

Abstract
The mechanism of permanganate-mediated dual C–H oxidation of complex diketopiperazines has been examined with density functional theory computations. The products of these oxidations are enabling intermediates in the synthesis of structurally diverse ETP natural products. We evaluated, for the first time, the impact of ion-pairing and aggregation states of the permanganate ion and counter-cations, such as bis(pyridine)-silver(I) (Ag+) and tetra-n-butyl ammonium (TBA+), on the C–H oxidation mechanism. The C–H abstraction occurs through an open shell singlet species, as noted previously, followed by O-rebound and a competing OH-rebound pathway. The second C–H oxidation proceeds with a second equivalent of oxidant with lower free energy barriers than the first C–H oxidation due to directing effects and the generation of a more reactive oxidant species after the first C–H oxidation. The success and efficiency of the second CH oxidation is found to be critically dependent on the presence of an ion-paired oxidant. We used the developed mechanistic knowledge to rationalize an experimentally observed oxidation pattern for C3-indole substituted diketopiperazine (+)-5 under optimal oxidation conditions: namely, the formation of diol (−)-6 as a single diastereomer and lack of the ketone products. We proposed two factors that may impede the ketone formation: (i) the conformational flexibility of the diketopiperazine ring, and (ii) hindrance of this site, making it less accessible to the ion-paired oxidant species.
Graphical Abstract
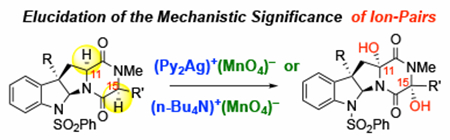
Introduction
Diketopiperazines are important building blocks for the synthesis of epipolythiodiketopiperazine (ETP) alkaloids,1 which are a diverse and structurally complex class of natural products.2–3 ETPs are of significant interest to the scientific community because of their synthetically challenging architecture and potent biological activities due to their unique polysulfane motif.4 Several strategies for converting complex diketopiperazines into the corresponding ETPs have been developed by the Movassaghi group.1, 5–10 A critical step in these ETP syntheses is the oxidation of the α-C–H bonds of diketopiperazine precursors with permanganate salts, bis(pyridine)-silver(I) permanganate (1)11 and tetra-n-butyl ammonium permanganate (2).12 This transformation is highly efficient and selective, providing di- and tetra-hydroxylation on monomeric and dimeric diketopiperazine substrates, respectively, to afford products as a single diastereomer with stereoretention at the α-position (e.g., (+)-3 → (+)-4 and (+)-5 → (−)-6, see Scheme 1).13 As such, this strategy has greatly increased the synthetic accessibility of complex ETP natural products, such as (+)-dideoxyverticilin A,6 and (+)-gliocladin B.14
Scheme 1.
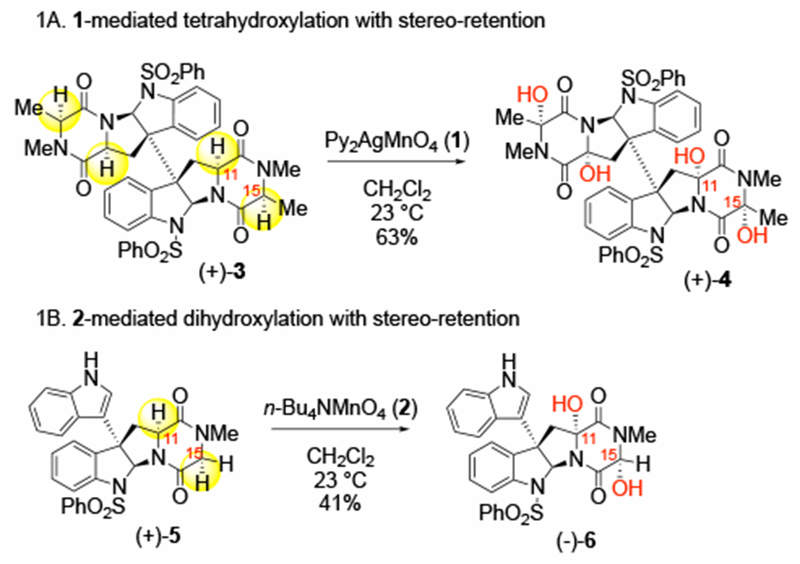
Representative hydroxylations of complex dimeric diketopiperazines enabling the synthesis of complex ETPs.
Extensive mechanistic studies of alkyl C–H oxidation by the permanganate ion have determined that the reaction proceeds through i) rate-limiting cleavage of the C–H bond either through hydrogen atom transfer (HAT) or hydride transfer, ii) coupling of the reduced Mn species and oxidized C center (i.e., radical or carbocation), which is often called the O-rebound step, and iii) hydrolysis of the resulting permanganate ester to alcohols (Figure 1).15–18 Mayer and coworkers have demonstrated that the character of the C–H cleavage step depends on the reaction environment: the C–H cleavage of toluene by potassium permanganate in water proceeds through a hydride transfer mechanism whereas the same process with 2 in toluene proceeds through a radical mechanism.17 The ability of the d0 MnVII permanganate ion—which has a closed-shell singlet ground state—to engage in HAT reactivity is attributed to the formation of strong O–H bonds in the reduced Mn species.16–19 It was also shown that the enthalpy of activation (ΔH‡) and reaction enthalpy (ΔH) for several aryl alkane substrates reacting with 2 fit a linear relationship (Evans-Polanyi)16 implicating bond dissociation energy (BDE) as a reasonable indicator of C–H bond reactivity within classes of substrates.
Figure 1.
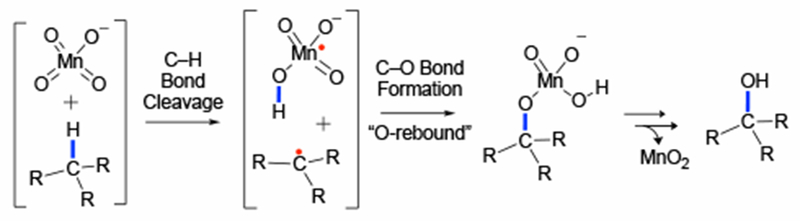
Previously proposed mechanism for alkyl C–H oxidation by permanganate ion. For the sake of simplicity, here we present only the hydrogen atom transfer (HAT) mechanism, while the reaction can also proceed via the hydride transfer pathway (see text).
However, this mechanism fails to explain several outcomes for permanganate mediated C–H oxidation in various complex diketopiperazine substrates, such as observation of partial oxidation (e.g., monohydroxylation), cases of either mono or double oxidation of methylenes, the impact of stereochemistry on the observed oxidation level, stereo-inversion (instead of typically observed stereo-retention), as well as the effect of the oxidant (e.g., 1 or 2) and solvent on the reaction outcomes. We therefore set out to evaluate the C–H oxidation mechanism for complex substrates in more details in order to gain insight into the source of these unexpected findings. Here, we use density functional theory (DFT) calculations to address the following questions: i) What is the mechanism for dual oxidation of the diketopiperazines? ii) How critical is the proximity of the counter-cation to the oxidant (i.e., through ion-pairing) and how do the counter-cations like bis(pyridine)-silver(I) (Py2Ag+) and tetra-n-butyl ammonium (TBA+) affect the reaction outcomes? and iii) What factors lead to mono-oxidation at the C15 position of (+)-5? (Scheme 1B) It is expected that expanding our atomistic-level understanding of the mechanism of this vital oxidation reaction to more complex settings will allow us to develop predictive models and will advance our ability for synthesis of valuable natural products from diketopiperazines.
Computational methodology.
Geometry optimizations and frequency calculations were performed with the Gaussian 09 suite of programs20 at the B3LYP-D3/[6-31G(d,p) + Lanl2dz (Mn)] level of theory (called as B3LYP-D3/BS1) with the corresponding Hay-Wadt effective core potentials21–23 for Mn and Grimme’s empirical dispersion-correction for B3LYP.24 Frequency analysis is used to characterize each minimum with zero imaginary frequencies and each transition state (TS) structure with only one imaginary frequency. Intrinsic reaction coordinate (IRC) calculations were performed for selected TSs to identify associated reactants and products.
Bulk solvent effects are incorporated in all calculations (optimization of geometries, frequency and energy calculations) at the self-consistent reaction field polarizable continuum model (IEF-PCM) level of theory and with dichloromethane (DCM) as the solvent.25–27 The final electronic energies were re-computed at the B3LYP-D3/[6-311+G(d,p) + LANL2DZ (Mn)] level of theory (called as B3LYP-D3/BS2) by utilizing the geometries optimized at the B3LYP-D3/BS1 level. Zero point energies, thermal corrections, and entropies for the free energy and enthalpy were calculated at the B3LYP-D3/BS1 level of theory and corrected to a solution standard state of 1M at 298.15 K.28 These corrections were then applied to the energies calculated at the B3LYP-D3/BS2 level to afford the free energy and enthalpy values discussed in the text.
Most of the TSs and intermediates on the singlet potential energy surface have a lower energy open-shell singlet electronic state. Some calculated triplet states have large spin contamination from the high-energy quintet electronic state.
Bond dissociation enthalpies (BDE) for C–H bonds are computed as BDE(Sub-C–H) = [H(Sub-C·) + H(H·)] − H(Sub-C–H),29 at the B3LYP-D3/BS2 level of theory (here, H stands for enthalpy). The enthalpy of the hydrogen atom, H(H·), was set to its exact value of −0.5 hartree.30 The computed BDE of the benzylic C–H bond of toluene at this level of theory is 87.5 kcal/mol, which is in reasonable agreement with experimental value (88.4 kcal/mol)31 and those previously computed (89.8 kcal/mol).29
Results and Discussion
Nature of the oxidant.
The electronic structure of permanganate ion (i.e. MnO4−) has been the subject of extensive analyses.32–35 In general, it is found that the triplet state of MnO4− is very high in energy relative to its closed shell singlet ground state (~51 kcal/mol experimentally).16 However, the B3LYP/6-311+G(d) calculations by Strassner and Houk provided a singlet-triplet energy difference of 20.8 kcal/mol for the permanganate ion.18 We have likewise found values in the range of ~17-35 kcal/mol with various DFT methods. (See SI) This indicates that popular DFT methods significantly underestimate the singlet-triplet energy splitting for the permanganate ion.36–37 Thus, in order to eliminate potential uncertainty with the DFT methods used in this study, we focus on relative trends in reactivity for C–H oxidation of diketopiperazine substrates rather than their absolute values.
