

Abstract
The purpose of this study is to evaluate the biodistribution properties of random copolymer-based core-crosslinked nanogels of various sizes and surface poly(ethylene glycol) composition. Systematic variations of near-IR labeled nanogels, comprising varying particle sizes (28–135 nm), PEG corona quantity (0–50 mole percent) and PEG length (PEG Mn 1000, 2000, and 5000), were prepared and injected in mice that had been subcutaneously implanted with MDA-MB-231-luc-D3H2LN human mammary carcinoma. In vivo biodistribution was obtained using Florescence Molecular Tomography imaging at 0, 6, 24, 48, and 72 hours post injection. Retention of total body probe and percentages of total injected dose in the tumor, liver, spleen, lungs, heart, intestine, and kidneys were obtained. Smaller nanogels (~30–40 nm) with a high PEG conjugation (~43–46 mole %) of Mn 2000 on their coronas achieved the highest tumor specificity with peak maximum 27% ID/g, a statistically significant propensity toward accumulation with 16.5% ID/g increase from 0–72 hours of imaging, which constitutes a 1.5-fold increase. Nanogels with greater tumor localization also had greater retention of total body probe over 72 hours. Nanogels without extensive PEGylation were rapidly excreted, even at similar sizes to PEGylated nanogels exhibiting whole body retention. Of all tissues, the liver had the highest %ID, however like other tissues displayed a monotonic decrease over time, suggesting nanogel clearance by hepatic metabolism. Ex vivo quantification of individual tissues from gross necropsy at 72 hours post injection generally correlated with the FMT analysis, providing confidence in tissue signal segmentation in vivo. The parameters determined to most significantly direct a nanogel to the desired tumor target can lead to improve effectiveness for nanogels as therapeutic delivery vehicles.
Keywords: polymeric nanogel, PEGylation, biodistribution, triple-negative breast cancer, near-infrared, florescence molecular tomography
TOC Graphic
INTRODUCTION
In recent years, the advancements in polymeric nanoparticle synthesis, formulation, and modification chemistries have advanced their use in diagnostics and therapeutics for various diseases, particularly cancer. Nanoparticles have the potential to improve the solubility, stability, and clearance profiles of therapeutics that are otherwise plagued with short half-life, and low efficacy and tolerated dose, poor tissue selectivity, and frequent administration.1, 2 Parameters such as their morphology, polymer composition, size, and surface chemistry can all impact their extent of nonspecific interaction and circulation residence time to substantially influence their biodistribution. A more detailed understanding of these features in relation to in vivo performance is required to improve the effectiveness of delivery to the region of interest, specifically solid tumors.
Nanoparticles have been under investigation following the discovery of the enhanced permeability and retention (EPR) effect, which showed increased tumor accumulation of macromolecules by extravasation through fenestrated blood vessels from 100 nm to 2 μM.3, 4 The poor lymphatic drainage of tumors also aids extracellular retention of contents.5 Various biological features that can impact biodistribution, such as accumulation in the liver and spleen due to leaky endothelium or renal clearance filtration of small particles (<10 nm).6, 7 Further, clearance through the reticuloendothelial system (RES), the body’s natural immune response to destroy foreign material, is a challenge for nanocarriers. Immune cells in the blood stream (monocytes, platelets, leukocytes, and dendritic cells), liver (Kupffer cells), lymph nodes (dendritic cells), and spleen (B cells) can all clear nanoparticles from circulation.8–12 The adsorption of plasma proteins, or opsonins, on the particle surface is a common pathway for uptake by immune cells.9, 13
Coating the nanocarrier surface with poly(ethylene glycol) (PEG), i.e. “PEGylation”, through grafting, conjugation, or adsorption has been shown to reduce their filtration and achieve extended blood circulation half-lives.14–17 The favorable results due to its inherent physiochemical properties of PEG to impart a hydrated corona which generates steric stabilization and reduces non-specific interactions with proteins.18, 19 Investigations on PEG length and density effects on either protein adsorption or biodistribution has led to some varied conclusions, likely heavily dependent on specific nanomaterial investigated and on particle size. However, in general, lengths of 1–5 kDa have been shown to be optimal in varied systems.13, 20–26 Protein adsorption deterrence with lower molecular weight PEG suffers from decreased chain flexibility. However, at higher molecular weight, chains can fold, leading to coils with loosely bound water molecules and a threshold length benefit. A more heavily hydrated random conformation of PEG chains is expected to best inhibit protein absorption due to the unfavorable entropy change from compressing this coating.23, 26
Currently the relationship between size and biodistribution of nanoparticles has some contrasting trends in results, likely due to nanomaterial differences.27–29 It is generally thought that the high curvature of smaller particles can reduce opsonization reactions and subsequent clearance by macrophages, leading to improved circulation time and higher tumor accumulation.22, 29, 30 Further, greater tumor penetration has been demonstrated with smaller particles.31 However, smaller particles have not always performed better in vivo.27, 28 The first polymeric micellar nanoparticle that has reached Phase II clinical trials in the Unites States is Genexol-PM, and ranges in size from 20 to 50 nm.32, 33
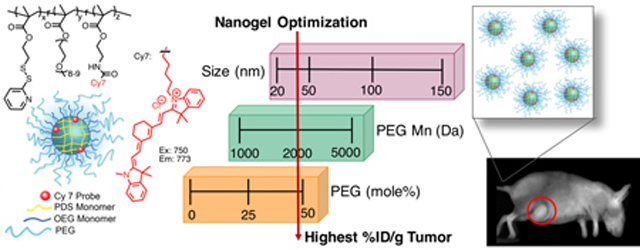
Our group has developed a versatile amphiphilic random copolymer-based nanogel that encompasses many of the desirable features of a drug delivery vehicle including particle size control, guest encapsulation, and selective release in cytosol.34–36 While extensive work has investigated biodistribution profiles using PEG containing block copolymers, to our knowledge relatively little work has investigated random copolymers that are extensively post-modified with PEG on their coronas. With the expectation that minor physicochemical differences can greatly impact the in vivo performance, we aim to systematically modify polymer nanoparticle size (20–135 nm), length of PEG conjugated to the surface (Mn of 1000, 2000, and 5000), and amount of PEG conjugated to the surface (0–50 mole %) to determine the parameters that can most significantly direct a nanogel to the desired tumor target, using triple negative human mammary carcinoma models.
EXPERIMENTAL
Materials:
Poly(ethylene glycol) methyl ether methacrylate (MW 475 g/mol, OEGMA), 2-aminoethyl methacrylate hydrochloride (AEMA), D,L-dithiothreitol (DTT), 4-cyano-4-(phenylcarbonothioylthio)pentanoic, cyanine 7 NHS ester (Cy7), poly(ethylene glycol) methyl ether thiol (average Mn 1000, 2000, and 5000), and other conventional reagents were obtained from commercial sources and without further purification, unless otherwise mentioned. AIBN (2,2’-azobis(2-methylpropionitrile) was purchased from Sigma Aldrich and purified by recrystallization. MDA-MB-231-luc-D3H2LN cells were obtained from Xenogen Corporation (Caliper Life Sciences). Pyridyl disulfide ethyl methacrylate (PDSMA) was prepared as previously reported.37 Polymers were synthesized using RAFT polymerization and purified by dialysis using a membrane with 3500 MWCO.
Measurements:
1H-NMR spectra were recorded on a 400 MHz Bruker NMR spectrometer using the residual proton resonance of the deuterated solvent as the internal standard. Polymer molecular weights were estimated by gel permeation chromatography (GPC, Waters) using THF as eluent at a flow rate of 1 mL/min by a refractive index detector compared to PMMA standard. Dynamic light scattering (DLS) measurements were conducted using a Malvern Nano Zetasizer. UV-visible absorption spectra were recorded on a Varian spectrophotometer (model EL 0112047). Near-IR probe was quantified using a NanoDrop 2000C spectrophotometer.
