

Abstract
Objective:
Alterations in extracellular matrix quantity and composition contribute to atherosclerosis, with remodeling of the subendothelial basement membrane to a fibronectin-rich matrix preceding lesion development. Endothelial cell interactions with fibronectin prime inflammatory responses to a variety of atherogenic stimuli; however, the mechanisms regulating early atherogenic fibronectin accumulation remain unknown. We previously demonstrated that oxidized LDL (oxLDL) promotes endothelial pro inflammatory gene expression by activating the integrin α5β1, a classic mediator of fibronectin fibrillogenesis.
Approach and Results:
We now show that oxLDL drives robust endothelial fibronectin deposition and inhibiting α5β1 (blocking antibodies, α5 knockout cells) completely inhibits oxLDL-induced fibronectin deposition. Consistent with this, inducible endothelial-specific α5 integrin deletion in ApoE knockout mice significantly reduces atherosclerotic plaque formation, associated with reduced early atherogenic inflammation. Unlike TGFβ-induced fibronectin deposition, oxLDL does not induce fibronectin expression (mRNA, protein) or the endothelial-to-mesenchymal transition phenotype. In addition, we show that cell-derived and plasma-derived fibronectin differentially affect endothelial function, with only cell-derived fibronectin capable of supporting oxLDL-induced VCAM-1 expression despite plasma fibronectin deposition by oxLDL. The inclusion of EIIIA and EIIIB domains in cell-derived fibronectin mediates this effect, as EIIIA/EIIIB knockout endothelial cells show diminished oxLDL-induced inflammation. Furthermore, our data suggests that EIIIA/EIIIB-positive cellular fibronectin is required for maximal α5β1 recruitment to focal adhesions and fibronectin fibrillogenesis.
Conclusions:
Taken together, our data demonstrate that endothelial α5 integrins drives oxLDL-induced fibronectin deposition and early atherogenic inflammation. Additionally, we show that α5β1-dependent endothelial fibronectin deposition mediates oxLDL-dependent endothelial inflammation and fibronectin fibrillogenesis.
Keywords: Fibronectin, Atherosclerosis, Oxidized LDL, Extracellular Matrix, Integrins
INTRODUCTION
During atherosclerotic plaque formation, remodeling of the subendothelial basement membrane to fibronectin-rich matrix promotes endothelial cell activation and early atherosclerotic plaque formation 1, 2. Naturally absent in healthy areas, provisional matrix proteins (i.e. fibronectin and fibrinogen) can be detected in atherosclerosis-prone regions concomitant with endothelial activation markers (ICAM-1, VCAM-1) but prior to monocyte recruitment, suggesting that subendothelial matrix remodeling is an early event in atherogenesis 1, 2. Furthermore, preventing fibronectin deposition at these sites significantly reduces plaque burden 2, 3, associated with reduced endothelial activation and monocyte recruitment. Cell culture studies show that the presence of a fibronectin matrix promotes nuclear factor-κ B (NF-κB) signaling and proinflammatory gene expression in response to classic atherogenic mediators, such as oxidized low density lipoproteins (oxLDL) and atheroprone hemodynamics 1, 4. Therefore, subendothelial matrix remodeling critically regulates early endothelial activation during the process of plaque formation.
Despite the well described role of fibronectin matrix deposition in endothelial activation, the mechanisms regulating fibronectin deposition in the context of atherosclerosis remains largely unknown. The fibronectin that accumulates in the subendothelial matrix during atherogenesis could arise from two sources, (1) the leak of circulating plasma fibronectin into the vessel wall or (2) the expression and deposition of cell-derived fibronectin by the endothelium. Fibronectin deletion in endothelial cells does not prevent fibronectin staining in models of atheroprone flow in vivo, suggesting that leak of plasma fibronectin is a major source of subendothelial fibronectin 5. However, the deletion of endothelial fibronectin induces a hemorrhage phenotype despite the presence of plasma fibronectin, suggesting that the source of fibronectin may critically affect endothelial function 5. Deposition of endothelial-derived fibronectin is a product of fibronectin expression and activation of the machinery for fibronectin fibrillogenesis, such as the integrin α5β1. Recent studies suggest that endothelial-to-mesenchymal transition (EndMT) is associated with endothelial fibronectin staining at atherosclerosis-prone sites in both mice and humans 6. While atheroprone flow patterns enhance fibronectin expression in vitro 7, 8, vascular regions exposed to atheroprone flow do not show fibronectin deposition in the absence of other atherogenic stimuli, such as hypercholesterolemia 1, 9. We previously demonstrated that oxLDL stimulates α5β1 integrin activation in endothelial cells in culture 4, suggesting that alterations in integrin function could contribute to fibronectin deposition. However, the role of oxLDL-mediated integrin activation in endothelial matrix remodeling remains unknown.
Although integrin αvβ3 classically mediates multiple aspects of cardiovascular disease, much less is known about α5β1 in atherosclerosis 10. Analysis of mRNA and protein isolated from atherosclerotic lesions and injured carotid arteries show increased α5 expression 11, 12. We have previously shown that inhibiting α5β1 prevents oxLDL-induced NF-κB activation, proinflammatory gene expression, and early atherosclerosis 4. In addition, α5−/+ mice and mice expressing an α5/α2 integrin chimera showed significantly reduced inflammation and atherosclerotic plaque size in atheroprone mice 13, 14. However, the influence of inhibiting α5 integrins on multiple cell types prevents these studies from assessing the role of α5 integrins in endothelial activation directly. For example, it has been demonstrated that inhibiting α5β1 in plaque macrophages alters macrophage migration, phagocytosis, and gene expression, which could certainly contribute to the decrease in atherosclerosis observed with systemic α5 inhibition 15–17. Therefore, we sought to assess endothelial α5 integrin signaling in atherogenic endothelial activation using endothelial culture and endothelial-specific knockout model systems.
Methods:
The authors declare that all supporting data are available within the article and its online supplementary files.
Endothelial Cell Culture and Transfections–
HAE cells (Lonza) were purchased at passage 3 (3 different donors) and maintained in MCDB 131 supplemented with 10% fetal bovine serum (FBS), 2mM glutamine, 10 U/mL penicillin (GIBCO), 100 µg/mL streptomycin (GIBCO), 60 µg/mL heparin sodium, and bovine brain extract (25 µg/mL) and were used between passages 6–10. Experiments were performed in MCDB-131 containing 0.5% FBS. HAECs at 75% confluency were transfected with SMARTpool siRNA oligos targeting fibronectin (50 nM) using Lipofectamine 2000 (Life Technologies) for 2.5 hours on two consecutive days and experiments were performed one day later. Mouse aortic endothelial cells (MAECs) were isolated from integrin α5f/fl mice (gift of Richard Hynes, MIT) as previously described. Briefly, aortic rings (3–5mM) were placed on polymerized Matrigel to induce endothelial sprouts. Sprouting cells were isolated, sorted for the endothelial marker CD105 using magnetic beads, and reversibly transformed using a retroviral temperature-sensitive large T-antigen. The α5 gene was deleted following adenoviral infection with GFP-Cre or GFP control viruses and sorting for GFP positive cells. The temperature-sensitive large T antigen allows MAEC expansion at 33°C with IFNγ, whereas moving cells to 37°C in the absence of IFNγ for >3 days abrogates large T antigen expression. EIIIA/EIIIB MAECS was isolated as previously described 18, 19. Briefly, aortic endothelial cells were derived from C57 background FN-EIIIAB−/− and FN-EIIIAB+/− littermates. The aorta was isolated from heparinized mice after perfusion via the left ventricle with PBS. Each aortic section was cleaned, filled with collagenase solution and tied at the ends before digestion in 20% fetal bovine serum DMEM. Crude endothelial cell isolate was flushed out into dishes coated with collagen I and grown in endothelial cell culture media 20% fetal bovine serum, 0.1mg/mL Endothelial cell growth supplement from bovine hypothalamus (Sigma) and 0.1mg/mL heparin with primocin antibiotic in DMEM. After 5–7 days, ~20K cells were sorted from each line by markers FITC-acetylated LDL+, Icam2+ and Cd31+, and then immortalized by TetOn-SV40. Cells were subsequently expanded in 1–2ug/mL doxycycline.