The next important question is the coordination environment and speciation of the permanganate oxidants in the reported oxidation reactions with 1 and 2. It is generally thought that the counter-ions associated with these oxidants (Ag+ and TBA+, respectively) expand the scope of permanganate oxidations by increasing solubility in organic solvents.38 Additionally, it is known that quaternary ammonium permanganates (including 2) form aggregates and/or ion-pairs in nonpolar solvents like dichloromethane.39 It is also known that Lewis acids such as BF3 can form strong complexes with permanganate that affect the rate of oxidation.40–41 We, therefore, became interested in assessing the role of the counter-cation on the reactivity and selectivity for C–H oxidation. For this purpose, we use five model oxidants in our analysis: (a) the “naked” MnO4− anion (N), (b) the [(Py2Ag)+–(MnO4)−] and [(Py2Ag)+–(MnO4)−]2 ion-pairs, i.e. monomeric (M1) and dimeric (D1) forms of 1, and (c) the [(n-Bu4N)+–(MnO4)−] and [(n-Bu4N)+−(MnO4)−]2 ion-pairs, i.e. monomeric (M2) and dimeric (D2) forms of 2. (Figure 2) One should emphasize that previous computational studies on the reactivity of the permanganate ion in C–H oxidation18, 42–43 and other reactions44–47 typically use N as the model oxidant and do not study the role of ion-pairs.
Figure 2.
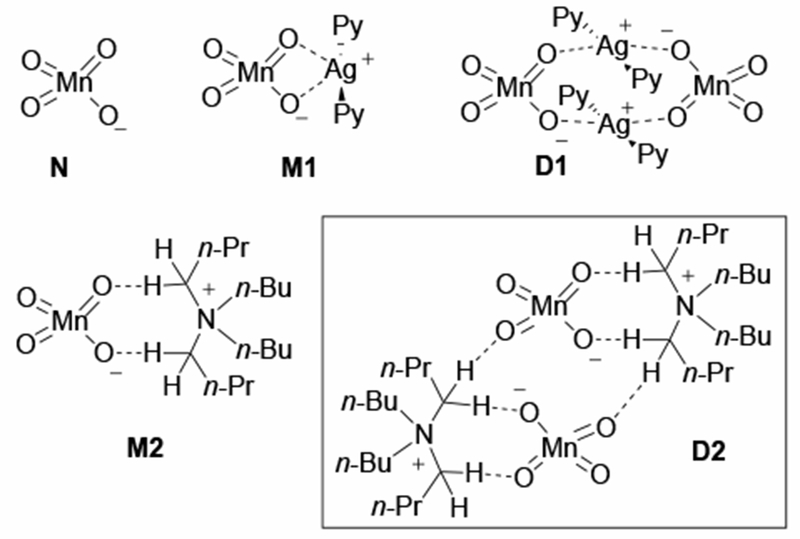
Model oxidants used in this study. Here, Py = pyridine.
Calculations presented in this paper show that the ion-complexation free energy for M1 and M2 are 10.9 and 6.2 kcal/mol, respectively. In these monomeric ion-pair species, either the silver atom of the Py2Ag+ counter-cation or the α-hydrogens of the TBA+ counter-cation directly interact with the permanganate oxygens (see Figure 2 and Supporting materials).48 The dimerization free energy for D1 and D2 are 18.1 and 12.8 kcal/mol, respectively. D1 has a sandwich-type structure with permanganate ions bridging the Py2Ag+ unit, whereas in D2 the permanganate ions are buried between two relatively flat TBA+ units (see Figure 2 and Supporting materials). The computed energetics indicate that aggregates (monomeric, dimeric or higher order) of ion-paired oxidants are likely to be present in the reaction mixture, at least, in non-polar solvents (such as dichloromethane). Therefore, herein we investigate the potential critical nature of the counter-cation and oxidant speciation on C–H oxidation by permanganate using computations for the first time.
Dual C–H oxidation of diketopiperazines.
We modeled the dimeric diketopiperazine substrate (+)-3 (see Scheme 1A) by replacing half of the dimer with a t-Bu group to simplify the calculations while maintaining the steric environment of the dimeric structure. The resulting model substrate, I, is given in Figure 3. Analysis of the optimized structure of I shows that the diketopiperazine ring adopts a boat conformation where the α-C–H bonds at C11 and C15 are in axial positions. Generally, the cis-amide bonds favor a planar ring structure, but proline-diketopiperazines, like those studied here, are known to favor a boat conformation.49
Figure 3.
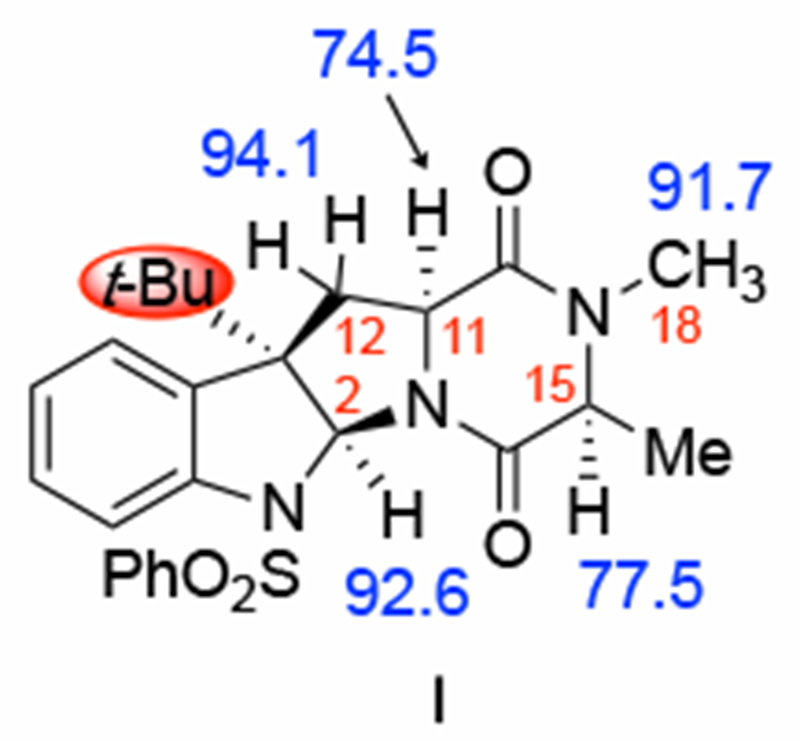
Model diketopiperazine I used for this mechanistic study and computed bond dissociation energies (BDEs) shown in blue and reported in kcal/mol.
We next assessed the strength of the unique C–H bonds in model species I by calculating their bond dissociation energies (BDEs). We find that the two weakest C–H bonds of I are C11–H (74.5 kcal/mol) and C15-H (77.5 kcal/mol), which is consistent with the experimentally observed oxidation pattern. The adjacent carbonyl and amide nitrogen groups stabilize the radical species through a capto-dative effect. While these computations indicate that the C11–H bond is weaker than the C15–H bond of model I, it is not known from experiments which position, C11 or C15, is oxidized first because no partial oxidation products are observed.
Comparison of the geometry and computed BDEs of the optimized structure of (+)-3 (obtained from the published crystal structure6) and I shows that the model accurately captures the features of the real compound. (See Supporting Materials.)
First C–H oxidation.
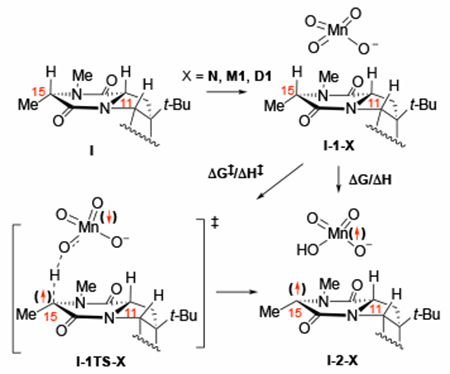
In studying the first C–H oxidation, we aimed to build upon the current understanding of the mechanism by emphasizing novelty and complexity introduced through the substrate and oxidants. Previous work18 utilizing “naked” oxidant N and simpler substrates, have identified two major steps of the reaction: C–H bond cleavage and C–O bond formation, which occurs via the fast O-rebound pathway (see Figure 1). Here, for our initial mechanistic study with I, we examine the mechanism and site-selectivity with oxidants X = N, M1, and D1. Furthermore, as a competing pathway to the previously reported the O-rebound mechanism for C–O bond formation, we also examined the OH-rebound mechanism. Thus, this section of the paper is designed not only to elucidate the mechanism of the dual C–H bond oxidation in complex model substrates, but also to identify the role of counter-cation (here we chose bis(pyridine)-silver(I), as an example) and aggregation states of the oxidant in the C–H oxidation reaction.
As mentioned above (and illustrated in Figure 4 for the case of X = N), the C–H oxidation reaction is initiated by coordination of the substrate I to oxidant (X) to form the pre-reaction complex I-1-X (see also scheme to Table 1. In our notations, roman numerals denote the model followed by the label for the structure on the potential energy surface and X to denote the oxidant). Calculations show that the permanganate ion, regardless of its aggregation state, has a closed-shell singlet ground state with higher-lying open-shell singlet and triplet states.50 As expected, association of singlet oxidant with substrate I also forms the pre-reaction complex I-1-X with the closed-shell singlet state. However, the following C–H cleavage transition state I-1TS-X has an open-shell singlet ground electronic state (i.e. HAT mechanism) with an α- and β-unpaired spins: its closed-shell singlet (i.e., hydride transfer mechanism) and triplet (i.e., radical mechanism with two unpaired α-spins) states are higher in energy. These findings allow us to conclude that the closed-shell and open-shell singlet state energy surfaces of the reaction cross before the C–H cleavage transition state (i.e. spin de-coupling occurs before the C–H bond breaks). Thus, these calculations support the C–H abstraction mechanism proposed by Mayer and coworkers.16, 19
Figure 4.