Synthesis and Formulation of NIR-labeled Nanogels:
Random copolymers p(OEGMA-co-PDSMA-co-AEMA) were synthesized by reversible addition-fragmentation chain-transfer (RAFT) polymerization to yield three polymers of Mn: 6.0 kDa with Ð: 1.5, Mn:13 kDa with Ð: 1.2, and Mn:22 kDa with Ð: 1.3, with monomer ratios for OEG:PDS:AE of 29:68:3, 28:70:2, and 27:69:4, respectively (see Supporting Information). The polymer was reacted with NIR probe, Cy7 NHS ester, to give covalently labeled polymer p(OEGMA-co-PDSMA-co-Cy7). The polymer was made into an aqueous particle suspension (10 mg/mL) using repeated cool and sonicate cycles until the solution appeared dissolved. Smaller aggregates (20–30 nm) were achieved at 25 °C sometimes requiring dilution (Table S1). To achieve aggregates, the polymer solutions were heated (to 40–50 °C), requiring the presence of sodium sulfate or sodium carbonate (1–15 mM) in some cases, until desired sizes of aggregate were obtained, similar to conditions previously reported.34, 38 The aggregates were chemically crosslinked using reducing agent DTT to lock their nanogel size under these solution conditions. Nanogels were then post-modified with poly(ethylene glycol)monomethyl ether thiol (mPEG thiol) (average Mn 1000, 2000, and 5000) using simple thiol-disulfide exchange using the remaining PDS groups of the nanogels. Typically, mPEG thiol was dissolved in a minimum volume water then reacted with crosslinked nanogel solution at PDS molar equivalencies of 1, 1.2, and 2, for Mn 1000, 2000 and 5000, respectively, and stirred for 24 hours. The conjugation extent was then determined using UV-vis absorption by quantification of pyridinethione absorbance at 343 nm as previously described.34, 35, 39 Dynamic light scattering experiments to obtain particle size were performed by using a digital correlator and goniometer with a light source operating at 514 nm. Final nanogel size measurements were obtained at 25 °C at a correlation time of 30 seconds. Dust was removed by filtering the solution through 0.45 μm polycarbonate filter. Final polymeric nanogel concentrations were calculated using initial feed, and final probe concentrations were determined using a NanoDrop 2000C spectrophotometer to obtain the absorbance of Cy7 (λmax: 750, ε: 199000).
Animals and Husbandry:
All animal work was conducted by Molecular Imaging, Inc. This experiment used female Harlan Beige SCID mice (Hsd:NIHS-LystbgFoxn1nuBtkxid), which lack T, B, and NK cells, for size and length PEG series nanogels, female Harlan Nude mice (Hsd:AthymicNude-Fox1nu), which lack T cells, for percent PEG series nanogels, and female Harlan Beige Nude XID mice (Hsd:NIHS-Lystbg-JFox1nuBtkxid) for small sixe high PEG series nanogels that were all 6–7 weeks old at the time of implantation. Animals were fed ad libitum (water and irradiated Harlan 2918.15 Rodent Diet) and housed inside Biobubble® Clean Rooms with Bed-O’Cobs™ bedding with an environment of 70±2 °F and 30–70% humidity. All measurements and imaging were conducted in the bubble environment with filtered high efficiency particulate air and 100 air changes per hour. All animal procedures were carried out under compliance with National Institutes of Health (NIH) guidelines and with Molecular Imaging, Inc.’s (AAALAC accredited and PHS assured facility) Animal Care and Use Committee approval.
Cell Preparation:
MDA-MB-231-luc-D3H2LN cells were grown in Minimal Essential Media (MEM) with Earle’s Balanced Salt Solution (EBSS) supplemented with 10% Fetal Bovine Serum (FBS), 1% penicillin-streptomycin-glutamine (PSG), 1% L-glutamine, 1% non-essential amino acids, and 1% sodium pyruvate. Cells were grown at 5% CO2 and 37 °C and, after expansion, cells (passage 5) were suspended using trypsin 0.25% and 2.21 mM EDTA in Hank’s Balanced Salt Solution (HBSS) then typsin was deactivated with complete growth media. Cells were counted using Trypan Blue exclusion and a hemacytometer then centrifuged (1,374 rpm, 8 min), decanted, then resuspended in serum-free media (2.5×107 cells/ml). On day 0, cells (5×106, 200 μL) with 50% Matrigel® were implanted subcutaneously low in the right flank of each animal using a 27-guage needle in the animals. Three thioglycolate cultures of tumor cells tested negative for gross bacterial contamination. Daily clinical observations were made and moribund or distressed animals or those bearing ulcerated, weeping, or excessive (>1 gram) tumors were euthanized.
Measurements and Endpoints:
Measurements were conducted using reported general principles.40–46 At the initiation of imaging (sample injection Day 0), animal body weights were recorded and tumor burden determined from caliper measurements of orthogonal length (L) and width (W) in mm, and using the prolate ellipsoid formula: (L × W2)/2 = tumor burden (mg). The primary endpoints for evaluation are total whole body probe signal and percent injected dose per gram of the tumor region of interest (%ID/g), and secondary endpoint are percent injected dose (%ID) for heart, lungs, liver, kidneys, spleen, and intestines. Percent injected dose is defined by the probe signal in the tissue divided by 1.15 fold the total whole body probe signal [%ID/(1.15× total whole probe signal)] to account for head mass, a common assumption.40 The percent injected dose per gram (%ID/g) is defined by %ID for the tumor region of interest (ROI) divided by the tumor weight in grams.
Treatment:
Following tumor implantation at Day 0, on Day 21 body weights were obtained and tumor burdens were determined from caliper measurements. For size series nanogels, the average SCID Beige mice weights (range of group means: 20.6– 21.2 g, all ≥18.0 g) and estimated tumor burden (all groups means: 343 mg, range: 312–396 mg) were in a well-matched range for the first day of treatment and imaging. For PEG length series nanogels, on Day 21 the average SCID Beige weights (range of group means: 19.4–20.4 g, all ≥17.8 g) and estimated tumor burden (all groups means: 316 mg, range: 312–323 mg) were in a well-matched range for the first day of treatment and imaging. For percent PEG series nanogels, the growth rate of tumors in the nude mice was slightly faster, with animal weights (range of group means: 20.9–22.6 g, all ≥19.0 g) and estimated tumor burden (all groups means: 377 mg, range: 363–404 mg) were in a well-matched range and, still within the historical norms for this model, ready for treatment and imaging at Day 19. Similarly, for small size high PEG series nanogels the beige nude mice on Day 19 were a little larger (range of group means: 20.6–22.4 g, all ≥19.3 g) and estimated tumor burden (all groups means: 294 mg, range: 283–301 mg) were in a well-matched range for the first day of treatment and imaging. Mice were triaged into groups so that the tumor burden was within 10% of the overall mean. Following weight and tumor burden measurements, mice were dosed once with a single injection of 100 μL nanogel solution in endotoxin free water for imaging on Day 0 (n=5 mice per nanogel solution were treated in each group).
Fluorescence Molecular Tomography:
Following sample injection, mice were individually imaged in vivo at 0, 6, 24, 48 and 72 hours by 3D FMT using the Perkin-Elmer FMT 2500TM LX Quantitative Tomography Imaging System. Just prior and throughout imaging mice were anesthetized with 2% isofluorane gas, then placed in the supine position in the imaging cassette, which was then was inserted to the heated (37 °C) docking system in the FMT imaging chamber. Prior to scanning the fluorescence scan region was manually positioned from the shoulders to base of tail for full body imaging. Total whole body signal was increased by 15% to account for body mass from non-imaged head.57 Specific regions of interest were scanned by manual positioning over the regions of interest using a medium source density (3 mm). Obtained images were analyzed using Perkin-Elmer TrueQuant software.
Ex Vivo Imaging:
Following final imaging at 72 hours, gross necropsy was performed and ex vivo images were acquired. On an IVIS 50 2D reflectance fluorescence images were obtained using the Indocyanine Green (ICG) filter set (ex:710–760 nm, em: 810–875 nm). Several hundred counts from each tissue were observable without saturation using 20 seconds of exposure and large binning off CCD camera chip, and image analysis was performed using Living Image software (Caliper Life Sciences, Hopkinton, MA). The signal intensities of each tissue were quantified by efficiency (radiance of subject/illumination intensity).
Assessment of Side Effects:
Throughout tumor growth and sample treatment animals were observed for clinical signs daily. Following treatment and euthanasia, or in the case of any inadvertent death, animals were necropsied for general assessment and specific organ toxicity including edema or enlargement. For each animal, the existence or absence of tumor metastases was recorded.
RESULTS AND DISCUSSION
Nanogel Synthesis and Characterization
To afford nanogels with defined size and surface composition we utilized our reported disulfide-based self-crosslinking nanogels as the scaffold.34–36 We aimed to formulate nanogels of different sizes (28–135 nm), corona PEG length (Mn 1000, 2000, and 5000), and extent of PEG functionalization (up to 50 mole%). Our polymeric nanogels were achieved using amphiphilic random copolymers that self-assemble into nanoscale aggregates with disulfide functionality that allows for both controlled guest entrapment and stimuli-responsive release. Redox-responsive guest release of nanoassemblies are of significant interest due to the relative concentrations of glutathione (GSH) in blood plasma (10 μM) compared to the cytosol (10 mM).47, 48 While stable encapsulation is observed at extracellular concentrations of GSH, these nanogels can release their therapeutic cargo following cellular internalization upon exposure to cytosolic concentration of GSH. Further, fine control in particle size has been demonstrated using polymer molecular weight34, as well as both temperature34 and Hofmeisterions38 due to the lower critical solution temperature (LCST) and relative hydration or dehydration behavior of PEG. In our current work, we expanded on this platform to include surface post-conjugation with PEGs of different length in a controlled and quantifiable manner.
