

Abstract
In this study we investigated lipolysis-triggered supersaturation and precipitation of a set of model compounds formulated in lipid-based formulations (LBFs). The purpose was to explore the relationship between precipitated solid form and inherent physicochemical properties of the drug. Eight drugs were studied after formulation in three LBFs, representing lipid-rich (extensively digestible) to surfactant-rich (less digestible) formulations. In vitro lipolysis of drug-loaded LBFs were conducted, and the amount of dissolved and precipitated drug was quantified. Solid form of the precipitated drug was characterized with polarized light microscopy (PLM) and Raman spectroscopy. A significant solubility increase for the weak bases in the presence of digestion products was observed, in contrast to the neutral and acidic compounds for which the solubility decreased. The fold-increase in solubility was linked to the degree of ionization of the weak bases and thus their attraction to free fatty acids. A high level of supersaturation was needed to cause precipitation. For the weak bases, the dose number indicated that precipitation would not occur during lipolysis; hence, these compounds were not included in further studies. The solid state analysis proved that danazol and griseofulvin precipitated in a crystalline form, while niclosamide precipitated as a hydrate. Felodipine and indomethacin crystals were visible in the PLM, whereas the Raman spectra showed presence of amorphous drug, indicating amorphous precipitation that quickly crystallized. The solid state analysis was combined with literature data to allow analysis of the relationship between solid form and the physicochemical properties of the drug. It was found that low molecular weight and high melting temperature increases the probability of crystalline precipitation, whereas precipitation in an amorphous form was favored by high molecular weight, low melting temperature, and positive charge.
Keywords: Poorly water-soluble drugs, lipid digestion, supersaturation, precipitation, solid state, physicochemical properties
Introduction
Contemporary biological targets have directed the drug discovery process toward drug candidates with increasing lipophilicity.1,2 This allows high potency but also results in poor water solubility. One strategy to overcome solubility limitations and enable efficient oral delivery of lipophilic drugs is to use lipid-based formulations (LBFs). In such delivery systems, the drug is typically predissolved in the formulation, thus overcoming the dissolution step in the gastrointestinal (GI) tract. After oral intake of an LBF, the included excipients mix with the intestinal fluids and aid the drug solubilization throughout the GI-tract. However, the lipids in the formulation are subjected to endogenous intestinal lipid processing (digestion) and bile dilution. During this process, the environment becomes more hydrophilic. Thus, the capability of keeping poorly water-soluble, lipophilic drugs dissolved generally decreases. Consequently, such drug compounds are challenged into a supersaturated state, thereby increasing the risk for drug precipitation.3,4
During intestinal transit, the drug is solubilized in a complex mixture of colloidal structures, containing the formulation components, endogenous bile salts, phospholipids, and cholesterol. Drug saturation and subsequent supersaturation is likely to occur along with the colloidal dispersion and digestion processes. Drug supersaturation may affect the free concentration of drug available for absorption in two different ways. The increased free concentration of drug could promote absorption through the higher thermodynamic activity5,6 or lead to drug precipitation, which may reduce absorption. However, recently it has been shown that precipitation is not always detrimental to absorption because precipitated drug may redissolve during GI transit. The redissolution ability is related to the solid form of the precipitated drug rather than to the extent of precipitated drug.7,8 In the event of amorphous precipitate, the drug is able to rapidly dissolve again due to its high-energy state, while the crystalline counterpart redissolves at a slower rate.9,10 As a result, research efforts have been directed toward understanding how LBF dispersion and digestion impact the complex interplay between drug supersaturation and precipitation.
