulations can yield diverse information regarding the binding of ligands to proteins and other macromolecules, as discussed further in the section on drug discovery below.
How can MD drive further experimental work?
A recent anecdote illustrates the increasing influence of simulation on experimental work.At the 2008 Keystone Symposium on GPCRs, no speaker mentioned computational approaches. At the 2018 version of the same meeting, a decade later, nearly half the speakers mentioned computational approaches, primarily MD simulations— including the first four speakers, who were all experimentalists.
Understanding how MD simulation can influence experimental work is complicated by the fact that much of the value of MD lies in its ability to probe molecular properties that are difficult or impossible to access through wet-lab experiments. In certain applications to ligand and protein design, simulations are used simply as a relatively inexpensive, though rough, filter for binding energy or stability in order to winnow down a large pool of candidates to a smaller one that can be tested experimentally (Chevalier et al., 2017; Hou et al., 2011; Wang et al., 2015). More frequently, however, simulations are used to generate a qualitative understanding of how a biomolecule or drug works. Usually, in such cases, no experiment is available that could provide all of the same information as the simulations. Experiments can, however, be designed to test specific predictions from these simulations in order to more broadly validate the simulation results. Perhaps even more importantly, simulations can generate hypotheses that lead to new experimental work. Table 1 lists a number of examples of simulations that influenced experimental work in various ways.
Table 1.
Examples of MD studies that influenced experimental design or interpretation.
| Study | Key MD Findings | Accompanying Experimental Validation |
Experimental Follow Up Studies & Validation |
|---|---|---|---|
| Ma et al., 2000 | Describes the transient interdomain motions during the GroEL allosteric cycle |
- | Cryo-EM (Ranson et al., 2001) |
| Beckstein et al.,2001 | Proposes a mechanism for hydrophobic gating in ion Channels |
- | Electrophysiology (Birkner et al., 2012) |
|
de Groot and Grubmüller, 2001; Tajkhorshid et al.,2002 |
Describes mechanism of water permeation through Aquaporin |
Mutagenesis and activity assays |
X-ray crystallography (Gonen et al., 2004; Tornroth-Horsefield et al., 2006) |
| Im and Roux, 2002a, b | Identifies how anions and cations travel down two separate pathways across the OmpF pore |
- | Anomalous x-ray diffraction (Dhakshnamoorthy et al., 2010) |
| Schames et al., 2004 | Identifies a previously unobserved binding site on HIV- Integrase |
- | Small molecule design, Pharmacokinetics ( Hazuda et al., 2004) |
| Freites et al., 2006 | Reveals that open state KvAP channel in a membrane environment resembles a water channel |
- | Fluorescence spectroscopy, neutron diffraction ( Krepkiy et al., 2009) |
| Cordero-Morales et al., 2007 | Development of a structural understanding of C-type inactivation of K+ channels |
- | X-ray crystallography, electrophysiology ( Cuello etal., 2010) |
| Arkin et al., 2007 | Development of an atomistic mechanism of an Na+/H+ Antiporter |
Mutagenesis and bacterial growth |
X-ray crystallography, electrophysiology ( Lee et al., 2013; Mager et al., 2011) |
|
Grabe et al., 2007; Vargas et al., 2011 |
Describes structural basis of voltage sensing through prediction of the resting state conformation of the Kv channel |
- | EPR, X-ray crystallography, electrophysiology,luminescence (Henrion et al.,2012; Kubota et al., 2017; Li et al., 2014) |
| Brannigan et al., 2008 | Describes a structural mechanism by which cholesterol binding stabilizes activation of nicotinic acetylcholine receptor |
- | X-ray crystallography, sequence analysis (Baier et al., 2011; Prevost et al., 2012) |
| Shi et al., 2008 | Identifies a second binding site in LeuT that helps to trigger release of Na+ and substrate |
Mutagenesis and binding assays |
X-ray crystallography and binding assays ( Quick et al., 2009) |
| Khafizov et al., 2012 | Identifies a second sodium binding site in the sodiumcoupled betaine transporter BetP |
X-ray crystallography,mutagenesis and binding assays, radiolabeling |
X-ray crystallography, electrophysiology ( Felts et al., 2014; Perez et al., 2014) |
| Dror et al., 2013 | Identifies binding sites, binding poses, and molecular mechanism for allosteric modulators of the M2 muscarinic acetylcholine receptor |
Mutagenesis and activity assays, small molecule design |
X-ray crystallography ( Kruse et al., 2013) |
| Li et al., 2013 | Identifies transient water-conducting but substrateoccluding states that are found across membrane transporters |
- | Mutagenesis, physiology (Erokhova et al., 2016; Zeuthen et al., 2016) |
| Ostmeyer et al., 2013 | Recovery from C-type inactivation is due to buried water molecules behind the selectivity filter |
Electrophysiology | X-ray crystallography ( Cuello et al., 2017) |
| Dror et al., 2015 | identifies the structural mechanism by which GPCRs stimulate G proteins | Protein engineering, DEER |
NMR (Goricanec et al., 2016) |
|
Hollingsworth et al., 2016; Hollingsworth and Poulos, 2015 |
Reveals that the electron donor protein Pdx favors binding to the open conformation of cytochrome P450cam |
Isothermal titration calorimetry (ITC) |
Resonance Raman Spectroscopy, mutagenesis and activity assays, DEER, (Batabyal et al., 2016; Batabyal et al., 2017; Liou et al., 2017) |
| Dawe et al., 2016 | Determination of a structural mechanism of activation for AMPA neurotransmitter-gated ion channels |
Electrophysiology ,X-ray crystallography |
Cryo-EM ( Twomey et al., 2016; Zhao et al., 2016) |
| Bae et al., 2016 | Identifies a hydrophobic region of TRPV1 that functions as a heat sensor |
NMR, electrophysiology mutagenesis andactivity assays |
Chimeric channel and activity assays (Zhang et al., 2018) |
| Bethel and Grabe, 2016 | Proposes a mechanism of lipid scrambling by TMEM16 scramblase |
- | Cryo-EM, mutagenesis, electrophysiology ( Jiang et al., 2017; Paulino et al., 2017) |
| Latorraca et al., 2017 | Determines the structural mechanism of substrate translocation in an alternating access transporter |
X-ray crystallography, mutagenesis and activity assays |
|
| Latorraca et al., 2018 | Reveals that arrestin can be activated through the binding of the GPCR core, the GPCR phosphorylated tail or both |
Fluorescence spectroscopy |
Mutagenesis, cellular imaging (Eichel et al., 2018) |
Experiments motivated by MD simulations generally take one of two forms. The first, and perhaps most obvious, involves experiments that directly probe structural properties. The experiments might involve actually solving a new structure (for example, of a protein with a different ligand bound, or of a mutant protein). Alternatively, the experiments might involve biophysical techniques that provide information on the structural ensemble or dynamics of a biomolecule, such as electron paramagnetic resonance (EPR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, fluorescence quenching (Figure 5), or hydrogen-deuterium exchange. These biophysical methods all report on changes in the chemical environment of a labeled residues. Some—such as double electron-electron resonance (DEER) spectroscopy, a form of EPR—can be used to derive probability distributions (histograms) of distances between two labeled residues.