Immunoblotting and Immunocytochemistry–
Cells were lysed by addition of 2X Laemmli buffer. Lysates were separated by SDS-PAGE gels and were transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA), and membranes were blocked in 5% nonfat dry milk before addition of primary antibodies. Antibodies used included rabbit anti-fibronectin (Sigma), rabbit anti-P-Smad2/3, rabbit anti-β-Tubulin, rabbit anti-GAPDH, rabbit anti-P-P65 (Ser 536), rabbit anti-P65 (Cell Signaling), rabbit anti-integrin α5, rabbit anti-integrin β1, rabbit anti-integrin β3, mouse anti-EDA-FN, mouse anti-GST (Santa Cruz), and rabbit anti-VCAM-1 (abcam) as described in the Supplemental Major Resources Table. Densitometry was performed using ImageJ software. Cells were fixed in formaldehyde, permeabilized, and stained. Briefly, cells were fixed for 20 minutes with 4% formaldehyde permeabilized for 10 minutes in 0.1% Triton X100. Cells were rinsed and blocked with 10% animal serum for at least one hour. Cells were then stained with primary antibodies for ~16–18 hours followed by addition of fluorochrome-tagged secondary antibodies (Life Technologies). Cells were rinsed and either counterstained for DAPI or 546-conjugated phalloidin. Stains were visualized on a Nikon Eclipse Ti inverted epifluorescence microscope equipped with a Photometrics CoolSNAP120 ES2 camera and the NIS Elements 3.00, SP5 imaging software. Cells were scored for dots per cell and at least 100 cells were counted per condition for each experiment.
Animals and Tissue Harvest-
The Louisiana State University Health Sciences Center-Shreveport Animal Care and Use Committee approved all animal protocols, and all animals were cared for according to the NIH Guide for the Care and Use of Laboratory Animals. ApoE−/− mice on the C57Bl/6J genetic background were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice that contained the α5fl/fl and αvfl/fl allele (a gift from Dr. Richard Hynes, MIT, Cambridge, MA) and mice that contained the vascular endothelial (VE)-cadherin CreERT2 transgene (a gift of Dr. Luisa Iruela-Arispe, UCLA, Los Angeles, CA), both on the C57Bl/6J background, were crossed with ApoE−/− mice. Inducible and endothelial cell-specific (iEC) α5 knockout (KO) mice (ApoE−/−, VE-cadherin CreERT2tg/?, α5fl/fl), iEC-αv KO mice (ApoE−/−, VE-cadherin CreERT2tg/?, αvfl/fl), iEC-α5/αv double KO mice (ApoE−/−, VE-cadherin CreERT2tg/?, α5fl/fl, αvfl/fl) and iEC-Control (ApoE−/−, VE-cadherin CreERT2tg/?) mice were treated with 1 mg/kg tamoxifen (Sigma-Aldrich, St. Louis, MO) via intraperitoneal injection every other day for five total injections to induce Cre expression and gene excision. Previous studies looking at treatment with α5 integrin inhibitors, global α5 transgenics, global α5 knockouts, and fibronectin knockouts all utilized male mice. Since this study sought to specifically assess the role of endothelial α5 in this regard, we utilized only males to be consistent with the previous literature 20. Eight to 10 week old mice were fed a high-fat, Western diet (TD 88137; Harlan-Teklad, Madison, WI) that contained 21% fat by weight (0.15% cholesterol and 19.5% casein without sodium cholate) for 2 or 8 weeks. Mice were then euthanized by pneumothorax under isoflurane anesthesia, and blood was collected. Total cholesterol, high-density lipoprotein cholesterol (Wako Bioproducts, Richmond, VA), and triglycerides (Pointe Scientific, Canton MI) were analyzed with commercially available kits. LDL cholesterol was calculated with the Friedewald equation. Hearts were then perfused with phosphate-buffered saline to remove residual blood from the circulation. The lungs were collected for enzymatic digestion and endothelial cell isolation by using magnetic beads coupled to ICAM2 antibodies (eBiosource). The left common carotid was collected and RNA isolation was performed by a TRIzol flush as previously described. Briefly, carotids were cleaned of perivascular adipose tissue and flushed with 150 mL TRIzol from an insulin syringe. The remaining media/adventitia were then placed in 150 mL TRIzol and sonicated to lyse the tissue. Samples were then frozen until analysis by qPCR. The aortic root, aorta, and carotid sinus were excised, placed in 4% phosphate-buffered saline buffered formaldehyde, and analyzed for plaque size and composition or immunostained. Quantification of plaque size was performed in concordance with the American Heart Association Scientific Statement on the design, reporting, and execution of animal atherosclerotic studies 21. Plaque size in the aorta, from the cusp to the renal arteries, was determined by Oil Red O staining and en face imaging, and quantification of plaque size was determined both for the entire aorta and for the atherosclerosis-prone aortic arch. Plaque size in the aortic root, innominate artery, and carotid sinus was quantified in multiple cross sections within each plaque-prone region as area inside the internal elastic laminae, as assessed by Movat Pentachrome staining.
LDL oxidation–
LDL (Intracel) was oxidized by dialysis in 1X PBS containing 13.8 µM Cu2SO4 for 3 days followed with 50 µM EDTA overnight and then for 4 hours the following day. This consistently displayed a relative electrophoretic mobility between 2 and 3, indicative of highly oxidized LDL. Oxidized LDL was stored under nitrogen gas and tested for endotoxin contamination using a chromogenic endotoxin quantification kit (Thermo Scientific).
Focal Adhesion Isolations-
Cells were plated on diluted Matrigel (includes 60% laminin, 30% collagen IV, 8% enactin, and low levels (pg/ml range) of growth factors) coated glass slides in low serum overnight. After treatments, cells underwent hypotonic shock using triethanolamine (2.5mM at pH 7.0) for 3 minutes. Cell bodies were subsequently removed by pulsed hydrodynamic force (Conair WaterPIK) at ~0.5cms from and ~90° to the surface of the slide scanning the entire length 3 times. Focal adhesions remaining bound to the slide were lysed in 2X Laemmli buffer and separated on SDS-PAGE gels.
Insoluble and Soluble Protein Isolation using DOC, by Immunocytochemistry or Western Blotting-
Cells were washed once in ice-cold 1X PBS then rinsed twice in Wash buffer 1 (3% Triton X-100 in 1XPBS) for 10 minutes each at mild agitation rates. Cells were then rinsed twice in Wash Buffer 2 (2% sodium deoxycholate, 50mM Tris-HCl, and pH 8.9) for 10 minutes each at mild agitation rates. Cells were then rinsed twice in 1X PBS for 10 minutes each at mild agitation rates. Cells were then fixed with 4% formaldehyde for 20 minutes followed by blocking with 10% animal serum. Cells were then immunostained as described elsewhere for fibronectin. Alternatively, this protocol can be adapted for Western blotting. Cells were washed in ice-cold 1X PBS then 1 mL of deoxycholate containing buffer (2% sodium deoxycholate, 20mM Tris-HCl at pH 8.8, 2mM PMSF, 2mM iodoacetic acid, and 2mM N-ethylmaleimide) was added for 10 minutes. Cells were scraped and collected in microcentrifuge tubes followed by passing lysates through a 25 gauge needle 5 times. Lysates were centrifuged at 15,000 RPMs for 15 minutes. Supernatant was collected as the soluble fraction. The remaining pellet was rinsed with DOC buffer and spun again. Buffer was removed and the pellet lysed in 2X Laemmli buffer. Lysate were separated on SDS-PAGE gels.
In vitro Permeability Assay and Shear Stress-
HAE cells were transfected with either 150nM a5 (SMARTPool siRNA; Dharmacon) or Mock control using Lipofectamine 3000 (Invitrogen). After 3h, the transfection reagent was removed and the cells were transfected again on the second day. Cells were used for the permeability assay after 12h of the second transfection. Endothelial cell permeability was assessed in a5 siRNA treated cells or controls as previously described 22. Briefly, cells (1×106) were plated on biotinylated gelatin coated slides (Corning) to confluence and the slides then assembled into a flow chamber to be subjected to disturbed flow as previously described23. In brief, oscillatory flow is generated using infusion withdrawal pump (±5dynes/cm2, 1Hz) with 1 dyne/cm2 forward flow superimposed by a peristaltic pump. After the cessation of flow, Streptavidin-Alex 647 (1:1000 in PBS, Invitrogen) was immediately added to the cells for 1min, then fixed in 4% formaldehyde. F-actin arrangement in static and shear exposed cells was visualized using 488-Alexa phalloidin (Invitrogen) according to manufacturer’s recommendation. Images were analyzed using NIS Elements software.
FACS Analysis-
Cells were removed from the surface using Accutase (Millipore). They were then blocked in 1% denatured albumin for 30 minutes. Cells were spun and incubated 1 × 106 cells/ml with FITC-labelled integrin α5 (Abcam) for 30 minutes and baseline was established with no antibody and IgG isotype control.
Quantitative PCR-
mRNA isolated from tissues and cultured endothelial cells was extracted with TRIzol (Life Technologies, Inc., Carlsbad, CA). Complimentary DNA was synthesized with the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative real-time PCR (qPCR) was performed with a Bio-Rad iCycler with the use of SYBR Green Master mix (Bio-Rad). Primers were designed with the online Primer3 software and then validated by sequencing the PCR products. Results were expressed as fold change by using the 2ΔΔCT method.