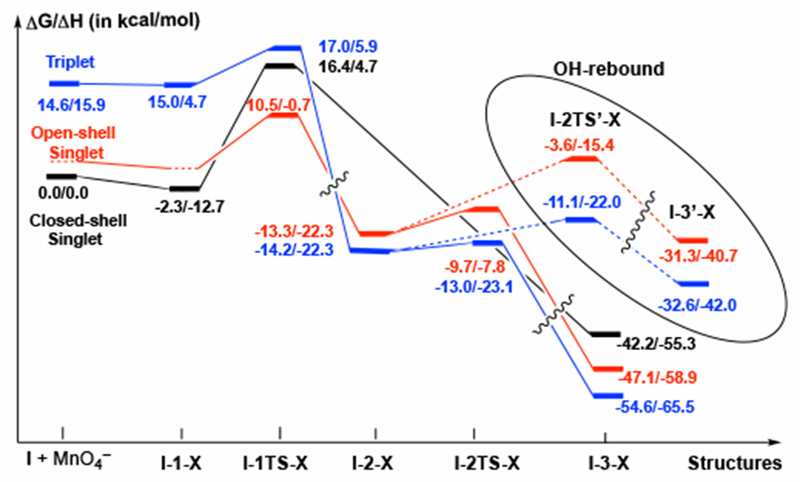
Schematic presentation (unscaled) of the free energy surface of the first C11–H oxidation. Relative energies are presented for the X = N oxidant. The calculated energies for the oxidants M1 and D1 are consistent and are given throughout the rest of this section.
Table 1.
The calculated energies for C–H abstraction at the C11 and C15 positions of I with model oxidants N, M1 and D1. For sake of simplicity, here the schematic reaction pathway is shown only for model oxidant N and for the C15–H oxidation.
| C11 | C15 | |||
|---|---|---|---|---|
| Ox. | ΔG‡/ΔH‡ | ΔG/ΔH | ΔG‡/ΔH‡ | ΔG/ΔH |
| N | 12.8/12.0 | −11.9/−9.5 | 12.0/10.4 | −8.5/−7.3 |
| M1 | 12.7/11.1 | −10.8/−10.6 | 10.1/8.0 | −9.1/−8.2 |
| D1 | 10.4/11.0 | −9.6/−7.0 | 11.3/12.1 | −6.7/−2.2 |
The product from I-1TS-X is a carbon centered radical and a reduced MnVI species HO-MnO3− (I-2-X). This intermediate has the triplet ground electronic state with energetically higher-lying open-shell singlet state. Examination of the energy surfaces suggest that spin-flip (i.e. singlet-triplet seam-of-crossing) is likely to occur either immediately after the transition state I-1TS-X or in the vicinity of the intermediate I-2-X. Regardless where the spin-flip occurs, this finding indicates that the lower energy triplet state I-2-X is likely the relevant intermediate at this stage of the reaction. On the closed-shell singlet surface, there is no intermediate like I-2-X, and the C–O bond formation via the O-rebound mechanism occurs without energy barrier.
Next, we discuss the important energy parameters for the site-selective (C11–H vs C15–H) C–H oxidation of I. In Table 1, we report the calculated barriers (ΔG‡/ΔH‡ I-1-X → I-1TS-X) and reaction energies (ΔG/ΔH, I-1-X → I-2-X) for C–H abstraction at both the C11 and C15 positions of I with the three model oxidants. In general, the presence of counter-cation (i.e. ion-paired oxidants) slightly lowers the C–H abstraction barriers relative to those with N. This can be attributed to the interaction between the Ag center and the permanganate oxygen that assumes spin during the reaction (Ag-O = 2.79 and 2.51 Å for M1 and D1 at C15, respectively) as shown in Figure 5.
Figure 5.
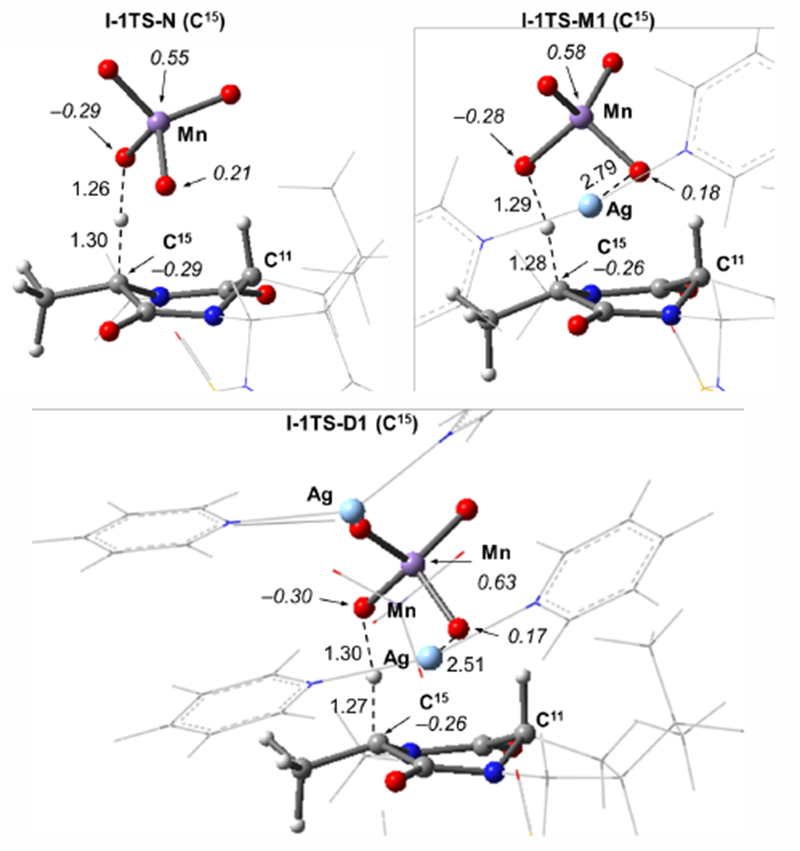
Optimized transition states for C–H abstraction at C15 (I-1TS-X) using the N, M1, and D1 model oxidants. Bond distances are in Å and Mulliken spin density values in |e| are shown in italics.
However, the trends based on the aggregation state are complex indicating the possibility of multiple steric and electronic contributions. The aggregation state of the oxidant also has a minor effect on the driving force of the reaction: the reaction with D1 is slightly less exergonic than that with N and M1.
However, the nature of the oxidant appears to have a larger effect on the site-selectivity of the reaction: With smaller oxidants N and M1, the oxidation at the C15 position is kinetically favored by 0.8 and 2.6 kcal/mol, respectively. In contrast, with D1, the oxidation at the C11 position is kinetically favored by 0.9 kcal/mol.
The patterns in site-selectivity are not intuitive as one might expect the larger oxidants to increasingly favor oxidation at the more accessible C–H bond at C15. However, the structure of the oxidant has an effect on the orientation of the MnO4− unit and consequently on its interactions with the substrate. For M1 and D1, the closest distance between the active MnO4− and the tert-butyl substituent at C3 is 2.29 and 2.40 Å, respectively. Therefore, the M1 oxidant is closer to the tert-butyl group of the substrate than D1 indicating the former produces worse steric interactions even though it is the smaller oxidant structure.
The computed reaction energies (Table 1) are consistent with the relative BDEs of C11–H and C15–H indicating that thermodynamics favor oxidation at C11. However, the computed barriers are not consistent with this trend indicating that there are factors based on the nature of oxidant and complexity of the substrate that can cause the kinetic trends to deviate from those expected from thermodynamics. This is an initial indication that information in addition to BDEs will be needed to fully understand the reactivity trends.
With the available computational data, it is difficult to make a definitive conclusion on which site (i.e. C11–H or C15–H) is oxidized first. However, it is clear that both sites should be reactive under the reaction conditions. For the consistency and simplicity of our discussion, below we present data for the rest of the reaction starting from oxidation at C15. The computational data for the reaction starting from C11 gives a consistent picture and is provided in the Supporting Information.
C–O bond formation.
We next examined C–O bond formation from the C–H abstraction product I-2-X (see Figure 4). This process may proceed via two competing pathways: (i) O-rebound pathway that involves coupling of the C15-radical center and HO–MnO3− through transition state 1-2TS-X to form a permanganate ester intermediate, I-3-X, and (ii) OH-rebound pathway that involves transfer of the OH group from HO–MnO3− to the C15-center through transition state 1-2TS’-X to form the alcohol product and a MnVO3− species (I-3’-X). (Figure 6) Assuming the triplet state I-2-X is the only relevant intermediate, both reactions occur entirely on the triplet energy surface although the open-shell singlet surface is only slightly higher in energy. (Figure 4)
Figure 6.