A second—and more common—approach for experiments motivated by simulationsinvolves non-structural techniques such as binding or functional assays. For example, if simulations indicate that a particular protein–ligand interaction is important for binding, one might mutate the relevant residues of the protein or alter the relevant moiety of the ligand and then examine the effect of these changes on ligand binding or ligand-induced protein activity (Dror et al., 2013; Hollingsworth et al., 2016; Mc Corvy et al., 2018) (Figure 3). If simulations indicate that a residues plays a particular mechanistic role in a protein’s function, one might mutate it and measure the effect on the protein’s functional properties (Fields et al., 2015; Latorraca et al., 2017) (Figure 4).
How can MD contribute to drug discovery?
Drug discovery provides a particularly interesting example of an area in which simulations can drive experiments (Borhani and Shaw, 2012; Durrant and McCammon, 2011). Recent advances in structural biology have led to structures for many key neuroscience drug discovery targets (e.g., GPCRs, ion channels, transporters, etc.). Fully exploiting the power of structure-based drug design for these and other targets requires taking the dynamic properties of these proteins into account.
MD simulation is particularly valuable in lead optimization, where one modifies a ligand to improve its efficacy or other properties. At a qualitative level, simulations can provide a variety of information to guide the ligand optimization process: simulations can be used to identify the key interactions a ligand makes with the binding pocket, to predict rearrangements of the binding pocket induced by a ligand, or to test and refine potential ligand poses (Spahn et al., 2017; Udier-Blagovic et al., 2003). In some cases, simulations of the full ligand-binding process can reveal the binding site and pose of a ligand (Dror et al., 2013; Dror et al., 2011b; Kappel et al., 2015; Shan et al., 2011). At a quantitative level, simulation-based methods provide substantially more accurate estimates of ligand binding affinities (free energies) than other computational approaches such as docking (Perez et al., 2016). Free energy perturbation and other “alchemical” methods, in which one ligand is gradually “transformed” into another through a series of simulations, generally offer the most accurate estimates of binding energies (Chodera et al., 2011). These methods are computationally expensive, however, and generally only reliable when computing relative binding energies between ligands that share a similar scaffold (Mobley and Dill, 2009; Wang et al., 2015). The Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) and Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) methods, which also use MD simulation but rely on continuum solvent models rather than an explicit representation of water, are substantially faster but less accurate (Hou et al., 2011).
MD may also be useful for virtual screening, where one selects an initial set of ligands predicted to bind a target. Traditional virtual screening is performed with docking software, using a single structure of a target protein (Shoichet, 2004). In reality, the binding pocket may be highly flexible, and docking to a single structure will thus lead to identification of only a subset of binders. Considering multiple possible structures identified by simulation can increase the diversity of binding ligands identified (Amaro et al., 2008; Lin et al., 2002).
The goal of many drug design projects—particularly for those targeting signaling receptors—is to find a ligand that not only binds to the target but achieves a particular signaling profile. One might wish to find a full agonist that strongly stimulates receptor activation and signaling, a partial agonist that stimulates signaling to a lesser degree, a neutral antagonist that does not signal on its own but blocks the body’s native agonists from binding, or an inverse agonist that reduces signaling below the basal (unliganded) levels. Achieving a given signaling profile requires that the drug stabilize specific conformational states of the receptor and thus specific conformational states of the binding pocket. An agonist, for example, stabilizes active states over inactive states. Designing such a ligand with confidence requires an understanding of how subtle conformational changes in the binding pocket lead to different signaling profiles. MD simulations may provide such information (Dror et al., 2011a; Huang et al., 2015). An area of great current interest in GPCR drug discovery is the design of biased ligands, which selectively stimulate certain signaling pathways but not others controlled by the same receptor (Kenakin and Christopoulos, 2013; Violin et al., 2014; Wacker et al., 2017a). Rational design of such ligands is even more of a challenge, requiring an understanding of the receptor conformations associated with different signaling pathways. MD simulations have proven useful in this regard as well (Latorraca et al., 2018; McCorvy et al., 2018; Nivedha et al., 2018).
Simulations may be particularly helpful in the design of allosteric drugs, which bind to a target at a site different from the native ligand. Such drugs are greatly sought after because they offer the potential to increase selectivity between closely related receptor subtypes, modulate the body’s natural signaling patterns, and achieve efficacy at targets otherwise deemed undruggable (Conn et al., 2009). Allosteric binding sites are often not evident from experimental structures, as their formation may depend on the presence of an appropriate ligand. Simulations have proven capable of both capturing such “cryptic” binding pockets in various proteins and discovering binding sites of known allosteric modulators, thus facilitating the design of new allosteric modulators (Bowman et al., 2015; Dror et al., 2013; Newman et al., 2012; Schames et al., 2004; Tan et al., 2012). Moreover, the effects of an allosteric drug generally depend on the manner in which it alters its target’s conformation. Enabling the rational design of allosteric drugs with desired effects requires deciphering the coupling of allosteric and orthosteric sites. Simulation has proven useful in this regard as well. In a recent proof-of-concept study, for example, we used a simulation-based approach to design chemical modifications that substantially altered an allosteric ligand’s functional effects at a GPCR (Dror et al., 2013; Figure 3).
