

Abstract
Macrophages are integral components of cardiac tissue and exert profound effects on the healthy and diseased heart. Paradigm shifting studies employing advanced molecular techniques have revealed significant complexity within these macrophage populations that reside in the heart. In this final of a 4-part review series covering the macrophage in cardiovascular disease, we review the origins, dynamics, cell surface markers, and respective functions of each cardiac macrophage subset identified to date, including in the specific scenarios of myocarditis and after myocardial infarction. Looking ahead, a deeper understanding of the diverse and often dichotomous functions of cardiac macrophages will be essential for the development of targeted therapies to mitigate injury and orchestrate recovery of the diseased heart. Moreover, as macrophages are critical for cardiac healing, they are an emerging focus for therapeutic strategies aimed at minimizing cardiomyocyte death, ameliorating pathological cardiac remodeling, and for treating heart failure and after myocardial infarction.
Keywords: Macrophage, Ontogeny, Heart Development, Coronary Angiogenesis, Heart Failure
Condensed Abstract:
Macrophages are integral components of cardiac tissue with profound effects on the healthy and diseased heart. Paradigm shifting studies have revealed significant complexity within macrophage populations that reside in the heart. In this final part of a 4-part review series we review the origins, dynamics, cell surface markers, and respective functions of each cardiac macrophage subset identified to date, including in myocarditis and after myocardial infarction. A deeper understanding of the diverse and often dichotomous functions of cardiac macrophages will be essential for the development of targeted therapies to mitigate injury and orchestrate recovery of the diseased heart.
In this review series, we have covered the basic macrophage biology in Part 1, macrophage biology in atherosclerosis in Part 2, and macrophage biology and imaging at the systemic level in Part 3. Here in Part 4 we address the role of the macrophage in the heart, both in normal homeostasis, and in myocarditis and myocardial infarction (MI).
Cardiac Macrophages in Health and Homeostasis
Introduction
Heart failure is an important cause of morbidity and mortality, and in parallel, it is recognized that innate immune system activation occurs in patients with heart failure, and is associated with adverse clinical outcomes (see review (1)). An important question which then arises is whether the immune system is directly responsible for disease progression, including pathological remodeling of the left ventricle (LV).
To that point, while it is accepted that neutrophils produce robust inflammatory responses and contribute to heart failure after acute ischemic injury (2–4), the exact roles of monocytes and macrophages continue to be debated. The literature is filled with seemingly contradictory reports that macrophages not only trigger damaging inflammatory responses, but also, that they mediate tissue repair and in some cases cardiac regeneration (5). This paradox is well established in models of ischemic cardiac injury, where macrophages in the post-MI heart drive robust inflammatory responses and pathological remodeling, as well as the resolution of inflammation, tissue repair, and coronary angiogenesis (6). One explanation is that distinct macrophage populations may mediate inflammatory and reparative macrophage behaviors (7). However, until recently, the exact identities of these proposed macrophage subsets were largely undefined.
In this review, we consider recent progress in elucidating macrophage populations resident within the heart, and discuss the functional relevance of these subsets, particularly as relevant to heart development, homeostasis, inflammation, myocarditis and after MI.
Macrophage Heterogeneity and Diversity
As highlighted throughout this review series, macrophages are found in all tissues and play important roles during development, adult physiologic homeostasis, tissue repair and remodeling, and immunity. However, as a challenge faced by the field, prior classification systems and definitions that rely on cell surface markers and inflammatory states (i.e., M1-M2) are increasingly understood to be imperfect, primarily because macrophages display significant plasticity and dynamic expression of cell surface and M1/M2 markers (see Part 1 review article). Furthermore, this heterogeneity has implications for the translation of studies across species. For example, the human equivalent to Ly6chigh monocytes was previously considered to be wCD14+CD16− monocytes, while the human equivalent to Ly6clow monocytes was thought to be CD14intCD16+ monocytes. However, as discussed in Part 1 of this series, studies using advanced profiling techniques have questioned this dogma, and the human equivalents of these populations remain to be clearly defined.
An alternative approach is to define macrophage subsets based on ontogeny or developmental lineage. Thus, recent studies have shown that many tissue-resident macrophages are established during embryonic development and persist into adulthood independent of blood monocyte input (8–11). Specifically, definitive macrophages are subdivided into embryonic (usually fetal liver) and adult-derived subsets, both of which transition through monocyte intermediates and express FLT3 as they differentiate (8,12). Based on these findings, a Flt3-Cre mouse was developed to track the progeny of definitive hematopoietic lineages (13). Using this lineage tracking tool, several studies identified primitive (Flt3-Cre negative) and definitive (Flt3-Cre positive) macrophages in a variety of tissues including the heart (8,12).
In addition to lineage tracking, transcription factors and surface markers may differ between macrophage lineages. For example, yolk sac-derived macrophages have a characteristic CX3CR1highF4/80high CD11blow phenotype, while definitive monocyte-derived macrophages display a CX3CR1intF4/80lowCD11bhigh phenotype (8,11,14). Collectively, these observations suggest that tissue resident macrophages are best defined by a combination of ontological origin, recruitment dynamics, and cell surface marker expression. In the following section we discuss how this approach has elucidated functionally distinct macrophage populations in the heart.
Cardiac Macrophage Populations
Resident cardiac macrophages represent 6–8% of the non-cardiomyocyte population in the healthy adult mouse heart and even larger fraction in the developing heart (15–17). Previously, it was thought that during homeostasis most cardiac macrophages were derived from circulating monocytes and represented a homogeneous population with M2 characteristics (18). Recently, it was shown that unlike other tissues such as the brain which contain a single dominant macrophage population (yolk sac-derived microglia), the heart contains several macrophage populations with discrete ontological origins including primitive yolk sac-derived macrophages, fetal monocyte-derived macrophages, and adult monocyte-derived macrophages (8,9,16). Each of these populations seeds the heart at distinct developmental stages and eventually co-exist within the adult heart.
Developing Heart
Macrophages are first evident in the mouse heart at E11.5 in association with the epicardium. These cells are derived from primitive yolk sac progenitors and are characterized by low surface expression of C-C chemokine receptor 2 (CCR2) and are referred to as CCR2− (19). They also express low levels of major histocompatibility Complex (MHC) class II. Mechanistically, yolk sac-derived CCR2− macrophages seed the heart and exist independent of monocyte input. Instead, they rely on instructive cues from the epicardium, with epicardial ablation impeding the recruitment of yolk sac-derived macrophages through an unclear signaling mechanism (20). Beginning at E13.5–14.5, yolk sac-derived CCR2− macrophages play a critical role in the development and maturation of the coronary system (described below).
Also beginning at E14.5, a population of CCR2+MHC-IIlow macrophages is recruited to the heart, and becomes associated with the endocardial surface (19). These cells are derived from definitive hematopoiesis (predominately fetal monocyte progenitors) and require monocyte input for their maintenance. The function of these cells is unknown as they appear to be dispensable for proper cardiac development.
Neonatal Heart
During the first week of life, the mouse heart contains a single yolk sac-derived CCR2− macrophage population that expands via local proliferation (21). Beginning 2 weeks after birth, a second population of CCR2− macrophages that are Flt3-Cre+ (definitive hematopoietic origin) enter the heart. This latter population is presumably derived from fetal monocytes based on their timing of entry, cell surface characteristics (CX3CR1int), and developmental origin (8,9). At this time-point, both primitive and definitive CCR2− macrophages are MHC-IIlow.
Adult Heart
During homeostasis, the adult mouse heart contains at least 3 macrophage subsets: CCR2MHC-IIlow, CCR2−MHC-IIhigh, and CCR2+MHC-IIhigh. Monocytes display a CCR2+MHC-IIlow cell surface phenotype (8,9,16,21). This classification system is supported by single-cell RNA sequencing data indicating that CCR2 and MHC-II expression are sufficient to define the major monocyte and macrophage populations within the naïve adult mouse heart (22).
CCR2−MHC-IIlow and CCR2−MHC-IIhigh macrophages are long-lived, derived from embryonic origins including primitive yolk sac and fetal monocyte progenitors, and are maintained independent of monocyte input through local proliferation (Figure 1). The mechanisms responsible for acquisition of MHC-IIhigh expression first observed at 3–4 weeks of age are unclear. Interestingly, during aging, more substantial contributions from circulating monocytes are observed, suggesting that monocytes may differentiate into CCR2− macrophages (21,23). CCR2+MHC-IIhigh macrophages are derived exclusively from blood monocytes and require ongoing monocyte input through a CCR2-dependent mechanism. CCR2+MHC-IIhigh macrophages are first evident in the heart at 3–4 weeks of age. The signaling events responsible for their initial recruitment remain unexplored.
Figure 1: Macrophage heterogeneity and functions in the adult mouse heart.
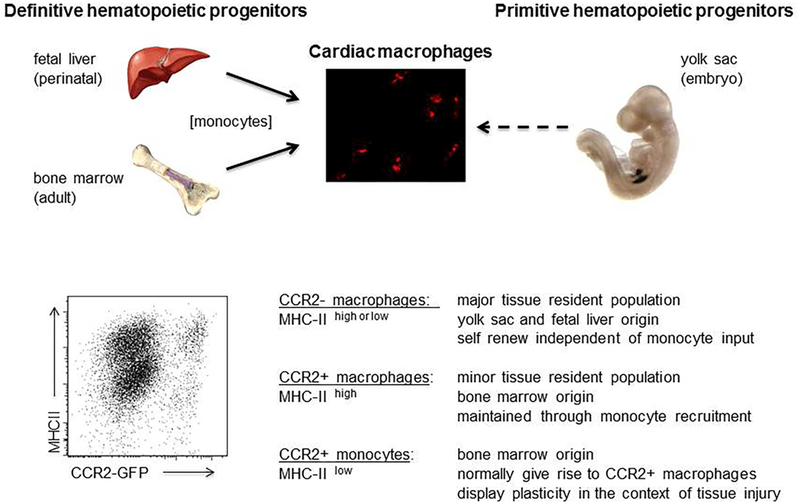
Top, Schematic depicting the developmental origins of cardiac macrophages. Bottom, Flow cytometry showing macrophage populations within the adult heart during homeostasis and the mechanisms by which each population is maintained. CCR2 expression was examined by measuring GFP fluorescence in Ccr2-GFP reporter mice. Adapted from (21).
