

Abstract
Low salt diet is beneficial in salt-sensitive hypertension but may provoke cardiovascular risk in patients with heart failure, diabetes, or other cardiovascular abnormalities because of endogenous renin-angiotensin system activation. PPARγ is a transcription factor which promotes an anti-oxidant pathway in the endothelium. We studied transgenic mice expressing a dominant-negative mutation in PPARγ selectively in the endothelium (E-V290M) to test the hypothesis that endothelial PPARγ plays a protective role in response to low salt-mediated renin-angiotensin system activation. Plasma renin and angiotensin-II were significantly and equally increased in all mice fed low salt for 6-weeks. Vasorelaxation to acetylcholine was not affected in basilar artery from E-V290M at baseline, but was significantly and selectively impaired in E-V290M after low salt. Unlike basilar artery, low salt was not sufficient to induce vascular dysfunction in carotid artery or aorta. Endothelial dysfunction in the basilar artery from E-V290M mice fed low salt was attenuated by scavengers of superoxide, inhibitors of NADPH oxidase, or blockade of the angiotensin-II AT1 receptor. Simultaneous AT1 and AT2 receptor blockade revealed that the restoration of endothelial function after AT1 receptor blockade was not a consequence of AT2 receptor activation. We conclude that interference with PPARγ in the endothelium produces endothelial dysfunction in the cerebral circulation in response to low salt-mediated activation of the endogenous renin-angiotensin system, mediated at least in part, through AT1 receptor activation and perturbed redox homeostasis. Moreover, our data suggest that the cerebral circulation may be particularly sensitive to inhibition of PPARγ activity and renin-angiotensin system activation.
Keywords: RAS, PPARγ, endothelial dysfunction, cerebral vascular dysfunction
Introduction
Cardiovascular diseases remain the leading cause of death in the United States despite advances in our understanding of its pathogenesis and treatment.1 Reduced dietary sodium intake is recommended as a prophylactic approach to decrease the risk of cardiovascular diseases.2 While a low-salt diet (LSD) is beneficial in the context of salt-sensitive hypertension, accumulating evidence suggests that reducing salt intake stimulates the endogenous renin-angiotensin system (RAS), induces insulin resistance, increases lipid levels and stimulates aldosterone and other stress hormones.3,4 Recent population-based studies indicate that salt restriction is associated with adverse health effects, particularly in patients with heart failure, diabetes, or other cardiovascular abnormalities.5–7 Nonetheless, the mechanisms underlying the detrimental effects associated with reduced salt intake in certain patient populations are unclear.
Abnormal activation of the RAS has been implicated in an array of cardiovascular disorders, including hypertension and stroke.8 A major regulatory system involved in maintenance of hemodynamic stability, the importance of the RAS is underscored by the sustained use of RAS blockers as primary treatments for hypertension.9 Further, angiotensin receptor blockers have been clinically shown to reduce the risk of fatal stroke.10 Similarly, ramipril, an angiotensin-converting enzyme inhibitor was effective in lowering the incidence of stroke in patients with a history of cerebrovascular disease.11,12 Endothelial dysfunction is believed to be an independent predictor of cardiovascular events and RAS blockers have been shown to restore endothelial function.13 To date, the physiological mechanisms underlying the complex interactions between RAS activation and endothelial dysfunction have not been fully elucidated.
Peroxisome proliferator-activated receptor-γ (PPARγ) is a ligand-activated transcription factor known to regulate anti-inflammatory and anti-oxidant signaling.14 Synthetic agonists of PPARγ have been reported to reduce blood pressure, improve insulin sensitivity, and exert vascular protection.15 In contrast, patients carrying an autosomal dominant mutation in PPARγ present with severe insulin resistance and early onset hypertension.16 Together, clinical and genetic evidence indicates a vital role for PPARγ in maintaining cardiovascular and metabolic homeostasis. To study the role of endothelial PPARγ, we generated mice (termed E-V290M) expressing a dominant-negative mutation in PPARγ selectively in the endothelium. E-V290M mice exhibit impaired vasodilation to acetylcholine (ACh) when fed a high fat diet or treated with sub-pressor dose of angiotensin-II, and exhibit increased inflammatory responses to cytokines.17–19 PPARγ regulates the actions of angiotensin-II in the vasculature and genetic interference with PPARγ specifically in the endothelium impaired the responses of the cerebral basilar artery to Ang-(1–7).20–22 Moreover, some PPARγ mutations were reported to cause RAS activation.23,24 Thus, experimental evidence suggests that endothelial PPARγ plays a role in modulating the effects of RAS on endothelial function. We tested the hypothesis that endothelial PPARγ protects against endothelial dysfunction induced by activation of the endogenous RAS. We provide evidence that genetic interference with PPARγ in the endothelium produces endothelial dysfunction in the cerebral circulation in response to LSD-mediated activation of the endogenous RAS. Imbalance in redox homeostasis and AT1 receptor activation were identified as potential mediators of these effects.
Methods
Details of the experimental procedures for blood pressure measurements, vascular function studies using wire- and pressurized myograph systems, use of inhibitors and receptor blockers, methods for RNA isolation and qPCR are presented in the expanded Methods section of the online-only data supplement. The data from this study are available from the corresponding author upon reasonable request.
Animals:
Adult male transgenic mice expressing a dominant-negative mutation in human PPARγ under the control of an endothelial-specific vascular cadherin promoter (E-V290M) was used as experimental models as reported.17 Age-and sex-matched non-transgenic (NT) littermates were used as controls. A separate study is currently ongoing specifically studying endothelial function in female E-V290M mice and in male and female offspring from E-V290M and NT mice undergoing arginine vasopressin-induced preeclampsia. NT and E-V290M mice were either fed TD.08290 (0.01–0.02% Na, Teklad) as LSD or standard chow (0.3% Na, NIH-31, Teklad) as normal salt diet. There was no difference in body or organ weights in response to diet or genotype (Table S1). Care of the mice met the standards set forth by the National Institutes of Health (NIH) guidelines for the care and use of experimental animals. All procedures were approved by The University of Iowa Animal Care and Use Committee.