The p(OEGMA-co-PDSMA-co-AEMA) random copolymers were synthesized by a reversible addition–fragmentation chain transfer (RAFT) copolymerization of monomers oligo(ethylene glycol) methyl ether methacrylate (OEGMA), pyridyl disulfide ethyl methacrylate (PDSMA), 2-aminoethyl methacrylate hydrochloride (AEMA) (Scheme 1). After polymerization, the polymers were purified by dialysis against dichloromethane to remove unreacted monomers, then characterized by NMR and GPC (Table 1). We used the characteristic resonances of the 2-pyridylthio moieties of PDS (δHa = 8.42, δHb = 7.63, δHc = 7.07 ppm), methoxy moieties of OEG (δH = 3.34, ppm), and methylene moieties of AE (δH = 3.78–3.46 ppm) to calculate the relative ratios of the monomer ratios for OEG:PDS:AE, which were similar for all copolymers. A range of different molecular weights were prepared, similar to those previously published, to access nanogels of various sizes.34 The number-average molecular weight (Mn) and dispersity (Đ = Mw/Mn) of the copolymers was evaluated by GPC. The AE moieties were then used as a handle to covalently conjugate the near IR probe, NHS ester functionalized cyanine 7 dye (Cy7). The reaction extent was determined to be nearly complete by evaluation of free amine using fluorescamine assay, signifying the p(OEGMA-co-PDSMA-co-Cy7) random copolymers contains ~3 mole % near IR dye (Figure S1).
Scheme 1.

(A) Synthetic scheme of p(OEGMA-co-PDSMA-co-Cy7) nanogel precursors; (B) schematic representation and corresponding reaction scheme of nanogel formulation by first crosslinking of PDS group by reducing agent DTT, then post-modification with poly(ethylene glycol) methyl ether thiol through thiol-disulfide exchange.
Table 1.
Characteristics of polymers prepared for nanogel formulation.
| Polymer | Mna | Ða | OEGb (Mole%) | PDSb (Mole%) | AEb (Mole%) |
|---|---|---|---|---|---|
| P1 | 6.0 K | 1.5 | 29 | 68 | 3 |
| P2 | 13 K | 1.2 | 28 | 70 | 2 |
| P3 | 22 K | 1.3 | 27 | 69 | 4 |
Estimated by GPC (THF) using PMMA as a standard.
Determined by NMR.
The NIR-labeled nanogels were formulated by first dispersing the p(OEGMA-co-PDSMA-co-Cy7) random copolymers (P1, P2 or P3) in the aqueous phase (10 mg/mL), then either diluting, heating, or mixing with sodium sulfate or sodium carbonate and monitored by DLS to manipulate the size of the self-assembled aggregates (Table S1). Once the desired size was achieved, the aggregates were locked under those specific solution conditions through intra-aggregate disulfide crosslink formation. The covalent crosslinking is achieved by addition of a stoichiometric amount of reducing agent DL-dithiothreitol (DTT), which generates the corresponding quantity of free thiols on PDS moieties in the aggregate interior, which then react with the remaining PDS moieties in the polymer chain. For these nanogels, specific and various crosslink densities were obtained (Table SI), which was quantified using the crosslinking reaction byproduct 2-pyridinethione’s characteristic absorption peak at 343 nm (Figure S2). The nanogel solution can then be manipulated (i.e. dilution, concentration, mild salt or heat treatment) without any consequence to the particle’s size.
Next, the remaining PDS handles of the nanogels were modified with mPEG thiols of Mn 1000, 2000, or 5000 through thiol-disulfide exchange reaction (Table 2). This post-modification was quantified by absorbance using the reaction byproduct 2-pyridinethione as previously demonstrated with post-modification with thiol ligands (Figure S2).35, 39 Prior to functionalization the nanogel zeta potential was −21 mV, and PEG conjugation did not appreciably change the surface charge with values of −15, −21, and −16 mV for PEG Mn 1000, 2000, and 5000, respectively (Figure S3). While not fully understood, several reports have found negative zeta potential of expectedly neutral PEG nanostructures.49–51 A high reaction extent was achievable with PEG Mn 1000 and 2000, ranging from ~80–97% remaining PDS groups, however it was more limited at ~68–72% with PEG Mn 5000 (Table S1). The PEG conjugation was carefully monitored to ensure that stoichiometric excess would not cause nanogel de-crosslinking. With PEG 5000, increasing stoichiometric excess did not result in higher conjugation extent, likely due to steric congestion, so we expect de-crosslinking is also an unlikely process. Even with lower conjugation extents, nanogels with PEG 5000 contained the highest total surface ethylene glycol. Final sizes of nanogels were obtained using DLS (Figure 1). Post-modification to the nanogels, particularly with Mn 5000, often increased smaller particles by as much as 10–20 nm in diameter, leading to some unavoidable size discrepancy in comparing smaller particles with differing PEG length. Five series of nanogels were prepared chronologically based on in vivo findings: size, length of PEG, percent PEG, and small size-high PEG (Table 2).
Table 2.
Size and corona properties of p(OEGMA-co-PDSMA-co-Cy7) nanogels.
| Series | Name | Diameter ± st dev (nm) (PDI) | PEG (Mn) | PEG (mole%) |
|---|---|---|---|---|
| Size | 28 nm | 28 ± 9 (0.35) | 1000 | 27 |
| Size | 50 nm | 50 ± 14 (0.27) | 1000 | 40 |
| Size | 80 nm | 80 ± 20 (0.12) | 1000 | 45 |
| Size | 135 nm | 135 ± 31 (0.12) | 1000 | 45 |
| Length PEG | 36 nm-PEG1K | 36 ± 10 (0.38) | 1000 | 51 |
| Length PEG | 56 nm-PEG2K | 56 ± 16 (0.29) | 2000 | 49 |
| Length PEG | 58 nm-PEG5K | 58 ± 17 (0.27) | 5000 | 37 |
| Length PEG | 78 nm-PEG1K | 78 ± 23 (0.10) | 1000 | 53 |
| Length PEG | 78 nm-PEG2K | 78 ± 22 (0.12) | 2000 | 51 |
| Length PEG | 79 nm-PEG5K | 79 ± 28 (0.15) | 5000 | 40 |
| Percent PEG | 49 nm 29% PEG | 49 ± 13 (0.29) | 2000 | 29 |
| Percent PEG | 44 nm 24% PEG | 44 ± 11 (0.20) | 2000 | 24 |
| Percent PEG | 42 nm 18% PEG | 42 ± 12 (0.29) | 2000 | 18 |
| Percent PEG | 34 nm 6% PEG | 34 ± 8 (0.16) | 2000 | 6 |
| Percent PEG | 31 nm 0% PEG | 31 ± 9 (0.32) | 2000 | 0 |
| Small Size High PEG | 36 nm 46% PEG | 36 ± 11 (0.40) | 2000 | 46 |
| Small Size High PEG | 35 nm 43% PEG | 35 ± 12 (0.43) | 2000 | 43 |
Figure 1.

Size distribution of (A) Size Series (B) Length PEG Series (C) Percent PEG Series (D) Small Size High PEG Series nanogels obtained by DLS measurements in water.
Tumor Cell Implantation and Growth
Triple negative breast cancer does not express the genes for estrogen receptor (ER), progesterone receptor (PRE), or human epidermal growth factor receptor 2 (Her2/neu).52 Aside from surgery and radiation, patient treatment commonly uses combinatorial chemotherapeutics including anthracyclines, taxanes, and platinum agents. Also, these patients are not candidates for endocrine therapies used to treat other more common forms of breast cancer.53 Triple negative breast cancers comprise 15–25% of breast cancer cases, and have shorter median relapses and lower survival times. Nanoparticle-based therapeutics are attractive for treatment of triple negative breast cancer, since they have the potential to increase target-specific delivery, weaken side effects, and are compatible with combinatorial therapy.
Cultured triple negative breast cancer cell line MDA-MB-231-luc-D3H2LN were implanted (5×106 cells, 50% Matrigel®) subcutaneously low in the right flank of each 6–7 week old animal on Day 0 and tumors grown until they reach the size of ~200–400 mg (target 300 mg). On Day 19–21, depending on tumor growth progress of mice model, the body weights were obtained and tumor burdens were determined from caliper measurements. The average weights and estimated tumor burdens were in a well-matched range for the first day of treatment and imaging for each series. Mice were triaged into groups (n=5 mice per group) with tumor burden within 10% overall mean, then dosed once with a 100 μL nanogel by intravenous injection for imaging on Day 0.