Simulations may also assist in the design of drugs with desired binding and dissociation kinetics, properties that have recently come to be recognized as critical to drug effectiveness and safety. The efficacy of ligands at certain targets, for example, correlates better with residence time than with binding affinity. A number of simulation studies have elucidated the factors that control binding and dissociation kinetics at various targets (Dror et al., 2011b; Schmidtke et al., 2011; Wacker et al., 2017b), providing a foundation for the rational design of ligands with faster or slower kinetics. Several recent studies have also demonstrated the use of MD-based methods to rank related ligands according to their dissociation rates (Dickson et al., 2017).
Practical considerations in using MD simulations
Actually performing an MD simulation is relatively straightforward. It requires a few choices: 1) Which computing hardware to use? GPUs have become a particularly attractive choice because they perform fast simulations at modest cost, but simulations are also run on supercomputers, which may be faster, as well as on traditional central processing units (CPUs), which may be more readily available. 2) Which force field to use? The most common choices are various versions of AMBER, CHARMM, and OPLS (Harder et al., 2016; Huang et al., 2017; Robustelli et al., 2018). These force fields all rely on similar functional forms, but each has certain strengths and weaknesses. For example, CHARMM36m and the complementary CHARMM General Force Field (CGenFF) have extensively optimized and validated parameters for proteins, lipids, and drug-like ligands (Huang et al., 2017; Klauda et al., 2010; Vanommeslaeghe and MacKerell Jr, 2012); the recently introduced A99SB-disp force field models disordered proteins particularly well (Robustelli et al., 2018); and OPLS3 may have the most extensively optimized ligand parameters, although their proprietary nature has generally precluded third-party evaluation (Harder et al., 2016). 3) Which software to use? Common choices include GROMACS, NAMD, AMBER, CHARMM, Desmond, and OpenMM (Abraham et al., 2015; Bowers et al., 2006; Brooks et al., 2009; Case et al., 2017; Eastman et al., 2017; Phillips et al., 2005). The AMBER and CHARMM software should not to be confused with AMBER and CHARMM force fields; most modern simulation software packages support multiple force fields. These software packages all perform similar computations but differ in how efficiently they map to various hardware and in supported features (e.g., enhanced sampling methods, temperature and pressure control schemes, support for coarse-grained simulations).
Before performing the simulation, one needs to prepare the molecular system by building in missing atoms (including hydrogen atoms, which are generally not resolved in crystal structures); adding in “solvent” molecules such as water, salt ions, and (for a membrane protein) lipids; and assigning force field parameters. Most of the common simulation software packages include some software for system preparation, and a number of recently introduced or improved software packages simplify the preparation process (Betz, 2017; Jo et al., 2008; Sastry et al., 2013).
The greater challenge is in deciding which simulations to perform (including which enhanced sampling techniques to use, if any) and, especially, in analyzing the results. Analyzing MD simulation results can be challenging for several reasons. These simulations produce a large amount of data: a typical simulation might track the positions and velocities of 100,000 atoms over billions of time steps. Identifying the most relevant and biologically important aspects of that data is challenging. In some cases, one is interested only in a particular well-defined quantity, such as the interaction energy between a ligand and a protein. However, in many cases—for example, when deciphering a functional me chanism—the most informative quantities and events are difficult to specify in advance.
Extracting maximally useful information from simulations requires interpreting them in light of all available experimental data for the molecular system under study (and, often, related systems as well). The analysis process generally demands a careful combination of visual analysis using molecular rendering software and quantitative analysis. A number of common analyses are “pre-packaged” in readily available so ftware, but most simulation projects benefit substantially from writing customized analysis programs or scripts, a task simplified by several analysis software frameworks (Abraham et al., 2015; McGibbon et al., 2015; Roe and Cheatham III, 2013; Skjærven et al., 2014).
When analyzing simulation results, one should keep in mind that the molecular systems being simulated—not only in simulation but also in real life—are chaotic, meaning that tiny perturbations in initial simulation conditions (e.g., the velocity of one water molecule) will often lead to substantially different simulation trajectories. One thus generally needs to perform multiple simulations under each condition. Often these simulations are initiated from the same atomic coordinates but with randomized initial velocities. To identify statistically significantdifferences in simulation results between conditions, one must compare variation between trajectories under different conditions to variation between trajectories under the same condition.
Both the design of MD simulations and the interpretation of their results should take into account the limitations of these simulations, several of which we highlight here. First, although the force fields employed in MD have improved substantially in recent years, they are inherently approximate (Lindorff-Larsen et al., 2012). Second, covalent bonds do not break or form during typical MD simulations, meaning that protonation states of titratable amino acid residues are fixed and must be set carefully at the beginning of a simulation (unless constant pH simulation approaches are employed (Goh et al., 2014), typically with a substantial increase in computational cost); the same is true for disulfide bonds. Third, an accurate simulation generally depends on the availability of an accurate experimental protein structure, or a good homology model, for use as an initial condition. Design of simulation studies is thus heavily influenced by the availability of experimental structures.
Finally, important biomolecular processes, including ligand binding and conformational change, often take place on timescales longer than those accessible by classical all-atom MD simulation. For systems with about 50,000 atoms (typical for a moderately sized, solvated protein), one GPU can currently simulate a microsecond in a few days. Specialized computing hardware, which can parallelize a simulation effectively across many computer chips, can increase simulation speed by at least an order of magnitude, though at substantially higher cost (Shaw et al., 2014). Using many GPUs to accelerate a single simulation is challenging, but Markov state modeling techniques can exploit many independent simulations to capture events that take place on longer timescales (Schwantes et al., 2014). Note that, regardless of the specific simulation methods employed, study design has a major impact on simulation timescalerequirements: for example, many equilibrium processes occur much more quickly in one direction than in the other, and one can exploit the principle of microscopic reversibility to study the forward process using simulations of the reverse process (Hertig et al., 2016).
In addition, a wide variety of enhanced sampling techniques allow simulations to capture longer-timescale events. These techniques employ a wide variety of strategies, such as: pulling a biomolecule from a desired initial conformation to a desired final conformation (e.g., targeted molecular dynamics (Schlitter et al., 1994)); pushing a simulation away from regions of conformational space it has already visited (e.g., metadynamics (Laio and Gervasio, 2008)); raising the effective temperature associated with certain degrees of freedom (e.g., replica exchange and temperature-accelerated molecular dynamics (Maragliano and Vanden-Eijnden, 2006; Sugita and Okamoto, 1999)); or altering the force field to reduce the height of energetic barriers (e.g., accelerated molecular dynamics (Hamelberg et al., 2004)). These techniques often prove very useful, particularly when certain reaction coordinates of interest can be specified in advance, but no single technique is a panacea for timescale limitations; different techniques are useful in different situations (Bernardi et al., 2015; Harpole and Delemotte, 2018). Enhanced sampling techniques can typically be tuned to access arbitrarily long timescales, but with an associated loss in accuracy (de Oliveira et al., 2006). Coarse-grained MD simulations, in which one particle represents a group of atoms rather than a single atom, can also extend accessible timescales by orders of magnitude (Marrink and Tieleman, 2013).