Statistical Analysis:
Results are expressed as mean ± SEM. Statistical evaluation of the data was performed using GraphPad Prism. Data were analyzed using one-way or two-way ANOVA with repeated measures as appropriate, followed by Bonferroni or Tukey post-hoc test as indicated in each figure legend. Differences were considered significant when P value was less than 0.05. Where appropriate individual data points are plotted in dot/whisker plots.
Results
Experimental and control mice were fed chow or LSD for 6-weeks. We measured circulating levels of renin and angiotensin peptides as an indicator of RAS activation. The levels of renin and angiotensin peptides were similar in NT and E-V290M mice fed chow (Figure 1A–B). The levels of both renin and angiotensin peptides in plasma were significantly and similarly elevated in both NT and E-V290M mice after LSD. Consistent with our previous reports, E-V290M mice exhibited similar systolic blood pressure as NT controls at baseline which did not change in mice fed LSD (Figure 1C).17,18
Figure 1. RAS Activation.
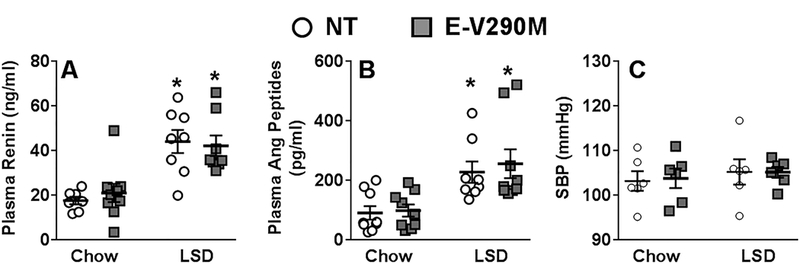
A) Circulating levels of renin measured by ELISA in NT and E-V290M mice (NT chow, n=7; E-V290M chow, n=9; NT LSD, n=8; E-V290M LSD, n=8). B) Plasma levels of angiotensin peptides measured by ELISA in NT and E-V290M mice (NT chow, n=9; E-V290M chow, n=9; NT LSD, n=8; E-V290M LSD, n=9). C) Systolic blood pressure measured using tail-cuff plethysmography in NT and E-V290M mice (n=6 per group). Systolic blood pressure was measured by tail cuff for 6 weeks following 3 weeks of training. Blood pressure was recorded 5 days per week for 6 weeks during the LSD. Data in all panels are presented as mean±SEM with individual data points shown and analyzed by two-way ANOVA. *P<0.05 vs. genotype-matched NT.
At baseline, carotid arteries from NT and E-V290M mice exhibited similar concentration-dependent vasodilation to ACh which were not affected by LSD (Figure 2A–B). Similarly, endothelium-independent dilation to SNP was not altered in LSD-fed E-V290M mice compared to other groups (Figure 2C–D). Contractile responses to KCl, angiotensin-II (Ang-II), serotonin (5-HT) and endothelin-1 (ET-1) were not altered in the carotid artery of E-V290M mice in response to activation of the endogenous RAS (Figure S1). Vascular function studies performed on the aorta from these mice yielded similar results (Figures S2 and S3).
Figure 2. Endothelial Function in Carotid Artery.
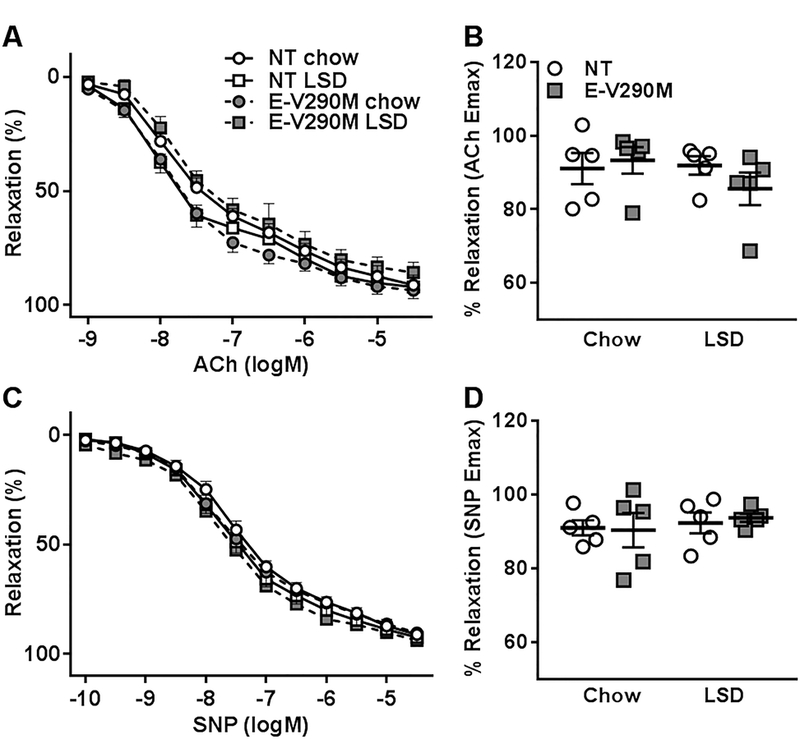
Dose-dependent relaxation was measured in the carotid arteries from NT and E-V290M mice fed chow or LSD. Cumulative concentration response curves for A) ACh and B) maximum relaxation to ACh (n=5 per group). Cumulative concentration response curves for C) SNP and D) maximum relaxation to SNP (n=5 per group). Data in all panels are presented as mean±SEM analyzed by two-way ANOVA with repeated measures.
Cerebrovascular dysfunction has been implicated in cardiovascular disorders such as stroke and neurological conditions such as dementia and Alzheimer’s disease.25 Thus, we sought to study the cerebral basilar arteries which have previously been shown to be particularly sensitive to PPARγ mutations.26 The baseline diameter of the basilar artery was not different between genotypes or in response to LSD (NT: chow 163.3±4.9 vs LSD 159.7±4.1µm; E-V290M: chow 174.5±11.4 vs LSD 173.9±5.5µm). Like larger vessels, responses of basilar artery to ACh were not different in NT and E-V290M mice fed standard chow (Figure 3A–B); and LSD-induced activation of RAS did not affect endothelium-dependent relaxation responses in the basilar artery of NT mice. In contrast, ACh-mediated vasodilation was significantly impaired in basilar artery from E-V290M mice fed LSD. Endothelium-independent relaxation to SNP in the basilar artery was not significantly affected by either RAS activation or PPARγ genotype (Figure 3C–D). Contraction to KCl was not altered in LSD-treated E-V290M mice (Figure 3E). There was a trend for decreased contraction to all doses of Ang-II in basilar artery from mice fed LSD, but there was no difference between genotypes (Figure 3F).