To date, a limited number of studies cover drug precipitation from LBFs under digestive conditions. In vitro studies of cinnarizine-loaded LBFs showed that the solid state of the precipitate was of amorphous character or in a molecular dispersion with digestion components.11,12 Different solid forms were observed when two other weak bases, carvedilol and loratadine, were exposed to digestion. Carvedilol precipitated in a crystalline form during dispersion (reflecting dilution in bile) but in an amorphous form during digestion, while loratadine precipitated in a crystalline form during both dispersion and digestion.13 The neutral drugs fenofibrate and danazol have been observed to precipitate in a crystalline form,14−16 while simvastatin (neutral) precipitated in an amorphous form.17 Moreover, when lipolysis-triggered precipitation was studied for tolfenamic acid, it was observed to precipitate in a crystalline form.15 Taken together, these results indicate that digestive conditions, drug inherent properties, and formulation excipients possibly influence the precipitation behavior. However, based on the few studies available it has been suggested that the solid form of the precipitate is related to ionization of the compound, where the hypothesis is that weakly basic drugs favor precipitation in a noncrystalline form, while acidic and neutral drugs precipitate in a crystalline form during in vitro digestion.18
Evidently, the solid form of the precipitated drug affects drug redissolution capability and thus absorption. Therefore, a better understanding of the mechanisms of drug precipitation during digestion and the reason for altered solid form is required since controlling the solid state of precipitated drug during digestion may present a valuable formulation strategy. Thus, in this work we aimed at studying supersaturation and precipitation behavior of a set of model compounds formulated in an LBF when subjected to in vitro lipolysis to explore the relationship between the solid form and drug inherent properties.
Experimental Section
Drug Compounds and Solvents
Cinnarizine, griseofulvin, haloperidol, indomethacin, ketoconazole, niclosamide, porcine pancreatin (8 × USP specifications activity), soybean oil (long-chain triglyceride), Cremophor EL (surfactant), and Carbitol (cosolvent) were purchased from Sigma-Aldrich (St. Louis, USA). Danazol was purchased from Toronto Research Chemicals, Inc. (Toronto, Canada), and felodipine was a gift from AstraZeneca (Mölndal, Sweden). Captex 355 (medium-chain triglyceride) and Capmul MCM EP (medium-chain mono-, di-, and triglyceride) were donated by Abitec (Janesville, USA), and Maisine 35–1 (long-chain mono-, di-, and triglyceride) was a gift from Gattefossé (Lyon, France). FaSSIF powder was bought from biorelevant.com (Croydon, UK). HPLC-solvents were bought from VWR International (Spånga, Sweden).
Data Set Selection, LBF Composition, and Drug Loading Capacity
In total, eight drugs were investigated in this work (Table 1). The data set was selected to have a diverse physicochemical profile and to include acidic, basic, and nonionizable drugs since ionization during lipolysis has been linked to solid form of the precipitate during digestion.12,13,18 Additionally, the weak bases were selected to display different extent of ionization at pH 6.5 (the pH used during in vitro lipolysis), to further elucidate the relationship between the extent of ionization and supersaturation and precipitation behavior, respectively.
Table 1. Physicochemical Properties of the Compoundsa.
| compound | Mw (g/mol) | A/B/N | pKa | logP | Tm (°C) |
|---|---|---|---|---|---|
| Danazol | 337.5 | N | na | 4.9 | 227 |
| Griseofulvin | 352.8 | N | na | 2.2 | 217 |
| Felodipine | 384.3 | N | na | 3.6 | 143 |
| Indomethacin | 357.8 | A | 3.9 | 4.2 | 160 |
| Niclosamide | 327.1 | A | 10.3 and 8.1 | 3.6 | 231 |
| Haloperidol | 375.9 | B | 8.6 | 3.9 | 151 |
| Cinnarizine | 368.6 | B | 7.5 | 5.5 | 119 |
| Ketoconazole | 531.4 | B | 3.4 and 6.3 | 3.9 | 146 |
Molecular weight (Mw), and the partition coefficient between octanol and water (logP) was calculated with DragonX 6.0.16 (Talete, Italy). pKa was adapted from the literature49 except for niclosamide, which was predicted with ADMET Predictor v7.1 (Lancaster, CA). Melting point (Tm) was determined with differential scanning calorimetry (see Experimental Section). Abbreviations: acid (A); base (B); neutral in the pH range 2–12 (N), not applicable (na).