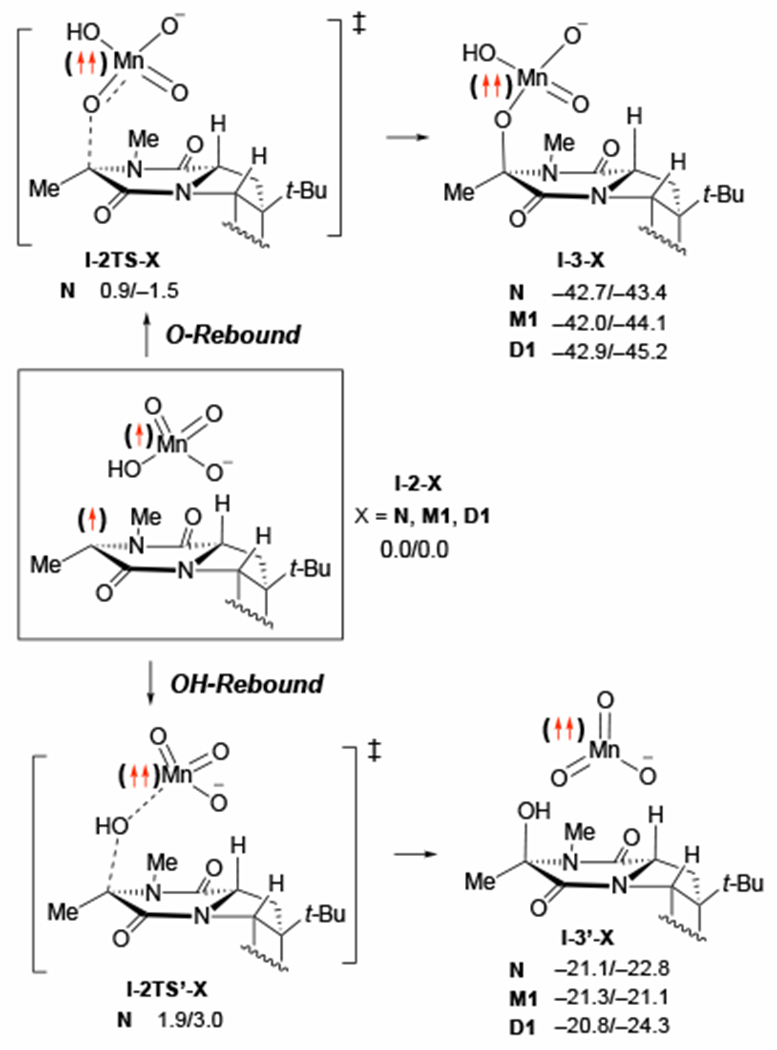
The examined O-rebound and OH-rebound pathways: energies are calculated relative to I-2-X and are given as ΔG/ΔH in kcal/mol. For sake of simplicity, here the schematic reaction pathway was shown only for model oxidant without counter-cation, i.e. for X = N.
With oxidant N, the computed free energy barrier for the O-rebound (at transition state I-2TS-N) is 0.9 kcal/mol, and the reaction is exergonic by 42.7 kcal/mol relative to I-2-N. On the other hand, the competing OH-rebound proceeds through a free energy barrier of 1.9 kcal/mol (at transition state I-2TS’-N) and is exergonic by 21.1 kcal/mol. (An apostrophe on the label for the structure indicates a pathway originating from OH-rebound while absence of an apostrophe indicates a pathway originating from O-rebound.) Thus, both O-rebound and OH-rebound are very fast processes. Unfortunately, all our attempts to locate both O-rebound and OH-rebound transition states with higher-order oxidants M1 and D1 failed.51 However, the thermodynamic trends are consistent across the examined oxidants: The O-rebound product I-3-X is thermodynamically favored by a significant margin regardless of the presence of the counter-cation (21.6, 20.7, and 22.1 kcal/mol for N, M1, and D1, respectively). This is likely because the three coordinate MnVO3− complex (X’, where N’ = MnO3−, M1’ = Py2AgMnO3, and D1’ = MnO4[Py2Ag]2MnO3) of I-3’-X is a high energy species. (Figure 6) Of course, I-3-X and I-3’-X species could, in general, rearrange to each other, but the equilibrium lies significantly toward I-3-X and the transformation requires a significant energy barrier (see Supporting Materials for more details). Therefore, we conclude that both the permanganate ester I-3-X and alcohol I-3’-X intermediates are possible products of the first oxidation, but they are not likely to be directly interconvertible to any significant degree through the pathway studied here.52 Practically, either of these mechanistic pathways is subject of reaction conditions, including the nature of the solvent and oxidant used, concentration of oxidant and temperature.
To summarize, the first C–H bond oxidation occurs at either activated C–H bond (i.e. C11–H and C15–H) of the diketopiperazine through an open-shell singlet transition state (i.e., radical pathway). Subsequent spin flip and fast O-rebound or OH-rebound lead to a new C–O bond in either the MnV permanganate ester I-3-X or alcohol I-3’-X intermediates, respectively. Inclusion of the counter-cation (i.e. the ion-paired oxidants) in the calculations has a slight stabilizing effect on the calculated free energy barriers of the first C–H abstraction (which are within 10.1-12.8 kcal/mol range depending on the nature of oxidants and positions of the activated C–H bonds).
Second C–H oxidation.
Thus, the products of the first C–H oxidation could be both permanganate ester I-3-X and alcohol I-3’-X intermediates, of which the former is thermodynamically more stable. Herein we examined the mechanisms of the second C–H bond oxidation from both intermediates of the first C–H oxidation and operate under the assumption (see above) that intermediates I-3-X and I-3’-X are not interconvertible. To maintain continuity with the previous section, we present here the results for oxidation of C11–H starting from the products I-3-X and I-3’-X of the C15–H oxidation. We also studied oxidation of C15–H starting from the product of the C11–H oxidation and found similar results. (See Supporting Materials.)
C11–H bond oxidation from the permanganate ester intermediate following O-rebound.
From the permanganate ester intermediate I-3-X, the C11–H bond oxidation could occur via two different pathways: i) intramolecular, i.e. with the MnV permanganate ester, and ii) intermolecular, i.e. with a second equivalent of MnVII permanganate. The computed barriers and reaction energies for these pathways and for N, M1 and D1 are provided in Figure 7.
Figure 7.
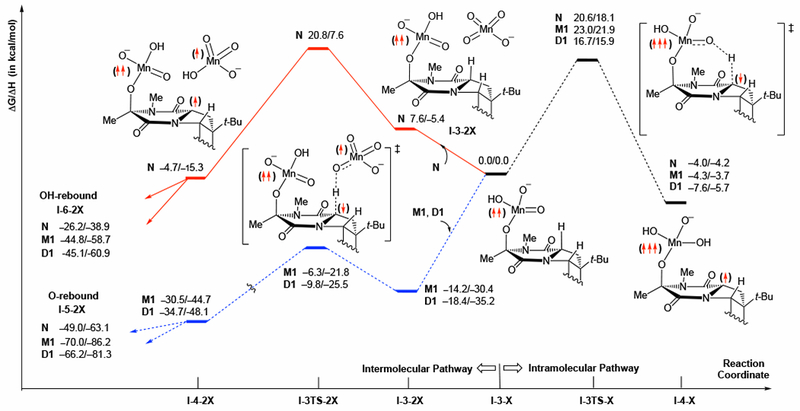
Schematic presentation intramolecular and intermolecular pathways of the C–H abstraction at the C15 position of the C11 permanganate intermediate I-3-X (i.e. the second C–H oxidation). All energies are calculated relative to the I-3-X + X dissociation limit. For simplicity, only the structures with N are depicted.
Briefly, we found that the ground electronic state of the reactant I-3-X is the triplet state and the C–H abstraction transition states I-3TS-X and I-3TS-2X (here, 2X indicates the utilization of two equivalents of oxidant) have triplet ground states with significant spin contamination from the quintet state (<S2> values are 2.6-2.8). Careful examination of the spin density for the spin-contaminated triplet C–H cleavage transition states shows that they are analogous to the open-shell singlet state above discussed for the first C–H oxidation. (Figure 8)
Figure 8.
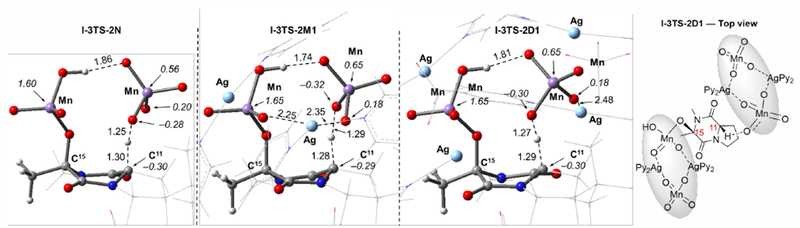
Optimized transition state structures for the second C–H abstraction through the intermodular pathway (I-3TS-2X) for the model oxidants N, M1, and D1. Bond distances (in Å) and Mulliken spin density values (in |e|) are shown in italics. Some parts of the counter-cations have been removed from the visual representation for clarity.
For the intramolecular C–H bond oxidation pathway, the aggregation state of the oxidant does not have much effect on the free energy barriers: the calculated free energy barriers at the transition state I-3TS-X are 20.6, 23.0, and 16.7 kcal/mol for N, M1, and D1, respectively. (Figure 7, right from the I-3-X) These values are all significantly higher than the barriers computed for the first C–H oxidation. Thus, if the dual oxidation of diketopiperazines 3 and 5 (Scheme 1) would occur via this mechanistic scenario, then we should expect formation of the partial oxidation product. However, partial oxidation products were not observed under optimal conditions. Therefore, we conclude that the intramolecular second C–H oxidation is not a likely pathway for the reaction, and it will not be discussed further.
Next, we examine the intermolecular pathway (see Figures 7 and 8). In this case, the aggregation state of the oxidant has a pronounced effect on the computed energetics. The calculated intermolecular oxidation of the second C–H bond with “naked” oxidant N requires 20.8 kcal/mol barrier, which is almost twice the barrier for the first C–H oxidation. Careful analysis of the energies (Figure 7, left from the I-3-X) and structures (Figure 8) reveals that the increased barrier for the second C–H oxidation is due in part to the unfavorable free energy of association of the second equivalent of N with the permanganate ester to form I-3-2N (ΔG = 7.6 kcal/mol) (see Figure S9 for contour plot of this structure showing no attractive interaction between the MnO4-units). However, inclusion of the counter-cation into the calculations produces favorable free energies of association for the second oxidant (ΔG = −14.2 and −18.4 kcal/mol for the formation of I-3-2M1 and I-3-2D1, respectively).53 This is likely due to the counter-cations mitigating electrostatic repulsion between the two negatively charged manganese species. The resulting computed free energy barriers for the second C–H activation are therefore much lower in the presence of the counter-cation (7.9 and 8.6 kcal/mol with the M1 and D1 oxidants, respectively). These barriers are considerably lower than those for the first C–H oxidation and are, therefore, consistent with the lack of partial oxidation products using cis-substituted diketopiperazines.1,54
To summarize, we found that C11–H bond (i.e. the second C–H) activation from the C15 permanganate ester I-3-X, (a) proceeds via the intermolecular pathway with free energy barriers which are lower than those reported for the first C–H bond activation, and (b) depends on the ion-pairing and aggregation state of the oxidant. Similar to the first C–H bond oxidation, I-4-2X can undergo O-rebound and OH-rebound to form di-MnV-permanganate ester I-5-2X and alcohol-ester I-6-2X intermediates, respectively (not shown in Figure 7: see supporting materials for more details). The di-MnV-permanganate ester products are more stable than alcohol-ester products although the formation of either product is possible. The formation of the alcohol-ester intermediates I-6-2X (relative to the I-3-X + X dissociation limit) is exergonic by 26.2, 44.8 and 45.1 kcal/mol for N, M1 and D1, respectively, while formation of the di-MnV-permanganate ester intermediates I-5-2X is exergonic by 49.0, 70.0 and 66.2 kcal/mol for N, M1 and D1, respectively,
The conversions of I-5-2X and I-6-2X to the diol (I”) as final product (see below Figures 9 and 10) and eventual formation of colloidal MnO2 — the known product of C–H oxidation by permanganate16 — require additional transformations that are not yet understood. We have considered a variety of potential transformations but have not been able to obtain experimental evidence to validate the details of these steps.