Figure 3. Vascular Function in Basilar Artery.
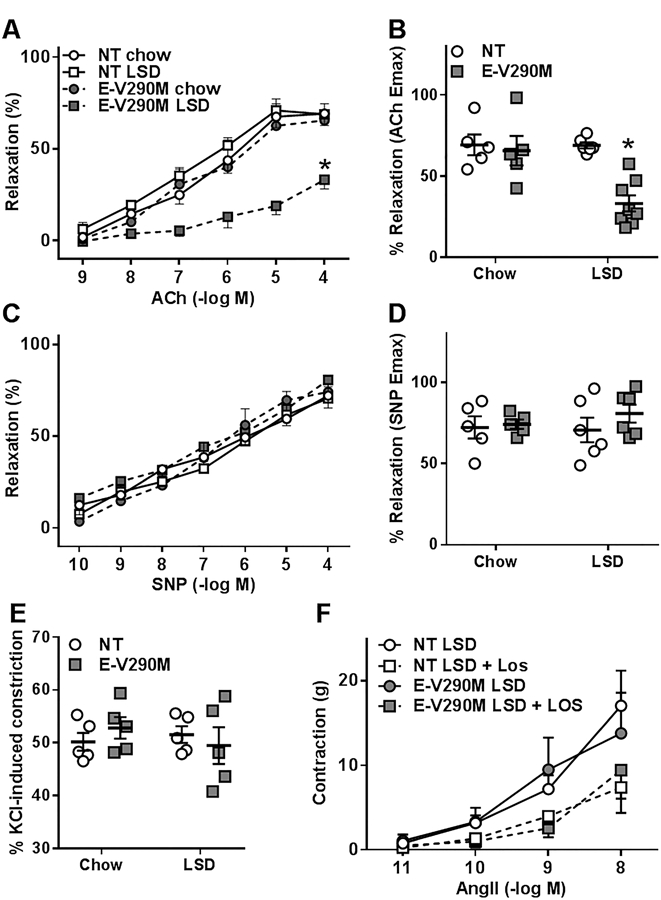
Dose-dependent relaxation was measured in the basilar arteries from NT and E-V290M mice fed chow or LSD. Arteries were pressurized at 60 mmHg and pre-constricted with thromboxane A2 mimetic (U46619). Cumulative concentration response curves for A) ACh and B) maximum relaxation to ACh (NT chow, n=5; E-V290M chow, n=5; NT LSD, n=6; E-V290M LSD, n=8). Cumulative concentration response curves for C) SNP and D) maximum relaxation to SNP (NT chow, n=5; E-V290M chow, n=5; NT LSD, n=6; E-V290M LSD, n=6). Data in panels A-D are presented as mean±SEM analyzed by two-way ANOVA with repeated measures. *P<0.05 E-V290M LSD vs. all other groups. Vasoconstrictive responses to E) Potassium chloride (n=5 per group) and F) Angiotensin II (n=5 per group) in the basilar artery are shown. Data in panels E-F are presented as mean±SEM analyzed by two-way ANOVA. P>0.05.
PPARγ is known to have antioxidant properties and we recently reported that interference with PPARγ in the endothelium induced Ang-II-mediated endothelial dysfunction through an oxidative stress-dependent mechanism.17 Therefore, we measured nitrotyrosine levels in the cerebral vessels from NT and E-V290M mice, as a marker of peroxynitrite generated from the reaction of nitric oxide with superoxide. 27 We observed that nitrotyrosine levels were significantly elevated in vessels from E-V290M mice fed a LSD, indicative of increased oxidative stress in the cerebral vasculature (Figure 4A). Consistent with this, endothelial function in the basilar artery was significantly improved in E-V290M mice incubated with the superoxide dismutase mimetic tempol (Figure 4B). In order to examine the underlying mechanism, we separately tested the effects of apocynin and VAS2870, two distinct inhibitors of NADPH oxidase. Both apocynin and VAS2870 had no effect on the relaxation responses to ACh in NT mice, but significantly improved endothelial function in the basilar artery from E-V290M mice fed LSD (Figure 4C–D). NADPH oxidase is comprised of a catalytic subunit Nox2 (gp91phox), p22phox, and the regulatory subunits p47phox, p67phox, p40phox and Rac.28 Of these regulatory subunits, phosphorylation of p47phox is known to be required for the activation of NADPH oxidase.29 Therefore, we measured the level of phosphorylated p47phox (P- p47phox) in the cerebral vessels of NT and E-V290M mice. Phosphorylated p47phox was significantly elevated in E-V290M mice fed a LSD compared to other groups, confirming NADPH oxidase activation and subsequent ROS generation (Figure 4E).
Figure 4. Endothelial Function in Basilar Artery: Role of Oxidative Stress.