In Vivo FMT Tissue Distribution
Fluorescence Molecular Tomography (FMT) is an in vivo whole body quantitative imaging modality that uses red to near-infrared (NIR) light in a wavelength range of 700–900 nm. The deep tissue penetration of light up to 5 cm thick allows for time-dependent and non-invasive characterization of small animal subjects treated with NIR fluorescent probes in a 3-dimensional manner with a resolution of 1–2 mm.54, 55 The minimal NIR auto-fluorescence from biological tissue allows for quantitative information and sub-picomolar sensitivity with the appropriate choice of probe with NIR excitation wavelength, high molar absorption coefficient and fluorescence quantum yield. Cy7 probe was chosen for covalent conjugation to nanogels for its NIR excitation (λmax: 750 nm, ε: 199000) and emission (773 nm).
In FMT imaging for nanogel samples, the total whole body fluorescence was obtained at time 0, when signal distribution is attributed to the animal vasculature, and then monitored over time to assess both clearance and changes in the tissue distribution, including extravasation into specific tissues. For all nanogels, the total body probe decreased over time, but was still detectable at 72 hours after administration (Figure 2, 3, and 4). The fluorescence values at the 0 hour imaging time point for each nanogel sample were normalized in their group average total whole body probe, which scanned shoulders to base of the tail, as 100% (Figure S5, S7, S8). The normalized percentages were calculated at each imaging time point, so that the percent retention of total body probe signal over 0–72 hours could be compared between nanogel samples, despite minor variation of fluorophore concentration between nanogel samples. Tissue-based quantification of fluorophore concentration was obtained by manual positioning and scanning over the regions of interest (ROI). Fluorescence signals were significant and localized to the various tissues, allowing for quantification in the tumor, liver, spleen, intestines, lung and heart. To normalize the obtained fluorescence values, the percent injected dose (%ID) in the tissue of the total body probe signal per imaging time point were reported. For the tumor ROI, the primary endpoints were the percent injected dose per gram (%ID/g) tumor and used as an indicator for percentage of nanogel that accumulates in the tumor.
Figure 2.

Size series quantitative in vivo (A) retention of total body probe over 72 hours; (B) %ID/g tumor over 72 hours; (C) tissue distribution for tumor, liver, spleen, intestines, lungs, heart, left and right kidney of 28 nm nanogel, (D) 50 nm nanogel, (E) 80 nm nanogel and (F) 135 nm nanogel following intravenous administration obtained by FMT imaging. Data are given as mean ± standard deviation (n = 5). *Tumor values are given %ID/g tumor to normalize for any variations in mass (all group mean estimated tumor burden: 343 mg, range: 312–396 mg).
Figure 3.

Length PEG series quantitative in vivo (A) retention of total body probe over 72 hours; (B) %ID/g tumor over 72 hours; (C) tissue distribution for tumor, liver, spleen, intestines, lungs, heart, left and right kidney of 36 nm-PEG1K nanogel, (D) 56 nm-PEG2K nanogel, (E) 58 nm-PEG5K nanogel, (F) 78 nm-PEG1K nanogel, (G) 78 nm-PEG2K nanogel and (H) 79 nm-PEG5K nanogel following intravenous administration obtained by FMT imaging. Data are given as mean ± standard deviation (n = 5, except n = 4 for 72 h measurement of 36 nm-PEG1K nanogel). *Tumor values are given %ID/g tumor to normalize for any variations in mass (all group mean estimated tumor burden: 316 mg, range: 312–323 mg).
Figure 4.

Quantitative in vivo (A) retention of total body probe and (B) %ID/g tumor of Percent PEG series (tumor burden all groups means: 377 mg, n = 5, range: 363–404 mg) and (C) retention of total body probe and (D) %ID/g tumor of Small Size High PEG series (tumor burden all groups means: 294 mg, n = 5, range: 283–301 mg) following intravenous administration. *Statistically significant (P<0.05) increase in tumor %ID/g from 0 hour of administration. Representative 2D fluorescent images (lateral view) of female Harlan Beige Nude XID mice with subcutaneously implanted MDA-MB-231-luc-D3H2LN cell line in the right flank injected with (E) 100 μL saline and (F) 100 μL 36 nm 46% PEG nanogel.
Size Series:
The 28, 50, and 80 nm nanogels showed a pattern of accumulation following injection over time to a maximum %ID/g tumor at 24 to 48 hours, followed by a decrease at 72 hours (Figure 2B). The 135 nm nanogel had a generally constant %ID/g tumor observed from 0–48 hours followed by an increase at 72 hours. From initial injection to nanogels peak maximum %ID/g, the group average tumor percent increased 7, 7, 5, and 4% for the 28, 50, 80, and 135 nm nanogel, respectively. In general, the observed accumulation trend correlated to a decrease in nanogel size, which all had a measurable, but non-significant, overall increase in %ID/g. This accumulation over time was observed despite a contrasting monotonic decrease in the whole body, liver, lungs, intestines, spleen, and heart (Figure 2B, C–F). This suggests that the nanogels exhibit a selective behavior towards accumulation in the tumor compared to normal tissues. The peak maximum %ID/g tumor was 12% (48 hours), 9% (48 hours) and 12% (24 hours), and 9% (72 hours) for nanogels 28nm, 50nm, 80nm, and 135 nm, respectively.
The retention of total body probe for all nanogels decreased over time, with generally the smaller particles with higher retention, similar to tumor accumulation trends (Figure 2B). The 28 nm nanogel had the highest percentage retention in total body probe over time, with 79% of total signal retained at 72 hours. The second, third, and fourth highest retention in probe at 72 hours was the 80, 50, and 135 nm nanogels with 73, 65, and 59% of total signal retained, respectively. It is worth mentioning that the near IR probe for this particle is covalently conjugated to the nanogel structure. The nanogel is GSH degradable, and therefore the intact nanogel and its redox-degraded products would contribute to the fluorescence signal. However, this covalent and non-stimuli-responsive conjugation allows us to track the fate of the polymeric-particle, rather than an encapsulated near IR probe that may leak and cause uncertain results.
While PEGylation of the nanogel can increase circulation time, the trend is consistent with the literature that, despite size and composition, the ultimate fate of nanoparticles is extensive accumulation in the RES organs.10–17, 56–60 The liver was consistently the tissue with the highest localization (Figure 2C–F). For the 28 nm nanogel the liver %ID was at a maximum at 45% 6 hours post injection, which deceased to 26% at 72 hours (Figure S5). For the 50 nm nanogel, the liver %ID was at a maximum at 42% 6 hours post injection, which deceased to 24% at 72 hours. The 80 nm nanogel was at a maximum liver %ID at 44% 6 hours post injection, which deceased to 27% at 72 hours. Lastly, the 135 nm nanogel liver %ID was at a maximum at 42% 0 hours post injection, which deceased to 23% at 72 hours, which was the highest rate of decreasing liver concentration. For all nanogels, the intestines, lungs, heart, and spleen showed a much lower %ID and similar general monotonic decrease over the time studied. The kidneys, showed minimal and virtually non-detectable %ID with < 1% for all nanogels and all imaging time points.
General results from this series suggest that smaller nanogels gave measurably greater tumor specificity with higher %ID/g, a greater tumor accumulation over time, and had higher total body retention over the time studied. However, the we were cautious to make far-reaching conclusions in this preliminary series due to sample variability (Table S2) and the statistical significance of differences in values of tumor accumulation were not deemed sufficient. These findings, however, motivated the further investigation of the 28 nm and 80 nm nanogels. Therefore, nanogels of a smaller and larger size were made (Figure S6), then decorated with different lengths of PEG: Mn 1000, 2000, and 5000, to obtain nanogels 36 nm-PEG1K, 56 nm-PEG2K, 58 nm-PEG5K, 78 nm-PEG1K, 78 nm-PEG2K, and 79 nm-PEG5K (Table 2). Notice that PEGylation of the smaller size particles caused a considerable diameter increase from Mn 1000 to Mn 5000, while similarly increasing the diameter of larger sized particles (Figure 1).
Length PEG Series:
Smaller nanogels showed a pattern of accumulation over time to a maximum %ID/g tumor at 48 hours, except the 36 nm-PEG1K nanogel which increased up to 72 hours, exhibiting a selective behavior towards accumulation the tumor (Figure 3B). This accumulation over time was observed despite a contrasting monotonic decrease in the whole body, liver, lungs, intestines, spleen, and heart (Figure 3B, C–F). An exception would be a small increase in the intestines in the smaller nanogels (36 nm-PEG1K, 56 nm-PEG2K, and 58 nm-PEG5K). Larger nanogel patterns were less clear, with maximum %ID/g tumor occurring at 72 hours for 78 nm-PEG1K and 6 hours for both 78 nm-PEG2K and 79 nm-PEG5K.