Following acute tissue injury such as MI, a dramatic shift occurs and resident macrophages are largely replaced by infiltrating CCR2+ monocytes and CCR2+ monocytederived macrophages (Central Illustration) (7,8,16,21,23). In general, infiltrating CCR2+ macrophages are thought to be pro-inflammatory and contribute to adverse remodeling through collateral myocardial injury, neutrophil recruitment, and inflammatory cytokine production (21,24). However, infiltrating monocytes likely assume a variety of cell fates that are influenced by environmental cues (covered in greater detail later in this article).
Central Illustration. Myocardial injury triggers a dramatic shift in macrophage composition.
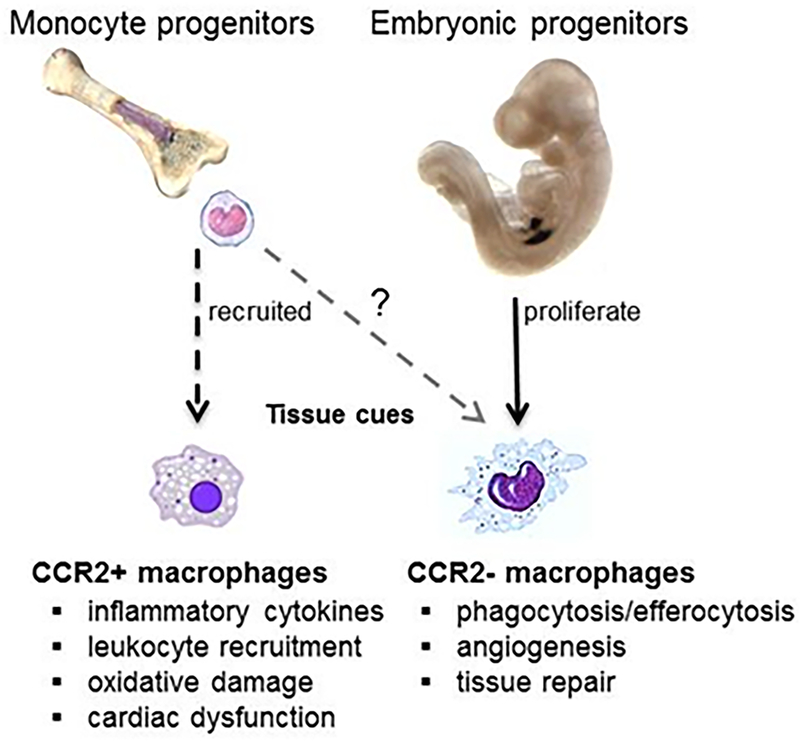
While tissue resident CCR2− macrophages expand through local proliferation, resident macrophage subsets are vastly outnumbered by recruited monocytes. Upon entering the heart, monocytes predominantly differentiate into CCR2+ macrophages with smaller contributions to other macrophage subsets (i.e., CCR2− macrophages). CCR2+ macrophages display a predominantly pro-inflammatory phenotype, while CCR2− macrophages exhibit a reparative/regenerative phenotype.
Cardiac Macrophages Functions
Shared Functions
Adult cardiac macrophages share a number of typical macrophage functions including the ability to sample the local microenvironment and engulf debris through micropinocytosis, phagocytosis, and efferocytosis (8,9,16,18). In addition, a recent report has implicated cardiac macrophages in atrial-ventricular (AV) conduction (22). The authors reported abundant CCR2− and CCR2+ macrophages within the AV node of the mouse and human heart. Within the AV node, resident cardiac macrophages appear to make direct contact with cardiomyocytes via gap junctions. Furthermore, cardiac macrophages in vitro were able to form spontaneous gap junctions with cultured cardiomyocytes and facilitated electrical conduction. In vivo, removal of cardiac resident macrophages or conditional deletion of connexin 43 in macrophages resulted in impaired AV conduction and in some instances heart block.
Functions of CCR2− macrophages
Yolk sac-derived CCR2− macrophages are essential mediators of coronary development (19). They are closely associated with the epicardium, epicardial-derived cells (precursors of coronary interstitial cells: pericytes and smooth muscle cells), and coronary endothelium during heart development. Removal of CCR2− macrophages in the embryo results in aberrant remodeling and maturation of the coronary arterial vasculature. Within this context, primitive CCR2− macrophages are selectively recruited to perfused coronary vasculature where they stimulate vessel growth and remodeling, at the expense of other vessels receiving minimal blood supply. Mechanistically, insulin-like growth factor (IGF) 1 and IGF2 are potential mediators by which primitive CCR2− macrophages regulate coronary remodeling (19).
Yolk sac-derived CCR2− macrophages are also critical regulators of cardiac tissue repair (21). Particularly in the neonatal heart and in certain species such as zebrafish, this function may be of major importance (15,25,26), although, it remains to be elucidated whether macrophage populations necessary for tissue regeneration in zebrafish and amphibians are derived from embryonic progenitors.
The functions of CCR2− macrophage subsets within the adult heart are less clear. Compared to CCR2+MHC-IIhigh macrophages, CCR2−MHC-IIlow and CCR2−MHC-IIhigh macrophages express fewer inflammatory mediators and have less capacity for inflammatory chemokine and cytokine expression (8,9,21). These observations suggest that CCR2−MHC-IIlow and CCR2−MHC-IIhigh subsets may retain a reparative phenotype. Consistent with this, CCR2MHC-IIlow and CCR2−MHC-IIhigh macrophages express IGF1 and are pro-angiogenic (19). Furthermore, compared to CCR2+ macrophages, resident CCR2− macrophages show enhanced capacity to phagocytose cardiomyocyte content (27), likely contributing to tissue homeostasis. To date, no differences between the functions of CCR2− macrophages derived from primitive versus definitive hematopoiesis have been observed. However, among the CCR2− subsets, only CCR2−MHC-IIhigh macrophages have the capacity to display antigen and stimulate T-cell responses (8,9).
Functions of CCR2+ Macrophages
CCR2+ cardiac macrophages are primarily derived from circulating monocytes and are enriched in proinflammatory genes, in particular the NLRP3 pathway, which leads to delivery of interleukin 1β (IL-1β) (21) - a proinflammatory cytokine and therapeutic target (28). While the exact functions of resident CCR2+MHC-IIhigh macrophages under steady state conditions are incompletely defined, their activation likely represents a proximate mechanism driving inflammation in the failing adult heart. This is supported by the observation that resident CCR2+MHC-IIhigh macrophages regulate myocardial neutrophil infiltration following ischemia-reperfusion injury. Mechanistically, resident CCR2+MHC-IIhigh macrophages are activated by mitochondrial DNA (presumably released from dying cardiomyocytes) through a toll-like receptor (TLR) 9-dependent pathway that results in the release of the neutrophil chemokines, chemokine (C-X-C motif) ligand (CXCL) 2 and CXCL5 (29). Whether resident CCR2+MHC-IIhigh macrophages similarly govern monocyte recruitment and macrophage ontogeny through a shared molecular pathway is not yet clear, but represents an attractive hypothesis. Similar to CCR2−MHC-IIhigh macrophages, CCR2+MHC-IIhigh macrophages have the capacity to display antigen and stimulate T-cell responses in vitro (8,9). Whether CCR2+MHC-IIhigh macrophages do so in vivo and whether this population displays differential capacity to stimulate effector and regulatory T-cell subsets remains to be seen.
The Macrophage, Myocardial Infarction and Cardiac Regeneration
In models of mammalian wound healing, immune cells are a major force in determining the ultimate quality of repair (30). Recent research has uncovered a central role for the innate immune system in the response to cardiac tissue damage, where successful tissue regeneration depends on cellular and signaling components of local inflammatory reactions. In the uninjured heart, tissue–resident immune cell populations participate in routine immune-surveillance, contribute to angiogenesis, and participate in signaling networks that modulate the local stromal environment to dampen tissue inflammation. Why then are mammalian hearts so prone to perturbed immune reactions that lead to detrimental inflammation and scarring after injury?
Clues can be gained from the central role played by macrophages in the resolution of cardiac injury in highly regenerative scenarios. The regenerative capacity of a tissue is influenced by regulatory networks orchestrated by local immune responses to tissue damage, with macrophages being a central component of the injury response, promoting healing and tissue remodeling through secretion of anti-inflammatory and angiogenic cytokines. Furthermore, as mentioned, the capacity for full cardiac regeneration of immature mammalian hearts is critically dependent on macrophage actions: the mouse neonatal heart harbors resident cardiac macrophages that generate minimal inflammation and promote post-MI cardiac recovery, where they influence neo-vascularization (15,21).
Variation in the interaction of macrophages with damaged tissues is a likely factor in the range of regenerative potential across the animal kingdom (26). Curiously, some closely related species exhibit robust cardiac regeneration, while others undergo regenerative failure. Among adult amphibians, several divergent salamander species are capable of cardiac regeneration, while more distantly related frogs fail to regenerate and heal cardiac injury with non-functional scar tissue (31). Among bony fishes, some teleost fish including the Zebrafish, Goldfish, and Giant danio feature robust regenerative potential (32–34), but those in the closely related Medaka group fail to regenerate (35).
While the immune landscape in many of these species remains to be fully characterized, studies of macrophage profiles in models of compromised versus effective repair suggest that retaining a balanced immune response is imperative for regenerative therapies to meet their full potential in the adult human heart.