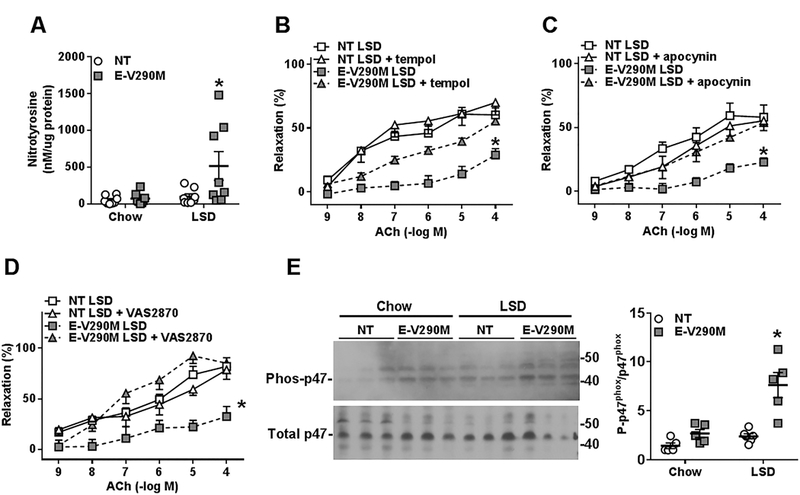
A) Nitrotyrosine levels measured by ELISA in the pooled cerebral vasculature of chow- and LSD-fed NT and E-V290M mice (n=8). Cumulative concentration response curves for B) ACh in basilar arteries from LSD-fed- NT (n=5) and E-V290M mice (n=7) with and without tempol (1mmol/L, 30 mins); C) ACh in basilar arteries from NT (n=5) and E-V290M mice (n=6) with and without apocynin (100 µmol/L, 30mins); D) ACh in basilar arteries from NT (n=5) and E-V290M mice (n=5) with and without VAS2870 (100 µmol/L, 30mins). Data in all panels are presented as mean±SEM analyzed by two-way ANOVA with repeated measures. *P<0.05 E-V290M LSD vs. all other groups. E) Western blot detecting phosphorylated P47phox (phos-p47phox) and total P47phox in pooled cerebral arteries (NT chow, n=5; E-V290M chow, n=5; NT LSD, n=6; E-V290M LSD, n=5). Quantification of the western blot data is also shown. Data were normalized to the average control value set to 1.0. Data in panel E are mean±SEM analyzed by two-way ANOVA. *P<0.05 E-V290M LSD vs. all other groups.
We measured the level of expression of genes related to oxidative stress in the cerebral vasculature by quantitative real time RT-PCR. The levels of expression of NADPH oxidase subunit Nox2, NF-κB subunit p65, eNOS, and the antioxidant enzymes SOD1, SOD3, and catalase were similar in the cerebral vessels from chow-fed NT and E-V290M mice (Figure 5). Expression of Nox2 was increased whereas expression of catalase and SOD3 were both decreased in basilar artery from E-V290M mice fed LSD. There were no differences in the expression levels of p65, eNOS or SOD1. Altogether, this suggests that oxidative stress in basilar artery from E-V290M mice fed LSD may be mediated through increased NADPH oxidase and reduced antioxidant gene expression.
Figure 5. Gene expression in cerebral vessels.
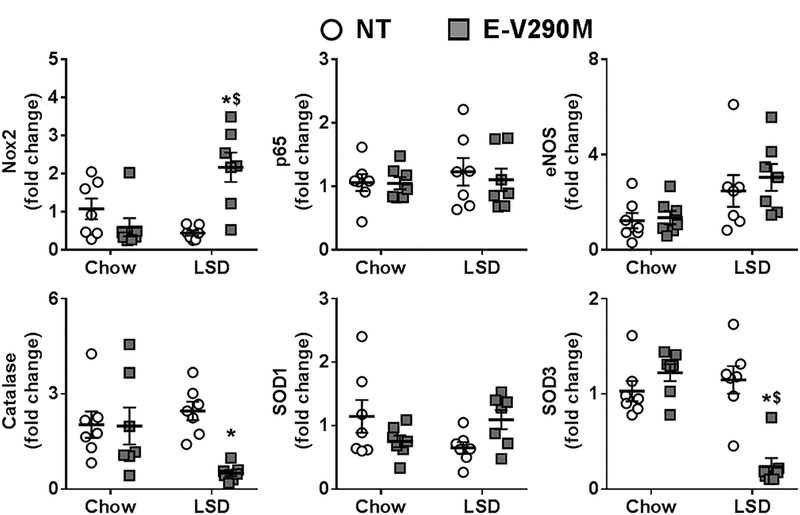
mRNA expression levels of the indicated genes in pooled cerebral vessels from NT and E-V290M mice fed a LSD or standard chow (n=7 per group). Data in all panels are presented as mean±SEM analyzed by two-way ANOVA. *P<0.05 vs NT LSD and $P<0.05 vs. E-V290M chow.
Since abnormal activation of the RAS is known to induce generation of ROS via an AT1 receptor-dependent mechanism, we hypothesized that the endothelial dysfunction observed in the basilar artery of E-V290M mice was mediated through AT1 receptors. To test this, we treated LSD-fed NT and E-V290M mice with Losartan for 3 weeks. Losartan treatment robustly increased plasma renin in NT and E-V290M mice, presumably due to relief of the classic negative feedback pathway (Figure 6A). Importantly, the increase in renin in response to losartan was the same in NT and E-V290M mice. There was a significant and similar reduction in systolic blood pressure in both losartan-treated LSD-fed NT and E-V290M mice compared to genotype-matched mice fed a LSD alone (Figure 6B). We then assessed vasodilation responses to ACh in the basilar artery following losartan administration. Replicating the data above, this independent cohort of E-V290M mice fed a LSD exhibited impaired vasodilation to ACh (Figure 6C). ACh-mediated relaxation was significantly improved and maximal relaxation to ACh was normalized in the basilar arteries from losartan-treated E-V290M mice fed LSD (Figure 6C–D), suggesting that the endothelial dysfunction induced by RAS activation is dependent on AT1 receptors. We next measured the expression of AT1 receptor mRNA in the cerebral arteries of these mice and we observed that regardless of diet, the levels of AT1 receptor mRNA were not different in the cerebral arteries of NT and E-V290M mice (Figure 6E). Thus, the improved endothelial function under AT1 receptor blockade in E-V290M mice fed LSD was not due to increased expression of AT1 receptor mRNA.
Figure 6. Endothelial Function in Basilar Artery: Role of AT1 and AT2 Receptors.