In general, the observed accumulation trend correlated to a decrease in nanogel size. The tumors showed a measurable, but non-significant, overall increase in %ID/g at variable times post-injection, followed by monotonic decreases to 72 hours. Over the time studied, the group average tumor percentage increased to 12, 20, 13, 4, 5, and 4% for the nanogels 36 nm-PEG1K, 56 nm-PEG2K, 58 nm-PEG5K, 78 nm-PEG1K, 78 nm-PEG2K, and 79 nm-PEG5K, respectively (Figure 3B). The peak maximum %ID/g tumor was 14% (72 hours), 22% (48 hours), 17% (48 hours), 6% (72 hours), 7% (6 hours), and 4% (6 hours) for nanogels 36 nm-PEG1K, 56 nm-PEG2K, 58 nm-PEG5K, 78 nm-PEG1K, 78 nm-PEG2K, 79 nm-PEG5K respectively. This generally also suggests that smaller nanogels (originally 28 nm) gave higher tumor specificity, with 2–3 fold the peak %ID/g obtained compared to larger nanogels.
The retention of total body probe for all nanogels decreased over time, with the smaller particles achieving nearly 2-fold the percent retention in total body probe compared to the larger particles, regardless of PEG length (Figure 3A). This was well-matched with tumor accumulation trends amongst nanogels. The 36 nm-PEG1K had the highest percentage retention in total body probe over time, with 82% of total signal retained at 72 hours. The 56 nm-PEG2K nanogel had the second highest percentage retention in total body probe over time, with 79% of total signal retained at 72 hours, and the 58 nm-PEG5K nanogel also had high retention with 75% of total signal retained at 72 hours. The 78 nm-PEG1K, 78 nm-PEG2K, and 79 nm-PEG5K nanogel had 35, 38, and 29% of total signal retained at 72 hours, respectively.
The tissue accumulation for these nanogels were slightly more variable. Generally, the liver showed the highest %ID, followed by the lungs (Figure 3C, E). Of the smaller nanogels, the 36 nm-PEG1K nanogel had a liver %ID of 53% 0 hours post injection, which deceased to 20% at 72 hours. The spleen and intestines for this nanogel remained relatively constant, while the lungs did not exhibit a clear pattern, and other tissues had a low %ID (Figure S7). Amongst all nanogels the kidneys again showed minimal and virtually non-detectable (< 1) %ID. The 56 nm-PEG2K nanogel had a liver %ID of 46% 6 hours post injection, which deceased monotonically to 22% at 72 hours. Some tissues did not show the same pattern of decrease over time. The lungs had an elevated percentage to 48 hours at 12%, and the intestines and spleen showed some increase (8% and 4%, respectively) to 48 hours. The 58 nm-PEG5K nanogel had a liver %ID of 49% 48 hours post injection and decreased to 32% at 72 hours, a greater decrease than other nanogels. The lungs for this nanogel also had an elevated percentage to 48 hours at 11%, as did the spleen (2%) to 48 hours, and the intestines (7%) to 72 hours.
For the larger nanogels, regardless of PEG length, the liver had considerable %ID decrease over time suggesting rapid clearance of these nanogels (Figure 3F–H). The 78 nm-PEG1K nanogel had a liver %ID of 59% 0 hours post injection and deceased to 16% at 72 hours (Figure S7). The intestines showed consistently low values over 72 hours, while the spleen showed generally low and decreasing %ID over the time studied. The 78 nm-PEG2K nanogel had a liver %ID of 34% 0 hours post injection, and decreased to 8% at 72 hours. The intestines showed some increase to 6 hours followed by decrease over 72 hours, the spleen was relatively constant. The 79 nm-PEG5K nanogel had a liver %ID of 66% 0 hours post injection, which deceased to 14% at 72 hours. The intestines showed some increase to 6 hours followed by decrease over 72 hours. These nanogels also showed generally low and decreasing %ID for the lungs, heart, and kidney.
The results suggested that smaller nanogels had greater tumor specificity with higher %ID/g tumor, propensity to accumulate over time, and retention of total body probe over the time studied. Consistent with literature findings, smaller particles have achieved long circulation time and high tumor accumulation,22, 29–32 attributed to high curvature and reduced opsonization.22, 29 It has also been demonstrated that smaller particles can enhance performance by greater penetration of poorly permeable tumors.31 The PEG2K decorated nanogels of both sizes performed slightly better with higher peak %ID/g compared to their 1K and 5K counterparts. We were therefore interested in further evaluating smaller particles decorated with different mole percent of PEG2K, to evaluate if a threshold advantage could be identified, using %ID/g tumor as the primary endpoint. Nanogels of 31 nm were made then decorated with PEG2K (0–29 mole percent) (Table 2).
Percent PEG Series:
Based on the general consistency of tissue profiles for previous nanogels, for this series only the total body probe and %ID/g tumor were obtained and calculated to establish relative nanogel performance. In this series, the whole total body fluorophore concentration was readily detected following administration, which then had 50% clearance and decrease to near-background levels by 24–48 hours (Figure 4A). With one nanogel, 34 nm 6% PEG, the whole-body fluorophore concentration reached background levels at 6 hours. The retention in total body probe over 72 hours was low, with 15, 15, 14, 3, and 15% observed for nanogels 49 nm 29% PEG, 44 nm 24% PEG, 42 nm 18% PEG, 34 nm 6% PEG, and 31 nm 0% PEG, respectively. Such a detrimental effect was attributed to these lower PEG percentages compared to previous systems.
Compared to previous nanogels tested, these nanogels had a highest %ID/g tumor immediately after injection or 6 hours post-injection with decrease to 72 hours post-administration (Figure 4B). This suggested no propensity towards tumor specificity or accumulation. The 49 nm 29% PEG nanogel had the peak %ID/g tumor at 0 hours with 15%, however one mouse introduced large variability in this time point measurement which normalized at subsequent time points. The %ID/g tumor then had a monotonic decrease to 3% at 72 hours. The peak %ID/g tumor then followed the trend in decrease in percent PEG, with nanogels 44 nm 24% PEG, 42 nm 18% PEG, 34 nm 6% PEG, and 31 nm 0% PEG obtaining peak %ID/g of 4, 3, 2, and 2%, respectively.
We expect that these very rapidly clearing and low tumor accumulating samples are highly correlated to their lower PEG percentages which are conjugated on their surface compared to previously tested samples. It was surprising to see a decrease of 10–20 mole% PEG could impart such a detrimental effect to these nanogels. Based on observed animal excretions this result is expected to be due to clearance through the renal system. This suggests up to 30% PEG does not fully coat the particles’ surface, and a higher conjugation extent is required to impart a hydrated corona and generate steric stabilization to reduce non-specific interactions with proteins. We therefore wanted to reexamine these smaller nanogels that would be comprised of even higher percentages of PEG2K conjugated to their surface. Two nanogels were prepared: 36 nm 46% PEG and 35 nm 43% PEG for FMT analysis (Table 2).
Small Size Higher PEG:
The 36 nm 46% PEG nanogel had the peak %ID/g tumor at 72 hours with 27% (Figure 4D). This nanogel had a statistically significant (P<0.05), monotonic increase in the tumor %ID/g from 11–27% from 0–72 hours post-administration, an additional accumulation of 17% over the study, which constitutes a 1.5-fold increase compared to the 0 hour tumor signal. Representative 2D fluorescent images of a mouse injected with saline (Figure 4E) and a mouse injected with the 36 nm 46% nanogel this group (Figure 4F) showed the increase in fluorescence intensity in the tumor ROI over time when treated with nanogel. This nanogel also had the highest retention in total body probe throughout the study, with 94% of total signal retained at 72 hours (Figure 4C). This signifies only 6% of the injected dose was lost through elimination.
The 35 nm 43% PEG nanogel had a peak %ID/g tumor at 6 hours with 28%, the highest maximum percentage achieved throughout this whole study (Figure 4D). This nanogel had a statistically significant (P<0.05), monotonic increase in the tumor %ID/g from 16–28% from 0–6 hours post-administration, an additional accumulation of 12% over 6 hours, which constitutes a 0.8-fold increase compared to the 0 hour tumor signal. The %ID/g tumor then monotonically decreased to a final 15% at 72 hours. This nanogel had less retention over the test period, with 70% of total whole body probe remaining at 72 hours (Figure 4C). In contrast to the 36 nm 46% PEG nanogel, this nanogel’s %ID/g tumor profile is similar to the total body profile over time. Because of this similar trend, is less clear for this nanogel whether there is tumor specificity. While the cause for this is unclear, the peak %ID/g tumor obtained for both highly PEGylated small nanogels were consistent.