It is important to note that while performing MD simulations has become relatively straightforward in recent years, using MD simulations to reach sound, high-impact conclusions remains decidedly nontrivial. To do high-quality, reliable work by MD, one must: (1) identify important biological questions that can be addressed by MD, (2) design appropriate simulations to answer these questions, (3) set up these simulations carefully, taking into account the relevant experimental and computational literature, (4) analyze the simulations meticulously, considering various sources of error that might affect the results as well as expected statistical fluctuation from one simulation to the next, (5) compare results to available experimental data and, when possible, design follow-up experiments to further validate the results. This requires a solid understanding of both the biological system of interest and the theoretical basis for molecular dynamics simulations. It also typically requires a substantial amount of iteration, with one round of simulation and analysis often suggesting additional simulations and further analysis.
Conclusion
We believe that the careful application of MD simulations in concert with complementary experimental methods currently represents an area of great opportunity in neuroscience and beyond. This opportunity will only grow as simulations become faster, cheaper, more widely accessible, and more accurate. Effectively applying simulations to molecular biology and drug discovery requires careful thinking about both experimental and computational data available and thus benefits from both broad expertise and interdisciplinary collaborations.
Acknowledgments
We thank Brian Kobilka, Oliver Beckstein, Mark Sansom, José Faraldo-Gómez, Naomi Latorraca, Robin Betz, Raphael Townshend, Joseph Paggi, A.J. Venkatakrishnan, Albert Pan, Stefano Piana, Paul Robustelli, Morten Jensen, Matthew Lewis, Alec Follmer, and Thomas Poulos, as well as the anonymous reviewers, for helpful discussion and valuable suggestions.
This work was supported by National Institutes of Health (NIH) grant R01GM127359 to R.O.D.and an NIH Biomedical Informatics postdoctoral fellowship to S.A.H. (T15-LM007033–33).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder BJ, and Wainwright TE (1957). Phase Transition for a Hard Sphere System. J Chem Phys 27, 1208–1209. [Google Scholar]
- Amaro RE, Baron R, and McCammon JA (2008). An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J Comput Aided Mol Des 22, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin IT, Xu H, Jensen MØ, Arbely E, Bennett ER, Bowers KJ, Chow E, Dror RO, Eastwood MP, Flitman-Tene R, et al. (2007). Mechanism of Na+/H+ antiporting. Science 317, 799–803. [DOI] [PubMed] [Google Scholar]
- Bae C, Anselmi C, Kalia J, Jara-Oseguera A, Schwieters CD, Krepkiy D, Won Lee C, Kim E-H, Kim JI, Faraldo-Gómez JD, and Swa rtz KJ (2016). Structural insights into the mechanism of activation of the TRPV1 channel by a membrane-bound tarantula toxin. eLife 5, e11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier CJ, Fantini J, and Barrantes FJ (2011). Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci Rep-Uk 1, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batabyal D, Lewis-Ballester A, Yeh SR, and Poulos TL (2016). A Comparative Analysis of the Effector Role of Redox Partner Binding in Bacterial P450s. Biochemistry 55, 6517–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batabyal D, Richards LS, and Poulos TL (2017). Effect of Redox Partner Binding on Cytochrome P450 Conformational Dynamics. J Am Chem Soc 139, 13193–13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstein O, Biggin PC, and Sansom MSP (2001). A hydrophobic gating mechanism for nanopores. Journal of Physical Chemistry B 105, 12902–12905. [Google Scholar]
- Bernardi RC, Melo MC, and Schulten K (2015). Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochimica et biophysica acta 1850, 872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berneche S, and Roux B (2001). Energetics of ion conduction through the K+ channel. Nature 414, 73–77. [DOI] [PubMed] [Google Scholar]
- Bethel NP, and Grabe M (2016). Atomistic insight into lipid translocation by a TMEM16 scramblase. Proceedings of the National Academy of Sciences of the United States of America 113, 14049–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz RM (2017, August 1). Dabble (Version v2.6.3). Zenodo 10.5281/zenodo.836914 [DOI] [Google Scholar]
- Birkner JP, Poolman B, and Kocer A (2012). Hydrophobic gating of mechanosensitive channel of large conductance evidenced by single-subunit resolution. Proceedings of the National Academy of Sciences of the United States of America 109, 12944–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borhani DW, and Shaw DE (2012). The future of molecular dynamics simulations in drug discovery. Journal of Computer-Aided Molecular Design 26, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, et al. (2006). Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE conference on Supercomputing (ACM), p. 84. [Google Scholar]
- Bowman GR, Bolin ER, Hart KM, Maguire BC, and Marqusee S (2015). Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proceedings of the National Academy of Sciences of the United States of America 112, 2734–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan G, Henin J, Law R, Eckenhoff R, and Klein ML (2008). Embedded cholesterol in the nicotinic acetylcholine receptor. Proceedings of the National Academy of Sciences of the United States of America 105, 14418–14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. (2009). CHARMM: The Biomolecular Simulation Program. Journal of Computational Chemistry 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, and Adams PD (2002). Molecular dynamics applied to X-ray structure refinement. Accounts Chem Res 35, 404–412. [DOI] [PubMed] [Google Scholar]
- Buch I, Giorgino T, and De Fabritiis G (2011). Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proceedings of the National Academy of Sciences of the United States of America 108, 10184–10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL, and Garcia KC (2015). Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Cerutti DS, Cheatham III TE, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Greene D, Homeyer N, et al. (2017). AMBER 2017 (University of California, San Francisco). [Google Scholar]
- Chevalier A, Silva DA, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam KH, Yao GR, et al. (2017). Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera JD, Mobley DL, Shirts MR, Dixon RW, Branson K, and Pande VS (2011). Alchemical free energy methods for drug discovery: progress and challenges. Curr Opin Struct Biol 21, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ, Tiwary R, Borrelli K, Feng SL, Miller EB, Abel R, Friesner RA, and Berne BJ (2016). Prediction of Protein Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. Journal of Chemical Theory and Computation 12, 2990–2998. [DOI] [PubMed] [Google Scholar]