Macrophage Functions in Damaged Cardiac Tissue
Resident tissue macrophages play distinct roles in stress responses, accumulating at sites of cardiac injury within five minutes (36), likely due to movement toward areas of damage. A diverse range of additional leukocytes is subsequently recruited (21,37–39). Following infiltration of neutrophils, which peak at the injury site after one day and release reactive oxidative species, monocytes/macrophages are the predominant leukocyte type within the injured heart from days 2–5 post injury (Figure 2) (7,39,40), with cell numbers gradually reaching basal levels thereafter (7,40). Sager et al (41) shed light on the pathways underlying this recruitment, showing that IL-1β is produced by the infarcted heart and leads to bone marrow production and release of neutrophils and Ly6chigh monocytes, which are then recruited to the injured heart.
Figure 2. Macrophage dynamics and phenotype during cardiac injury.
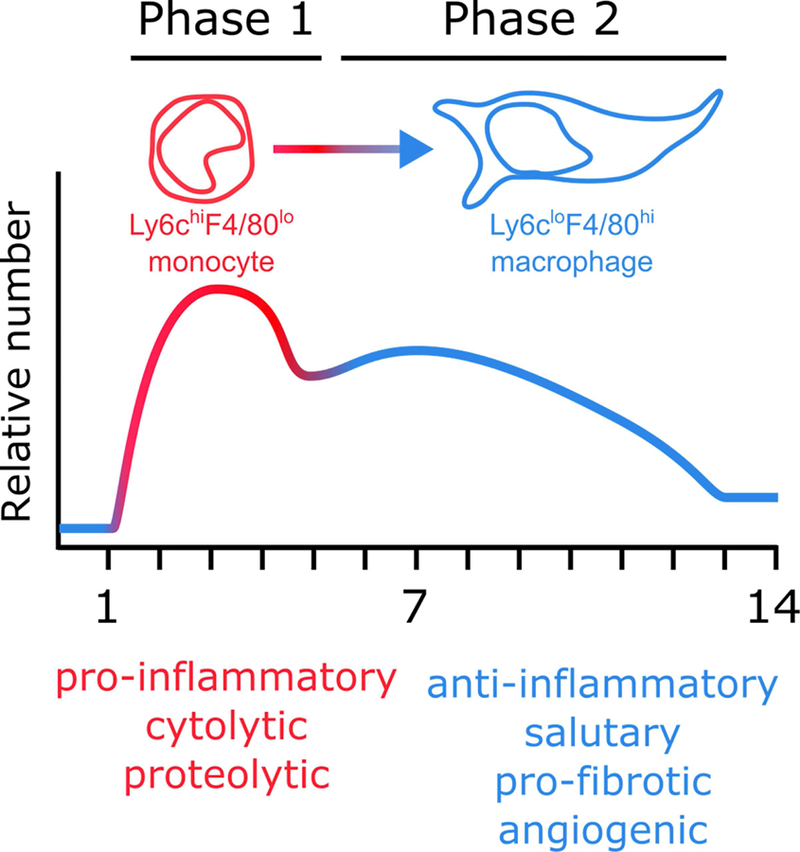
During phase 1 or the ‘inflammatory’ phase of the injury response, Ly6chigh monocytes are the predominant monocyte/macrophage population. During phase 2, or the ‘reparative’ phase, Ly6clow macrophages are the most prevalent. Figure based on data from (7,39,40) and adapted from (40).
In response to cardiac injury, recruited macrophages produce a sequential spectrum of cytokines, chemokines, matrix metalloproteinases, and growth factors exerting both pro- and anti- inflammatory effects. In addition, resident and monocyte-derived macrophages are decorated with an array of pattern recognition receptors that detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). DAMPs are comprised of exogenous molecular signatures such as PAMPs, or alarmins which are endogenous molecular signatures arising from tissue damage. To detect these DAMPs, macrophages express TLRs, mannose receptors, purinergic receptors and surface molecules that mediate phagocytosis, such as CD36. Following MI, macrophage pattern recognition receptors sense key alarmins such as HMGB1, ATP and other nucleotides, extracellular matrix components such as hyaluronon and fibronectin fragments, and dead cells (42–44). While much of resident macrophage activity likely involves non-phlogistic phagocytosis (phagocytosis that does not signal inflammation), the alarmins generated in the infarcted heart from apoptotic and necrotic cardiomyocytes elicit a pro-inflammatory response from both resident macrophages and infiltrating monocytes. As an example of this regulatory system and how DAMPs are critical following cardiac injury, King et al (45) have shown that MI causes the release of danger signals including double stranded DNA from dying cells. Infiltrating cells including macrophages sense the DNA, leading to activation of the transcription factor IRF3 (interferon regulatory factor 3) and subsequent induction of type I interferon production. Attenuated post-MI IRF3-dependent signaling reduced cardiac inflammation and improved cardiac function and survival, suggesting this may be a target for therapeutic cardioprotection (45).
Post-MI macrophage depletion in both neonatal and adult hearts results in impaired tissue remodeling, with decreased cardiac fibrosis and functional decrement, including cardiac thrombus formation (15,46). A key function of macrophages in the cardiac lesion is clearance of tissue debris, and in particular efferocytosis—the phagocytosis of dead cells. Recent work has demonstrated that while macrophage function goes beyond phagocytosis, efferocytosisdependent signaling is important for almost all aspects of macrophage activity in the injured heart. In multiple cardiac injury models, macrophage depletion results in the inability to clear tissue debris and dead cells including necrotic cardiomyocytes (15,46,47). The targeting of genes that support macrophage phagocytosis such as MerTK and Mfge8 results in impaired tissue healing and functional decline after cardiac injury, demonstrating that tissue debris clearance is an essential aspect of healing following cardiac injury (27,48,49).
Efferocytosis after MI also stabilizes the myocardial wall to prevent wall rupture, with the loss of SR-A, another gene implicated in macrophage tissue clearance activity (50), increasing post-MI cardiac rupture (51). This is at least partly due to increased proteolytic activity through MMP9, which is induced by increased levels of post-MI tumor necrosis factor (TNF)-α (51,52). Indeed, MMP9 deficiency decreases the incidence of cardiac rupture (53), and also increases CD36-dependent macrophage phagocytosis (54). Macrophages also express transforming growth factor (TGF)-β, a key factor inducing myofibroblast differentiation and myocardial fibrosis (55), which is enhanced by MerTK-dependent efferocytosis following ischemic injury (27).
Macrophages also participate in post-MI vascular bed regeneration. In this context, macrophages may physically mediate vascular anastomosis or regulate angiogenesis by paracrine means (56–58). Macrophage depletion following cardiac injury leads to a reduction in vascular endothelial growth factor A (VEGFa), and a coinciding reduction in cardiac capillary density (47). Loss of MerTK and Mfge8 results in blunted VEGFa production (48). Moreover, macrophage-restricted genetic ablation of VEGFa, in LysM-Cre:VEGFafl/fl mice, leads to impaired heart function and angiogenesis (48). Furthermore, VEGFa is likely to also act on nonendothelial cells. In non-cardiac models of tissue injury, VEGFa overexpression augments the recruitment of circulating monocytes to injured tissues and promotes a pro-angiogenic macrophage phenotype (59).
The regulation of transient extracellular matrix and matrix-derived signals is important in mediating cardiomyocyte proliferation in regenerative species such as salamander and zebrafish (see review (60)). In macrophage-depleted salamanders, expression of extracellular matrix components is suppressed, correlating with regenerative failure (25,61). Thus, evidence is mounting that macrophages play a critical role in regulation of the extracellular matrix microenvironment in regeneration by secretion of matrix degrading enzymes, and through interactions with local mesenchymal cells (62). However in the salamander cardiac cryo-injury model, macrophage depletion derails regeneration despite the persistence of cardiomyocyte proliferation (25). In the mouse neonate, major transcriptional shifts in the leukocyte population following MI may provide clues to the local inflammatory responses providing a permissive environment (63), however macrophage-depleted neonatal mouse hearts fail to regenerate despite normal cardiomyocyte proliferation (15). These findings indicate that current clinical efforts into promoting cardiomyocyte proliferation (reviewed in (64)) may be necessary but not sufficient, and that modulation of the macrophage-dependent stromal environment is critical for promoting effective scar-free cardiac repair.
Monocyte and Macrophage Dynamics Following Cardiac Injury
Findings in regenerative organisms such as the zebrafish and salamander implicate the timing and profile of innate immune cell recruitment to injured tissues as a critical factor in regenerative success. Salamanders require macrophages to regenerate limbs and hearts after injury (25,61) and a similar requirement is observed in zebrafish fins and heart (65,66). The timing of macrophage invasion post injury is accelerated relative to mammalian injury, featuring a rapid switch to anti-inflammatory signaling (61). Liposomal clodronate depletion of macrophages in regenerating salamanders or zebrafish structures has identified an early requirement for macrophage infiltration. When macrophage engagement is delayed, scar formation prevents tissue regeneration (25,61,65). Studies comparing the response to cardiac damage in zebrafish with Medaka, a related teleost lacking cardiac regenerative capacity (35), have revealed that the timing of inflammatory resolution may be critical for successful regeneration. These studies demonstrated that the infiltration kinetics of Medaka neutrophils, and their lack of early and effective clearance and resolution by macrophages, led to poor regeneration that could be enhanced by macrophage stimulation (65). In addition, as reviewed in Part 2 of this review series, failure of resolution of inflammation is a key aspect leading to atherosclerosis progression. In further support of this concept, additional studies have pinpointed the regulation of inflammation via macrophages and TNFα as a key factor for zebrafish fin regeneration (67). Furthermore, yet another study highlighted the critical role of macrophages in dampening the IL-1β response to injury from epithelial cells that leads to apoptosis of regenerative progenitors (68).