The results again suggested smaller nanogels performed better in both tumor specificity with high %ID/g, accumulation over time, and retention of total body probe over time, with prerequisite that is contains extensive PEGylation. Consistent with other studies, particles bearing PEG on their coronas of at least 2000 Mn have and improved circulation or tumor accumulation.12, 30, 60 which has also been demonstrated in the literature to reduce protein binding.23–25 These results were consistent with general conclusions from the size series and the PEG length series, and suggests that a PEG2K-modification has thus far imparted the greatest advantage in tumor specificity.
Liver Localization:
The nanogels evaluated for full tissue profiles (size series and length PEG series) had the highest %ID in the liver. For all nanogels, throughout 72 hours the liver displayed a monotonic decrease in probe concentration over time. In all cases the liver localization at initial injection was frequently above 40% (Figure 2, 3). In the case of smaller particles, the %ID in the liver decreased to ~20–30% over 72 hours, however with larger particles they decreased to as little as ~10% (Figure S5, S7). The pattern of highest accumulation in the liver with slow monotonic decrease suggests that the nanogels are being cleared through the liver through hepatic uptake and not through kidney filtration. Nanoparticles can become sequestered in one of the RES organs following surface absorption of proteins leading to opsonization and removal from the blood stream. Despite PEG improvements on circulation, it has been shown that RES clearance is often significant even in optimized PEGylated systems, with >50% of the ID residing in the liver and spleen after 48 hours.56–59 An additional contributor to this observation is that the noncontiguous vasculature in the liver with fenestrae pore size (50–100 nm) allows nanoparticles to penetrate their endothelial wall and nonspecifically accumulate.28
Other Tissue Localization:
The second highest tissue of localization at initial imaging was the lungs, while the spleen, intestines, and heart showed less though detectable and resolvable %ID (Figure 2, 3). The kidneys, however, showed minimal and virtually non-detectable %ID with < 1% for all nanogels and all imagining time points. Very little observed kidney localization indicates hepatic metabolism and not renal elimination as the mechanism of clearance. Also, relatively little spleen localization was observed, which is not always the case in polymeric nanoparticles.27 The considerable lung localization and low splenic accumulation may indicate that lung macrophages may contribute to particle uptake,61 but also may be a result of the mechanical deformability of these heavily PEGylated particles.62 “Softer” deformation-prone nanoparticles and hydrogels with high water content have been shown to reduce accumulation in the spleen compared to those with hydrophobic or tightly crosslinked cores.27, 63, 64 Further, recent microfluidic blood capillary models have shown that deformability of particles may aid in transport through small capillaries like those in the lung.62 The tissue profile obtained with these nanogels may likewise be impacted by the deformable parameter of these particles.
Toxicity
Injection of 100 μL of all samples were well-tolerated and throughout imaging no abnormal side effects were observed. For one mouse treated with the 28 nm nanogel sample (size series), during necropsy an edema in the abdominal cavity was observed. One mouse treated with 36 nm-PEG1K nanogel and one mouse treated with 56 nm-PEG2K nanogel had an enlarged spleen and tumor-like growth on the pancreas (PEG length series). Separate mice treated with 56 nm-PEG2K, 78 nm-PEG1K, and 78 nm-PEG2K also had enlarged spleens. During necropsy, one mouse treated with 36 nm 46% PEG nanogel (small size high PEG series) was observed as having an enlarged spleen. For all other mice used throughout this study, no unusual observations were made during necropsy.
Ex Vivo Tissue Distribution
The ex vivo quantification of the fluorescence signal in individual tissue homogenates from gross necropsy 72 hours post injection were reasonably consistent with the in vivo FMT analysis (Figure 5). Comparing across tissues of mice treated with the PEG length nanogel series, ex vivo homogenates showed the highest intensity in the liver in all groups, followed by the tumor and intestines. Plots of in vivo versus ex vivo results for tissues gave reasonable correlation for the smaller nanogels that had higher retention after 72 hours, with R2 values of 0.93, 0.89, and 0.89 for nanogels 36 nm-PEG1K, 56 nm-PEG2K, and 58 nm-PEG5K, respectively (Figure S9). The larger nanogels, 78 nm-PEG1K, 78 nm-PEG2K, and 79 nm-PEG5K, which had been more greatly eliminated by 72 hours, were more variable with R2 values of 0.70, 0.76, and 0.34, respectively. The discrepancies for these samples of ex vivo and in vivo results for these appear to be from the intestines, while the liver, tumor, spleen, lungs, heart, and kidney fluorescence intensities were in reasonable agreement. The greater excretion and lower signal intensity of these larger nanogels may influence the error observed from FMT and homogenate comparison. Nevertheless, ex vivo findings provided additional confidence in the reliability of tissue signal segmentation by in vivo FMT. FMT distribution has been shown to well correlated with ex vivo results,65, 66 however performance can be impacted by tissue optical properties and depth.67, 68 Live imaging can be used in combination with X-ray computed tomography, magnetic resonance imaging, or diffuse optical tomography to improve either structural imaging or optical property accuracy.69–71
Figure 5.

Length PEG series nanogels (A) ex vivo quantification of tissue fluorescence efficiency (radiance of subject/illumination intensity) normalized by sample probe concentration following final imaging and gross necropsy at 72 hours, quantified using filter set: ex: 710–760 nm, em: 810–875 nm, and (B) in vivo FMT comparison at 72 hours. Data are given as mean ± standard deviation (n = 5, except n = 4 for in vivo 72 h measurement of 36 nm-PEG1K nanogel).
CONCLUSION
In summary, NIR-labeled nanogels of various size and PEG content were formulated and exhibited discernable performance trends by in vivo FMT imaging. Using the model of triple negative human mammary carcinomas (cell line MDA-MB-231-luc-D3H2LN), we determined that smaller nanogels (~30 nm) with a high PEG conjugation (~50 mole %) of Mn 2000 length performed better in both tumor specificity with highest maximum %ID/g, a greater accumulation over time, and greater retention of total body probe. These conclusions were supported by the consistency of smaller particles with extensive PEG conjugation outperforming those that were larger or less PEGylated. Nanogels without extensive PEGylation were excreted faster, even at similar sizes to PEGylated nanogels which achieved greater total body retention and tumor specificity. Nanogels had the highest %ID in the liver throughout the time studied, however like other tissues displayed a monotonic decrease over time suggesting the nanogels are being cleared by hepatic metabolism and not through the kidneys. The %IDs observed in RES organs are consistent with reported findings on other optimized PEGylated nanomaterials.56–59 The ex vivo quantification of individual tissues from gross necropsy at 72 hours post injection generally showed trends that correlated with the FMT analysis, providing additional confidence in the reliability of tissue signal segmentation in vivo. Injection of all samples were well-tolerated and throughout imaging no abnormal side effects were observed. During necropsy, among the 85 mice used throughout this study, some observations included one mice with an edema in the abdominal cavity, five mice with enlarged spleens, two mice with a tumor-like growth on the pancreas. For all other mice, no unusual observations were made during necropsy. Overall, we determine here that minor physical and chemical differences in these nanogels greatly impacted in vivo performance. Given that the key versatility of these nanogels arises from the fact that the self-assembly strategy offers enormous tunability in the nanoparticle, these findings will guide the design of next generation of nanogels as therapeutic delivery vehicles for cancer therapy.
Supplementary Material
Acknowledgment.
We thank Johnson & Johnson for support and Dr. Edward Lawson (Janssen Pharma) for thoughtful discussions. We also thank NIGMS of the NIH (GM-065255) and the U.S. Army Research Office, whose basic science funding support provided the basis for the development of this technology platform. The authors wish to thank Tracey Woolliscroft, Christopher Bull, Deepa Balagurunathan, Lisa Repke, Patrick McConville, Erin Trachet, John L. Chunta, Deanne Lister, Scott Wise at Molecular Imaging, Inc., for performing all animal work.
Footnotes
Supporting Information Available: Synthetic experimental procedures, nanogel formulation details, statistics, and FMT results for individual tissues is supplied as Supporting Information.