Three LBFs, representatives of different types in the lipid formulation classification system,19 were assembled for this study; IIIA containing long-chain lipids (IIIA-LC), IIIB containing medium-chain lipids (IIIB-MC), and IV containing surfactant and cosolvent (Figure 1). The LBFs were prepared by preheating the excipients to 37 °C (except Maisine 35–1) and weighing them into vials in predefined fractions (% w/w). Maisine 35–1 was heated to 70 °C (as recommended by the manufacturer) to fully melt and blend before mixing with the other formulation components. Determination of maximum drug loading capacity (i.e., the saturated drug concentration) in the LBFs was performed by following a previously published protocol.20 Drug was added in excess to ∼700 mg of LBF, and the vials were vortexed and placed on a shaker (37 °C). At predetermined time points (24, 48, 72 h), the vials were centrifuged (37 °C, 2800g, 30 min), after which 20–30 mg of the supernatant was transferred to a 5 mL volumetric flask and diluted with methanol (IIIB-MC and IV) or isopropanol (IIIA-LC). The samples were further diluted in 96-well plates (UV-Star, Greiner, USA), and drug concentration determined at compound specific wavelengths (Tecan, Safire2, Austria). Equilibrium solubility was considered to be reached when two consecutive sample points differed by ≤10%.
Figure 1.
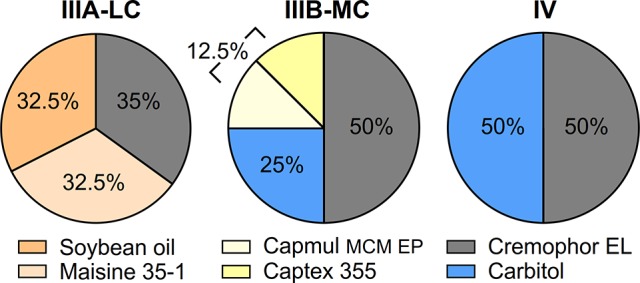
Assembled type IIIA-LC, IIIB-MC, and IV lipid-based formulations for this study. The percent of excipient corresponds to % w/w.
In Vitro Lipolysis Experiments
The in vitro lipolysis experiments was carried out as described by Williams and colleagues.21 A temperature controlled vessel containing digestion medium (37 °C) with a pH-stat (iUnitrode), coupled to a dosing unit was applied (Metrohm 907 Titrando, Switzerland). The digestion medium consisted of a buffer (pH 6.5) containing 2 mM Tris-maleate, 1.4 mM CaCl2·2H2O, and 150 mM NaCl supplemented with FaSSIF powder (3.0 mM sodium taurocholate and 0.75 mM lecithin). Approximately 12−24 h prior to an experiment, FaSSIF powder was added into the buffer (pH 6.5), stirred (1–2 h), and kept in room temperature. The following day (∼1 h prior to the experiment), the digestion medium was heated to 37 °C. Pancreatic enzyme extract was prepared by mixing 1.2 g of porcine pancreatin, 6 mL of buffer, and 20 μL of 5 M NaOH in a 12 mL tube and centrifuged at 5 °C and 2144g for 15 min. The pancreatic enzyme was tested to have an activity of ∼33 TBU/mg equal to ∼6600 TBU/mL extract, which resulted in complete digestion (Supporting Information, Figure S1). At the start of the experiment, 45 mL of the digestion medium (37 °C) and 1.25 g of LBF (preheated to 37 °C) were added and allowed to mix for 10 min (450 rpm). During this dispersion phase, the pH was manually adjusted to 6.5 ± 0.05. Digestion was initiated by addition of 4.44 mL of pancreatin enzyme extract. In the end of the dispersion (after sampling), the medium volume was adjusted to ∼40.5 mL, to keep the volume to 45 mL at the start of the digestion. To maintain the pH at 6.5 in spite of the release of ionized free fatty acids (FFAs), 0.2 M (IIIA-LC and IV) or 0.6 M (IIIB-MC) NaOH was automatically titrated from the dosing unit until the experiment was terminated at 60 min.