Figure 9.
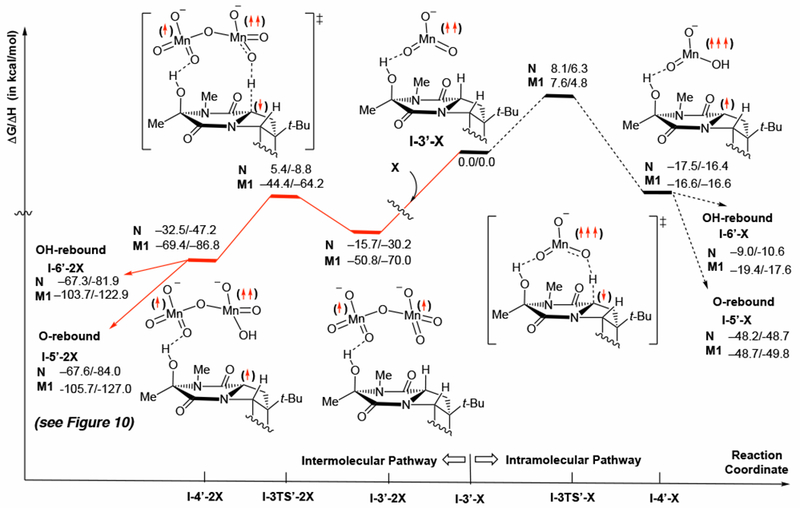
Schematic presentation of the intramolecular and intermolecular pathways for C11–H abstraction starting from the C15 alcohol intermediate I-3’-X. All energies are calculated relative to the I-3’-X + X dissociation limit. For simplicity, only the structures with N are depicted.
Figure 10.
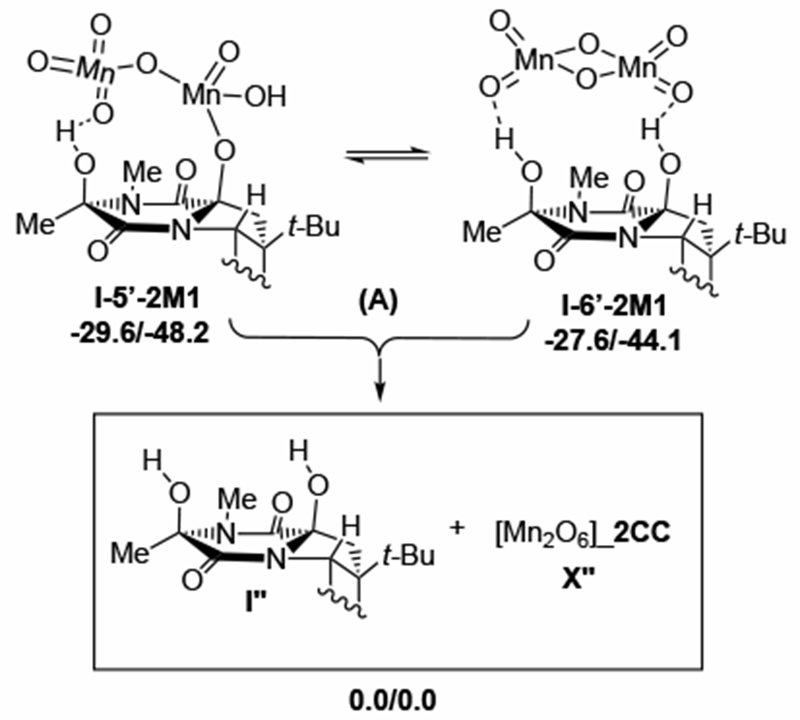
Schematic presentation of the O-rebound, I-5’-2X, and OH-rebound I-6’-2X, intermediates of the intermolecular pathway for the second C–H oxidation at the C11 position starting from the C15 alcohol intermediate I-3’-X (see Figure 9 for more details). Counter-cations (CC = Py2Ag+) are not shown for clarity. Relative energies, as ΔG/ΔH, are given in kcal/mol.
C11–H bond oxidation from the alcohol intermediate following OH-rebound.
While the second oxidation of a permanganate ester by another equivalent of oxidant, guided by ion-pairing with cation, does explain the formation of only diol, we also explored the possibility that the alcohol is formed in the first step by OH rebound. From the alcohol intermediate, I-3’-X, C11–H abstraction, like that from the permanganate ester intermediate I-3-X (reported above), may proceed via either i) intramolecular mechanism with the MnV byproduct X’ (where N’ = MnO3− and M1’ = Py2AgMnO3) as an oxidant, or ii) intermodular mechanism with a second equivalent of oxidant (X). Our data to this point show consistent results using M1 and D1 for the first C–H oxidation, as well as for the second C–H oxidation through the permanganate ester intermediate I-3-X (Figure 7), so for efficiency of computation and simplicity of presentation from here on we only report calculations with M1.
The computed potential energy surfaces of the C11–H oxidation via these two mechanistic pathways are presented in Figure 9. Starting from I-3’-X, intramolecular C–H abstraction by the MnV byproduct X’ requires 8.1 and 7.6 kcal/mol free energy barrier at the transition state I-3TS’-X and is exergonic by 17.5 and 16.6 kcal/mol for N’ and M1’, respectively. As in all the C–H abstraction processes described here, the lowest energy electronic state for the C11–H activation transition states, I-3TS’-X, is a triplet state that is highly spin-contaminated by the quintet state, while the triplet and quintet electronic states of the C11–H activation product I-4’-X are energetically almost degenerated.
From the C11–H activation product, I-4’-X, O-rebound and OH-rebound lead to the alcohol-ester I-5’-X and diol I-6’-X intermediates, respectively. Calculations show that O-rebound leading to I-5’-X is more exergonic than the OH-rebound leading to I-6’-X by ~39 and ~30 kcal/mol for X = N and M1, respectively. Subsequent coordination of another oxidant (X) leads to the manganese byproduct (X”) and diol (I”) discussed above.
As shown in Figure 9 (left-hand side), the intramolecular mechanism competes with an intermolecular mechanism that is initiated from the same alcohol intermediate, I-3’-X, by the coordination of a second equivalent oxidant X. Calculations show that coordination of X to I-3’-X is exergonic for both N and M1. Furthermore, reaction follows via the similar intermediates and transition states for both oxidants. However, as observed in the intermolecular pathway from the permanganate ester (through I-3-2X), the presence of the counter-cation has a remarkable effect on the energy of associating two Mn species: In this case, the presence of the counter-cation brings over 35 kcal/mol additional stabilization to the formation of I-3’-2X. Because of this large difference in energy and for simplicity of our presentation, we only discuss intermolecular mechanism of the reaction of I-3’-M1 with M1, while we also include all related data for the reaction of I-3’-N with N in Figure 9.
As calculations show, upon coordination of oxidant M1 to the alcohol intermediate I-3’-M1, the M1 and M1’ units combine and form a highly stable [Py2Ag]2[Mn2O7] fragment (M1”’) within the intermediate I-3’-2M1. This process is exergonic by 50.8 kcal/mol. Analysis of the Mulliken spin density of the lowest energy triplet state of I-3’-2M1 shows that one spin is located on each Mn-centers suggesting the MnVI-MnVI nature of the [Py2Ag]2[Mn2O7] fragment. From this intermediate C11–H abstraction occurs with a similarly low free energy barrier (calculated relative to the intermediate I-3’-2M1), by 6.4/5.8 kcal/mol, and is exergonic by 18.6/16.8 kcal/mol.
The direct product of the C11–H abstraction is intermediate I-4’-2M1, where the reduced (O)3Mn-O-Mn(O)2(OH) inorganic core is hydrogen bonded to the mono-hydroxylated substrate. Once again, the C–H abstraction product can undergo O-rebound or OH-rebound to form alcohol-ester and diol products, I-5’-2M1 and I-6’-2M1, respectively (see Figure 10). Surprisingly, the O-rebound and OH-rebound products are energetically close: These processes are found to be exergonic by 36.3 and 34.3 kcal/mol, respectively. To understand this finding, we closely analyzed the geometries of I-5’-2M1 and I-6’-2M1. In the alcohol-ester complex I-5’-2M1, two Mn-centers are bridged with only one oxo-center, while the I-6’-2M1 they are bridged with two oxo-centers (see Supporting Materials and Figure 10). Since product complexes I-5’-2M1 and I-6’-2M1 are close in energy, it is likely they can rearrange to each other. Calculations show that the dissociation of the manganese byproduct (M1”) from I-6’-2M1 requires 27.6/44.1 kcal/mol energy.