- Coleman JA, Green EM, and Gouaux E (2016). X-ray structures and mechanism of the human serotonin transporter. Nature 532, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, and Lindsley CW (2009). Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales JF, Jogini V, Lewis A, Vasquez V, Cortes DM, Roux B, and Perozo E (2007). Molecular driving forces determining potassium channel slow inactivation. Nat Struct Mol Biol 14, 1062–1069. [DOI] [PubMed] [Google Scholar]
- Cuello LG, Cortes DM, and Perozo E (2017). The gating cycle of a K+ channel at atomic resolution. Elife 6, e28032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, Pan AC, Gagnon DG, Dalmas O, Cordero-Morales JF, Chakrapani S, Roux B, and Perozo E (2010). Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature 466, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe GB, Musgaard M, Aurousseau MRP, Nayeem N, Green T, Biggin PC, and Bowie D (2016). Distinct Structural Pathways Coordinate the Activation of AMPA Receptor-Auxiliary Subunit Complexes. Neuron 89, 1264–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot BL, and Grubmüller H (2001). Water pe rmeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science 294, 2353–2357. [DOI] [PubMed] [Google Scholar]
- de Oliveira CAF, Hamelberg D, and McCammon JA (2006). On the application of accelerated molecular dynamics to liquid water simulations. Journal of Physical Chemistry B 110, 22695–22701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, and Dobson CM (2005). Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. Journal of the American Chemical Society 127, 476–477. [DOI] [PubMed] [Google Scholar]
- Delemotte L, Tarek M, Klein ML, Amaral C, and Treptow W (2011). Intermediate states of the Kv1.2 voltage sensor from atomistic molecular dynamics simulations. Proceedings of the National Academy of Sciences of the United States of America 108, 6109–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakshnamoorthy B, Raychaudhury S, Blachowicz L, and Roux B (2010). Cation-selective pathway of OmpF porin revealed by anomalous X-ray diffraction. J Mol Biol 396, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson A, Tiwary P, and Vashisth H (2017). Kinetics of Ligand Binding Through Advanced Computational Approaches: A Review. Curr Top Med Chem 17, 2626–2641. [DOI] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, and Shaw DE (2011a). Activation mechanism of the beta2-adrenergic receptor. Proceedings of the National Academy of Sciences of the United States of America 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, et al. (2013). Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299. [DOI] [PubMed] [Google Scholar]
- Dror RO, Mildorf TJ, Hilger D, Manglik A, Borhani DW, Arlow DH, Philippsen A, Villanueva N, Yang Z, Lerch MT, et al. (2015). Structural basis for nucleotide exchange in heterotrimeric G proteins. Science 348, 1361–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, and Shaw DE (2011b). Pathway and mechanism of drug binding to G-protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America 108, 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant JD, and McCammon JA (2011). Molecular dynamics simulations and drug discovery. BMC Biol 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao YT, Beauchamp KA, Wang LP, Simmonett AC, Harrigan MP, Stern CD, et al. (2017). OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS computational biology 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, Jullie D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita JB, Dror RO, and von Zastrow M (2018). Catalytic activation of beta-arrestin by GPCRs. Nature 557, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erokhova L, Horner A, Ollinger N, Siligan C, and Pohl P (2016). The Sodium Glucose Cotransporter SGLT1 Is an Extremely Efficient Facilitator of Passive Water Transport. J Biol Chem 291, 9712–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts B, Pramod AB, Sandtner W, Burbach N, Bulling S, Sitte HH, and Henry LK (2014). The two Na+ sites in the human serotonin transporter play distinct roles in the ion coupling and electrogenicity of transport. J Biol Chem 289, 1825–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Leiro R, and Scheres SH (2016). Unravelling biological macromolecules with cryo-electron microscopy. Nature 537, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields JB, Hollingsworth SA, Chreifi G, Heyden M, Arce AP, Magana-Garcia HI, Poulos TL, and Tobias DJ (2015). “Bind and Crawl” Association Mechanism of Leishmania major Peroxidase and Cytochrome c Revealed by Brownian and Molecular Dynamics Simulations. Biochemistry 54, 7272–7282. [DOI] [PubMed] [Google Scholar]
- Fields JB, Nemeth-Cahalan KL, Freites JA, Vorontsova I, Hall JE, and Tobias DJ (2017). Calmodulin Gates Aquaporin 0 Permeability through a Positively Charged Cytoplasmic Loop. J Biol Chem 292, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freites JA, Tobias DJ, and White SH (2006). A voltage-sensor water pore. Biophys J 91, L90–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh GB, Hulbert BS, Zhou HQ, and Brooks CL (2014). Constant pH molecular dynamics of proteins in explicit solvent with proton tautomerism. Proteins-Structure Function and Bioinformatics 82, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen T, Sliz P, Kistler J, Cheng Y, and Walz T (2004). Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature 429, 193–197. [DOI] [PubMed] [Google Scholar]
- González A, Perez-Acle T, Pardo L, and Deupi X (2011). Molecular Basis of Ligand Dissociation in beta-Adrenergic Receptors. Plos One 6, e23815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goricanec D, Stehle R, Egloff P, Grigoriu S, Pluckthun A, Wagner G, and Hagn F (2016). Conformational dynamics of a G-protein alpha subunit is tightly regulated by nucleotide binding. Proceedings of the National Academy of Sciences of the United States of America 113, E3629–E3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabe M, Lai HC, Jain M, Jan YN, and Jan LY (2007). Structure prediction for the down state of a potassium channel voltage sensor. Nature 445, 550–553. [DOI] [PubMed] [Google Scholar]
- Groban ES, Narayanan A, and Jacobson MP (2006). Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS computational biology 2, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Shrivastava IH, Amara SG, and Bahar I (2009). Molecular simulations elucidate the substrate translocation pathway in a glutamate transporter. Proceedings of the National Academy of Sciences of the United States of America 106, 2589–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelberg D, Mongan J, and McCammon JA (2004). Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J Chem Phys 120, 11919–11929. [DOI] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu CJ, Reboul M, Xiang JY, Wang LL, Lupyan D, Dahlgren MK, Knight JL, et al. (2016). OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. Journal of Chemical Theory and Computation 12, 281–296. [DOI] [PubMed] [Google Scholar]