Drug Solubility in Lipolysis Medium
Drug solubility in the dispersion and digestion medium was determined by performing “blank” lipolysis according to the protocol described above. No drug was added to the LBF, i.e., a placebo formulation was digested, and the resulting lipolysis medium was sampled (1 mL) in the end of the dispersion phase (10 min) and the digestion phase (60 min). Collected samples were treated with 5 μL/mL lipase inhibitor (0.5 M 4-bromophenyl boronic acid in methanol) to inhibit further lipolysis, followed by centrifugation (37 °C, 21,000g, 15 min) to separate the pancreatic extract from the lipolysis medium. Next, 900 μL of the supernatant was transferred to tubes containing excess of crystalline compound (2–7 mg). The samples were vortexed and placed on a shaker in an incubator (37 °C). After 1–2 h of incubation, pH was measured and adjusted to 6.5 if needed. After 5 and 24 h, the samples were centrifuged (37 °C, 21,000g, 15 min), and the supernatant was sampled and quantified for drug with an HPLC (1290 Infinity with a Zorbrax Eclipse XDB-C18 column 4.6 × 100 mm, Agilent Technologies, USA). All samples were diluted 10-fold in acetonitrile and a second time in a compound-specific mobile phase (2–20-fold) prior to analysis. The analytical conditions can be found in the Supporting Information (Table S1). Solubility in dispersion and digestion medium was defined as the mean value of the 24 h triplicate samples, the 5 h sample was used as a reference to ensure that equilibrium solubility was reached.
Dose Number and Level of Drug Loading
Dose number (Do) calculations were performed for each drug–LBF pair to approximate the amount of drug required for precipitation to occur during the lipolysis experiment (eq 1).
| 1 |
where M0 is the dose of the compound (mg), V0 is the volume (mL), and Cs is the equilibrium solubility in the medium used (mg/mL).22 For the Do-calculations, the dose at 80% of maximum drug loading capacity was used (Table 2), the volume of digestion medium was 45 mL, and solubility in the digestion medium is presented in Figure 2. Overall, a high level of drug loading, corresponding to a Do > 5 when exposed to the lipolysis, was targeted to increase the probability of drug supersaturation and precipitation during in vitro lipolysis. This level of supersaturation was selected since previous studies have observed lipolysis-triggered precipitation at concentrations around 3-fold greater than the solubility.15,23 Based on these calculations we deemed it unlikely for precipitation to occur when Do < 1; therefore, compounds with such low Do values were not included in further experiments.
Table 2. Determined maximum drug loading capacity in IIIA-LC, IIIB-MC, and IV (37 °C), and Dose Number at 80% Drug Load (eq 1)a.
| IIIA-LC |
IIIB-MC |
IV |
||||||
|---|---|---|---|---|---|---|---|---|
| compound | loading (mg/g) | Do 80% | loading (mg/g) | lipolysis drug load (mg/g) | lipolysis Dob | Do 80% | loading (mg/g) | Do 80% |
| Danazol | 16.4 (0.7) | 4.9 | 38.8 (1.4) | 15.5 | 4.8 | 9.5 | 50.1 (1.2) | 13.5 |
| Griseofulvin | 2.8 (0.1) | 1.4 | 8.1 (0.0) | 6.5 | 2.9 (5.5) | 2.9 | 12.4 (0.5) | 4.6 |
| Felodipine | 75.4 (4.7) | 6.8 | 176.2 (2.1) | 140.7 | 9.1 | 9.1 | 218.0 (9.3) | 11.1 |
| Indomethacin | 31.9 (1.1) | 0.7 | 84.7 (3.5) | 67.8 | 1.6 (4.8) | 1.6 | 121.3 (4.2) | 2.4 |
| Niclosamide | 13.6 (0.4) | 14.5 | 50.9 (1.6) | 8.3 | 11.1 | 54.6 | 80.7 (4.5) | 55.8 |
| Haloperidol | 3.9 (1.2) | 0.0 | 17.7 (1.2) | nd | nd | 0.1 | 21.5 (2.1) | 0.4 |
| Cinnarizine | 34.5 (3.2) | 0.2 | 41.8 (0.7) | nd | nd | 0.5 | 43.2 (1.7) | 2.1 |
| Ketoconazole | 9.7 (1.2) | 0.2 | 23.0 (1.0) | nd | nd | 0.5 | 29.9 (1.5) | 1.4 |
Loading is provided as mean with standard deviation within brackets. For IIIB-MC, the applied drug load and experimental dose number during in vitro lipolysis are also shown.