This discussion shows that, in general, the second C–H bond oxidation initiated from either the MnV permanganate ester I-3-X (i.e. O-rebound product of the first C–H oxidation) or alcohol I-3’-X (i.e. OH-rebound product of the first C–H oxidation) intermediates proceeds via intermolecular mechanisms with free energy barriers in the range of ~5-9 kcal/mol, which are significantly lower than those computed for the first C–H oxidation. Critically, the close contact ion-paired oxidant (in contrast to the “naked” ion N, i.e. solvent separated ion-pair), is important for facilitating the second C–H oxidation regardless of the starting point. If the reaction starts from the permanganate ester intermediate I-3-X, the acceleration of the second C–H oxidation may be due to stabilizing interactions between the oxidants leading to a directing effect. In contrast, if the reaction starts from the alcohol intermediate I-3’-X, the second equivalent of MnVII permanganate captures the high energy MnVO3− species and forms a highly reactive di-nuclear MnVI-MnVI inorganic species that facilitates the C–H abstraction process.55
To summarize, we found that: (a) The first C–H oxidation of diketopiperazine by permanganate (regardless of its aggregation state) proceeds with 10-13 kcal/mol activation free energy barriers, and results in a permanganate ester I-3-X and alcohol I-3’-X intermediates through the O-rebound and OH-rebound pathways, respectively; (b) the second C–H bond oxidation initiated from either the MnV permanganate ester I-3-X or alcohol I-3’-X intermediates proceeds via the intermolecular mechanism with lower free energy barriers than those computed for the first C–H oxidation, (c) The close proximity of the counter-cation to the oxidant in the ion-pair is critical in facilitating this second C–H oxidation and leads to a dual oxidation of diketopiperazine regardless of the reaction pathway.
Substrate oxidation patterns.
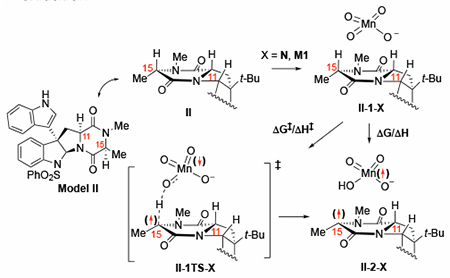
With this mechanistic understanding in hand for dual C–H oxidation, we begin efforts to systematically study substituted diketopiperazines that produce unexpected oxidation patterns. Many such substrates have an indole at the C3 position. We, therefore, used model substrate (model II, see figure to Table 2) to investigate the impact of indole substitution at the C3 position relative to the tert-butyl substitution of I.
Table 2.
The calculated energies for C–H abstraction at the C11 and C15 positions of II with model oxidants N and M1. For sake of simplicity, here the schematic reaction pathway is shown only for model oxidant N and for the C11–H oxidation
| C11 | C15 | |||
|---|---|---|---|---|
| Ox. | ΔG‡/ΔH‡ | ΔG/ΔH | ΔG‡/ΔH‡ | ΔG/ΔH |
| N | 12.3/12.6 | −10.3/−9.0 | 11.6/11.4 | −4.7/−2.5 |
| M1 | 11.9/11.6 | −11.7/−11.2 | 10.1/9.2 | −8.8/−5.2 |
The calculated barriers (ΔG‡/ΔH‡) and reaction energies (ΔG/ΔH) for C–H abstraction at the C11 and C15 positions of II with the naked (N) and ion-paired (M1) model oxidants are shown in Table 2. (See Supporting Materials for BDEs and data with D1.) The observed differences in reactivity between I and II is small, indicating that the t-Bu to indole substitution at the C3 position has no appreciable impact of the dual oxidation in these diketopiperazine models. Again, we found slightly lower free energy barriers with M1 than N, suggesting that the counter-cation may some effect on stabilizing the first C–H abstraction TS.
Di- vs. tri-oxidation.
Next, we explored the unexpected oxidation pattern upon the reaction of C3-indole substituted diketopiperazine (+)-5 and oxidant 2 (see Scheme 1B). It is evident that the existence of two unique hydrogens at C15 in (+)-5 increases the complexity of the oxidation compared to the models I and II and creates conditions in which diastereomeric product formation could occur. In addition, the resulting alcohol can be further oxidized to the ketone. However, previous experiments by the Movassaghi group 14 showed that the reaction of (+)-5 with 2 yields diol (–)-6 as a single diastereomer, with stereoretention of the singly oxidized C11–H and C15–H bonds. This result suggests that 2 is not capable of resulting in over-oxidation to the triketopiperazine. We explored how this outcome is achieved.
Because of the relatively small size of (+)-5, we used it directly in our computational modelling (called model III, see Figure 11). For the sake of expanding our understanding of the role of the counter-cation in the reaction, as well as consistency with the experimental conditions, we extended our modeling and also included oxidant 2 (i.e., M2, Figure 2), which has a TBA+ counter-cation, in the calculation of the C–H oxidation barriers of III.
Figure 11.
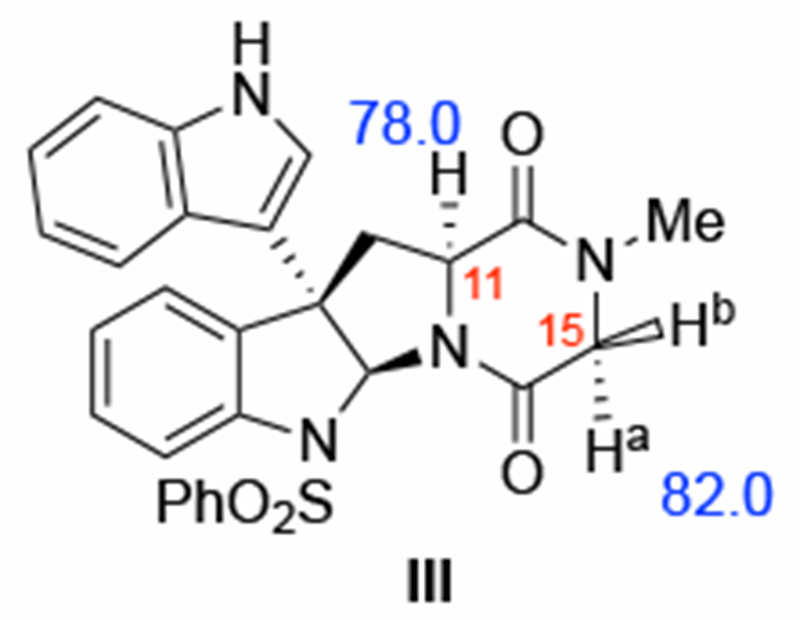
Model diketopiperazine III used for (+)-5 and computed bond dissociation energies (BDEs, in kcal/mol) shown in blue.
The computed BDEs for the C11–H and C15–H bonds (both C15–Ha and C15–Hb)56 of III are 78.0 and 82.0 kcal/mol, respectively. These values indicate that the α-C–H bonds of III are stronger than those of I (74.7 and 77.7 kcal/mol, respectively) and II (75.8 and 78.5 kcal/mol, respectively, see Supporting Material). Comparison of the calculated C11–H and C15–H BDEs for the II and III shows that methyl-to-hydrogen substitution at the C15 position increases these BDEs by 2.3 and 3.7 kcal/mol, respectively. The large difference at C15 is expected based on the direct effect of the methyl group in stabilizing the resulting radical intermediate. The increased BDE at C11 indicates that the substitution pattern at C15 methyl group also has a wider effect on the diketopiperazine ring.
The calculated barriers (ΔG‡/ΔH‡) and reaction energies (ΔG/ΔH) for C–H abstraction of the C11–H, C15–Ha, and C15–Hb bonds (see Figure 11) of III with the N, M1 and M2 oxidants are shown in Figure 12. We first analyze the data with a “naked” oxidant N as a reference and then analyze the data with M1 and M2 to examine the effect of the counter-cations.
Figure 12.
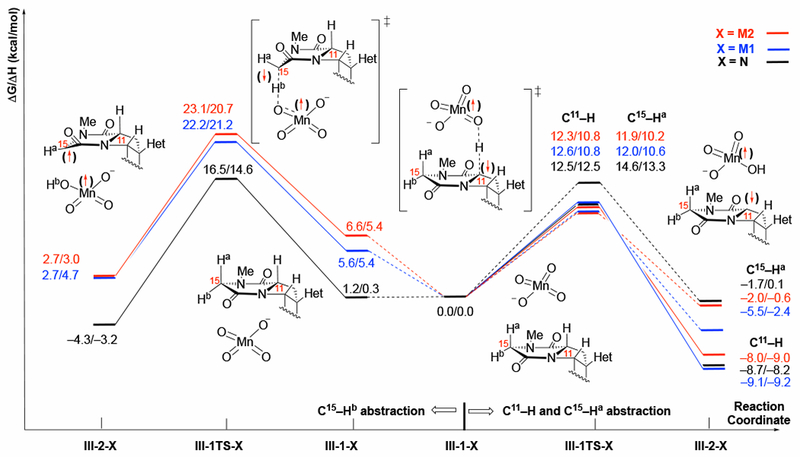
Schematic presentation of the pathways for C–H abstraction of the C11–H, C15–Ha and C15–Hb bonds of III with model oxidants N, M1 and M2. All energies are reported as ΔG/ΔH and are calculated relative to the III-1-X for the a face (C11–H, C15–Ha). For simplicity, only the structures with N are depicted. We also omitted structures associated with the C15–Ha abstraction.
For this substrate, reactive C–H bonds exist on both faces of the diketopiperazine: We define these as the a face, which has the C11–H and C15–Ha bonds, and the b face, which has the C15–Hb bond. These correspond with the convex and concave faces of the proline-diketopiperazine structure, respectively. Association of the naked oxidant, N, with the b face is only slightly less favorable than with the a face (ΔG/ΔH = 1.2/0.3 kcal/mol, Figure 12, black line). The barriers and reaction energies for the subsequent C–H abstractions are consistent with the analysis of the computed BDEs in Figure 11. Compared with the tertiary center of C11, the free energy barriers at the secondary center of C15 are higher by 2.1 and 4.0 kcal/mol for the C15–Ha, and C15–Hb bonds, respectively. Careful examination of the C–H abstraction transition state structure for the C15–Hb bond reveals a conformational change in the diketopiperazine ring as the source of the 1.9 kcal/mol increase in the barrier relative to the C15–Ha bond. In order to align the breaking C–H bond with the π orbitals of the amide, the diketopiperazine must undergo a half chair flip to put the C15–Hb bond in an axial conformation as shown in Figure 13. As shown in this figure, the diketopiperazine ring is in a higher energy chair conformation in the C15–Hb abstraction transition state: This is evidenced by the non-eclipsing dihedrals, C15–N–C(=O)–C11 and C15–C(=O)–N–C11 are −21 and 28 deg., respectively, which are characteristic of a chair conformation in six-membered rings. On the other hand, the transition state structure for C15–Ha (also C11–H) abstraction has more eclipsing dihedrals, C15–N–C(=O)–C11 and C15–C(=O)–N–C11 are 2 and 19 deg., respectively, which are characteristic of a boat conformation. It is known that the boat conformation is more stable than the chair conformation for proline-diketopiperazines.48
Figure 13.