Statistical analysis-
Statistical comparisons between groups were performed using GraphPad Prism software. Data was tested for Normality (Kolmogorov-Smirnov test) and data that passed the Normality assumption was analyzed using Student’s T-test, one-way ANOVA with Newman-Keuls post-test or two-way ANOVA with Bonferroni post-tests. Data that failed the Normality assumption were analyzed by using the non-parametric Mann-Whitney U test and the Kruskal-Wallis test with post-hoc analysis. Error bars indicate standard error.
RESULTS
Integrin α5 mediates oxLDL-induced fibronectin deposition
Since oxLDL activates endothelial α5β1 integrins 4, we tested whether oxLDL could also elicit fibronectin deposition in human aortic endothelial cells (HAECs). Cells plated on basement membrane proteins for approximately 28 hours were treated with oxLDL for the final 6 or 24 hours to stimulate α5β1 integrins, and fibronectin deposition into the insoluble matrix fraction was assessed following extraction of the deoxycholate (DOC)-soluble cell-associated fibronectin. Immunocytochemistry and western blot analysis for fibronectin reveal that oxLDL treatment causes rapid and robust fibronectin deposition at 6 and 24 hours after treatment (Fig. 1 A and B). Fibronectin deposition by oxLDL is apparent at concentrations as low as 20 μg/ml and is maximal between 50–100 μg/ml (Fig. SIa). This induction of fibronectin deposition was similar to the levels observed in response to the classic pro-fibrotic growth factor transforming growth factor β (TGFβ) (Fig. SIb), suggesting that oxLDL is a potent regulator of fibronectin deposition. To assess the specific role of α5β1 in oxLDL-induced fibronectin deposition, we assessed oxLDL-induced matrix remodeling in immortalized mouse aortic endothelial cells (MAECs) lacking α5. While α5 wildtype (α5 WT, α5fl/fl expressing GFP) MAECs show significant fibronectin deposition upon oxLDL treatment, α5 knockout (α5 KO, α5fl/fl expressing GFP-Cre) MAECs show significantly reduced fibronectin deposition associated with diminished fibronectin levels and reduced fibronectin fibril length (Figure 1C–E). Furthermore, we demonstrate that two distinct α5 blocking antibodies (P1D6, SNAKA52) completely prevents oxLDL-induced fibronectin deposition in HAECs (Fig. 1 F).
Figure 1. α5β1 mediates oxLDL-induced fibronectin deposition.
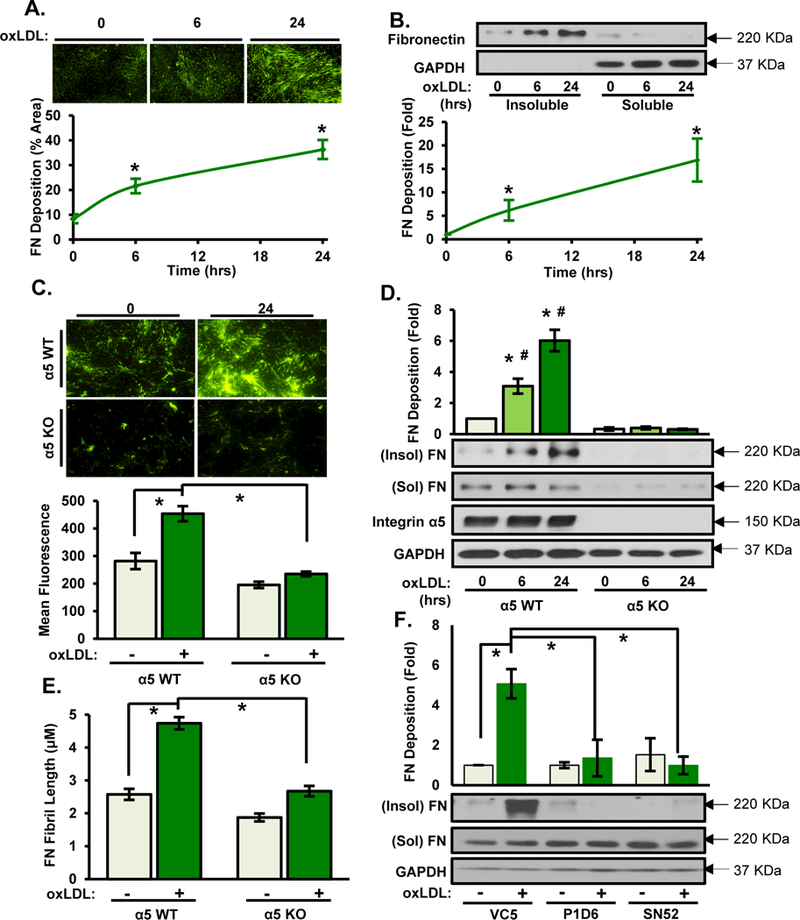
A/B) Human aortic endothelial cells (HAECs) were treated with oxLDL (100 μg/ml) for indicated times. Deoxycholate (DOC)-soluble and insoluble fractions were collected and assessed by (A) immunostaining for fibronectin or (B) Western blotting. Representative Western blots and images are shown (n=4). C-E) Conditionally immortalized mouse aortic endothelial cells (MAECs) either wildtype (WT) or knockout (KO) for α5 integrin were treated with oxLDL for 6 or 24 hrs and fibronectin deposition assessed as described in A/B. Representative Western blots and images are shown (n=4). (E) Length of fibronectin fibrils in (C) was assessed (n=4). F) HAECs were treated with either α5 non-blocking (VC5) or α5 function-blocking antibodies (P1D6, SNAKA52 (SN52)) at 10 μg/mL for 1hr and then treated with oxLDL. DOC-insoluble fibronectin deposition was assessed. Representative Western blots are shown (n=4). Values are means±SE. * p<0.05 compared with no treatment condition. # p<0.05 compared with α5 KO cells.
Previous studies demonstrated that atheroprone hemodynamics contributes to altered fibronectin deposition through enhanced TGFβ signaling, resulting in an endothelial-to-mesenchymal (EndMT) transition that promotes fibronectin deposition 6. Since oxLDL and TGFβ show similar patterns of fibronectin deposition (Fig. SIb), we next tested if oxLDL-induced TGFβ signaling and EndMT to mediate fibronectin deposition. However, oxLDL failed to activate the classic TGFβ-dependent transcription factors Smad2/3, as assessed by Smad2/3 phosphorylation (Fig. SIc–d). However, TGFβ-induced fibronectin deposition was similarly reduced in α5 KO MAECs (Fig. SIe), consistent with a critical role for α5 integrins in endothelial fibronectin deposition. Additionally, oxLDL failed to induce the expression of classic EndMT marker genes, such as vimentin, smooth muscle actin (SMA), SM22, Myh11, leiomodin, and Notch3 (Fig. SIIa), and oxLDL did not increase endothelial SMA staining (Fig. SIIb). Finally, qRT-PCR analysis indicates that oxLDL does not increase fibronectin expression (Fig. SIIc), suggesting that oxLDL drives fibronectin deposition through activation of the fibronectin deposition machinery rather than through induction of fibronectin expression.
Endothelial-specific α5 deletion abrogates atherogenic inflammation and early atherosclerosis
To assess the role of endothelial α5 integrins in early atherogenic inflammation, ApoE knockout mice that express the tamoxifen-inducible, endothelial cell-specific Cre transgene (iEC-control) were crossed with mice containing a floxed α5 integrin allele (iEC-α5 KO) to allow for tamoxifen-dependent α5 deletion in endothelial cells (Fig. 2 A). We confirmed the endothelial-specific deletion of α5 by analyzing mRNA isolated from the common carotid intima and media/adventitia (Fig. 2 B) and by Western blotting analysis of lung endothelial cells isolated from iEC-Control and iEC-α5 KO mice (Fig. 2 C). iEC-α5 KO mice show significant reduction of VCAM-1 expression in the atheroprone aortic arch after 2 weeks high fat diet feeding, suggesting atherogenic inflammation in reduced in these mice (Fig. 2 D). Paradoxically, iEC-α5 KO mice did show enhanced endothelial fibronectin staining in atheroprone regions, and to a lesser extent the atheroprotective regions, of the aortic arch (Fig. 2E). Validity of these staining patterns were verified using controls for non-immune IgG and secondary antibody alone (Fig. SIII). Previous studies showed that deleting fibronectin from endothelial cells similarly enhanced endothelial fibronectin staining 5, suggesting that preventing fibronectin deposition at atheroprone sites may enhance vascular permeability. Consistent with this model, deposition of fibrinogen, a plasma protein not expressed in endothelial cells, was similarly enhanced in iEC-α5 KO mice (Fig. SIVa), and α5 KO MAECs showed enhanced permeability in cell culture models (Fig. SIVb). While αvβ3 has been proposed to promote fibronectin deposition in the absence of α5β1, deletion of endothelial αv alone did not affect endothelial fibronectin staining, and deletion of both α5 and αv in the endothelial layer showed increased fibronectin deposition similar to the α5 endothelial KO alone (Fig. SV). Therefore, the enhanced fibronectin staining in the endothelial α5 KO is not likely due to compensation by another fibronectin-binding integrin.