The value within the brackets is the apparent Do during the digestion after spike of a concentrated stock-solution (see Dose Number and Level of Drug Loading). Abbreviations: maximum drug loading capacity (loading), dose number (Do).
Figure 2.

Drug solubility (expressed on a log-scale) in dispersion and digestion media (37 °C, pH 6.5). Overall, the neutral and acidic drugs display higher solubility in dispersion compared to digestion media. The opposite pattern is observed for the basic drugs, for which the solubility increases when the lipids are digested into free fatty acids. The difference in solubility between dispersion and digestion media was statistically significant (p < 0.05) for all drug–LBF pairs except for haloperidol in the type IV formulation. Abbreviation: Aqueous (AQ)
Lipolysis of Loaded LBFs, Collection, and Quantification of Drug
Among the LBFs composed for this study, IIIB-MC is digested to the greatest extent (Figure S1) and is also the lipid-rich LBF with highest Do (Table 2). This LBF was therefore selected as the model formulation for the lipolysis experiments of drug supersaturation and precipitation behavior (Figure 1). Prepared IIIB-MC were loaded with drug by weighing required amount of drug (Table 2) and formulation into vials. The vials were vortexed and placed on a shaker at 37 °C until all drug had dissolved. Subsequently, the drug-loaded IIIB-MCs were digested (following the protocol above), the extent of drug precipitation was quantified, and the solid form of the drug precipitate was evaluated. For these analyses, samples of 1 mL were removed during the in vitro lipolysis after 5 and 10 min of dispersion and 5, 30, and 60 min of digestion. After centrifugation, the aqueous phase was transferred to a new tube leaving the pellet in the vial. To dissolve the precipitated drug, 1 mL of acetonitrile was added to the pellet followed by mixing and centrifugation (22 °C, 21,000g, 15 min). Prior to HPLC analysis, the aqueous phase and the pellet phase were diluted in acetonitrile and compound-specific mobile phase (5–200-fold), see Supporting Information (Table S1).
Solid State Characterization
Polarized Light Microscopy
The solid form of the acquired pellet after 60 min of digestion was characterized using an Olympus BX51 microscope (Olympus, Japan) equipped with crossed polarizing filters. Directly at termination of the lipolysis experiment, the pellet was carefully transferred to a microscope slide and images recorded. Using the same technique crystalline drug, “blank” pellet after lipolysis of placebo LBF and “blank” pellet spiked with crystalline drug (as received from the manufacturer) were examined as references.
Raman Spectroscopy
In addition to the microscopy examination, the pellet was investigated with a Raman spectrometer (Rxn-2 Hybrid, Kaiser Optical System Inc., USA), with a laser wavelength of 785 nm and laser power of 400 mW. A fiber-optic PhAT probe was used, and the spectra were monitored in the range 100–1890 cm–1. Spectra were also collected for crystalline drug and “blank” pellet after lipolysis of placebo LBF. All spectra were baseline corrected by standard normal variate. Additionally, the spectra of the “blank” IIIB-MC was used to correct for the background caused by residues from the lipolysis in the pellet samples (e.g., pancreatic enzyme).