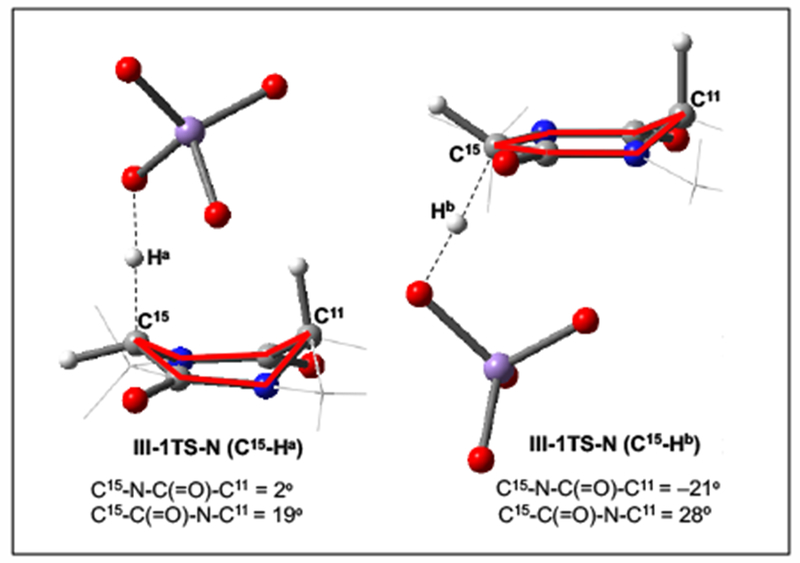
Optimized C–H abstraction transition state structures for the C15–Ha and C15–Hb bonds of III with the model oxidant N (III-1TS-N). The important dihedral angles are reported in deg. and highlighted in red. Some parts of the structures have been removed from the visual representation for clarity.
Thus, for III, the C15–Hb bond is intrinsically less kinetically reactive than the C15–Ha bond due to the requirement that the diketopiperazine ring adopt a higher energy conformation in the transition state. This effect likely contributes to the lack of oxidation at this position in the reaction of (+)-5 and 2, but the magnitude of the effect suggests there may also be other factors.
For the oxidant 1 (Figure 12, X = M1, blue line), we observe a remarkable lowering of the oxidation barrier for the C15–Ha bond so that both positions on the a face are oxidized at about the same rate despite the differences in bond strength indicated in the BDEs and reaction energies. This suggests that the ion-pairing in oxidant is more effective at stabilizing the TS for C15–Ha cleavage, which is also consistent with our findings on the reaction of substrate I and oxidant M1, presented above (see Table 1). Very similar trends are also observed in the presence of the TBA+ counter-cation of oxidant 2 (Figure 12, X = M2, red line).
In striking contrast to the “naked” oxidant N, coordination of M1 (i.e. oxidant with a Py2Ag+ counter-cation) to the b face of III is endergonic by 5.6 kcal/mol. However, the intrinsic barrier for oxidation of the C15–Hb bond (i.e., calculated from the pre-reaction complex) by oxidant M1 is 16.6 kcal/mol, which is very similar to the intrinsic barrier for oxidant N at the same position. Likewise, coordination of M2 (i.e. oxidant with a TBA+ counter-cation) to the b face of III is endergonic by 6.6 kcal/mol, while having no effect on the intrinsic barrier for C–H abstraction (16.5 kcal/mol). These results indicate that the presence of the ion-paired oxidant raises the overall barrier for oxidation of the C15–Hb bond by impeding the oxidant’s access to it. (See Supporting Materials for the geometries.) These findings indicate that the counter-cation impedes access of the oxidant to the C–H bond on the more hindered b face of III that makes oxidation of the C15–Ha and C15–Hb bonds highly selective.57
Thus, the computations provide evidence for two major contributing factors for the lack of over-oxidation at C15 in (+)-5: i) increased inherent kinetic stability due to required deviation from the lowest energy conformation of the diketopiperazine ring, and ii) impeded approach of the oxidant to more hindered faces of the substrate.
Conclusions.
We have examined the mechanism of permanganate-mediated dual C–H oxidation of complex diketopiperazines with density functional theory. We evaluated the impact of ion-pairing and aggregation states of the permanganate and counter-cation on theses oxidation for the first time. We explored the commonly used counter-cations, bis(pyridine)-silver(I) (Ag+) and tetra-n-butyl ammonium (TBA+).
We confirmed the previously proposed the C–H abstraction followed O-rebound mechanism leading to a permanganate ester intermediate, but also found that there can be competing OH-rebound pathway leading to the alcohol product.
For dual oxidation of diketopiperazine, we found that: (a) the first C–H oxidation by permanganate (regardless of the nature of the oxidant) proceeds with free energy barriers of ~10-13 kcal/mol, and can result in either permanganate ester or alcohol intermediates; (b) the second C–H bond oxidation initiated from either intermediate proceeds via an intermolecular mechanism (involving a second equivalent of oxidant) with lower free energy barriers than those computed for the first C–H oxidation, (c) the presence of the counter-cation (i.e. ion-paired oxidant species) is critical for success of the second C–H oxidation.
We rationalized an experimentally observed oxidation pattern for C3-indole substituted diketopiperazine (+)-5 by oxidant 2: Under optimal conditions, the secondary center at C15 of substrate (+)-5 is oxidized stereoselectively to produce the cis-diol diketopiperazine (−)-6.14 Further oxidation at C15 is likely avoided because of: (i) increased inherent kinetic stability due to required deviation from the lowest energy conformation of the diketopiperazine ring, and (ii) impeded approach of the oxidant to the more hindered face of the substrate because of the larger size of the ion-paired oxidant.
We expect this work will assist in the development of effective oxidants and in oxidation patterns for complex substrates such as those we have investigated.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1700982). We gratefully acknowledge the NSF MRI-R2 grant (CHE-0958205) and the use of the resources of the Cherry Emerson Center for Scientific Computation at Emory University. M.M. acknowledges financial support by NIH-NIGMS (GM089732). B.M.N. thanks the National Science Foundation for a graduate fellowship
Footnotes
Supporting Information
Includes: (1) Full data for the permanganate ions (oxidants) used in this paper, (2) additional data for oxidation of substrates I, II and III, (3) Energies of all calculated structures, (4) Contour plot and electrostatic potential map of intermediates I_3_2N and I_3_2M1, and (5) cartesian coordinates of all calculated structures. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interests.
References
- 1.Kim J; Movassaghi M Biogenetically-Inspired Total Synthesis of Epidithiodiketopiperazines and Related Alkaloids. Acc. Chem. Res 2015, 48, 1159–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser D; Weber HP; Sigg HP Isolation and Structure Elucidation of Chaetocin. Helv. Chim. Acta 1970, 53, 1061–1073. [DOI] [PubMed] [Google Scholar]
- 3.Katagiri K; Sato K; Hayakawa S; Matsushi T; Minato H Verticillin a, a New Antibiotic from Verticillium-Sp. J. Antibiot 1970, 23, 420–422. [DOI] [PubMed] [Google Scholar]
- 4.Jiang CS; Guo YW Epipolythiodioxopiperazines from Fungi: Chemistry and Bioactivities. Mini-Rev. Med. Chem 2011, 11, 728–745. [DOI] [PubMed] [Google Scholar]
- 5.Kim J; Movassaghi M Biogenetically Inspired Syntheses of Alkaloid Natural Products. Chem. Soc. Rev 2009, 38, 3035–3050. [DOI] [PubMed] [Google Scholar]
- 6.Kim J; Ashenhurst JA; Movassaghi M. Total Synthesis of (+)-11,11’-Dideoxyverticillin A. Science 2009, 324, 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim J; Movassaghi M General Approach to Epipolythiodiketopiperazine Alkaloids: Total Synthesis of (+)-Chaetocins a and C and (+)-12,12 ‘-Dideoxychetracin A. J. Am. Chem. Soc 2010, 132, 14376–14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyer N ; Morrison KC ; Kim J; Hergenrother PJ; Movassaghi M Synthesis and Anticancer Activity of Epipolythiodiketopiperazine Alkaloids. Chem. Sci 2013, 4, 1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coste A; Kim J; Adams TC; Movassaghi M Concise Total Synthesis of (+)-Bionectins a and C. Chem. Sci 2013, 4, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams TC; Payette JN; Cheah JH;Movassaghi M Concise Total Synthesis of (+)-Luteoalbusins a and B. Org. Lett 2015, 17, 4268–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Firouzabadi H; Vessal B; Naderi M Bispyridinesilver Permanganate [Ag(C5H5N)2]MnO4 - an Efficient Oxidizing Reagent for Organic Substrates. Tetrahedron Lett 1982, 23, 1847–1850. [Google Scholar]
- 12.Sala T; Sargent MV Tetrabutylammonium Permanganate - Efficient Oxidant for Organic Substrates. J. Chem. Soc., Chem. Commun 1978, 253–254. [Google Scholar]
- 13.With the exception of the unique DKP oxidation related to synthesis of bionectins, (ref. 9) all other DKP oxidations examined to date have proceeded with stereoretention.(ref. 1)
- 14.Boyer N; Movassaghi M Concise Total Synthesis of (+)-Gliocladins B and C. Chem. Sci 2012, 3, 1798–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brauman JI; Pandell AJ Mechanism of Permanganate Oxidation of Tertiary Hydrogen to Hydroxyl. J. Am. Chem. Soc 1970, 92, 329–335. [Google Scholar]
- 16.Gardner KA; Kuehnert LL; Mayer JM Hydrogen Atom Abstraction by Permanganate: Oxidations of Arylalkanes in Organic Solvents. Inorg. Chem 1997, 36, 2069–2078. [DOI] [PubMed] [Google Scholar]
- 17.Gardner KA; Mayer JM Understanding C-H Bond Oxidations - H-Center-Dot and H-Transfer in the Oxidation of Toluene by Permanganate. Science 1995, 269, 1849–1851. [DOI] [PubMed] [Google Scholar]