Figure 2. Endothelial-specific deletion of α5 integrins abrogates atherogenic endothelial activation.
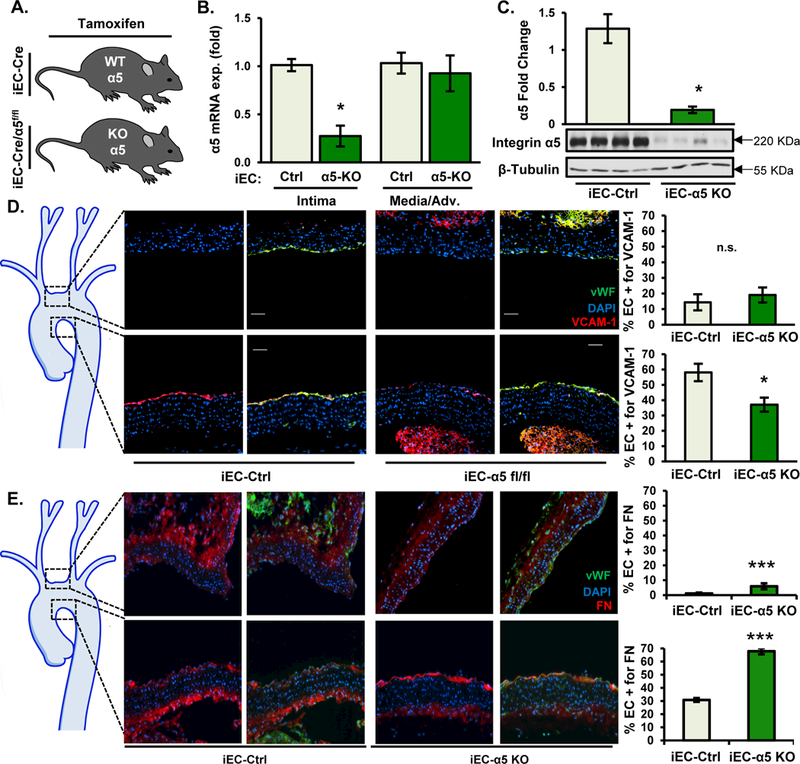
A) iEC-Ctrl (VE-Cadherin-CreERT2tg/?, ApoE−/−) and iEC-α5 KO (α5flox/flox,VE-Cadherin-CreERT2tg/?, ApoE−/−) mice were treated with tamoxifen (1 mg in peanut oil for 5 consecutive days) and fed a Western Diet for 2 weeks. B) α5 expression in the carotid intima and media was assessed by qRT-PCR following the TRIZol flush method. n=6. C) Lung endothelial cells were isolated from post-tamoxifen-treated mice and α5 expression was assessed by immunoblotting. n=4. D/E) 2 weeks after Western Diet feeding, mice were euthanized, and transverse sections of the aortic arch were assessed by immunohistochemistry for (D) VCAM-1 (red) and (E) Fibronectin (red). Endothelial cells (EC) were identified by staining for CD31 (green), and DAPI (blue) was used to show nuclear staining. Representative images are shown (n=6). Images were taken at ×20. Analysis of EC positivity for VCAM-1 or fibronectin staining in the upper curvature and lesser curvature of the aortic arch was performed using NIS elements software. n=6 mice per group. Values are means±SE. *p<0.05 and ***p<0.001 compared with iEC-Control.
After 8 weeks high fat diet, iEC-α5 KO mice show no differences in plasma lipid levels (Fig. SVI) but show significantly reduced atherosclerotic plaque area as assessed by oil red O staining of the aorta (Fig. 3A) and by cross-sectional analysis of the aortic root (Fig. 3B), innominate artery (Fig. 3C), and carotid sinus (Fig. 3D) 21. Consistent with a role in endothelial activation, loss of endothelial α5 expression reduces macrophage accumulation in both the aortic root and innominate arteries (Fig. 3 E and F). Previous studies show that loss of plasma fibronectin reduces smooth muscle accumulation and fibrous cap size 2. Since loss of α5 reduces fibronectin assembly, we examined total smooth muscle cell area and collagen content in the more advanced plaques of the aortic root. Our results show that endothelial α5 deletion does not negatively impact smooth muscle content (Fig. 3 G) or collagen content in these plaques (Fig. 3 H).
Figure 3. Abrogation of early atherosclerosis in α5 endothelial specific knockout without compromising smooth muscle recruitment.
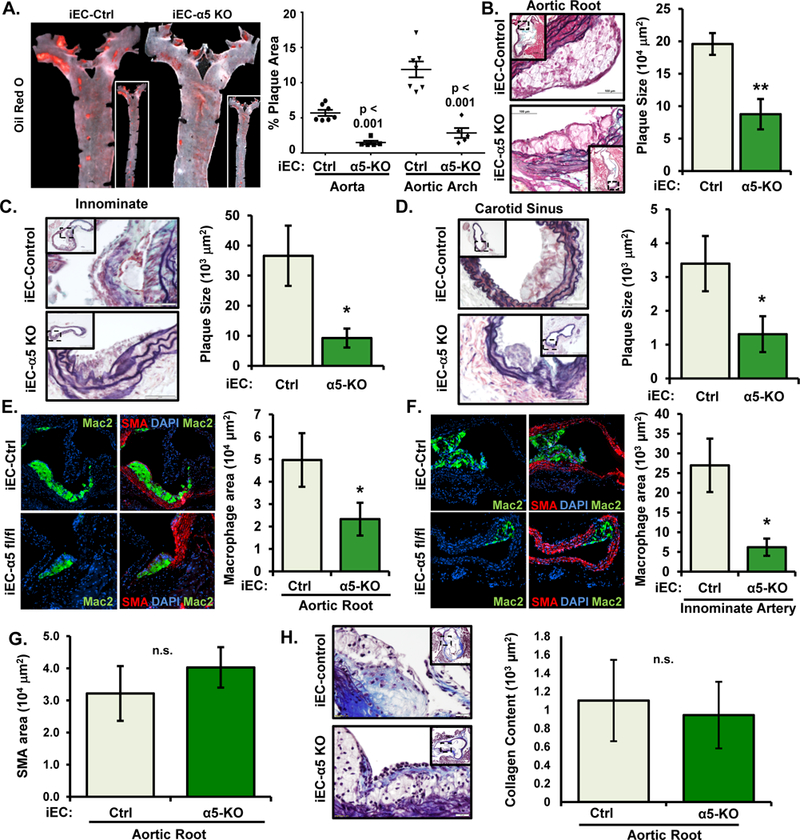
A-D) After 8 weeks Western Diet, atherosclerosis in iEC-Ctrl and iEC-α5 KO mice was assessed by (A) Oil Red O staining in the aorta and by (B-D) Movat Pentachrome staining in the (B) aortic root, (C) innominate artery, and (D) carotid sinus. Representative images are shown (n=5–7). E/F) Macrophage area (Mac2-positive, green) and smooth muscle area (smooth muscle actin (SMA)-positive, red) was determined by immunohistochemistry in the (E) aortic root and (F) innominate artery. Representative images are shown (n=5–7). G/H) No changes were observed in (G) smooth muscle (SMA-positive) area or (H) collagen content of the aortic root. n=6–7. * p<0.05 and ** p<0.01 compared with iEC-Control. n.s.=not significant.
Cell-derived, not plasma-derived, fibronectin mediates VCAM-1 expression induced by oxLDL
We and others have shown that endothelial cell interactions with fibronectin tune the response to atherogenic mediators to increase proinflammatory gene expression 24. While α5 may be important for some of these responses, several groups have shown an important role for other fibronectin-binding integrins, such as αvβ3, in endothelial activation 25, 26. Therefore, the loss of endothelial VCAM-1 staining in iEC-α5 KO knockout mice despite the presence of fibronectin in the endothelial cell layer suggests that cell-derived and plasma-derived fibronectin have different roles in endothelial activation. To assess this, we depleted fibronectin from HAECs (siRNA) and from the cell culture media (fibronectin-depleted serum, Fig. SVII) and assessed oxLDL-induced VCAM-1 expression. While fibronectin depletion blunted both baseline and oxLDL-induced VCAM-1 expression (Fig. 4 A), addition of plasma fibronectin was unable to rescue this phenotype. In contrast, addition of cell-derived fibronectin isolated from human foreskin fibroblasts successfully restored oxLDL-induced VCAM-1 expression (Fig. 4 B). Since the primary difference between plasma-derived and cell-derived fibronectin is the inclusion of the additional fibronectin type III repeats EIIIA and EIIIB in cell-derived fibronectin, we assessed the role of EIIIA and EIIIB in oxLDL-induced inflammation using MAECs isolated from EIIIA/EIIIB knockout mice. Consistent with a critical role for cell-derived fibronectin in this response, oxLDL induced VCAM-1 expression in EIIIA/EIIIB wildtype (EIIIA/B WT) but not EIIIA/EIIIB knockout (EIIIA/B KO) MAECs (Fig. 4 C). Consistent with reduced VCAM-1 expression, oxLDL- induced NF-κB activation was significantly suppressed in HAECs depleted of cellular fibronectin (fibronectin siRNA) and in EIIIA/EIIIB knockout MAECs (Fig. 4D–E). Taken together, these results suggest that oxLDL-induced NF-κB activation and VCAM-1 expression are mediated by endothelial cell-derived fibronectin.