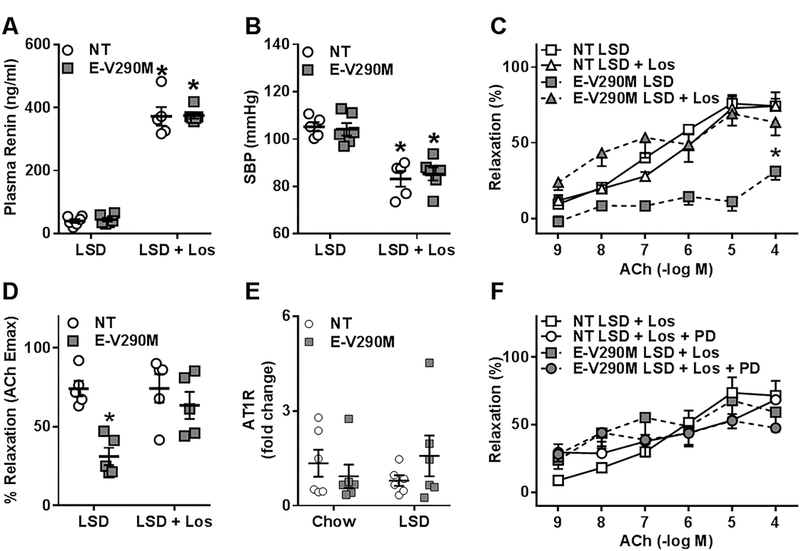
A) Circulating levels of renin measured by ELISA in LSD-fed NT and E-V290M mice in response to Losartan administration (NT LSD, n=6; E-V290M LSD, n=6, NT LSD+Los, n=5; E-V290M LSD+Los, n=6), *P<0.05 LSD vs. LSD + Los. B) Systolic blood pressure measured using tail-cuff plethysmography in LSD-fed NT and E-V290M mice in response to Losartan administration (NT LSD, n=5; E-V290M LSD, n=6, NT LSD+Los, n=5; E-V290M LSD+Los, n=6), *P<0.05 LSD vs. LSD + Los. C) Cumulative concentration response curves for ACh and D) maximum relaxation to ACh in basilar arteries from LSD-fed- NT (n=5) and E-V290M mice (n=5) ± losartan. E) mRNA expression levels of the AT1R gene in pooled cerebral vessels from NT and E-V290M mice fed a LSD or standard chow (n=6 per group). F) Cumulative concentration response curves for ACh in basilar arteries from LSD-fed- NT (n=4) and E-V290M mice (n=4) + losartan ± PD123319. Data in all panels are presented as mean±SEM analyzed by two-way ANOVA (A, E) or two-way ANOVA with repeated measures (B, C, D, F). *P<0.05 E-V290M LSD vs. all other groups.
AT1 receptor antagonism has been previously shown to exert protective effects in the vasculature through activation of AT2 receptors.30 Therefore, we investigated if the restoration of endothelial function in the cerebral vasculature from losartan-treated E-V290M mice was the result of AT1 receptor blockade or due to concomitant AT2 receptor activation. Thus, we examined relaxation responses to ACh in the basilar artery from NT and E-V290M mice treated with both losartan and PD123319, an AT2 receptor antagonist (Figure 6F). The improvement in ACh-induced relaxation observed upon AT1 receptor blockade was not affected by simultaneous blockade of AT2 receptors, suggesting that the restoration of endothelial function in the basilar artery of E-V290M mice was not a consequence of AT2 receptor activation.
Discussion
The salient findings of the present study are that: a) genetic interference with PPARγ in the endothelium did not affect endothelial function in cerebral basilar arteries from mice fed chow, but augmented the effects of LSD-mediated endogenous RAS activation causing endothelial dysfunction, b) the effects on endothelial function in response to PPARγ mutation and activation of RAS after a six-week LSD regimen were minimal in carotid arteries and aorta, c) endothelial dysfunction in the basilar artery of E-V290M mice was prevented by a scavenger of superoxide or inhibitors of NADPH oxidase, d) RAS-induced impairment in endothelial function in the basilar artery was dependent on AT1 receptor activation, and e) improvement in endothelial function upon AT1 receptor blockade was not a consequence of AT2 receptor activation. Together, these findings highlight the effect of endogenous RAS activation particularly on the cerebral vasculature and provide novel insights into the mechanisms by which endothelial PPARγ protects against endothelial dysfunction induced by the normal activation of the RAS caused in response to salt restriction.
The RAS plays a key role in the regulation of fluid balance and electrolyte homeostasis in the body. Individual components of the RAS have been well studied, and their importance has been documented in preclinical and clinical models of cardiovascular diseases.31 A major factor regulating endogenous RAS activity is salt intake, and high salt consumption has been widely associated with increased risk of cardiovascular diseases such as hypertension.32 In the United States, sodium intake exceed recommended amounts across all age groups and race. Limiting sodium intake to 2300 mg/day has been a consistent dietary recommendation for decades.33 Recent propositions of a further reduction in daily sodium consumption to 1500 mg/day have been made (DASH dietary pattern); nevertheless the feasibility and benefits of these reductions have been questioned and are still debatable at least among specific population sub-groups who are at risk of developing cardiovascular disorders. In clinics, sodium restriction has been effective in patients with salt-sensitive hypertension.34 This has led to a popular notion that reducing salt intake is beneficial for cardiovascular diseases in general. However, recent epidemiological studies reported that salt restriction is associated with adverse effects in patients with cardiovascular abnormalities and diabetes.5–7 This may be due to the activation of the endogenous RAS as higher plasma renin levels have been previously associated with myocardial infarction and increased mortality and morbidity in patients with coronary artery disease.35 Consistent with this, we show that a LSD induced the endogenous RAS and notably, this induction was similar irrespective of the PPARγ genotype. As previously reported by our lab, interference with PPARγ in the endothelium through targeted expression of a dominant-negative mutation in PPARγ has no effect under baseline conditions, but results in oxidative stress and endothelial dysfunction in cerebral basilar arteries in response high fat diet, sub-pressor doses of Ang-II, and most recently in aging.17,18,22 Our previous studies combined with the current study imply that resistance vessels may be more sensitive to the loss of PPARγ activity and compared to conduit arteries when the mice are also subjected to another cardiovascular stressor. The actual mechanism for this difference remains unclear. Nevertheless, this data underscores the critical role played by endothelial PPARγ in protecting against accelerated cerebral vascular dysfunction induced by a host of cardiovascular risk factors including sodium restriction.