While the injured mammalian heart accumulates high numbers of monocytes and macrophages, these cells are only present transiently (Figure 2). Within two hours of injury, resident macrophages begin to die (16). In contrast, infiltrating monocytes and macrophages have a cardiac residence period of ~20 hours (69). After MI, mammalian monocytes and macrophages follow a bi-phasic response to injury, transiting from a pro-inflammatory toward an anti-inflammatory phenotype at later stages (7,40) (Figure 3). In the inflammatory phase (days 1–4 post-MI), CCR2+Ly6chigh monocytes are the dominant monocytic population and exhibit a pro-inflammatory phenotype, secreting factors such as IL-1β, IL-6 and TNFα (40). These monocytes also produce proteolytic factors and undertake extensive phagocytosis. In the reparative phase (days 5–14), monocytes differentiate to F4/80high macrophages, exhibit an antiinflammatory phenotype by secretion of factors such as IL-10 and TGF-β (40), and promote angiogenesis and scar formation by secretion of factors such as VEGFa in addition to TGF-β (40). Although macrophage transition from proinflammatory to a reparative phentoype has been classically defined as M1-to-M2 polarization, macrophages in the post-MI heart produce mediators that lie outside the M1/M2 spectrum, again highlighting the complexity of macrophage subtypes and their dependence on the specific stimulus/environment (70).
Figure 3. Signaling circuitry promoting monocyte and macrophage accumulation in the injured heart.
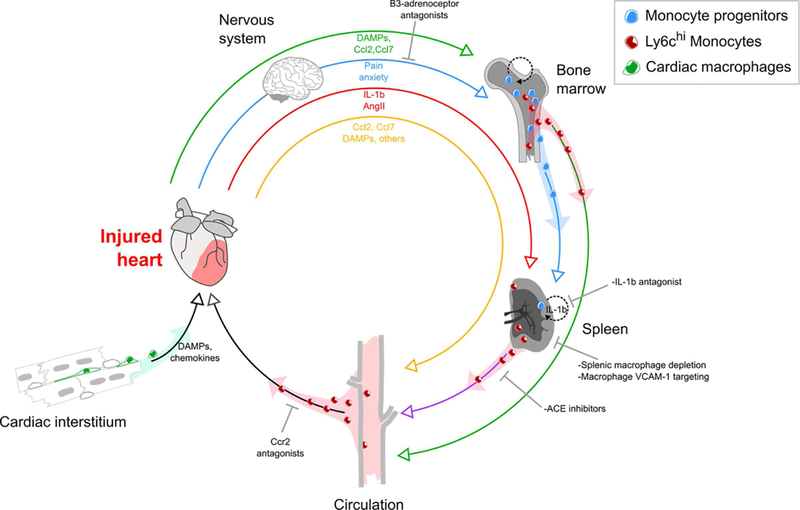
(Green pathway) Circulating DAMPs and inflammatory mediators mobilize bone marrow hematopoietic progenitors and monocytes. Monocytes enter the circulation, whereas progenitors migrate to the spleen and support splenic monocytopoiesis. Monocytes from the bone marrow and spleen eventually arrive at the injured heart. (Blue pathway) Pain and anxiety activate sympathetic pathways suppressing hematopoietic progenitor retention factors in the bone marrow leading to further progenitor cell release. (Red pathway) The injured heart directly signals to the spleen and induces splenic monocytopoiesis and hematopoietic progenitor cell proliferation by IL-1β and AngII dependent mechanisms. (Yellow pathway) Inflammatory mediators and DAMPs signal directly to recruit monocytes. Adapted from (157).
Origins of Monocytes and Macrophages at Cardiac Injury Sites
In the injured neonatal heart, an embryonic lineage of resident macrophages induces minimal inflammation, instead promoting cardiomyocyte proliferation and coronary angiogenesis (15,21). Conversely in the adult, in addition to local immune cell activation, cardiac stress induces a robust systemic inflammatory response (71,72), leading to an increased number of circulating monocytes that support the monocyte/macrophage demands of the injured myocardium (Figure 3). In steady state and depending upon the subset, mouse monocytes have a lifespan of 1–2 days (11,73,74). In contrast, the major human monocyte subset to infiltrate sites of injury—classical monocytes (CD14+CD16−)—have a lifespan of ~3 days (75). The increased demand for monocytes and macrophages in adult MI is associated with increased monocyte production in the bone marrow, likely stimulated by circulating DAMPs arising from the injured myocardium (76). While other pathways are also of importance (45), a key stimulus for emigration of monocytes from the bone marrow is CCR2 signaling (77). After tissue injury or pathogen-induced inflammation, CCL2 (monocyte chemoattractant protein-1) is produced locally by a wide range of cells including endothelial cells, fibroblasts and smooth muscle cells (78). In addition, bone marrow endothelial cells produce CCL2 following MI (79), while CCR2+ hematopoietic progenitors proliferate and increase the production of CCR2+ monocytes.
The spleen also serves as an ‘emergency reservoir’ that may contribute up to half of all monocytes that enter the cardiac lesion. In the spleen, monocytes reside in clusters of 20–50 cells (80) and respond to angiotensin II, which increases in circulation after MI (81). Angiotensin II induces splenic monocytes to undergo chemokinesis and enter the circulation where they eventually migrate to the injured heart (81). Splenic monocytopoiesis is an important factor in maintaining this monocyte reservoir. In response to MI, splenic progenitors undergo proliferation which is dependent on IL-1β signaling (69). Inhibition of sympathetic nervous system activity by β3-aderoceptor antagonists leads to a decrease in circulating hematopoietic progenitors and splenic monocytopoiesis (71), supporting the concept that splenic monocytopoiesis is enabled by colonization of the spleen by bone marrow-derived hematopoietic progenitors (71). Following MI, sympathetic nervous system activity inhibits bone marrow expression of stem cell retention factors CXCL12, angiopoietin and stem cell factor, permitting medullary egression of progenitors (71). In contrast, there is a concomitant increase in expression of stem cell factor and vascular cell adhesion molecule (VCAM)-1 (an additional stem cell retention factor) in the spleen, to accommodate splenic engraftment and retention of stem cells (71,82). Intriguingly, retention of hematopoietic progenitors through regulation of the bone marrow stem cell niche is dependent upon macrophages (82,83).
Targeting Monocytes and Macrophages for Treating MI
A large body of experimental research has demonstrated that modulation of monocytes is beneficial for cardiac recovery (Online Table 1). Beneficial outcomes have been achieved by interdicting CCR2 signaling, either by ablating Ccr2 or Ccl2 genes or by targeting key inflammatory mediators supporting monocyte recruitment. Conversely, mice that have increased levels of circulating monocytes, such as in ApoE−/− mutants, have impaired cardiac recovery after MI (84). In the clinical setting, increased levels of classical monocytes (CD14+CD16− monocytes) correlate with adverse outcomes following acute MI (85–91).
While no clinical trials have addressed the specific issue of whether targeting monocytes is beneficial post-MI, approaches that adventitiously impair monocyte function and their mobilization are now in routine use, specifically, the administration of angiotensin converting enzyme (ACE) inhibitors and beta-blockers. Indeed, administration of these antagonists within the first 24 hours following MI forms part of the recommended treatment guidelines for STelevation MI (92). Moreover, while many clinical trials dampening inflammation have yielded negative results (93,94), the IL-1β inhibitor Canakinumab was recently shown to reduce cardiovascular events in high risk patients with atherosclerosis (28).
These examples notwithstanding, reducing or depleting monocytes post-MI may have unintended consequences, including risk of prolonging cardiac inflammation due to insufficient clearance of tissue debris (i.e., failed resolution of inflammation). While it has not been directly proven that monocyte depletion compromises cardiovascular repair, a study of veterans who lost their spleens due to injury found that this group suffered increased mortality from ischemic heart-disease (95), suggesting that significant reduction in monocytes during the course of MI may be deleterious. Notably, monocyte depletion would also reduce salutary macrophages that are differentiated from monocytes at later injury phases (40) and impairment of tissue macrophage phagocytic activity negatively correlates with healing after MI (96).
A more compelling case can be made for boosting anti-inflammatory macrophage numbers and activity as a potential treatment following MI, primarily because this should enhance tissue stabilization, clearance of tissue debris and cardiac healing. However, expanding the anti-inflammatory cardiac macrophage population without affecting circulating monocytes may be difficult, since increased macrophage numbers after MI are dependent on infiltrating Ly6chigh monocyte precursors. One approach may be to exploit the intrinsic capacity of cardiac resident macrophages to divide, both in a resting state (8,16,97) or after MI (98). An alternate approach would involve accelerating the transition of infiltrating monocytes to an antiinflammatory phenotype, using key mediators such as Nr4a1 (40) or the transcription factor IRF5 (99). The validity of this approach is supported by a recent study of neutrophil depletion in mice, which unexpectedly perturbed the functionality of M2-like macrophages in the post-MI heart, leading to increased fibrosis, worsened cardiac function and heart failure (3). Furthermore, experimental bone marrow transplantation in mice elevated levels of reparative M2-like macrophages in the heart, with enhanced cardiomyocyte survival, neovascularization and improved post-MI remodeling (100). Future therapeutic strategies will benefit from advancing our understanding of the molecular networks underpinning cardiac macrophage phenotypes and dynamics, using single-cell transcriptomic analyses, and gene editing of macrophages and their progenitors for promoting a regenerative response to cardiac injury.
Monocytes, Macrophages and Myocarditis
Myocarditis – Background
The evolutionary role of inflammation is to defend tissue against pathogens while retaining the ability to promote beneficial, reparative programs after injury. Defensive mechanisms (e.g., leukocyte recruitment, cytotoxic cell death) and changes in environmental cues (cytokine production, metabolic signals, etc.) can disrupt basic homeostatic mechanisms that govern specialized organ functions, such as within the heart, and in turn, set in motion a cascade that leads to pathological tissue remodeling and heart failure. Here we review the pathogenesis of myocarditis and the role of the cardiac macrophage in modulating heart failure development. Given that loss of all macrophages during myocarditis can lead to significant mortality (101), understanding their precise role and gaps in the literature is critical to move the field forward (Figure 4).
Figure 4. The role of macrophages in viral myocarditis.