References
- 1.). Safra T; Muggia F; Jeffers S; Tsao-Wei DD; Groshen S; Lyass O; Henderson R; Berry G; Gabizon A Pegylated liposomal doxorubicin (doxil): Reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann. Oncol 2000, 11, 1029–1033. [DOI] [PubMed] [Google Scholar]
- 2.). Fassas A; Buffels R; Kaloyannidis P; Anagnostopoulos A Safety of high-dose liposomal daunorubicin (daunoxome) for refractory or relapsed acute myeloblastic leukaemia. Br. Jo. Haematol 2003, 122, 161–163. [DOI] [PubMed] [Google Scholar]
- 3.). Matsumura Y; Maeda H A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- 4.). Maeda H; Nakamura H; Fang J The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev 2013, 65, 71–79. [DOI] [PubMed] [Google Scholar]
- 5.). Bae YH; Park K Targeted drug delivery to tumors: myths, reality and possibility. J. Controlled Release 2011, 153, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.). Li S-D; Huang L Pharmacokinetics and Biodistribution of Nanoparticles. Mol. Pharmaceutics, 2008, 5 (4), 496–504. [DOI] [PubMed] [Google Scholar]
- 7.). Venturoli D; Rippe B Ficoll and dextran vs. globular proteins as probes for testing glomerular permselectivity: effects of molecular size, shape, charge and deformability. Am. J. Physiol 2005, 288, F605–F613. [DOI] [PubMed] [Google Scholar]
- 8.). Romberg B; Hennink WE; Storm G Sheddable coatings for long-circulating nanoparticles. Pharm. Res 2008, 25, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.). Moghimi SM; Hunter AC; Murray JC Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. ReV 2001, 53, 283–318. [PubMed] [Google Scholar]
- 10.). Vittaz M; Bazile D; Spenlehauer G; Verrecchia T; Veillard M; Puisieux F; Labarre D Effect of PEO surface density on long-circulating PLA-PEO nanoparticles which are very low complement activators. Biomaterials 1996, 17, 1575–1581 [DOI] [PubMed] [Google Scholar]
- 11.). Owens DE III; Peppas NA Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm 2006, 307, 93–102. [DOI] [PubMed] [Google Scholar]
- 12.). Liu Y; Hu Y; Huang L Influence of polyethylene glycol density and surface lipid on pharmacokinetics and biodistribution of lipid-calcium-phosphate nanoparticles. Biomaterials, 2014, 35, 3027–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.). Gref R; Lück M; Quellec P; Marchand M; Dellacherie E; Harnisch S; Blunk T; Müller RH ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. 2000, 18, 301–313. [DOI] [PubMed] [Google Scholar]
- 14.). Verbaan FJ; Oussoren C; Snel CJ; Crommelin DJ; Hennink WE; Storm G Steric stabilization of poly(2-(dimethylamino)ethyl methacrylate)-based polyplexes mediates prolonged circulation and tumor targeting in mice. J. Gene Med 2004, 6, 64–75. [DOI] [PubMed] [Google Scholar]
- 15.). Vader P; van der Aa LJ; Engbersen JF; Storm G; Schiffelers RM Physicochemical and Biological Evaluation of siRNA Polyplexes Based on PEGylated Poly(amido amine)s. Pharm. Res 2012, 29(2), 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.). Crownover E; Duvall CL; Convertine A; Hoffman AS; Stayton PS RAFT-synthesized Graft Copolymers that Enhance pH-dependent Membrane Destabilization and Protein Circulation Times. J. Controlled Release 2011, 155, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.). Perry JL; Reuter KG; Kai MP; Herlihy KP; Jones SW; Luft JC; Napier M; Bear JE; DeSimone JM PEGylated PRINT Nanoparticles: The Impact of PEG Density on Protein Binding, Macrophage Association, Biodistribution, and Pharmacokinetics. Nano Lett, 2012, 12 (10), 5304–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.). Molineux G PEGylation: engineering improved pharmaceuticals for enhanced therapy. Cancer Treat Rev. 2002, 28 (Suppl. A), 13–16. [DOI] [PubMed] [Google Scholar]
- 19.). Moribe K; Maruyama K Reviews on PEG coated liposomal drug carriers. Drug Delivery Syst. 2001, 16 (3), 165–171. [Google Scholar]
- 20.). Jeon SI; Andrade JD Protein-surface interactions in the presence of olyethylene oxide: Effect of protein size. J. Colloid Interf. Sci 1991, 142, 159–166. [Google Scholar]
- 21.). Jeon SI; Lee JH; Andrade JD; De Gennes PG Protein-surface interactions in the presence of olyethylene oxide: Simplified theory. J. Colloid Interf. Sci 1991, 142, 149–158. [Google Scholar]
- 22.). Fang C; Shi B; Pei YY; Hong MH; Wu J; Chen HZ In vivo tumor targeting of tumor necrosis factor-R-loaded stealth nanoparticles: Effect of MePEG molecular weight and particle size. Eur. J. Pharm. Sci 2006, 27, 27–36. [DOI] [PubMed] [Google Scholar]
- 23.). Gombotz WR; Guanghui W; Horbett TA; Hoffman AS Protein adsorption to poly(ethylene oxide) surfaces. J. Biomed. Mater. Res 1991, 25, 1547–1562. [DOI] [PubMed] [Google Scholar]
- 24.). Stolnik S; Dunn SE; Garnett MC; Davies MC; Coombes AGA; Taylor DC; Irving MP; Purkiss SC; Tadros TF; Davis SS; Illum L Surface Modification of Poly(lactide-co-glycolide) Nanospheres by Biodegradable Poly(lactide)-Poly(ethylene glycol) Copolymers. Pharm. Res 1994, 11(12), 1800–1808. [DOI] [PubMed] [Google Scholar]
- 25.). Bergström K; Holmberg K; Safranj A; Hoffman AS; Edgell MJ; Kozlowski A; Hovanes BA; Harris JM Reduction of fibrinogen adsorption on PEG-coated polystyrene surfaces. J. Biomed. Mater. Res 1992, 26(6), 779–790. [DOI] [PubMed] [Google Scholar]
- 26.). Bergström K; Osterberg E; Holmberg K; Hoffman AS; Schuman TP; Kozlowski A; Harris JM Effects of branching and molecular weight of surface-bound poly(ethylene oxide) on protein rejection. J Biomater Sci Polymer Edn 1994, 6, 123–132. [DOI] [PubMed] [Google Scholar]
- 27.). Yang Z; Leon J; Martin M; Harder JW; Zhang R; Liang D; Lu W; Tian M, Gelovani JG; Qiao A; Li C Pharmacokinetics and biodistribution of near-infrared fluorescence polymeric nanoparticles. Nanotechnology 2009, 20, 165101–16511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.). Liu D; Mori A; Huang L Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1- containing liposomes. Biochim. Biophys. Acta 1992, 1104, 95–101. [DOI] [PubMed] [Google Scholar]
- 29.). Nagayama S; Ogawara K; Fukuoka Y; Higaki K; Kimura T Time-dependent changes in opsonin amount associated on nanoparticles alter their hepatic uptake characteristics. Int. J. Pharm 2007, 342, 215–221. [DOI] [PubMed] [Google Scholar]
- 30.). Kim TY; Kim DW; Chung JY; Shin SG; Kim SC; Heo DS; Kim NK; Bang YK Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin. Cancer. Res 2004, 10, 3708–3716. [DOI] [PubMed] [Google Scholar]
- 31.). Cabral H; Matsumoto Y; Mizuno K; Chen Q; Murakami M; Kimura M; Terada Y; Kano MR; Miyazono K; Uesaka M; Nishiyama N; Kataoka K Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nature Nanotechnology, 2011, 6, 815–823. [DOI] [PubMed] [Google Scholar]
- 32.). Kim DW; Kim SY; Kim HK; Kim SW; Shin SW; Kim JS; Park K; Lee MY Heo, D. S. Multicenter phase II trial of Genexol-PM, a novel Cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol 2007, 18, 2009–2014. [DOI] [PubMed] [Google Scholar]
- 33.). Lee KS; Chung HC; Im SA; Park YH; Kim CS; Kim SB; Rha SY; Lee MY; Ro J Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res. Treat 2008, 108, 241–250. [DOI] [PubMed] [Google Scholar]
- 34.). Ryu JH; Chacko RT; Jiwpanich S; Bickerton S; Babu RP; Thayumanavan S Self-Cross-Linked Polymer Nanogels: A Versatile Nanoscopic Drug Delivery Platform. J. Am. Chem. Soc 2010, 132, 17227–17235. [DOI] [PubMed] [Google Scholar]
- 35.). Ryu JH; Jiwpanich S; Chacko R; Bickerton S; Thayumanavan S Surface-Functionalizable Polymer Nanogels with Facile Hydrophobic Guest Encapsulation Capabilities. J. Am. Chem. Soc 2010, 132, 8246–8247. [DOI] [PubMed] [Google Scholar]