Figure 4. EIIIA/EIIIB-containing fibronectin mediates oxLDL-mediated proinflammatory gene expression.
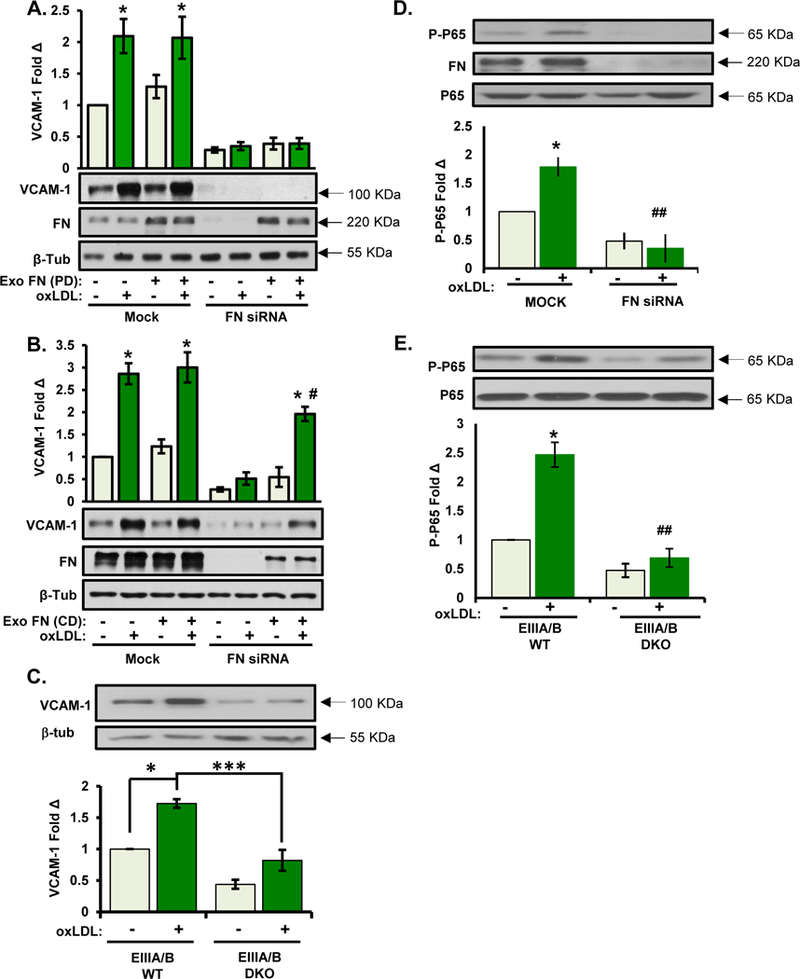
A/B) HAECs were transfected with FN siRNA and treated with oxLDL for 6 hours in the presence of either (A) plasma fibronectin or (B) cell-derived fibronectin (from human foreskin fibroblasts). Cells were lysed and analyzed for VCAM-1 expression by Western blotting. Representative Western blots are shown (n=4). C) MAECs isolated from EIIIA/EIIIB WT and EIIIA/EIIIB DKO mice were treated with oxLDL for 6 hrs, and VCAM-1 expression was assessed by Western blotting. Representative Western blots are shown (n=4). D) oxLDL-induced NF-κB activation was assessed in (D) HAECs transfected with FN siRNA or in (E) EIIIA/EIIIB WT and EIIIA/EIIIB KO MAECs. NF-κB activation was assessed by measuring phosphorylation (Ser536) of the p65 subunit. Representative Western blots are shown (n=3–4). Values are means ± SE. *p<0.05 and ***p<0.001 compared with no treatment condition. # p<0.05, ## p<0.01 compared with FN siRNA on BM or EIIIA/B DKO.
Ablation of cell-derived fibronectin limits oxLDL-induced fibronectin fibrillogenesis
The requirement for cell-derived fibronectin could be due to specific EIIIA/EIIIB receptors, alterations in the cell’s ability to deposit plasma fibronectin, or changes in the presentation of the integrin-binding sites by EIIIA/EIIIB inclusion. Since cell-derived fibronectin does not stimulate VCAM-1 expression in the absence of oxLDL (Fig. 4 B), the direct activation of EIIIA/EIIIB receptors is unlikely. To evaluate the role of cell-derived fibronectin in oxLDL-induced fibronectin assembly, HAECs plated on basement membrane proteins were stimulated with oxLDL and deposition of plasma fibronectin (488-labeled plasma fibronectin added to cell culture medium) and cell-derived fibronectin (positive for the alternatively spliced EIIIA site) were assessed by confocal microscopy. We observed that oxLDL treatment enhances both plasma-derived and cell-derived fibronectin staining in the subendothelial matrix (Fig. 5 A). To assess whether cell-derived fibronectin regulates plasma-fibronectin assembly, we measured deposition of plasma-derived fibronectin in HAECs treated with fibronectin siRNA to specifically deplete cell-derived fibronectin. While plasma fibronectin staining following oxLDL treatment didn’t change with endothelial fibronectin knockdown (Fig. 5 B and C), the lack of cell-derived fibronectin significantly limited the size of the fibronectin fibrils that formed (Fig. 5 D). Moreover, EIIIA/EIIIB KO MAECs, which still express fibronectin lacking these alternative domains, showed a similar abrogation of fibronectin assembly in response to oxLDL (Fig. 5 E), suggesting that it is the inclusion of the EIIIA/EIIIB domains rather than retained ability to express fibronectin that regulates endothelial fibronectin deposition.
Figure 5. Ablation of cell-derived fibronectin limits oxLDL-induced fibronectin fibrillogenesis.
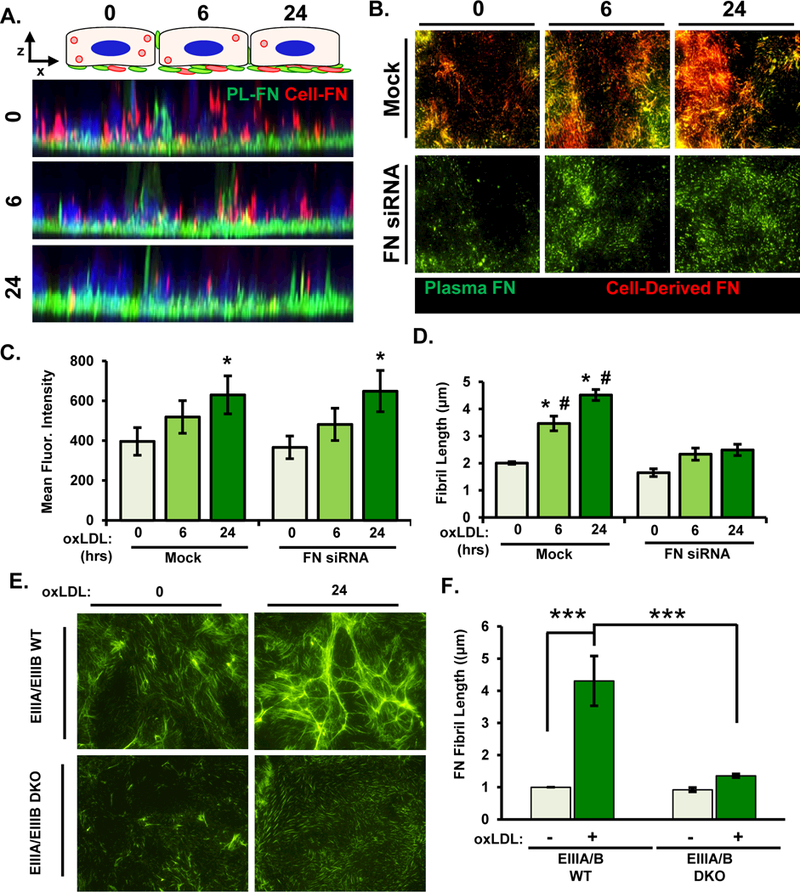
A) Alexa488-labeled plasma fibronectin was added to HAECs in culture media overnight, and cells were treated with oxLDL for 6 or 24 hours. Z-stack projections of Alexa488-labeled plasma fibronectin (green) and EIIIA-FN (546nM, red) were assessed by confocal microscopy. Representative images are shown (n=3). B-D) HAECs were transfected with FN siRNA and were treated with oxLDL and labelled fibronectin as described in (A). After immunostaining for EIIIA-FN, fibers were visualized by epifluorescence microscopy and quantified for (C) mean fluorescent intensity of plasma fibronectin and (D) fibril length. Representative images are shown (n=4). E/F) EIIIA/EIIIB WT and EIIIA/EIIIB KO MAECs were treated with oxLDL for 24 hrs and fibronectin fibril length was assessed by immunocytochemistry. Representative images are shown (n=4). Values are means±SE. * p< 0.05 compared to 0 hr timepoint. # p<0.05 compared to FN siRNA.