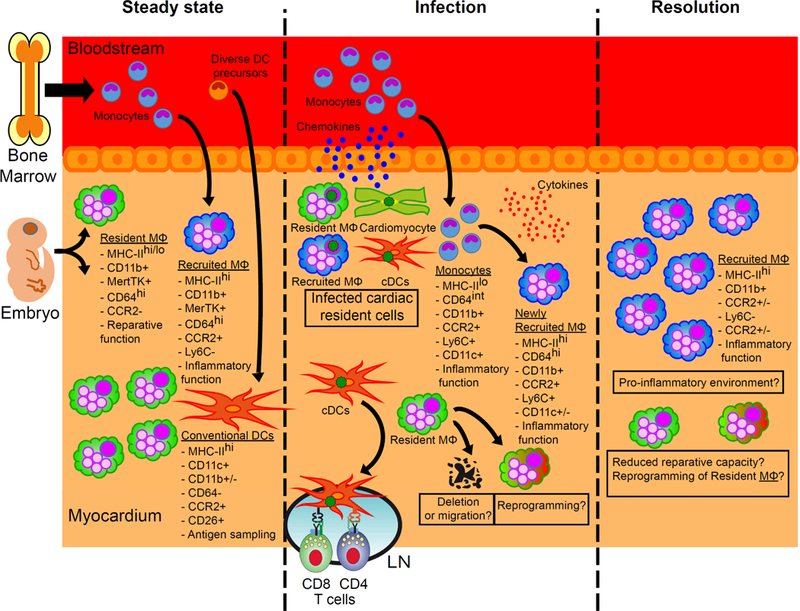
In homeostasis, two ontologically different populations of macrophages (MΦ), with different functions, reside in the heart. Upon viral infection, the resident cells of the myocardium produce chemokines that promote the recruitment of monocytes, which in turn differentiate into macrophages with proinflammatory functions. However, besides the fact that their numbers recede, little is known about the role of resident macrophages during and after infection, and many questions remain (see boxes). The difficulty in distinguishing these different populations using (significantly overlapping) surface markers adds to the challenges faced in addressing these questions.
Infectious Myocarditis
The most common cause of myocarditis is viral infection. Indeed, several studies suggest that we are underestimating the impact of viral myocarditis and development of heart failureassociated with viral infections (102,103). For example, viral genomes can be detected within the myocardium of 35–70% of patients with dilated or chronic cardiomyopathy, suggesting that chronic cardiac disease may develop from previous myocarditis (104,105). Interestingly, while the presence of viral genomes within the myocardium associates with cardiac dysfunction, the presence of virus does not predict progression to cardiomyopathy (104–106). Since a precise diagnosis is rarely achieved in viral myocarditis, our current clinical knowledge is largely based on the association of a prior infectious exposure (i.e. viral prodrome), unexplained LV dysfunction and correlative clinical investigations (systemic markers of inflammation and cardiac imaging).
Autoimmune Myocarditis
Autoimmune myocarditis results from dysregulated activation of innate or adaptive immunity. Some autoimmune syndromes, such as systemic lupus erythematosus or inflammatory bowel disease, can present with a cardiac component (107). Giant cell myocarditis and cardiac sarcoidosis are rare forms of inflammation-driven myocarditis with specific histological features, including the formation of multinucleated giant cells within the myocardium presumed to be macrophages (108–111).
Animal models have been developed to mimic autoimmune myocarditis, referred to as Experimental Autoimmune Myocarditis (EAM). These models are typically based on immunization with cardiac antigens and a strong adjuvant or antigen presenting cells, such as dendritic cells (DCs) loaded with cardiac antigens in order to promote an autoreactive adaptive immune response (112).
Some evidence suggests that under certain circumstances infectious agents may lead to the activation of autoreactive T cell clones that promote heart failure after full clearance of the primary infection in animal models (113,114). In clinical studies, it has been difficult to demonstrate an autoimmune component, with recent trials in viral myocarditis showing little beneficial effect of corticosteroids in the prevention of heart failure and dilated cardiomyopathy (115).
Toxic Myocarditis
The increasing clinical use of disease modifying pharmaceuticals to treat recalcitrant diseases, such as cancer or autoimmunity, has been associated with an increasing number of patients with cardiotoxicity (116,117). A wide range of substances, including heavy metals and several immunomodulatory agents, can promote cardiac injury through a variety of mechanisms (107), including direct cardiomyocyte injury (e.g. doxorubicin) and indirect cardiac tissue damage secondary to inflammation. More recently, immune check-point inhibitors, which release inhibitory signals on activated cytotoxic CD8+ T cells, have been associated with potentially lethal fulminant myocarditis in a small subset of patients secondary to massive myocardial infiltration of CD8+ T cells (118).
Macrophages and Myocarditis – Direct Evidence Versus Inference
The majority of published studies have focused on viral and autoimmune myocarditis and, to a lesser extent, doxorubicin-induced myocarditis. It will be in this experimental context that we will examine macrophage dynamics and functions. In all myocarditis models, there is a clear expansion of macrophage numbers within the myocardium (119–121), presumably driven by monocyte influx. Genetic fate mapping has, within the context of viral myocarditis in mouse models, confirmed the massive infiltration of circulating monocytes, and also identified a loss of resident CCR2− reparative macrophages during the acute infectious period (119). While embryonic-derived cardiac macrophages have been tracked during viral myocarditis, their exact functions remain elusive. It is known that those cardiac macrophage subsets that express high levels of MHC-II are capable of processing and presenting antigen to CD4+ T cells ex vivo (8). However, the role of antigen presentation by resident immune cells to recruited T cells in vivo is not clear. By extrapolation, neonatal cardiac macrophages, which lack MHC-II expression, are less proinflammatory (produce less TNF-α and IL-1β), and promote more reparative functions (cardiomyocyte and endothelial proliferation) as compared to cardiac macrophages isolated from injured adult animals (15,21).
Broad differences exist in the character of the innate immune response to cardiac stress when comparing different physiological stressors. For example, while the resident macrophage population is lost following viral myocarditis, it increases in number during hypertensive challenge (8). MI and hypertensive stress both promote significant neutrophil infiltration, while there is a surprising lack of neutrophil infiltration in viral myocarditis (7,119,121). Thus, conclusions about macrophage functions made in one model may not necessarily apply to other systems.
Cardiac Macrophages and the Confounding Role of Cardiac Dendritic Cells
Any discussion about the role of tissue macrophages is incomplete without understanding whether macrophages and DCs can be efficiently delineated, in particular during inflammation. Recent studies have developed a framework to identify and separate out cardiac DCs from cardiac macrophages, both of which can express DC markers such as CD11c and MHC-II (119,122). Conventional DCs can be distinguished from macrophages through their expression of the conventional DC lineage transcription factor ZBTB46 (123,124), and a grouping of additional cell surface markers (most importantly CD26) (119,122,125).
Until recently, the ability to specifically deplete conventional DCs was lacking, and the main system to deplete DCs relied on the Cd11c-DTR strain. Depletion of DCs during viral myocarditis (using encephalomyocarditis virus [EMCV-D]) leads to significant mortality, while use of the more specific Zbtb46-DTR does not, but leads to impaired generation of antigen specific CD8+ T cells and cardiac dysfunction, suggesting that additional cell types, such as macrophages, could also be targeted by CD11c-dependent cell depletion (101,119). Thus, the appropriate selection of cell surface markers, transcription factors and targeting strategies that utilize a contemporary immunologic understanding are required to study the individual roles of macrophages and DCs.
Monocyte versus Macrophage Functions in Myocarditis
Studies have attempted to ascertain the role of macrophages in myocarditis through the use of clodronate liposomes, which deplete circulating blood monocytes and differentially deplete resident cardiac macrophages (depending on the formulation). Using the EMCV viral myocarditis model, clodronate-mediated depletion was associated with a pronounced increase in mortality (101), while CVB3 infection led to increased viral titres, a variable increase in mortality, and reduced late cardiac fibrosis in surviving animals by depletion of cardiac macrophages that produced the pro-fibrotic cytokine galectin-3 (101,126). In contrast to viral infection where the immune system presumably promotes a combination of immune-protective and immune-damaging effects, the immune response in EAM appears to be entirely pathologic as it is directed at self-antigens. Clodronate-liposome administration in the chronic phase of EAM, and not in the acute phase, led to improved cardiac function and decreased adverse remodeling (127). As an important consideration, the use of clodronate-liposomes leads to difficulties in data interpretation, since depleting blood monocytes prior to their recruitment versus depletion of all monocytes/macrophages would likely have very different effects. Nevertheless, the data appear to suggest that in viral myocarditis, while monocytes and macrophages may promote cardiac fibrosis their net effect is protective; however, in autoimmune models, activation promotes chronic heart failure. Additional studies using targeted approaches are required to fully understand the role of recruited monocytes versus resident macrophages in these settings.
Monocytes can differentiate into either macrophages or DCs, which are an essential part of protective immunity. Deletion of the receptor of CX3CL1 (fractalkine) or CX3CR1, worsens acute CVB3-induced myocarditis (128). CX3CR1 appears to act on monocyte differentiation into macrophages, macrophage survival, and is also important in vascular patrolling behavior of Ly6clow monocytes, which are processes that may be activated following viral infection (129,130). Interfering with CCR5 or its ligand CCL5, led to increased cardiac tissue injury (131); whereas interfering with CCL2 or CCL3-mediated recruitment ameliorated acute CVB3 infection (132–134), suggesting that CCL2, CCL3 and CCL5, despite their known ability to target monocytes, possess non-overlapping roles. In autoimmune myocarditis, preferential monocyte targeting appears to be beneficial – a finding supported by siRNA-mediated inhibition of CCR2 on peripheral monocytes (121). Preventing the recruitment of monocytes by chemokine neutralization (CCL2, CCL3) or deletion of chemokine receptors (CCR2-deficient mice which are blood monocyte deficient; or CCR5) has been shown to ameliorate EAM (121,135). While CCR2 inhibition is classically associated with inhibiting monocyte influx, CCR2 is expressed on cardiac DC subsets and is specifically required for DC homing to cardiac tissue, but not other tissue beds (119). Thus, care must be used when assigning cellular targets when chemokines are targeted.