- Harpole TJ, and Delemotte L (2018). Conformational landscapes of membrane proteins delineated by enhanced sampling molecular dynamics simulations. Bba-Biomembranes 1860, 909–926. [DOI] [PubMed] [Google Scholar]
- Hazuda DJ, Anthony NJ, Gomez RP, Jolly SM, Wai JS, Zhuang LH, Fisher TE, Embrey M, Guare JP, Egbertson MS, et al. (2004). A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proceedings of the National Academy of Sciences of the United States of America 101, 11233–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrion U, Renhorn J, Borjesson SI, Nelson EM, Schwaiger CS, Bjelkmar P, Wallner B, Lindahl E, and Elinder F (2012). Tracking a complete voltage-sensor cycle with metal-ion bridges. Proceedings of the National Academy of Sciences of the United States of America 109, 8552–8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertig S, Latorraca NR, and Dror RO (2016). Revealing Atomic-Level Mechanisms of Protein Allostery with Molecular Dynamics Simulations. PLoS computational biology 12, e1004746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger D, Masureel M, and Kobilka BK (2018). Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 25, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth SA, Batabyal D, Nguyen BD, and Poulos TL (2016). Conformational selectivity in cytochrome P450 redox partner interactions. Proceedings of the National Academy of Sciences of the United States of America 113, 8723–8728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth SA, and Poulos TL (2015). Molecular dynamics of the P450cam-Pdx complex reveals complex stability and novel interface contacts. Protein Sci 24, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou TJ, Wang JM, Li YY, and Wang W (2011). Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. Journal of Chemical Information and Modeling 51, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmuller H, and MacKerell AD (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, et al. (2015). Structural insights into micro-opioid receptor activation. Nature 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im W, and Roux B (2002a). Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J Mol Biol 322, 851–869. [DOI] [PubMed] [Google Scholar]
- Im W, and Roux B (2002b). Ions and counterions in a biological channel: a molecular dynamics simulation of OmpF porin from Escherichia coli in an explicit membrane with 1 M KCl aqueous salt solution. J Mol Biol 319, 1177–1197. [DOI] [PubMed] [Google Scholar]
- Jensen MØ, Jogini V, Borhani DW, Leffler AE, Dror RO, and Shaw DE (2012). Mechanism of voltage gating in potassium channels. Science 336, 229–233. [DOI] [PubMed] [Google Scholar]
- Jiang T, Yu K, Hartzell HC, and Tajkhorshid E (2017). Lipids and ions traverse the membrane by the same physical pathway in the nhTMEM16 scramblase. Elife 6, e28671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, and Im W (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Kappel K, Miao Y, and McCammon JA (2015). Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q Rev Biophys 48, 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M, and McCammon JA (2002). Molecular dynamics simulations of biomolecules. Nature structural biology 9, 646–652. [DOI] [PubMed] [Google Scholar]
- Kenakin T, and Christopoulos A (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12, 205–216. [DOI] [PubMed] [Google Scholar]
- Khafizov K, Perez C, Koshy C, Quick M, Fendler K, Ziegler C, and Forrest LR (2012). Investigation of the sodium-binding sites in the sodium-coupled betaine transporter BetP. Proceedings of the National Academy of Sciences of the United States of America 109, E3035–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandogin J, and Brooks CL 3rd (2007). Linking folding with aggregation in Alzheimer’s beta-amyloid peptides. Proceedings of the National Academy of Sciences of the United States of America 104, 16880–16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, and Pastor RW (2010). Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. Journal of Physical Chemistry B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, et al. (2018). Structure of the mu-opioid receptor-Gi protein complex. Nature 558, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepkiy D, Mihailescu M, Freites JA, Schow EV, Worcester DL, Gawrisch K, Tobias DJ, White SH, and Swartz KJ (2009). Structure and hydration of membranes embedded with voltage-sensing domains. Nature 462, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, et al. (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Durek T, Dang B, Finol-Urdaneta RK, Craik DJ, Kent SB, French RJ, Bezanilla F, and Correa AM (2017). Mapping of voltage sensor positions in resting and inactivated mammalian sodium channels by LRET. Proceedings of the National Academy of Sciences of the United States of America 114, E1857–E1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laio A, and Gervasio FL (2008). Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep Prog Phys 71, 126601. [Google Scholar]
- Latorraca NR, Fastman NM, Venkatakrishnan AJ, Frommer WB, Dror RO, and Feng L (2017). Mechanism of Substrate Translocation in an Alternating Access Transporter. Cell 169, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, and Dror RO (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kang HJ, von Ballmoos C, Newstead S, Uzdavinys P, Dotson DL, Iwata S, Beckstein O, Cameron AD, and Drew D (2013). A two-domain elevator mechanism for sodium/proton antiport. Nature 501, 573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M, and Lifson S (1969). Refinement of Protein Conformations Using a Macromolecular Energy Minimization Procedure. Journal of Molecular Biology 46, 269–279. [DOI] [PubMed] [Google Scholar]
- Li J, Shaikh SA, Enkavi G, Wen PC, Huang Z, and Tajkhorshid E (2013). Transient formation of water-conducting states in membrane transporters. Proceedings of the National Academy of Sciences of the United States of America 110, 7696–7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wanderling S, Paduch M, Medovoy D, Singharoy A, McGreevy R, Villalba-Galea CA, Hulse RE, Roux B, Schulten K, et al. (2014). Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain. Nat Struct Mol Biol 21, 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Swanson JM, Madsen JJ, Hong M, DeGrado WF, and Voth GA (2016). Acid activation mechanism of the influenza A M2 proton channel. Proceedings of the National Academy of Sciences of the United States of America 113, E6955–E6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifson S, and Warshel A (1968). Consistent Force Field for Calculations of Conformations Vibrational Spectra and Enthalpies of Cycloalkane and N-Alkane Molecules. J Chem Phys 49, 5116–5129. [Google Scholar]