In humans, activation of PPARγ lowers arterial blood pressure and dominant-negative PPARγ mutations cause impaired PPARγ function and hypertension.15,16 This strongly supports the physiological significance of PPARγ and exemplifies the severe consequences for cardiovascular events when PPARγ signaling is impaired. The use of mice expressing human disease-causing mutations increases the relevance to human diseases and provides greater mechanistic insights into the protective role of PPARγ in the endothelium because we can manipulate PPARγ signaling in a cell-specific manner. We view this as a strength of the current study. PPARγ is normally expressed in the endothelium and smooth muscle where it has been shown to exhibit anti-inflammatory and anti-oxidant properties.17 Previous studies have linked dysregulation of the RAS and oxidative stress with cerebrovascular dysfunction associated with aging and cardiovascular diseases.36 Ang-II has been shown to activate pro-oxidant and pro-inflammatory signaling that generates reactive oxygen species and causes vascular diseases.37 Our data that mice expressing dominant-negative PPARγ in the endothelium exhibited oxidative stress-dependent endothelial dysfunction in response to sodium restriction expands this concept. Further, consistent with previous reports of increased NADPH oxidase components in these mice; incubation of the basilar artery from E-V290M mice with apocynin and VAS2870 restored endothelial function.17 Although our data suggest a role for superoxide generated primarily by NADPH oxidase, the involvement of other sources of superoxide and even other reactive oxygen species such as peroxides cannot be ruled out. In this light, the decrease in expression of antioxidant enzymes catalase and SOD3 in the basilar artery of E-V290M mice suggests a role for an impaired antioxidant defense mechanism in response to RAS activation. The fact that catalase and SOD3 are PPARγ target genes further highlights the significance of PPARγ in the regulation of endothelial function and provides mechanistic insights into cerebral endothelial dysfunction induced by RAS activation.18,38
In addition to oxidative stress, previous studies in animal models exhibiting impaired vascular function associated with RAS activation have illustrated the significance of AT1 receptors in mediating endothelial dysfunction.39 Most of the detrimental effects of Ang-II on vascular beds have been attributed to its actions via the AT1 receptor. In the cerebral circulation, circulating and locally produced Ang-II acts via AT1 receptors in cerebral arteries and capillaries, and is known to participate in the regulation of cerebral blood flow and cerebrovascular function.40 Chronic activation of the AT1 receptor in the cerebral vasculature has been shown to contribute to inflammation which in turn leads to cerebrovascular remodeling and impaired cerebrovascular function.41,42 Furthermore, administration of losartan, a selective and competitive inhibitor of AT1 receptors, has been previously shown to improve vascular reactivity in cerebral arteries in preclinical models associated with vascular oxidative stress.43 Interestingly, our data that impaired vasodilation to acetylcholine in the basilar artery of LSD-treated E-V290M mice was improved by losartan is consistent with these findings. The efficacy of losartan treatment in this study was validated by the substantial elevation in circulating renin levels and the lowered blood pressure in LSD-fed NT and E-V290M mice. Importantly, blood pressure was equally lowered in NT and E-V290M mice suggesting the improvement in endothelial function by AT1 receptor blockade was not a consequence of decreased blood pressure. Moreover, our data suggests a potential mechanism by which endothelial PPARγ exerts protective effects on cerebral endothelial function by combating the pro-oxidant effects induced by elevated AT1 receptor activation under salt restricted conditions. Although blockade of AT1 receptor is beneficial in improving endothelial function in E-V290M mice, it is worth mentioning that RAS blocking drugs may be harmful in excessively salt-restricted cardiovascular patients who have high plasma renin levels as the elevation in circulating levels of renin might be required to prevent hypotension, hyperkalemia and reduced glomerular filtration rates.44,45 These results highlight the potential detrimental effects of excessive and chronic AT1 receptor activation, resulting from sodium restriction in the cerebral vascular function. Nevertheless, we recognize that the very low level of sodium in the diet used in this study may cause abnormally low sodium balance (severe sodium depletion) and may represent a weakness. Thus, additional studies with intermediate levels of sodium restriction may be warranted.
Angiotensin-receptor blockers such as losartan antagonize Ang-II at its AT1 receptor but do not affect the AT2 receptor subtype when administered in recommended concentrations. AT2 receptors have unique and predominantly oppositional effects than that of AT1 receptors on blood vessels.46 During stress-related events, such as vascular injury and even sodium depletion, AT2 receptors are believed to be upregulated to oppose the pathologic effects mediated by the AT1 receptor subtype.47 Therefore, we investigated if the losartan-mediated improvement in endothelial dysfunction observed in the E-V290M was a result of AT2 receptor activation. Our data illustrating improved endothelial relaxation to acetylcholine in the basilar artery of E-V290M mice upon simultaneous blockade of AT1 and AT2 receptors rule out a plausible protective effect mediated by AT2 receptor activation in the cerebral arteries under low sodium conditions.
Perspectives
Although limiting dietary sodium consumption is beneficial in salt-sensitive hypertension, several lines of evidence suggest that it causes serious health consequences in certain patient populations, mostly as a result of off-target effects such as endogenous RAS activation. Herein, our data indicates that in conditions with impaired PPARγ activity, which can occur in obesity, diabetes and other associated cardiovascular abnormalities, RAS activation induced by a low sodium diet might increase the susceptibility to endothelial dysfunction, particularly in smaller cerebral arteries.
Recent results from the SPRINT (Systolic Blood Pressure Intervention Trial) suggest that reductions in diastolic blood pressure offers strong health benefits in non-diabetic individuals who are at risk for increased cardiovascular morbidities.48 In contrast, patients with type-2 diabetes were reported to have an increased cardiovascular mortality that was paradoxically associated with lower excretion of urinary sodium and alarmingly, the highest mortality rate was observed in diabetics with the lowest sodium intake.49 Although these recent reports are highly specific to certain subpopulations, it is indeed understandable that these high-risk patient populations are the ones where such aggressive dietary interventions are prescribed. Further studies in this area may facilitate therapeutic approaches that could be used to prevent the off-target deleterious effects of sodium restriction in these populations.
Supplementary Material
Novelty and Significance:
What Is New?
A low salt diet (LSD) activates the endogenous RAS in circulation.