Associative Studies of Macrophage Activation and Myocarditis Outcomes
Anti-viral immune responses are initiated and modulated by sensing systems able to detect PAMPs and DAMPs in a variety of cell types, including macrophages. Enteroviral (singlestranded RNA Picornaviruses) infection, such as with EMCV or CVB3, is associated with the release of double-stranded RNA, a common intermediate of viral replication and a TLR3 ligand. Hence, deficiency of TLR3 or its molecular adaptor TRIF is associated with fulminant myocarditis (136–141). Interestingly, TLR3 polymorphisms may also confer susceptibility to myocarditis and dilated cardiomyopathy in patients (142). In contrast, whole-body deletion of the TLR adaptor molecule MyD88, and its downstream signaling molecule IRAK4, ameliorates myocarditis and inflammation (131,143). IRAK4 limits the expression of CCR5 and its ligands preventing the recruitment of protective CCR5+ monocytes – a pathway active in macrophages but not conventional or plasmacytoid DCs. TLR4 signaling seems to play a role in controlling the induction of deleterious pro-inflammatory cytokines, such as IL-1β or IL-18 (144). TIM-3, an inhibitory receptor found on macrophages and DCs, prevents exaggerated inflammatory responses, with blockade leading to worsened CVB3 myocarditis associated with an expansion of cardiac macrophage and neutrophil populations (145).
Progression from acute CVB3 myocarditis to chronic inflammation is dependent on viral persistence (146,147). Hence, an effective, but limited, immune response is important to prevent chronic myocarditis. Several studies have shown that in resistant mouse strains, early type I interferon or interferon-γ production promotes viral clearance via nitric oxide production (148–150) and concurrent early induction of antigen-presentation machinery (151). In contrast, in susceptible strains, delayed and exaggerated proinflammatory cytokine and inducible-nitric oxide synthase production by macrophages was associated with increased cardiac damage (152). Hence, persistent infection and an ineffectual, delayed and deleterious inflammatory response could contribute to macrophage mediated cardiac injury.
Macrophage Activation in Doxorubicin-Induced Myocarditis
The cardio-toxic effects of doxorubicin are well recognized. In correlative studies, TLR4 and TLR2 deficiency attenuated doxorubicin cardiotoxicity in mice, suggesting a role for innate immune cells in the disease (153,154). In contrast, TLR2 blockade with neutralizing antibodies reduced the inflammatory response and subsequent adverse cardiac remodeling, while TLR4 blockade exacerbated cardiac tissue damage (155). Importantly, these studies did not differentiate between TLRs expressed in cardiomyocytes, macrophages or other immune cells. Kobayashi et al. demonstrated that NLRP3-induced production of IL-10 by macrophages is an innate mechanism that protects against severe doxorubicin cardiotoxicity (120). Nevertheless, knowledge of the precise role of macrophages in chemotherapy-induced myocarditis is limited.
Conclusions and Future Directions on Cardiac Macrophages
A new paradigm has emerged in which various populations of cardiac macrophages, which include recruited and resident cells with distinct lineages/subsets, have specific functions in cardiac homeostasis and disease. However, numerous challenges remain. A critical hurdle will be to determine whether the human heart contains similar macrophage populations as observed in the mouse. Furthermore, while it is clear that the human heart contains an abundance of macrophages that are involved in myocarditis and MI, many questions remain. Indeed, a recent report provided initial evidence that the human failing heart contains CCR2− and CCR2+ macrophage subsets that are functionally analogous to mouse CCR2− and CCR2+ macrophages. Intriguingly, CCR2+ macrophage abundance was associated with persistent LV systolic dysfunction and LV remodeling following mechanical unloading, suggesting that CCR2+ macrophages contribute to heart failure pathogenesis (156). Nevertheless, the exact roles for these cells in myocarditis, MI, and chronic heart failure remain to be determined.
As an exciting development, contemporary immunological techniques have provided a framework to separately investigate the roles of recruited monocytes, resident macrophage subsets and cardiac DCs in cardiac homeostasis and disease. By utilizing lineage-specific genetic tools that can separate the roles of these subsets, the ability to perform a precise immunologic dissection of the innate and adaptive immune systems is likely to yield major mechanistic progress. In turn, a deeper understanding of the functions of each macrophage subset should facilitate the development of strategies to target inflammatory responses and improve the heart’s intrinsic capacity for tissue repair. The future of cardiac immunology is indeed bright, and rich with new possibilities for therapeutic innovation.
Supplementary Material
Acknowledgements:
Kory Lavine is supported by funding from the NIH/NHLBI K08 HL123519, R01 HL138466, and R01 HL139714, Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CHII2015-462, CH-II-2017-628), Foundation of Barnes-Jewish Hospital (8038-88), Burroughs Foundation Welcome Fund, and Longer Life Foundation (2016-004). Benjamin Kopecky is supported by the Physician Scientist Training Program at Washington University School of Medicine in St Louis. Slava Epelman is supported by funding from the Canadian Institutes of Health Research, March of Dimes, Heart and Stroke Foundation of Canada, the Peter Munk Cardiac Centre, and the Ted Rogers Centre for Heart Research. Xavier Clemente-Casares holds a Canadian Institutes of Health Research Banting Fellowship. Jason Kovacic acknowledges research support from the National Institutes of Health (R01HL130423), the American Heart Association (14SFRN20490315; 14SFRN20840000) and The Leducq Foundation (Transatlantic Network of Excellence Award). Alexander Pinto is supported by funding from the American Heart Association (17IRG33270004). James Godwin is funded by a COBRE award from NIH-NIGMS (P20GM104318).
Abbreviations
- CCL
CC chemokine ligand
- CCR
CC chemokine Receptor
- CD11b
Cluster of Differentiation 11b
- CD206
Cluster of Differentiation 206
- CVB3
Coxsackievirus B3
- CXCL
chemokine (C-X-C motif) ligand
- CXCR1
CXC chemokine Receptor 1
- DAMP
damage-associated molecular patterns
- DC
Dendritic cell
- EAM
Experimental Autoimmune Myocarditis
- EMCV-D
Encephalomyocarditis virus
- IGF
Insulin-like growth factor
- IL
Interleukin
- IRF3
Interferon regulatory factor 3
- MHC
Major Histocompatibility Complex
- PAMP
pathogen-associated molecular patterns
- TGF-β
transforming growth factor-β
- TLR
Toll-like receptor
- TNFα
tumor necrosis factor -α
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors declare no conflicts of interest.
References
- 1.Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 2015;116:1254–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jolly SR, Kane WJ, Hook BG, Abrams GD, Kunkel SL, Lucchesi BR. Reduction of myocardial infarct size by neutrophil depletion: effect of duration of occlusion. Am Heart J 1986;112:682–90. [DOI] [PubMed] [Google Scholar]
- 3.Horckmans M, Ring L, Duchene J et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 2017;38:187–197. [DOI] [PubMed] [Google Scholar]
- 4.Litt MR, Jeremy RW, Weisman HF, Winkelstein JA, Becker LC. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation 1989;80:1816–27. [DOI] [PubMed] [Google Scholar]
- 5.Glaros T, Larsen M, Li L. Macrophages and fibroblasts during inflammation, tissue damage and organ injury. Front Biosci (Landmark Ed) 2009;14:3988–93. [DOI] [PubMed] [Google Scholar]
- 6.Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res 2014;102:240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nahrendorf M, Swirski FK, Aikawa E et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 2007;204:3037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epelman S, Lavine KJ, Beaudin AE et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity 2014;41:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakubzick C, Gautier EL, Gibbings SL et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 2013;39:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yona S, Kim KW, Wolf Y et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013;38:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoeffel G, Chen J, Lavin Y et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015;42:665–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyer SW, Schroeder AV, Smith-Berdan S, Forsberg EC. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell 2011;9:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashimoto D, Chow A, Noizat C et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013;38:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aurora AB, Porrello ER, Tan W et al. Macrophages are required for neonatal heart regeneration. J Clin Invest 2014;124:1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heidt T, Courties G, Dutta P et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res 2014;115:284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinto AR, Ilinykh A, Ivey MJ et al. Revisiting Cardiac Cellular Composition. Circ Res 2016;118:400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinto AR, Paolicelli R, Salimova E et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One 2012;7:e36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ Res 2016;118:1498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stevens SM, von Gise A, VanDusen N, Zhou B, Pu WT. Epicardium is required for cardiac seeding by yolk sac macrophages, precursors of resident macrophages of the adult heart. Dev Biol 2016;413:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavine KJ, Epelman S, Uchida K et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A 2014;111:16029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hulsmans M, Clauss S, Xiao L et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017;169:510–522 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molawi K, Wolf Y, Kandalla PK et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med 2014;211:2151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiraishi M, Shintani Y, Shintani Y et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest 2016;126:2151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godwin JW, Debuque R, Salimova E, Rosenthal NA. Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regen Med 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godwin JW, Pinto AR, Rosenthal NA. Chasing the recipe for a pro-regenerative immune system. Semin Cell Dev Biol 2017;61:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeBerge M, Yeap XY, Dehn S et al. MerTK Cleavage on Resident Cardiac Macrophages Compromises Repair After Myocardial Ischemia Reperfusion Injury. Circ Res 2017;121:930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ridker PM, Everett BM, Thuren T et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 29.Li W, Hsiao HM, Higashikubo R et al. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forbes SJ, Rosenthal N. Preparing the ground for tissue regeneration: from mechanism to therapy. Nat Med 2014;20:857–69. [DOI] [PubMed] [Google Scholar]