- Lin JH, Perryman AL, Schames JR, and McCammon JA (2002). Computational drug design accommodating receptor flexibility: The relaxed complex scheme. Journal of the American Chemical Society 124, 5632–5633. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, and Vendruscolo M (2005). Simultaneous determination of protein structure and dynamics. Nature 433, 128–132. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Maragakis P, Piana S, Eastwood MP, Dror RO, and Shaw DE (2012). Systematic Validation of Protein Force Fields against Experimental Data. Plos One 7, e32131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Dror RO, and Shaw DE (2011). How fast-folding proteins fold. Science 334, 517–520. [DOI] [PubMed] [Google Scholar]
- Liou SH, Myers WK, Oswald JD, Britt RD, and Goodin DB (2017). Putidaredoxin Binds to the Same Site on Cytochrome P450cam in the Open and Closed Conformation. Biochemistry 56, 4371–4378. [DOI] [PubMed] [Google Scholar]
- Liu YF, Ke M, and Gong HP (2015). Protonation of Glu(135) Facilitates the Outward-to-Inward Structural Transition of Fucose Transporter. Biophysical Journal 109, 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Sigler PB, Xu Z, and Karplus M (2000). A dynamic model for the allosteric mechanism of GroEL. J Mol Biol 302, 303–313. [DOI] [PubMed] [Google Scholar]
- Mager T, Rimon A, Padan E, and Fendler K (2011). Transport mechanism and pH regulation of the Na+/H+ antiporter NhaA from Escherichia coli: an electrophysiological study. J Biol Chem 286, 23570–23581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hubner H, et al. (2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragliano L, and Vanden-Eijnden E (2006). A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem Phys Lett 426, 168–175. [Google Scholar]
- Marrink SJ, and Tieleman DP (2013). Perspective on the Martini model. Chem Soc Rev 42, 6801–6822. [DOI] [PubMed] [Google Scholar]
- McCammon JA, Gelin BR, and Karplus M (1977). Dynamics of Folded Proteins. Nature 267, 585–590. [DOI] [PubMed] [Google Scholar]
- McCorvy JD, Butler KV, Kelly B, Rechsteiner K, Karpiak J, Betz RM, Kormos BL, Shoichet BK, Dror RO, Jin J, and Roth BL (2018). Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat Chem Biol 14, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang L-P, Lane TJ, and Pande VS (2015). MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophysical journal 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor DL (2007). The neurobiologist’s guide to structural biology: A primer on why macromolecular structure matters and how to evaluate structural data. Neuron 54, 511–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirjalili V, and Feig M (2013). Protein Structure Refinement through Structure Selection and Averaging from Molecular Dynamics Ensembles. Journal of Chemical Theory and Computation 9, 1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley DL, and Dill KA (2009). Binding of small-molecule ligands to proteins:”what you see” is not always “what you get”. Structure 17, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AH, Beuming T, Banala AK, Donthamsett P, Pongetti K, LaBounty A, Levy B, Cao JJ, Michino M, Luedtke RR, et al. (2012). Molecular Determinants of Selectivity and Efficacy at the Dopamine D3 Receptor. Journal of Medicinal Chemistry 55, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, and Hall CK (2004). Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proceedings of the National Academy of Sciences of the United States of America 101, 16180–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivedha AK, Tautermann CS, Bhattacharya S, Lee S, Casarosa P, Kollak I, Kiechle T, and Vaidehi N (2018). Identifying Functional Hotspot Residues for Biased Ligand Design in G-Protein-Coupled Receptors. Molecular pharmacology 93, 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostmeyer J, Chakrapani S, Pan AC, Perozo E, and Roux B (2013). Recovery from slow inactivation in K+ channels is controlled by water molecules. Nature 501, 121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulino C, Neldner Y, Lam AKM, Kalienkova V, Brunner JD, Schenck S, and Dutzler R (2017). Structural basis for anion conduction in the calcium-activated chloride channel TMEM16A. Elife 6, e26232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez A, Morrone JA, Simmerling C, and Dill KA (2016). Advances in free-energy-based simulations of protein folding and ligand binding. Current opinion in structural biology 36, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C, Faust B, Mehdipour AR, Francesconi KA, Forrest LR, and Ziegler C (2014). Substrate-bound outward-open state of the betaine transporter BetP provides insights into Na+ coupling. Nat Commun 5, 4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. Journal of Computational Chemistry 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevost MS, Sauguet L, Nury H, Van Renterghem C, Huon C, Poitevin F, Baaden M, Delarue M, and Corringer PJ (2012). A locally closed conformation of a bacterial pentameric proton-gated ion channel. Nat Struct Mol Biol 19, 642–649. [DOI] [PubMed] [Google Scholar]
- Provasi D, Artacho MC, Negri A, Mobarec JC, and Filizola M (2011). Ligand-Induced Modulation of the Free-Energy Landscape of G Protein-Coupled Receptors Explored by Adaptive Biasing Techniques. PLoS computational biology 7, e1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Winther AM, Shi L, Nissen P, Weinstein H, and Javitch JA (2009). Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proceedings of the National Academy of Sciences of the United States of America 106, 5563–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson NA, Farr GW, Roseman AM, Gowen B, Fenton WA, Horwich AL, and Saibil HR (2001). ATP-bound states of GroEL captured by cryo-electron microscopy. Cell 107, 869–879. [DOI] [PubMed] [Google Scholar]
- Raval A, Piana S, Eastwood MP, Dror RO, and Shaw DE (2012). Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins-Structure Function and Bioinformatics 80, 2071–2079. [DOI] [PubMed] [Google Scholar]
- Robustelli P, Piana S, and Shaw DE (2018). Developing a molecular dynamics force field for both folded and disordered protein states. Proceedings of the National Academy of Sciences of the United States of America 115, E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DR, and Cheatham III TE (2013). PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. Journal of chemical theory and computation 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013). Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. Journal of Chemical Theory and Computation 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, and Sherman W (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Schames JR, Henchman RH, Siegel JS, Sotriffer CA, Ni H, and McCammon JA (2004). Discovery of a novel binding trench in HIV integrase. J Med Chem 47, 1879–1881. [DOI] [PubMed] [Google Scholar]
- Schlitter J, Engels M, and Kruger P (1994). Targeted Molecular-Dynamics - a New Approach for Searching Pathways of Conformational Transitions. Journal of Molecular Graphics 12, 84–89. [DOI] [PubMed] [Google Scholar]
- Schmidtke P, Luque FJ, Murray JB, and Barril X (2011). Shielded Hydrogen Bonds as Structural Determinants of Binding Kinetics: Application in Drug Design. Journal of the American Chemical Society 133, 18903–18910. [DOI] [PubMed] [Google Scholar]