Previous studies indicate that loss of cell-derived fibronectin results in decreased staining of α5 integrins in focal adhesions, potentially accounting for decreased fibronectin assembly after oxLDL treatment and decreased proinflammatory gene expression 27. Therefore, we examined if fibronectin deletion in HAECs altered α5β1 recruitment to focal adhesions. HAECs were treated with oxLDL for 6 hours and the focal adhesion fraction was isolated. Our results demonstrate that fibronectin knockdown decreases α5β1 levels within focal adhesions (Fig. 6A and B), whereas α5 surface levels remain unchanged (Fig. SVIII). Similarly, oxLDL enhanced α5β1 adhesions in HAECs as assessed by immunocytochemistry for α5 and active β1 integrins (12G10 antibody, gift of Dr. Dr. Martin Humphries, Univ. of Manchester, Manchester, England), and depletion of cell-derived fibronectin significantly reduced α5 and β1 integrin staining at focal/fibrillar adhesions (Fig. 6 C). Utilizing oxLDL- treated EIIIA/EIIIB DKO MAECs, we demonstrated that preventing EIIIA/EIIIB inclusion in cell-derived fibronectin blunts oxLDL-induced α5 focal adhesion formation as assessed by western blotting (Fig. 7 A and B) and immunostaining (Fig. 7 C). Consistent with this, rescuing fibronectin-depleted HAECs with cell-derived fibronectin, but not plasma fibronectin, supports oxLDL-induced α5 focal adhesion formation (Fig. 7 D). Together, these data suggest that cell-derived fibronectin regulates α5β1 adhesion formation to promote fibronectin assembly and inflammation in response to oxLDL.
Figure 6. Deletion of fibronectin blunts incorporation α5 in focal adhesions in response to oxLDL.
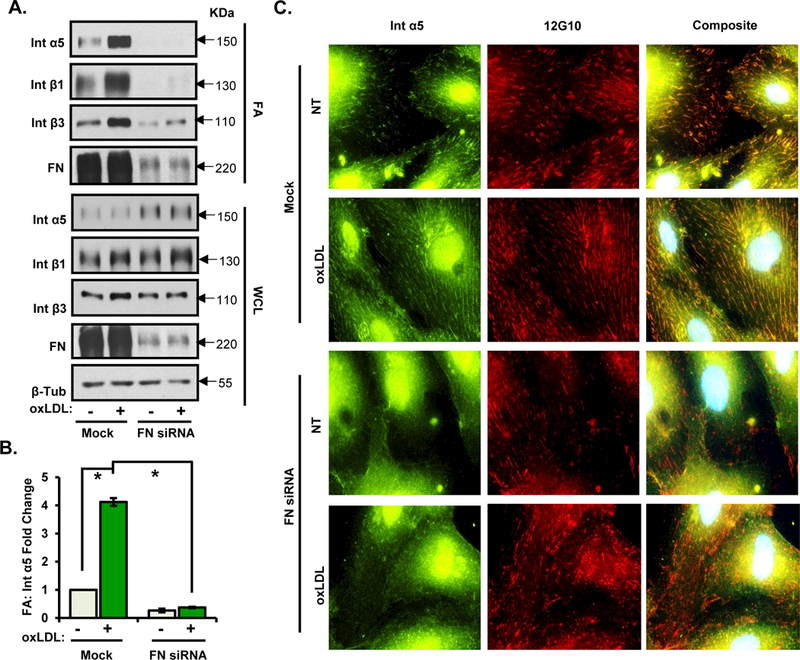
A/B) HAECs were transfected with FN siRNA, treated with oxLDL for 6 hrs, and subjected to focal adhesion isolation (by hydrodynamic force). The isolated focal adhesion fraction and whole cell lysate were analyzed by Western blotting. Representative Western blots are shown (n=3). C) HAECs were transfected with FN siRNA and treated with oxLDL for 6 hours. Cells were fixed and immunostained for α5 and 12G10. Representative images are shown (n=3). Values are means ±SE. *p<0.05 compared with no treatment condition.
Figure 7. EIIIA/EIIIB inclusions promote oxLDL-induced α5 localization in focal adhesions.
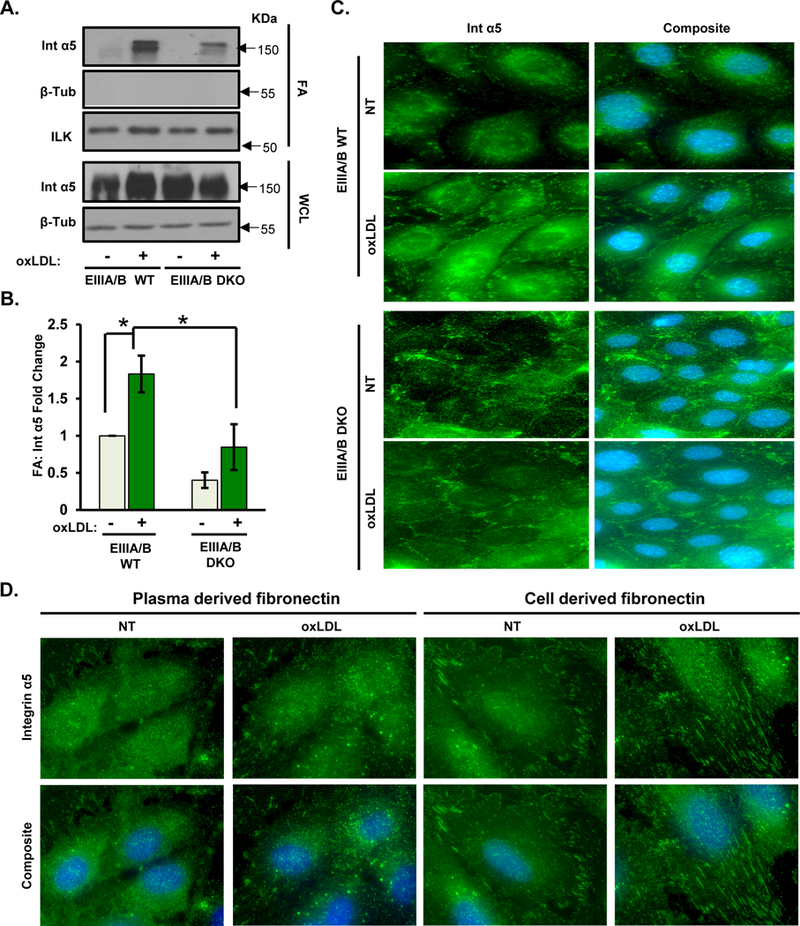
A/B) MAECs isolated from EIIIA/EIIIB WT and EIIIA/EIIIB KO mice were treated with oxLDL for 6 hrs. Focal adhesion fractions and whole cell lysates were analyzed by Western blotting. Representative Western blots are shown (n=4). C) EIIIA/EIIIB WT and EIIIA/EIIIB DKO cells were treated with oxLDL for 6 hrs, followed by fixation and immunostaining for α5. Representative images are shown (n=4). D) HAECs were transfected with FN siRNA, plated on either cell-derived or plasma fibronectin, treated with oxLDL for 6 hrs, and analyzed by immunostaining for α5. Representative images are shown (n=4). Values are means±SE. *P<0.05 compared with no treatment condition.
DISCUSSION
While fibronectin deposition contributes to endothelial activation in early atherogenesis, the mechanisms regulating fibronectin deposition remain poorly characterized. Although previous studies have shown that disturbed flow patterns regulate endothelial fibronectin expression, fibronectin is absent in regions of disturbed flow in the absence of other stimuli, such as hypercholesterolemia or hyperglycemia 1, 2, 9. We previously demonstrated that oxLDL drives activation of the fibronectin-binding integrin α5β1, suggesting that oxLDL may promote early fibronectin deposition in atherosclerosis 4. Here, we show that oxLDL drives a robust increase in endothelial fibronectin deposition and define a critical role for α5β1 integrins in this response. OxLDL-induced α5β1 signaling on fibronectin has also been shown to increase endothelial NF-κB activation, VCAM-1 expression, and monocyte adhesion. Our data demonstrate that endothelial-specific deletion of α5 integrins blunts VCAM-1 expression during early atherogenesis and limits atherosclerotic plaque formation, characterized by reduced macrophage accumulation in plaque. However, this decrease in inflammation did not correlate with reduced fibronectin content in the vessels, presumably due to enhanced leak of plasma fibronectin into the vessel wall consistent with previous reports 5, 27. We further show that only cell-derived fibronectin mediates oxLDL-induced proinflammatory responses, whereas plasma-derived fibronectin lacks these proinflammatory properties. We further narrowed the proinflammatory properties of cell-derived fibronectin to the presence of the EIIIA/EIIIB alternatively spliced domains and showed that the presence of EIIIA/EIIIB domains was required for efficient fibronectin fibrillogenesis in response to oxLDL. Loss of EIIIA/EIIIB-containing fibronectin prevented α5β1 recruitment to focal and fibrillary adhesions, consistent with a role for these domains in the presentation of fibronectin integrin-binding sites 28–31.