- 31.Uygur A, Lee RT. Mechanisms of Cardiac Regeneration. Dev Cell 2016;36:362–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grivas J, Haag M, Johnson A et al. Cardiac repair and regenerative potential in the goldfish (Carassius auratus) heart. Comp Biochem Physiol C Toxicol Pharmacol 2014;163:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lafontant PJ, Burns AR, Grivas JA et al. The giant danio (D. aequipinnatus) as a model of cardiac remodeling ad regeneration. Anat Rec (Hoboken) 2012;295:234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science 2002;298:2188–90. [DOI] [PubMed] [Google Scholar]
- 35.Ito K, Morioka M, Kimura S, Tasaki M, Inohaya K, Kudo A. Differential reparative phenotypes between zebrafish and medaka after cardiac injury. Dev Dyn 2014;243:1106–15. [DOI] [PubMed] [Google Scholar]
- 36.Jung K, Kim P, Leuschner F et al. Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ Res 2013;112:891–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frangogiannis NG, Perrard JL, Mendoza LH et al. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation 1998;98:687–98. [DOI] [PubMed] [Google Scholar]
- 38.Somasundaram P, Ren G, Nagar H et al. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J Pathol 2005;205:102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zouggari Y, Ait-Oufella H, Bonnin P et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med 2013;19:1273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hilgendorf I, Gerhardt LM, Tan TC et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res 2014;114:1611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sager HB, Heidt T, Hulsmans M et al. Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation 2015;132:1880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol 2016;16:177–92. [DOI] [PubMed] [Google Scholar]
- 43.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 2010;48:504–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huebener P, Abou-Khamis T, Zymek P et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol 2008;180:2625–33. [DOI] [PubMed] [Google Scholar]
- 45.King KR, Aguirre AD, Ye YX et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med 2017;23:1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frantz S, Hofmann U, Fraccarollo D et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J 2013;27:871–81. [DOI] [PubMed] [Google Scholar]
- 47.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol 2007;170:818–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howangyin KY, Zlatanova I, Pinto C et al. Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase and Milk Fat Globule Epidermal Growth Factor 8 Coordinately Improve Remodeling After Myocardial Infarction via Local Delivery of Vascular Endothelial Growth Factor. Circulation 2016;133:826–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wan E, Yeap XY, Dehn S et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res 2013;113:1004–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Platt N, Gordon S. Is the class A macrophage scavenger receptor (SR-A) multifunctional? - The mouse’s tale. J Clin Invest 2001;108:649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsujita K, Kaikita K, Hayasaki T et al. Targeted deletion of class A macrophage scavenger receptor increases the risk of cardiac rupture after experimental myocardial infarction. Circulation 2007;115:1904–11. [DOI] [PubMed] [Google Scholar]
- 52.Irwin MW, Mak S, Mann DL et al. Tissue expression and immunolocalization of tumor necrosis factor-alpha in postinfarction dysfunctional myocardium. Circulation 1999;99:1492–8. [DOI] [PubMed] [Google Scholar]
- 53.Heymans S, Luttun A, Nuyens D et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 1999;5:1135–42. [DOI] [PubMed] [Google Scholar]
- 54.DeLeon-Pennell KY, Tian Y, Zhang B et al. CD36 Is a Matrix Metalloproteinase-9 Substrate That Stimulates Neutrophil Apoptosis and Removal During Cardiac Remodeling. Circ Cardiovasc Genet 2016;9:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 1998;101:890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cattin AL, Burden JJ, Van Emmenis L et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015;162:1127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fantin A, Vieira JM, Gestri G et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010;116:829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C. Macrophages and angiogenesis. J Leukoc Biol 1994;55:410–22. [DOI] [PubMed] [Google Scholar]
- 59.Avraham-Davidi I, Yona S, Grunewald M et al. On-site education of VEGF-recruited monocytes improves their performance as angiogenic and arteriogenic accessory cells. J Exp Med 2013;210:2611–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Godwin J, Kuraitis D, Rosenthal N. Extracellular matrix considerations for scar-free repair and regeneration: insights from regenerative diversity among vertebrates. Int J Biochem Cell Biol 2014;56:47–55. [DOI] [PubMed] [Google Scholar]
- 61.Godwin JW, Pinto AR, Rosenthal NA. Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A 2013;110:9415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mescher AL. Macrophages and fibroblasts during inflammation and tissue repair in models of organ regeneration. Regeneration (Oxf) 2017;4:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quaife-Ryan GA, Sim CB, Ziemann M et al. Multicellular Transcriptional Analysis of Mammalian Heart Regeneration. Circulation 2017;136:1123–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cahill TJ, Choudhury RP, Riley PR. Heart regeneration and repair after myocardial infarction: translational opportunities for novel therapeutics. Nat Rev Drug Discov 2017;16:699–717. [DOI] [PubMed] [Google Scholar]
- 65.Lai SL, Marin-Juez R, Moura PL et al. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Petrie TA, Strand NS, Yang CT, Rabinowitz JS, Moon RT. Macrophages modulate adult zebrafish tail fin regeneration. Development 2014;141:2581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nguyen-Chi M, Laplace-Builhe B, Travnickova J et al. TNF signaling and macrophages govern fin regeneration in zebrafish larvae. Cell Death Dis 2017;8:e2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hasegawa T, Hall CJ, Crosier PS et al. Transient inflammatory response mediated by interleukin-1beta is required for proper regeneration in zebrafish fin fold. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leuschner F, Rauch PJ, Ueno T et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 2012;209:12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nahrendorf M, Swirski FK. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ Res 2016;119:414–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dutta P, Courties G, Wei Y et al. Myocardial infarction accelerates atherosclerosis. Nature 2012;487:325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujiu K, Shibata M, Nakayama Y et al. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat Med 2017;23:611–622. [DOI] [PubMed] [Google Scholar]
- 73.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med 1968;128:415–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Furth R, Diesselhoff-den Dulk MC, Mattie H. Quantitative study on the production and kinetics of mononuclear phagocytes during an acute inflammatory reaction. J Exp Med 1973;138:1314–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patel AA, Zhang Y, Fullerton JN et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med 2017;214:1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi C, Jia T, Mendez-Ferrer S et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity 2011;34:590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011;11:762–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 2009;29:313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dutta P, Sager HB, Stengel KR et al. Myocardial Infarction Activates CCR2(+) Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015;16:477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Swirski FK, Nahrendorf M, Etzrodt M et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009;325:612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leuschner F, Panizzi P, Chico-Calero I et al. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ Res 2010;107:1364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dutta P, Hoyer FF, Grigoryeva LS et al. Macrophages retain hematopoietic stem cells in the spleen via VCAM-1. J Exp Med 2015;212:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chow A, Lucas D, Hidalgo A et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 2011;208:261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Panizzi P, Swirski FK, Figueiredo JL et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol 2010;55:1629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bauters A, Ennezat PV, Tricot O et al. Relation of admission white blood cell count to left ventricular remodeling after anterior wall acute myocardial infarction. Am J Cardiol 2007;100:182–4. [DOI] [PubMed] [Google Scholar]
- 86.Leers MPG, Stockem C, Ackermans D et al. Intermediate and nonclassical monocytes show heterogeneity in patients with different types of acute coronary syndrome. Cytometry A 2017;91:1059–1067. [DOI] [PubMed] [Google Scholar]
- 87.Maekawa Y, Anzai T, Yoshikawa T et al. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction:a possible role for left ventricular remodeling. J Am Coll Cardiol 2002;39:241–6. [DOI] [PubMed] [Google Scholar]
- 88.Mariani M, Fetiveau R, Rossetti E et al. Significance of total and differential leucocyte count in patients with acute myocardial infarction treated with primary coronary angioplasty. Eur Heart J 2006;27:2511–5. [DOI] [PubMed] [Google Scholar]
- 89.Tsujioka H, Imanishi T, Ikejima H et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol 2009;54:130–8. [DOI] [PubMed] [Google Scholar]
- 90.van der Laan AM, Hirsch A, Robbers LF et al. A proinflammatory monocyte response is associated with myocardial injury and impaired functional outcome in patients with STsegment elevation myocardial infarction: monocytes and myocardial infarction. Am Heart J 2012;163:57–65 e2. [DOI] [PubMed] [Google Scholar]
- 91.Zaliaduonyte-Peksiene D, Simonyte S, Lesauskaite V et al. Left ventricular remodelling after acute myocardial infarction: impact of clinical, echocardiographic parameters and polymorphism of angiotensinogen gene. J Renin Angiotensin Aldosterone Syst 2014;15:286–93. [DOI] [PubMed] [Google Scholar]
- 92.American College of Emergency P, Society for Cardiovascular A, Interventions et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;61:e78–140. [DOI] [PubMed] [Google Scholar]
- 93.O’Donoghue ML, Glaser R, Cavender MA et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA 2016;315:1591–9. [DOI] [PubMed] [Google Scholar]
- 94.Investigators S, White HD, Held C et al. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med 2014;370:1702–11. [DOI] [PubMed] [Google Scholar]
- 95.Robinette CD, Fraumeni JF Jr., Splenectomy and subsequent mortality in veterans of the 1939–45 war. Lancet 1977;2:127–9. [DOI] [PubMed] [Google Scholar]