- 36.). Jiwpanich S; Ryu JH; Bickerton S; Thayumanavan S Noncovalent Encapsulation Stabilities in Supramolecular Nanoassemblies, J. Am. Chem. Soc 2010, 132, 10683–10685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.). Ghosh S; Basu S; Thayumanavan S Simultaneous and Reversible Functionalization of Copolymers for Biological Applications. Macromolecules 2006, 39, 5595–5597. [Google Scholar]
- 38.). Li L; Ryu JH; Thayumanavan S Effect of Hofmeister Ions on the Size and Encapsulation Stability of Polymer Nanogels. Langmuir, 2013, 29 (1), 50–55. [DOI] [PubMed] [Google Scholar]
- 39.). Ventura J Eron SJ González-Toro DC; Raghupathi K; Wang F; Hardy JA; Thayumanavan S Reactive Self-Assembly of Polymers and Proteins to Reversibly Silence a Killer Protein. Biomacromolecules, 2015, 16(10), 3161–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.). Vasquez KO; Casavant C; Peterson JD Quantitative Whole Body Biodistribution of Fluorescent-Labeled Agents by Non-Invasive Tomographic Imaging. PLoS ONE, 2011, 6(6), e20594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.). Schabel F; Griswold D; Laster W; Corbett T; Lloyd H Quantitative evaluation of anticancer agent activity in experimental animals. Pharmac. Ther. A 1977, 1, 411–435. [Google Scholar]
- 42.). Corbett T; Griswold D; Roberts B; Peckham J; Schabel F; Evaluation of single agents and combinations of chemotherapeutic agents in mouse colon carcinomas. Cancer, 1977, 40(5), 2660–2690. [DOI] [PubMed] [Google Scholar]
- 43.). Schabel F; Griswold D; Corbett T; Laster R; Mayo J; Lloyd H Testing therapeutic hypotheses in mice and man: Observations on the therapeutic activity against advanced solid tumors of mice treated with anticancer drugs that have demonstrated or potential clinical utility for treatment of advanced solid tumors of man. Methods in Cancer Research 1979, 17, 3–51. [Google Scholar]
- 44.). Plowman J, Dykes D, Hollingshead M, Simpson-Herren L, and Alley M. Human tumor xenograft models in NCI drug development. In: Anticancer drug development guide: preclinical screening, clinical trials, and approval. Teicher (ed) Humana Press Inc. 1993. [Google Scholar]
- 45.). Corbett T; Valeriote F; LoRusso P; Polin L; Panchapor C; Pugh S; White K; Knight J; Demchik L; Jones J; Jones L; Lowichik N; Biernat L; Foster B; Wozniak A; Lisow L; Valdivieso M; Baker L; Leopold W; Sebolt J; Bissery M; Mattes K; Dzubow J; Rake J; Perni R; Wentland M; Coughlin S; Shaw JM; Liversidge G; Liversidge E; Bruno J; Sarpotdar P; Moore R; Patterson G Tumor models and the discovery and secondary evaluation of solid tumor active agents. Int. J. Pharmacognosy, 1995, 33, 102–122. [Google Scholar]
- 46.). Corbett T; Polin L; Roberts BJ; Lawson AJ; Wilbur R; Leopold WR; White K; Kushner J; Paluch J; Hazeldine S; Moore R; Rake J; Horwitz JP Transplantable Syngeneic Rodent Tumors: Solid Tumors of Mice Tumor Models in Cancer Research, 2002, Humana Press, 41–71. [Google Scholar]
- 47.). Saito G; Swanson JA; Lee KD Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv. Drug Delivery Rev 2003, 55, 199–215. [DOI] [PubMed] [Google Scholar]
- 48.). Meister A; Anderson ME Glutathione. Ann. Rev. Biochem 1983, 52, 711–760. [DOI] [PubMed] [Google Scholar]
- 49.). Li L; Raghupathi K; Yuan C; Thayumanavan S Surface charge generation in nanogels for activated cellular uptake at tumor-relevant pH. Chem. Sci 2013, 4, 3654–3360. [Google Scholar]
- 50.). Radovic-Moreno AF; Lu TK; Puscasu VA; Yoon CJ; Langer R; Farokhzad OC Surface Charge-Switching Polymeric Nanoparticles for Bacterial Cell Wall-Targeted Delivery of Antibiotics. ACS Nano 2012, 6, 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.). Bahadur KCR; Xu P Multicompartment Intracellular Self-Expanding Nanogel for Targeted Delivery of Drug Cocktail. Adv. Mater 2012, 24, 6479–6483. [DOI] [PubMed] [Google Scholar]
- 52.). Foulkes WD; Smith IE; and Reis-Filho JS Triple-negative breast cancer. N. Engl. J. Med 2010, 363, 1938–1948. [DOI] [PubMed] [Google Scholar]
- 53.). Hudis CA; Gianni L Triple-Negative Breast Cancer: An Unmet Medical Need. The Oncologist. 2011, 16, 1–11. [DOI] [PubMed] [Google Scholar]
- 54.). Kovar JL; Simpson MA; Schutz-Geschwender A; Olive DM A systematic approach to the development of fluorescent contrast agents for optical imaging of mouse cancer models. Anal Biochem. 2007, 367, 1–12. [DOI] [PubMed] [Google Scholar]
- 55.). Weissleder R A clearer vision for in vivo imaging. Nat Biotechnol. 2001, 19, 316–317. [DOI] [PubMed] [Google Scholar]
- 56.). Perrault SD; Walkey C; Jennings T; Fischer HC; Chan WC Mediating tumor targeting efficiency of nanoparticles through design. Nano letters. 2009, 9, 1909–1915. [DOI] [PubMed] [Google Scholar]
- 57.). Alexis F; Pridgen E; Molnar LK; Farokhzad OC Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008, 5, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.). Nahrendorf M; Keliher E; Marinelli B; Waterman P; Feruglio PF,; Fexon L; Pivovarov M; Swirski FK; Pittet MJ; Vinegoni C; Weissleder R Hybrid PET-optical imaging using targeted probes. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 7910–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.). Choi CH; Alabi CA; Webster P; Davis ME Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 1235–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.). Wang J; Bai R; Yang R; Liu J; Tang J; Liu Y; Li J; Chai Z; Chen C Size- and surface chemistry-dependent pharmacokinetics and tumor accumulation of engineered gold nanoparticles after intravenous administration. Metallomics, 2015, 7, 516–524. [DOI] [PubMed] [Google Scholar]
- 61.). Blanco E; Shen H; Ferrari M Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature Biotechnology, 2015, 33, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.). Cui J; Björnmalm M; Liang K; Xu C; Best JP; Zhang X; Caruso F Super-soft hydrogel particles with tunable elasticity in a microfluidic blood capillary model. Adv. Mater 2014, 26, 7295–7299. [DOI] [PubMed] [Google Scholar]
- 63.). Zhang L; Cao Z; Li Y; Ella-Menye J-R; Bai T; Jiang S Softer zwitterionic nanogels for longer circulation and lower splenic accumulation. ACS Nano, 2012, 6, 6681–6686. [DOI] [PubMed] [Google Scholar]
- 64.). Merkel TJ; Chen K; Jones SW; Pandya AA; Tian S; Napier ME; Zamboni WE; DeSimone JM The effect of particle size on the biodistribution of low-modulus hydrogel PRINT particles. J. Control. Release, 2012, 162, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.). Hage C; Gremse F; Griessinger CM; Maurer A; Hoffmann SHL; Osl F; Pichler BJ; Kiessling F; Scheuer W; Pöschinger T Comparison of the Accuracy of FMT/CT and PET/MRI for the Assessment of Antibody Biodistribution in Squamous Cell Carcinoma Xenografts. J. Nucl. Med 2017. [DOI] [PubMed] [Google Scholar]
- 66.). Vasquez KO; Casavant C; Peterson JD Quantitative Whole Body Biodistribution of Fluorescent-Labeled Agents by Non-Invasive Tomographic Imaging. PLoS One, 2011, 6, e20594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.). Ale A; Ermolayev V; Herzog E; Cohrs C; de Angelis MH; Ntziachristos V FMT-XCT: in vivo animal studies with hybrid fluorescence molecular tomography–X-ray computed tomography. Nature Methods, 2012, 9, 613–622. [DOI] [PubMed] [Google Scholar]
- 68.). Jacques SL; Pogue BW Tutorial on diffuse light transport. J. Biomed. Opt 2008, 13, 041302. [DOI] [PubMed] [Google Scholar]
- 69.). Stuker F; Ripoll J; Rudin M Fluorescence Molecular Tomography: Principles and Potential for Pharmaceutical Research. Pharmaceutics, 2011, 3, 229–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.). Tan Y; Jiang H Diffuse optical tomography guided quantitative fluorescence molecular tomography. Appl Opt. 2008, 47, 2011–2016. [DOI] [PubMed] [Google Scholar]
- 71.). Tan Y; Jiang H DOT guided fluorescence molecular tomography of arbitrarily shaped objects. Med. Phys 2008, 35, 5703–5707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.