While fibronectin deposition is seen only in regions of turbulent blood flow, these sites are largely devoid of fibronectin in the absence of atherogenic risk factors, such as hypercholesterolemia or hyperglycemia 1, 9. Furthermore, laminar flow significantly inhibits fibronectin deposition, whereas fibronectin deposition is only mildly enhanced by disturbed flow patterns compared to static culture conditions 8, 9. While disturbed flow patterns have been shown to promote fibronectin expression through NF-κB, β-catenin, and TGFβ-induced Smad2 signaling 6–8, treatment with oxLDL failed to induce fibronectin expression at either the mRNA or protein level (Fig. SIIC, Fig. 4). Similarly, oxLDL did not induce the EndMT transition previously associated with fibronectin deposition at atherosclerosis-prone sites 6. Rather, treatment with oxLDL drives α5β1 activation, fibrillary adhesion formation, and α5β1-dependent fibronectin deposition in the absence of altered fibronectin expression. Endothelial cells express multiple fibronectin-binding integrins (α5β1, αvβ3, α5β1), and embryonic endothelial-specific deletion of α5 integrins does not compromise developmental angiogenesis. However, knockout of both α5 and αv integrins results in extensive defects in remodeling of the vessels and heart, suggesting that the fibronectin-binding integrins are functionally redundant in development 32. While endothelial-specific deletion of α5 integrins paradoxically enhances endothelial fibronectin staining (Fig. 2E), this effect may be due to enhanced leak of plasma fibronectin into the vessel at these sites, since the plasma protein fibrinogen shows a similar staining pattern and endothelial knockdown of α5 enhances permeability in culture (Fig. SIV). Interestingly, our data show that only endothelial deletion of α5 integrins enhances fibronectin staining, whereas αv knockout did not show the same pattern (Fig. SV), suggesting that the enhanced fibronectin staining following endothelial α5 deletion is not due to compensation by other fibronectin-binding integrins.
Fibronectin has been ascribed multiple roles in the pathogenesis of atherosclerotic plaque formation. In the subendothelial matrix, fibronectin regulates proinflammatory endothelial activation in response to shear stress and oxLDL, and inhibiting fibronectin deposition or endothelial fibronectin-binding integrins (α5β1, αvβ3) prevents atherogenic endothelial activation 2–4, 14, 25. Our data suggests that α5β1 may play multiple roles in fibronectin-associated inflammation, as it is implicated in both fibronectin-dependent proinflammatory signaling 4, 13, 14 and oxLDL-dependent fibronectin deposition. Although inhibiting fibronectin-binding integrins reduces atherogenic inflammation, mice deficient in plasma fibronectin and mice globally deficient in EIIIA inclusion both show reduced smooth muscle incorporation into the atherosclerotic plaque 2, 33, suggesting an important role for fibronectin in plaque stability. However, only αvβ3 inhibitors, and not α5β1 inhibitors, showed a similar reduction in plaque smooth muscle content 4, 25, and neither αv nor α5 deletion in endothelial cells altered smooth muscle content. Taken together, these data suggest that inhibiting α5β1 integrins may reduce plaque-associated inflammation without negatively affecting plaque stability.
Fibronectin deposition contributes to immune cell recruitment in various ways. Previous studies, and our data here, demonstrate that fibronectin deposition into the subendothelial matrix increases endothelial cell activation by multiple atherogenic stimuli 1–3, 34. Modified LDL was previously reported to induce apical presentation of fibronectin containing the alternatively spliced connecting segment (CS-1) domain 35. CS-1 provides direct interaction sites for α4β1-bearing leukocytes, conceivably enhancing leukocyte interactions with the blood vessel wall and contributing to atherosclerotic plaque development. However, it was later shown ex vivo that only a minority of monocyte interactions with the endothelium occur through CS-1 fibronectin 36. Moreover, our results examining z-stack projections show both plasma-derived and cell-derived fibronectin only at intercellular, intracellular, or subendothelial spaces. Depletion of endothelial fibronectin (fibronectin knockdown and depletion of serum fibronectin) reduced both baseline and oxLDL-induced VCAM-1 expression (Fig. 4). However, only rescue with cell-derived fibronectin rescued oxLDL-induced VCAM-1 expression. Furthermore, oxLDL failed to induce VCAM-1 in endothelial cells lacking the EIIIA and EIIIB alternatively spliced domains found in cell-derived fibronectin but absent from plasma fibronectin, suggesting a key role for the EIIIA/EIIIB domain in the oxLDL-induced inflammatory response 37, 38.
While EIIIA and EIIIB-containing fibronectin significantly contributes to plaque formation 39–41, the link between these two remain undefined. EIIIA contains additional binding sites for integrins α4β1 and α9β1 42, however these integrins are not expressed in arterial endothelial cells 25. EIIIA has also been shown to bind to toll like receptor 4 (TLR4), and deletion of TLR4 reduces atherosclerosis in mice with constitutive inclusion of EIIIA 39. However, ApoE knockout mice are prone to endotoxemia 43, 44, and these studies ignore the potential anti-inflammatory effect of TLR4 deletion on exacerbated atherosclerosis by other ligands. Furthermore, TLR4 activation is sufficient to induce VCAM-1 expression, and treatment with cell-derived fibronectin was not sufficient to induce VCAM-1 expression in the absence of oxLDL (Fig. 4). Rather, our data support the hypothesis that EIIIA/EIIIB inclusion affects the interaction between fibronectin and its integrin receptors 28–31, since endothelial fibronectin knockdown and endothelial cells lacking the EIIIA/EIIIB domains both show reduced α5β1 incorporation into focal and fibrillar adhesions. This enhancement in α5β1 signaling likely mediates proinflammatory responses to EIIIA/EIIIB in endothelial cells, as oxLDL-induced inflammation requires α5β1-dependent NF-κB activation through a focal adhesion kinase-dependent pathway 4.
Supplementary Material
Highlights:
-
-
Fibronectin enhances atherogenic endothelial activation.
-
-
The atherogenic mediator oxidized LDL induces robust fibronectin deposition through α5β1 integrins in endothelial cells.
-
-
Endothelial-specific α5 deletion limits atherogenic inflammation and early atherosclerosis.
-
-
Cell-derived fibronectin, not plasma fibronectin, shows proinflammatory properties by enhancing α5β1 integrin signaling.
Acknowledgements:
None.
Source of Funding:
This work was supported by National Heart, Lung, and Blood Institute R01 HL098435, HL133497, and GM121307 (to A.W.O.), by an American Heart Association Pre-doctoral Fellowship 14PRE18660003 (to A.Y.J.), and by a Malcolm Feist Cardiovascular Research Endowment Pre-doctoral Fellowship (to Z.A.Y.) and Post-doctoral Fellowship (to. M.A.).
Nonstandard Abbreviations:
- ApoE
apolipoprotein E
- EIIIA
alternative exon EIIIA (EDA) of fibronectin
- EIIIB
alternative exon EIIIB (EDB) of fibronectin
- EndMT
endothelial-to-mesenchymal transition
- FN
fibronectin
- HAEC
human aortic endothelial cell
- ICAM-1
intercellular adhesion molecule-1
- MAEC
mouse aortic endothelial cell
- NF-κB
nuclear factor κ-B
- OxLDL
oxidized low -density lipoprotein
- VCAM-1
vascular cell adhesion molecule-1
Footnotes
Disclosures:
The authors declare no conflicts.