- Schwantes CR, McGibbon RT, and Pande VS (2014). Markov models for long-timescale biomolecular dynamics. J Chem Phys 141, 090901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn HM, and Thiel W (2009). QM/MM Methods for Biomolecular Systems. Angew Chem Int Edit 48, 1198–1229. [DOI] [PubMed] [Google Scholar]
- Shan YB, Kim ET, Eastwood MP, Dror RO, Seeliger MA, and Shaw DE (2011). How Does a Drug Molecule Find Its Target Binding Site? Journal of the American Chemical Society 133, 9181–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw DE, Deneroff MM, Dror RO, Kuskin JS, Larson RH, Salmon JK, Young C, Batson B, Bowers KJ, Chao JC, et al. (2008). Anton, a special-purpose machine for molecular dynamics simulation. Communications of the ACM 51, 91–97. [Google Scholar]
- Shaw DE, Grossman JP, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, et al. (2014). Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis (IEEE Press), pp. 41–53. [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, and Javitch JA (2008). The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell 30, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet BK (2004). Virtual screening of chemical libraries. Nature 432, 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjærven L, Yao XQ, Scarabelli G, and Grant BJ (2014). Integrating protein structural dynamics and evolutionary analysis with Bio3D. Bmc Bioinformatics 15, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow CD, Nguyen H, Pande VS, and Gruebele M (2002). Absolute comparison of simulated and experimental protein-folding dynamics. Nature 420, 102–106. [DOI] [PubMed] [Google Scholar]
- Spahn V, Del Vecchio G, Labuz D, Rodriguez-Gaztelumendi A, Massaly N, Temp J, Durmaz V, Sabri P, Reidelbach M, Machelska H, et al. (2017). A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 355, 966–969. [DOI] [PubMed] [Google Scholar]
- Stone JE, Hallock MJ, Phillips JC, Peterson JR, Luthey-Schulten Z, and Schulten K (2016). Evaluation of Emerging Energy-Efficient Heterogeneous Computing Platforms for Biomolecular and Cellular Simulation Workloads. Ieee Sym Para Distr, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y, and Okamoto Y (1999). Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett 314, 141–151. [Google Scholar]
- Suomivuori CM, Gamiz-Hernandez AP, Sundholm D, and Kaila VRI (2017). Energetics and dynamics of a light-driven sodium-pumping rhodopsin. Proceedings of the National Academy of Sciences of the United States of America 114, 7043–7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajkhorshid E, Nollert P, Jensen MØ, Miercke LJW, O’Connell J, Stroud RM, and Schulten K (2002). Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science 296, 525–530. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Kato HE, Koyama M, Ito J, Kamiya M, Hayashi S, Maturana AD, Deisseroth K, Ishitani R, and Nureki O (2015). Molecular Dynamics of Channelrhodopsin at the Early Stages of Channel Opening. Plos One 10, e0131094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YS, Sledz P, Lang S, Stubbs CJ, Spring DR, Abell C, and Best RB (2012). Using Ligand-Mapping Simulations to Design a Ligand Selectively Targeting a Cryptic Surface Pocket of Polo-Like Kinase 1. Angew Chem Int Edit 51, 10078–10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornroth-Horsefield S, Wang Y, Hedfalk K, Johanson U, Karlsson M, Tajkhorshid E, Neutze R, and Kjellbom P (2006). Structural mechanism of plant aquaporin gating. Nature 439, 688–694. [DOI] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Mitra K, Frank J, and Schulten K (2008). Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, and Sobolevsky AI (2016). Elucidation of AMPA receptor-stargazin complexes by cryo-electron microscopy. Science 353, 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udier-Blagovic M, Tirado-Rives J, and Jorgensen WL (2003). Validation of a model for the complex of HIV-1 reverse transcriptase with nonnucleoside inhibitor TMC125. Journal of the American Chemical Society 125, 6016–6017. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, and MacKerell AD Jr (2012). Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. Journal of chemical information and modeling 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas E, Bezanilla F, and Roux B (2011). In search of a consensus model of the resting state of a voltage-sensing domain. Neuron 72, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Crombie AL, Soergel DG, and Lark MW (2014). Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci 35, 308–316. [DOI] [PubMed] [Google Scholar]
- Wacker D, Stevens RC, and Roth BL (2017a). How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, et al. (2017b). Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389 e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, et al. (2015). Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc 137, 2695–2703. [DOI] [PubMed] [Google Scholar]
- Watanabe A, Choe S, Chaptal V, Rosenberg JM, Wright EM, Grabe M, and Abramson J (2010). The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature 468, 988–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, and Shea JE (2013). Structural Similarities and Differences between Amyloidogenic and Non-Amyloidogenic Islet Amyloid Polypeptide (IAPP) Sequences and Implications for the Dual Physiological and Pathological Activities of These Peptides. PLoS computational biology 9, e1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuthen T, Gorraitz E, Her K, Wright EM, and Loo DDF (2016). Structural and functional significance of water permeation through cotransporters. Proceedings of the National Academy of Sciences of the United States of America 113, E6887–E6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Jara-Oseguera A, Chang TH, Bae C, Hanson SM, and Swartz KJ (2018). Heat activation is intrinsic to the pore domain of TRPV1. Proceedings of the National Academy of Sciences of the United States of America 115, E317–E324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao GP, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning JY, Ahn J, Gronenborn AM, Schulten K, Aiken C, and Zhang PJ (2013). Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497, 643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Chen S, Yoshioka C, Baconguis I, and Gouaux E (2016). Architecture of fully occupied GluA2 AMPA receptor-TARP complex elucidated by cryo-EM. Nature 536, 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HE, Kim YS, Paggi JM, Evans KE, Allen WE, Richardson C, Inoue K, Ito S, Ramakrishnan C, Fenno LE, Yamashita K, Hilger D, Lee SY, Berndt A, Shen K, Kandori C, Dror RO, Kobilka BK and Deisseroth K (2018). Structural mechanisms of selectivity and gating in anion channelrhodopsins. Nature doi: 10.1038/s41586-018-0504-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl LS, Fowler PW, Sansom MS and Beckstein O Flexible gates generate occluded intermediates in the transport cycle of LacY. J. Mol. Biol 2014; 426: 735–751 [DOI] [PMC free article] [PubMed] [Google Scholar]