- 96.Dehn S, Thorp EB. Myeloid receptor CD36 is required for early phagocytosis of myocardial infarcts and induction of Nr4a1-dependent mechanisms of cardiac repair. FASEB J 2018;32:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pinto AR, Godwin JW, Chandran A et al. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging (Albany NY) 2014;6:399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sager HB, Hulsmans M, Lavine KJ et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res 2016;119:853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Courties G, Heidt T, Sebas M et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol 2014;63:1556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Protti A, Mongue-Din H, Mylonas KJ et al. Bone marrow transplantation modulates tissue macrophage phenotype and enhances cardiac recovery after subsequent acute myocardial infarction. J Mol Cell Cardiol 2016;90:120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McCartney SA, Vermi W, Lonardi S et al. RNA sensor-induced type I IFN prevents diabetes caused by a beta cell-tropic virus in mice. J Clin Invest 2011;121:1497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Elamm C, Fairweather D, Cooper LT. Pathogenesis and diagnosis of myocarditis. Heart 2012;98:835–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mahfoud F, Gartner B, Kindermann M et al. Virus serology in patients with suspected myocarditis: utility or futility? Eur Heart J 2011;32:897–903. [DOI] [PubMed] [Google Scholar]
- 104.Kuhl U, Pauschinger M, Noutsias M et al. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005;111:887–93. [DOI] [PubMed] [Google Scholar]
- 105.Bock CT, Klingel K, Kandolf R. Human parvovirus B19-associated myocarditis. N Engl J Med 2010;362:1248–9. [DOI] [PubMed] [Google Scholar]
- 106.Lotze U, Egerer R, Gluck B et al. Low level myocardial parvovirus B19 persistence is a frequent finding in patients with heart disease but unrelated to ongoing myocardial injury. J Med Virol 2010;82:1449–57. [DOI] [PubMed] [Google Scholar]
- 107.Caforio AL, Pankuweit S, Arbustini E et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013;34:2636–48, 2648a-2648d. [DOI] [PubMed] [Google Scholar]
- 108.Litovsky SH, Burke AP, Virmani R. Giant cell myocarditis: an entity distinct from sarcoidosis characterized by multiphasic myocyte destruction by cytotoxic T cells and histiocytic giant cells. Mod Pathol 1996;9:1126–34. [PubMed] [Google Scholar]
- 109.Saeki M, Takahashi-Iwanaga H, Iwanaga T et al. Morphological analysis of multinucleated giant cells occurred in experimental autoimmune myocarditis. Tohoku J Exp Med 1994;172:195–204. [DOI] [PubMed] [Google Scholar]
- 110.Theaker JM, Gatter KC, Brown DC, Heryet A, Davies MJ. An investigation into the nature of giant cells in cardiac and skeletal muscle. Hum Pathol 1988;19:974–9. [DOI] [PubMed] [Google Scholar]
- 111.Theaker JM, Gatter KC, Heryet A, Evans DJ, McGee JO. Giant cell myocarditis: evidence for the macrophage origin of the giant cells. J Clin Pathol 1985;38:160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Myers JM, Fairweather D, Huber SA, Cunningham MW. Autoimmune myocarditis, valvulitis, and cardiomyopathy. Curr Protoc Immunol 2013;Chapter 15:Unit 15 14 1–51. [DOI] [PMC free article] [PubMed]
- 113.Fairweather D, Rose NR. Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods 2007;41:118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huber SA. Autoimmunity in coxsackievirus B3 induced myocarditis. Autoimmunity 2006;39:55–61. [DOI] [PubMed] [Google Scholar]
- 115.Chen HS, Wang W, Wu SN, Liu JP. Corticosteroids for viral myocarditis. Cochrane Database Syst Rev 2013:CD004471. [DOI] [PMC free article] [PubMed]
- 116.Jain V, Bahia J, Mohebtash M, Barac A. Cardiovascular Complications Associated With Novel Cancer Immunotherapies. Curr Treat Options Cardiovasc Med 2017;19:36. [DOI] [PubMed] [Google Scholar]
- 117.Santoni M, Guerra F, Conti A et al. Incidence and risk of cardiotoxicity in cancer patients treated with targeted therapies. Cancer Treat Rev 2017;59:123–131. [DOI] [PubMed] [Google Scholar]
- 118.Johnson DB, Balko JM, Compton ML et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med 2016;375:1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Clemente-Casares X, Hosseinzadeh S, Barbu I et al. A CD103(+) Conventional Dendritic Cell Surveillance System Prevents Development of Overt Heart Failure during Subclinical Viral Myocarditis. Immunity 2017;47:974–989 e8. [DOI] [PubMed] [Google Scholar]
- 120.Kobayashi M, Usui F, Karasawa T et al. NLRP3 Deficiency Reduces Macrophage Interleukin-10 Production and Enhances the Susceptibility to Doxorubicin-induced Cardiotoxicity. Sci Rep 2016;6:26489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Leuschner F, Courties G, Dutta P et al. Silencing of CCR2 in myocarditis. Eur Heart J 2015;36:1478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Van der Borght K, Scott CL, Nindl V et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep 2017;18:3005–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Meredith MM, Liu K, Darrasse-Jeze G et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med 2012;209:1153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Satpathy AT, Kc W, Albring JC et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med 2012;209:1135–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guilliams M, Dutertre CA, Scott CL et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016;45:669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jaquenod De Giusti C, Ure AE, Rivadeneyra L, Schattner M, Gomez RM. Macrophages and galectin 3 play critical roles in CVB3-induced murine acute myocarditis and chronic fibrosis. J Mol Cell Cardiol 2015;85:58–70. [DOI] [PubMed] [Google Scholar]
- 127.Wu L, Ong S, Talor MV et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J Exp Med 2014;211:1449–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Muller I, Pappritz K, Savvatis K et al. CX3CR1 knockout aggravates Coxsackievirus B3induced myocarditis. PLoS One 2017;12:e0182643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Auffray C, Fogg D, Garfa M et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007;317:666–70. [DOI] [PubMed] [Google Scholar]
- 130.Carlin LM, Stamatiades EG, Auffray C et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013;153:362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Valaperti A, Nishii M, Liu Y et al. Innate immune interleukin-1 receptor-associated kinase 4 exacerbates viral myocarditis by reducing CCR5(+) CD11b(+) monocyte migration and impairing interferon production. Circulation 2013;128:1542–54. [DOI] [PubMed] [Google Scholar]
- 132.Cook DN. The role of MIP-1 alpha in inflammation and hematopoiesis. J Leukoc Biol 1996;59:61–6. [DOI] [PubMed] [Google Scholar]
- 133.Shen Y, Xu W, Chu YW, Wang Y, Liu QS, Xiong SD. Coxsackievirus group B type 3 infection upregulates expression of monocyte chemoattractant protein 1 in cardiac myocytes, which leads to enhanced migration of mononuclear cells in viral myocarditis. J Virol 2004;78:12548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shen Y, Zhang FQ, Wei X. Truncated monocyte chemoattractant protein-1 can alleviate cardiac injury in mice with viral myocarditis via infiltration of mononuclear cells. Microbiol Immunol 2014;58:195–201. [DOI] [PubMed] [Google Scholar]
- 135.Goser S, Ottl R, Brodner A et al. Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation 2005;112:3400–7. [DOI] [PubMed] [Google Scholar]
- 136.Abston ED, Coronado MJ, Bucek A et al. Th2 regulation of viral myocarditis in mice: different roles for TLR3 versus TRIF in progression to chronic disease. Clin Dev Immunol 2012;2012:129486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hardarson HS, Baker JS, Yang Z et al. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am J Physiol Heart Circ Physiol 2007;292:H251–8. [DOI] [PubMed] [Google Scholar]
- 138.Riad A, Westermann D, Zietsch C et al. TRIF is a critical survival factor in viral cardiomyopathy. J Immunol 2011;186:2561–70. [DOI] [PubMed] [Google Scholar]
- 139.Richer MJ, Lavallee DJ, Shanina I, Horwitz MS. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS One 2009;4:e4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sesti-Costa R, Francozo MCS, Silva GK, Proenca-Modena JL, Silva JS. TLR3 is required for survival following Coxsackievirus B3 infection by driving T lymphocyte activation and polarization: The role of dendritic cells. PLoS One 2017;12:e0185819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weinzierl AO, Szalay G, Wolburg H et al. Effective chemokine secretion by dendritic cells and expansion of cross-presenting CD4-/CD8+ dendritic cells define a protective phenotype in the mouse model of coxsackievirus myocarditis. J Virol 2008;82:8149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gorbea C, Makar KA, Pauschinger M et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J Biol Chem 2010;285:23208–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fuse K, Chan G, Liu Y et al. Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3-induced myocarditis and influences type I interferon production. Circulation 2005;112:2276–85. [DOI] [PubMed] [Google Scholar]
- 144.Fairweather D, Yusung S, Frisancho S et al. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol 2003;170:4731–7. [DOI] [PubMed] [Google Scholar]
- 145.Frisancho-Kiss S, Nyland JF, Davis SE et al. Cutting edge: T cell Ig mucin-3 reduces inflammatory heart disease by increasing CTLA-4 during innate immunity. J Immunol 2006;176:6411–5. [DOI] [PubMed] [Google Scholar]
- 146.Klingel K, Hohenadl C, Canu A et al. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci U S A 1992;89:314–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Klingel K, Schnorr JJ, Sauter M, Szalay G, Kandolf R. beta2-microglobulin-associated regulation of interferon-gamma and virus-specific immunoglobulin G confer resistance against the development of chronic coxsackievirus myocarditis. Am J Pathol 2003;162:1709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Henke A, Zell R, Ehrlich G, Stelzner A. Expression of immunoregulatory cytokines by recombinant coxsackievirus B3 variants confers protection against virus-caused myocarditis. J Virol 2001;75:8187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jarasch N, Martin U, Kamphausen E, Zell R, Wutzler P, Henke A. Interferon-gammainduced activation of nitric oxide-mediated antiviral activity of macrophages caused by a recombinant coxsackievirus B3. Viral Immunol 2005;18:355–64. [DOI] [PubMed] [Google Scholar]
- 150.Lowenstein CJ, Hill SL, Lafond-Walker A et al. Nitric oxide inhibits viral replication in murine myocarditis. J Clin Invest 1996;97:1837–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jakel S, Kuckelkorn U, Szalay G et al. Differential interferon responses enhance viral epitope generation by myocardial immunoproteasomes in murine enterovirus myocarditis. Am J Pathol 2009;175:510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li K, Xu W, Guo Q et al. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ Res 2009;105:353–64. [DOI] [PubMed] [Google Scholar]
- 153.Nozaki N, Shishido T, Takeishi Y, Kubota I. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation 2004;110:2869–74. [DOI] [PubMed] [Google Scholar]
- 154.Riad A, Bien S, Gratz M et al. Toll-like receptor-4 deficiency attenuates doxorubicininduced cardiomyopathy in mice. Eur J Heart Fail 2008;10:233–43. [DOI] [PubMed] [Google Scholar]
- 155.Ma Y, Zhang X, Bao H et al. Toll-like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS One 2012;7:e40763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bajpai G, Schneider C, Wong N et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 2018. [DOI] [PMC free article] [PubMed]
- 157.Libby P, Nahrendorf M, Swirski FK. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum”. J Am Coll Cardiol 2016;67:1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.