PPARγ in the endothelium exerts protection against augmented endothelial dysfunction induced by endogenous RAS activation, particularly in cerebral basilar arteries.
Endothelial PPARγ exerts protective effects on cerebral endothelial function by combating the pro-oxidant effects induced at least in part by elevated AT1 receptor activation.
What Is Relevant?
This work is relevant to our understanding of the detrimental effects of sodium restriction in certain subpopulations, such as diabetes and obesity where PPARγ may be impaired.
Endothelial PPARγ impairment may predispose these patient populations to endothelial dysfunction as a consequence of low-salt diet mediated activation of the RAS.
Summary
Genetic interference with PPARγ activity specifically in the vascular endothelium induces accelerated endothelial dysfunction in the cerebral resistance arteries in response to sodium restriction-mediated activation of the endogenous RAS.
Cerebral endothelial dysfunction is due to oxidative stress and was AT1 receptor dependent but not AT2 receptor dependent.
Loss of the protective effects of PPARγ in the endothelium may predispose certain patients who are at risk for cardiovascular morbidities and mortalities associated with sodium restriction to vascular complications.
Acknowledgements
The authors thank Bill Paradee, Norma Sinclair, JoAnne Schwarting, and Patricia Yarolem for genotyping mice. Transgenic mice were generated at the University of Iowa Genome Editing Facility supported in part by grants from the National Institutes of Health (NIH) and from the Roy J. and Lucille A. Carver College of Medicine.
Funding Source
This work was supported through research grants from the National Institutes of Health (NIH) to C.D.S. (HL084207, HL125603, HL131689), and grants from the American Heart Association to C.D.S. (15SFRN23480000). Dr. Jing Wu is supported by an American Heart Association Postdoctoral Fellowship (17POST33660685). The authors gratefully acknowledge the generous research support of the Roy J. Carver Trust.
Footnotes
Disclosures
None
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics−−2015 update: a report from the American Heart Association. Circulation 2015;131:e29–e322. [DOI] [PubMed] [Google Scholar]
- 2.Whelton PK, Appel LJ, Sacco RL, et al. Sodium, blood pressure, and cardiovascular disease: further evidence supporting the American Heart Association sodium reduction recommendations. Circulation 2012;126:2880–2889. [DOI] [PubMed] [Google Scholar]
- 3.Shao W, Seth DM, Prieto MC, Kobori H, Navar LG. Activation of the renin-angiotensin system by a low-salt diet does not augment intratubular angiotensinogen and angiotensin II in rats. Am J Physiol Renal Physiol 2013;304:F505–F514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg R, Williams GH, Hurwitz S, Brown NJ, Hopkins PN, Adler GK. Low-salt diet increases insulin resistance in healthy subjects. Metabolism 2011;60:965–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas MC, Moran J, Forsblom C, Harjutsalo V, Thorn L, Ahola A, Wadén J, Tolonen N, Saraheimo M, Gordin D, Groop P- H. The Association Between Dietary Sodium Intake, ESRD, and All-Cause Mortality in Patients With Type 1 Diabetes. Diabetes Care 2011;34:861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stolarz-Skrzypek K, Kuznetsova T, Thijs L, et al. Fatal and nonfatal outcomes, incidence of hypertension, and blood pressure changes in relation to urinary sodium excretion. JAMA 2011;305:1777–1785. [DOI] [PubMed] [Google Scholar]
- 7.O’Donnell M, Mente A, Rangarajan S, et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med 2014;371:612–623. [DOI] [PubMed] [Google Scholar]
- 8.Re RN. Mechanisms of Disease: local renin–angiotensin–aldosterone systems and the pathogenesis and treatment of cardiovascular disease. Nature Clinical Practice Cardiovascular Medicine 2004;1:42–47. [DOI] [PubMed] [Google Scholar]
- 9.Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2017;71:1269–1324. [DOI] [PubMed] [Google Scholar]
- 10.Dahlof B, Devereux RB, Kjeldsen SE, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002;359:995–1003. [DOI] [PubMed] [Google Scholar]
- 11.Bosch J, Yusuf S, Pogue J, Sleight P, Lonn E, Rangoonwala B, Davies R, Ostergren J, Probstfield J. Use of ramipril in preventing stroke: double blind randomised trial. British Medical Journal 2002;324:699–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P, Anderson C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547–1559. [DOI] [PubMed] [Google Scholar]
- 13.Volpe M, Iaccarino G, Vecchione C, Rizzoni D, Russo R, Rubattu S, Condorelli G, Ganten U, Ganten D, Trimarco B, Lindpaintner K. Association and cosegregation of stroke with impaired endothelium-dependent vasorelaxation in stroke prone, spontaneously hypertensive rats. J Clin Invest 1996;98:256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sigmund CD. Endothelial and vascular muscle PPARgamma in arterial pressure regulation: lessons from genetic interference and deficiency. Hypertension 2010;55:437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005;366:1279–1289. [DOI] [PubMed] [Google Scholar]
- 16.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O’Rahilly S. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999;402:880–883. [DOI] [PubMed] [Google Scholar]
- 17.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circulation Research 2008;103:654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu C, Lu KT, Mukohda M, Davis DR, Faraci FM, Sigmund CD. Interference with PPARgamma in endothelium accelerates angiotensin II-induced endothelial dysfunction. Physiol Genomics 2016;48:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mukohda M, Stump M, Ketsawatsomkron P, Hu C, Quelle FW, Sigmund CD. Endothelial PPAR-gamma provides vascular protection from IL-1beta-induced oxidative stress. Am J Physiol Heart Circ Physiol 2016;310:H39–H48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benkirane K, Viel EC, Amiri F, Schiffrin EL. Peroxisome proliferator-activated receptor gamma regulates angiotensin II-stimulated phosphatidylinositol 3-kinase and mitogen-activated protein kinase in blood vessels in vivo. Hypertension 2006;47:102–108. [DOI] [PubMed] [Google Scholar]