References
- 1.Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates nf-kappab activation by flow: A potential role in atherosclerosis. J Cell Biol 2005;169:191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rohwedder I, Montanez E, Beckmann K, Bengtsson E, Duner P, Nilsson J, Soehnlein O, Fassler R. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol Med 2012;4:564–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J. Fibronectin is an important regulator of flow-induced vascular remodeling. Arterioscler Thromb Vasc Biol 2009;29:1074–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yurdagul A Jr., Green J, Albert P, McInnis MC, Mazar AP, Orr AW. Alpha5beta1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol 2014;34:1362–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy PA, Hynes RO. Alternative splicing of endothelial fibronectin is induced by disturbed hemodynamics and protects against hemorrhage of the vessel wall. Arterioscler Thromb Vasc Biol 2014;34:2042–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 2015;125:4514–4528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feaver RE, Gelfand BD, Wang C, Schwartz MA, Blackman BR. Atheroprone hemodynamics regulate fibronectin deposition to create positive feedback that sustains endothelial inflammation. Circ Res 2010;106:1703–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gelfand BD, Meller J, Pryor AW, Kahn M, Bortz PD, Wamhoff BR, Blackman BR. Hemodynamic activation of beta-catenin and t-cell-specific transcription factor signaling in vascular endothelium regulates fibronectin expression. Arterioscler Thromb Vasc Biol 2011;31:1625–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green J, Yurdagul A Jr., McInnis MC, Albert P, Orr AW. Flow patterns regulate hyperglycemia-induced subendothelial matrix remodeling during early atherogenesis. Atherosclerosis 2014;232:277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev 2013;93:1317–1542 [DOI] [PubMed] [Google Scholar]
- 11.Cai WJ, Li MB, Wu X, Wu S, Zhu W, Chen D, Luo M, Eitenmuller I, Kampmann A, Schaper J, Schaper W. Activation of the integrins alpha 5beta 1 and alpha v beta 3 and focal adhesion kinase (fak) during arteriogenesis. Mol Cell Biochem 2009;322:161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickering JG, Chow LH, Li S, Rogers KA, Rocnik EF, Zhong R, Chan BM. Alpha5beta1 integrin expression and luminal edge fibronectin matrix assembly by smooth muscle cells after arterial injury. Am J Pathol 2000;156:453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun X, Fu Y, Gu M, Zhang L, Li D, Li H, Chien S, Shyy JY, Zhu Y. Activation of integrin alpha5 mediated by flow requires its translocation to membrane lipid rafts in vascular endothelial cells. Proc Natl Acad Sci U S A 2016;113:769–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yun S, Budatha M, Dahlman JE, Coon BG, Cameron RT, Langer R, Anderson DG, Baillie G, Schwartz MA. Interaction between integrin alpha5 and pde4d regulates endothelial inflammatory signalling. Nat Cell Biol 2016;18:1043–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abshire MY, Thomas KS, Owen KA, Bouton AH. Macrophage motility requires distinct alpha5beta1/fak and alpha4beta1/paxillin signaling events. J Leukoc Biol 2011;89:251–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vernon-Wilson EF, Aurade F, Brown SB. Cd31 promotes beta1 integrin-dependent engulfment of apoptotic jurkat t lymphocytes opsonized for phagocytosis by fibronectin. Journal of leukocyte biology 2006;79:1260–1267 [DOI] [PubMed] [Google Scholar]
- 17.Jun HK, Lee SH, Lee HR, Choi BK. Integrin alpha5beta1 activates the nlrp3 inflammasome by direct interaction with a bacterial surface protein. Immunity 2012;36:755–768 [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T. A simple method of isolating mouse aortic endothelial cells. J Atheroscler Thromb 2005;12:138–142 [DOI] [PubMed] [Google Scholar]
- 19.Astrof S, Crowley D, Hynes RO. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev Biol 2007;311:11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from atvb council. Arteriosclerosis, thrombosis, and vascular biology 2018;38:292–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R, American Heart Association Council on Arteriosclerosis T, Vascular B, Council on Basic Cardiovascular S. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the american heart association. Arterioscler Thromb Vasc Biol 2017;37:e131–e157 [DOI] [PubMed] [Google Scholar]
- 22.Dubrovskyi O, Birukova AA, Birukov KG. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab Invest 2013;93:254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Funk SD, Yurdagul A Jr., Green JM, Jhaveri KA, Schwartz MA, Orr AW. Matrix-specific protein kinase a signaling regulates p21-activated kinase activation by flow in endothelial cells. Circ Res 2010;106:1394–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yurdagul A Jr., Finney AC, Woolard MD, Orr AW. The arterial microenvironment: The where and why of atherosclerosis. Biochem J 2016;473:1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Green J, Yurdagul A Jr., Albert P, McInnis MC, Orr AW. Alphavbeta3 integrins mediate flow-induced nf-kappab activation, proinflammatory gene expression, and early atherogenic inflammation. Am J Pathol 2015;185:2575–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Luo JY, Li B, et al. Integrin-yap/taz-jnk cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016 [DOI] [PubMed]
- 27.Cseh B, Fernandez-Sauze S, Grall D, Schaub S, Doma E, Van Obberghen-Schilling E. Autocrine fibronectin directs matrix assembly and crosstalk between cell-matrix and cell-cell adhesion in vascular endothelial cells. J Cell Sci 2010;123:3989–3999 [DOI] [PubMed] [Google Scholar]
- 28.Balza E, Sassi F, Ventura E, Parodi A, Fossati S, Blalock W, Carnemolla B, Castellani P, Zardi L, Borsi L. A novel human fibronectin cryptic sequence unmasked by the insertion of the angiogenesis-associated extra type iii domain b. International journal of cancer. Journal international du cancer 2009;125:751–758 [DOI] [PubMed] [Google Scholar]
- 29.Carnemolla B, Leprini A, Allemanni G, Saginati M, Zardi L. The inclusion of the type iii repeat ed-b in the fibronectin molecule generates conformational modifications that unmask a cryptic sequence. The Journal of biological chemistry 1992;267:24689–24692 [PubMed] [Google Scholar]
- 30.Manabe R, Ohe N, Maeda T, Fukuda T, Sekiguchi K. Modulation of cell-adhesive activity of fibronectin by the alternatively spliced eda segment. The Journal of cell biology 1997;139:295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner CJ, Badu-Nkansah K, Hynes RO. Endothelium-derived fibronectin regulates neonatal vascular morphogenesis in an autocrine fashion. Angiogenesis 2017;20:519–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Flier A, Badu-Nkansah K, Whittaker CA, Crowley D, Bronson RT, Lacy-Hulbert A, Hynes RO. Endothelial alpha5 and alphav integrins cooperate in remodeling of the vasculature during development. Development 2010;137:2439–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pulakazhi Venu VK, Uboldi P, Dhyani A, Patrini A, Baetta R, Ferri N, Corsini A, Muro AF, Catapano AL, Norata GD. Fibronectin extra domain a stabilises atherosclerotic plaques in apolipoprotein e and in ldl-receptor-deficient mice. Thromb Haemost 2015;114. [DOI] [PubMed]
- 34.Yurdagul A Jr., Chen J, Funk SD, Albert P, Kevil CG, Orr AW. Altered nitric oxide production mediates matrix-specific pak2 and nf-kappab activation by flow. Mol Biol Cell 2013;24:398–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shih PT, Elices MJ, Fang ZT, Ugarova TP, Strahl D, Territo MC, Frank JS, Kovach NL, Cabanas C, Berliner JA, Vora DK. Minimally modified low-density lipoprotein induces monocyte adhesion to endothelial connecting segment-1 by activating beta1 integrin. J Clin Invest 1999;103:613–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res 2000;87:153–159 [DOI] [PubMed] [Google Scholar]
- 37.Ffrench-Constant C, Hynes RO. Alternative splicing of fibronectin is temporally and spatially regulated in the chicken embryo. Development 1989;106:375–388 [DOI] [PubMed] [Google Scholar]
- 38.Pankov R, Yamada KM. Fibronectin at a glance. Journal of cell science 2002;115:3861–3863 [DOI] [PubMed] [Google Scholar]
- 39.Doddapattar P, Gandhi C, Prakash P, Dhanesha N, Grumbach IM, Dailey ME, Lentz SR, Chauhan AK. Fibronectin splicing variants containing extra domain a promote atherosclerosis in mice through toll-like receptor 4. Arteriosclerosis, thrombosis, and vascular biology 2015 [DOI] [PMC free article] [PubMed]
- 40.Tan MH, Sun Z, Opitz SL, Schmidt TE, Peters JH, George EL. Deletion of the alternatively spliced fibronectin eiiia domain in mice reduces atherosclerosis. Blood 2004;104:11–18 [DOI] [PubMed] [Google Scholar]
- 41.Babaev VR, Porro F, Linton MF, Fazio S, Baralle FE, Muro AF. Absence of regulated splicing of fibronectin eda exon reduces atherosclerosis in mice. Atherosclerosis 2008;197:534–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L. The eiiia segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. The Journal of biological chemistry 2002;277:14467–14474 [DOI] [PubMed] [Google Scholar]
- 43.de Bont N, Netea MG, Demacker PN, Verschueren I, Kullberg BJ, van Dijk KW, van der Meer JW, Stalenhoef AF. Apolipoprotein e knock-out mice are highly susceptible to endotoxemia and klebsiella pneumoniae infection. Journal of lipid research 1999;40:680–685 [PubMed] [Google Scholar]
- 44.Rensen PC, Oosten M, Bilt E, Eck M, Kuiper J, Berkel TJ. Human recombinant apolipoprotein e redirects lipopolysaccharide from kupffer cells to liver parenchymal cells in rats in vivo. The Journal of clinical investigation 1997;99:2438–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.