- 18.Strassner T; Houk KN Mechanism of Permanganate Oxidation of Alkanes: Hydrogen Abstraction and Oxygen “Rebound”. J. Am. Chem. Soc 2000, 122, 7821–7822. [Google Scholar]
- 19.Saouma CT; Mayer JM Do Spin State and Spin Density Affect Hydrogen Atom Transfer Reactivity? Chem. Sci 2014, 5, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaussian 09, Revision E.01, Frisch MJ;Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J ; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam MJ; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas ö.; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian, Inc., Wallingford CT, 2009. [Google Scholar]
- 21.Hay PJ; Wadt WR Abinitio Effective Core Potentials for Molecular Calculations - Potentials for the Transition-Metal Atoms Sc to Hg. J. Chem. Phys 1985, 82, 270–283. [Google Scholar]
- 22.Hay PJ; Wadt WR Abinitio Effective Core Potentials for Molecular Calculations - Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys 1985, 82, 299–310. [Google Scholar]
- 23.Wadt WR; Hay PJ Abinitio Effective Core Potentials for Molecular Calculations - Potentials for Main Group Elements Na to Bi. J. Chem. Phys 1985, 82, 284–298. [Google Scholar]
- 24.Grimme S; Antony J; Ehrlich S; Krieg H A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 25.Cances E; Mennucci B; Tomasi J A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys 1997, 107, 3032–3041. [Google Scholar]
- 26.Mennucci B; Tomasi J Continuum Solvation Models: A New Approach to the Problem of Solute’s Charge Distribution and Cavity Boundaries. J. Chem. Phys 1997, 106, 5151–5158. [Google Scholar]
- 27.Scalmani G; Frisch MJ Continuous Surface Charge Polarizable Continuum Models of Solvation. I. General Formalism. J. Chem. Phys 2010, 132, 114110–114124. [DOI] [PubMed] [Google Scholar]
- 28.Essafi S; Tomasi S; Aggarwal VK; Harvey JN Homologation of Boronic Esters with Organolithium Compounds: A Computational Assessment of Mechanism. J. Org. Chem 2014, 79, 12148–12158. [DOI] [PubMed] [Google Scholar]
- 29.Nam PC; Nguyen MT; Chandra AK The C-H and Alpha(C-X) Bond Dissociation Enthalpies of Toluene, C6H5-CH2x (X = F, CI), and Their Substituted Derivatives: A DFT Study. J. Phys. Chem. A 2005, 109, 10342–10347. [DOI] [PubMed] [Google Scholar]
- 30.DiLabio GA; Pratt DA; LoFaro AD; Wright JS Theoretical Study of X-H Bond Energetics (X = C, N, O, S): Application to Substituent Effects, Gas Phase Acidities, and Redox Potentials. J. Phys. Chem. A 1999, 103, 1653–1661. [Google Scholar]
- 31.Berkowitz J; Ellison GB; Gutman D Three Methods to Measure Rh Bond Energies. J. Phys. Chem 1994, 98, 2744–2765. [Google Scholar]
- 32.Blasse G Struct. Bonding (Berlin) 1980, 42, 1–42. [Google Scholar]
- 33.Viste A; Gray HB Electronic Structure of Permanganate Ion. Inorg. Chem 1964, 3, 1113–1123. [Google Scholar]
- 34.Seidu I; Krykunov M; Ziegler T Applications of Time-Dependent and Time-Independent Density Functional Theory to Electronic Transitions in Tetrahedral D(0) Metal Oxides. J. Chem. Theory Comput 2015, 11, 4041–4053. [DOI] [PubMed] [Google Scholar]
- 35.Sharma P; Truhlar DG; Gagliardi L Active Space Dependence in Multiconfiguration Pair-Density Functional Theory. J. Chem. Theory Comput 2018, 14, 660–669. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh A; Taylor PR High-Level Ab Initio Calculations on the Energetics of Low-Lying Spin States of Biologically Relevant Transition Metal Complexes: First Progress Report. Curr. Opin. Chem. Biol 2003, 7, 113–124. [DOI] [PubMed] [Google Scholar]
- 37.Yang TH; Quesne MG; Neu HM; Reinhard FGC; Goldberg DP; de Visser SP Singlet Versus Triplet Reactivity in an Mn(V)-Oxo Species: Testing Theoretical Predictions against Experimental Evidence. J. Am. Chem. Soc 2016, 138, 12375–12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dash S; Patel S; Mishra BK Oxidation by Permanganate: Synthetic and Mechanistic Aspects. Tetrahedron 2009, 65, 707–739. [Google Scholar]
- 39.Karaman H; Barton RJ; Robertson BE; Lee DG Preparation and Properties of Quaternary Ammonium and Phosphonium Permanganates. J. Org. Chem 1984, 49, 4509–4516. [Google Scholar]
- 40.Du HX; Lo PK; Hu ZM; Liang HJ; Lau KC; Wang YN; Lam WWY; Lau TC Lewis Acid Activated Oxidation of Alcohols by Permanganate. Chem. Commun 2011, 47, 7143–7145. [DOI] [PubMed] [Google Scholar]
- 41.Lam WWY; Yiu SM; Lee JMN; Yau SKY; Kwong HK; Lau TC; Liu D; Lin ZY BF3-Activated Oxidation of Alkanes by MnO4-. J. Am. Chem. Soc 2006, 128, 2851–2858. [DOI] [PubMed] [Google Scholar]
- 42.Klajman K; Ciepielowski G; Kamiński R; Adamczyk P; Paneth P Resolving Discrepancy between Theory and Experiment in 4-Nitrotoluene Oxidation. J. Phys. Chem. A 2017, 121, 6638–6645. [DOI] [PubMed] [Google Scholar]
- 43.Adamczyk P; Wijker RS; Hofstetter TB; Paneth P A Dft Study of Permanganate Oxidation of Toluene and Its Ortho-Nitroderivatives. J. Mol. Model. 2014, 20, 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiberg KB; Wang Y.-g.; Sklenak S; Deutsch C; Trucks G Permanganate Oxidation of Alkenes. Substituent and Solvent Effects. Difficulties with MP2 Calculations. J. Am. Chem. Soc 2006, 128, 11537–11544. [DOI] [PubMed] [Google Scholar]
- 45.Collman JP; Slaughter LM; Eberspacher TA; Strassner T; Brauman JI Mechanism of Dihydrogen Cleavage by High-Valent Metal Oxo Compounds: Experimental and Computational Studies. Inorg. Chem 2001, 40, 6272–6280. [DOI] [PubMed] [Google Scholar]
- 46.Jayaraman A; East ALL The Mechanism of Permanganate Oxidation of Sulfides and Sulfoxides. J. Org. Chem 2012, 77, 351–356. [DOI] [PubMed] [Google Scholar]
- 47.Ess DH Distortion, Interaction, and Conceptual DFT Perspectives of MO4-Alkene (M = Os, Re, Tc, Mn) Cycloadditions. J. Org. Chem 2009, 74, 1498–1508. [DOI] [PubMed] [Google Scholar]
- 48.Fry AJ Computational Studies of Ion Pairing. 10. Ion Pairing between Tetrabutylammonium Ion and Inorganic Ions: A General Motif Confirmed. J. Org. Chem 2015, 80, 3758–3765. [DOI] [PubMed] [Google Scholar]
- 49.Fischer PM Diketopiperazines in Peptide and Combinatorial Chemistry. J. Pept. Sci 2003, 9, 9–35. [DOI] [PubMed] [Google Scholar]
- 50.In addition, we find that the ion-pairing does not significantly affect the description of the electronic states of the permanganate ion: The singlet-triplet energy difference for M1 and M2 are 15.7 and 14.1 kcal/mol, respectively.
- 51.All our attempts to locate both O-rebound and OH-rebound transition states with higher-order oxidants M1 and D1 led either to the pre-reaction complexes or the O-rebound and OH-rebound products, respectively.
- 52.Based on the computational data, we cannot fully rule out alternative pathways that may stabilize or entirely bypass the high energy MnVO3− intermediate en route to a putative hydroxylated diketopiperazine intermediate.
- 53.Calculations show that in intermediates I-3-2N, I-3-2M and I-3-2D oxidant species only electrostatically interact with each other. Their isomers with a [−OMn(O)2(OH)-O-Mn(O)3] core are energetically less stable regardless of the nature of oxidant X.
- 54.A trans-substituted isomer of diketopiperazine 3 was found to afford a partial oxidation product (ref. 6), possibly due to absence of any directing effects after C15-oxidation.
- 55.We also find that it is possible for the second C–H abstraction to occur directly via MnVO3− in the case if this reaction occurs from the alcohol intermediate I-3’-X. Even in this case, the presence of counter-cation is required for formation of the final products.
- 56.Formally, the BDEs for C15–Ha and C15–Hb are the same because upon cleavage they both result in the same C15 radical.
- 57.Interestingly, the coordination free energy for M2 to the b face is higher than for M1 by 1.0 kcal/mol, while the coordination enthalpy is equal. While difficult to fully realize in the calculations performed here, this finding suggests a potentially key difference in the reactivity of 1 and 2. The inherent reactivity of 1 and 2 appears to be very similar. However, it is likely that the entropic component of the calculations is much less accurate (i.e., more severely underestimated) for the more flexible TBA+ counter-cation of 2 than the Ag+ counter-cation of 1. This may give the appearance of similar reactivity in calculations based on only one conformation of the oxidant. However, in line with empirical observations of the reactivity of the two oxidants (i.e., 1 > 2), we might expect 2 to be generally less reactive due to a greater entropic penalty for its association with the substrate
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.