- 21.Takeda K, Ichiki T, Tokunou T, Funakoshi Y, Iino N, Hirano K, Kanaide H, Takeshita A. Peroxisome proliferator-activated receptor gamma activators downregulate angiotensin II type 1 receptor in vascular smooth muscle cells. Circulation 2000;102:1834–1839. [DOI] [PubMed] [Google Scholar]
- 22.De Silva TM, Hu C, Kinzenbaw DA, Modrick ML, Sigmund CD, Faraci FM. Genetic Interference With Endothelial PPAR-gamma (Peroxisome Proliferator-Activated Receptor-gamma) Augments Effects of Angiotensin II While Impairing Responses to Angiotensin 1–7. Hypertension 2017;70:559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, Lascols O, Capeau J, Caron-Debarle M. Peroxisome proliferator-activated receptor-gamma mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler Thromb Vasc Biol 2013;33:829–838. [DOI] [PubMed] [Google Scholar]
- 24.Sigmund CD. A clinical link between peroxisome proliferator-activated receptor gamma and the renin-angiotensin system. Arterioscler Thromb Vasc Biol 2013;33:676–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iadecola C, Davisson RL. Hypertension and Cerebrovascular Dysfunction. Cell Metabolism 2008;7:476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Silva TM, Modrick ML, Ketsawatsomkron P, Lynch C, Chu Y, Pelham CJ, Sigmund CD, Faraci FM. Role of peroxisome proliferator-activated receptor-gamma in vascular muscle in the cerebral circulation. Hypertension 2014;64:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiter CD, Teng RJ, Beckman JS. Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J Biol Chem 2000;275:32460–32466. [DOI] [PubMed] [Google Scholar]
- 28.Meijles DN, Fan LM, Howlin BJ, Li JM. Molecular insights of p47phox phosphorylation dynamics in the regulation of NADPH oxidase activation and superoxide production. J Biol Chem 2014;289:22759–22770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J Biol Chem 2003;278:12094–12100. [DOI] [PubMed] [Google Scholar]
- 30.Wassmann S, Czech T, van Eickels M, Fleming I, Bohm M, Nickenig G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation 2004;110:3062–3067. [DOI] [PubMed] [Google Scholar]
- 31.Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system--an endocrine and paracrine system. Endocrinology 2003;144:2179–2183. [DOI] [PubMed] [Google Scholar]
- 32.Juraschek SP, Woodward M, Sacks FM, Carey VJ, Miller ER, 3rd, Appel LJ. Time Course of Change in Blood Pressure From Sodium Reduction and the DASH Diet. Hypertension 2017;70:923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeSalvo KB, Olson R, Casavale KO. Dietary guidelines for americans. JAMA 2016;315:457–458. [DOI] [PubMed] [Google Scholar]
- 34.Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER, 3rd, Simons-Morton DG, Karanja N, Lin PH. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med 2001;344:3–10. [DOI] [PubMed] [Google Scholar]
- 35.Alderman MH, Ooi WL, Cohen H, Madhavan S, Sealey JE, Laragh JH. Plasma renin activity: a risk factor for myocardial infarction in hypertensive patients. Am J Hypertens 1997;10:1–8. [DOI] [PubMed] [Google Scholar]
- 36.De Silva TM, Faraci FM. Effects of angiotensin II on the cerebral circulation: role of oxidative stress. Front Physiol 2012;3:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. CircRes 1994;74:1141–1148. [DOI] [PubMed] [Google Scholar]
- 38.Khoo NK, Hebbar S, Zhao W, Moore SA, Domann FE, Robbins ME. Differential activation of catalase expression and activity by PPAR agonists: implications for astrocyte protection in anti-glioma therapy. Redox Biol 2013;1:70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Modrick ML, Didion SP, Sigmund CD, Faraci FM. Role of oxidative stress and AT1 receptors in cerebral vascular dysfunction with aging. Am J Physiol Heart Circ Physiol 2009;296:H1914–H1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J, Pavel J, Macova M, Yu ZX, Imboden H, Ge L, Nishioku T, Dou J, Delgiacco E, Saavedra JM. AT1 receptor blockade regulates the local angiotensin II system in cerebral microvessels from spontaneously hypertensive rats. Stroke 2006;37:1271–1276. [DOI] [PubMed] [Google Scholar]
- 41.Paulson OB, Waldemar G. Role of the local renin-angiotensin system in the autoregulation of the cerebral circulation. Blood Vessels 1991;28:231–235. [DOI] [PubMed] [Google Scholar]
- 42.Ito T, Yamakawa H, Bregonzio C, Terron JA, Falcon-Neri A, Saavedra JM. Protection against ischemia and improvement of cerebral blood flow in genetically hypertensive rats by chronic pretreatment with an angiotensin II AT1 antagonist. Stroke 2002;33:2297–2303. [DOI] [PubMed] [Google Scholar]
- 43.Papadopoulos P, Tong XK, Imboden H, Hamel E. Losartan improves cerebrovascular function in a mouse model of Alzheimer’s disease with combined overproduction of amyloid-beta and transforming growth factor-beta1. J Cereb Blood Flow Metab 2017;37:1959–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sealey JE, Alderman MH, Furberg CD, Laragh JH. Renin-angiotensin system blockers may create more risk than reward for sodium-depleted cardiovascular patients with high plasma renin levels. Am J Hypertens 2013;26:727–738. [DOI] [PubMed] [Google Scholar]
- 45.Kurtz TW. When blockade of the renin-angiotensin system becomes a two-edged sword. Am J Hypertens 2013;26:721–722. [DOI] [PubMed] [Google Scholar]
- 46.Siragy HM. The role of the AT2 receptor in hypertension. Am J Hypertens 2000;13:62s–67s. [DOI] [PubMed] [Google Scholar]
- 47.Janiak P, Pillon A, Prost JF, Vilaine JP. Role of angiotensin subtype 2 receptor in neointima formation after vascular injury. Hypertension 1992;20:737–745. [DOI] [PubMed] [Google Scholar]
- 48.Wright JT Jr., Williamson JD, Whelton PK, et al. A Randomized Trial of Intensive versus Standard Blood-Pressure Control. N Engl J Med 2015;373:2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ekinci EI, Clarke S, Thomas MC, Moran JL, Cheong K, MacIsaac RJ, Jerums G. Dietary salt intake and mortality in patients with type 2 diabetes. Diabetes Care 2011;34:703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.