Preparation of Amorphous Drug
If the solid state analysis of the pellet phase (post digestion) indicated presence of amorphous drug, the crude crystalline drug was amorphized through melt quenching. The pure crystalline material was melted in an oven to 10–20 °C above the melting point, followed by immediate cooling on dry ice and ethanol. Conversion to the amorphous form was verified by analyzing the material with PLM and differential scanning calorimetry (DSC), as described in the below section. Raman spectra of the crude amorphous material was collected to allow for comparison with spectra of the pellet and the crude crystalline material.
Differential Scanning Calorimetry
The melting temperature (Tm), crystallinity, and purity of the model compounds (as received by the manufacturer) were determined by conventional DSC (TA Instrument Co., USA). After melt quenching of crystalline drug, DSC was also used to verify conversion into the amorphous form. The instrument was calibrated using indium (Tm = 156.59 °C and heat of fusion, Hf = 28.57 J/g) and purged with 50 mL/min of nitrogen. The pure crystalline or amorphous material were analyzed using conventional DSC. One to five milligrams of sample was weighed into an aluminum pan and sealed with an aluminum lid containing pin holes. The thermal analysis started with equilibration at 0 °C, after which the sample was heated 10 °C/min to 20–30 °C above the expected Tm. The onset of Tm is reported from the resulting thermogram.
Statistics and Multivariate Data Analysis
All data handling was carried out in Excel, whereas visualization and statistical analysis were performed in GraphPad Prism 7.0 (Graphpad Software Inc., USA). An unpaired parametric t test was used to statistically analyze differences between solubility in dispersion and digestion media. Data were expressed as the mean (n = 3) ± standard deviation (sd). A difference was considered statistically significant when p ≤ 0.05. Multivariate data analysis (MVA) (Simca 15, Umetrics, Sweden) in the form of projections to latent structures discriminant analysis (PLS-DA) was applied to identify trends between solid form of precipitated drug during lipolysis and physicochemical properties. For the PLS-DA, drug physicochemical properties that previously have been related to the solid form of drug precipitate and/or glass-forming ability were selected, such as ionization, melting point, and molecular weight. The included variables (Supporting Information Table S2) were molecular weight (g/mol), acid/base/neutral compound (A/B/N), if the compound was ionized during lipolysis or not (yes/no), Tm (°C), and glass-forming classification (glass-former (GF), nonglassformer (nGF)). The glass-forming classification was based on previous studies in which compounds were classified based on their behavior when undergoing heat–cool–heat cycles in the DSC.24,25 In those studies the drugs were classified in class I (nGF), class II (GFs crystallizing above the glass-transition temperature (Tg)), and class III (GFs with no sign of crystallization above Tg), while in this work we simplified the classification nGF (class I) and GF (class II and III).
From our study five compounds (danazol, griseofulvin, felodipine, indomethacin, and niclosamide) were included in the PLS-DA. To extend the data set an additional seven compounds studied elsewhere were added; carvedilol,13 cinnarizine,11 fenofibrate,15 halofantrine,26 loratidine,13 simvastatin,17 and tolfenamic acid.15 These compounds have been reviewed by Khan and colleagues,18 and our inclusion criterion was that the compound should have been studied in an LBF under digestive conditions. The response variable (y) for the 12 included compounds in the PLS-DA were either amorphous (A) or crystalline (C). Compounds that were found to precipitate in an amorphous–crystalline mixture in this work were classified as amorphous, arguing that most likely the compound precipitated in an amorphous form and quickly crystallized. Likewise, hydrates were included in the crystalline group.
Results
Maximum Drug Loading Capacity
The eight studied drugs (Table 1) displayed a trend of improved loading capacity with increased amount of hydrophilic excipients (Figure 1 and Table 2), consistent with previous studies.21,27 The equilibrium solubility for this compound data set ranged 2 orders of magnitude, from 2.8 to 218.0 mg/g in the LBFs. The maximum drug loading capacity was further used to calculate the Do (eq 1) to decide on a suitable loading for the in vitro lipolysis of drug-loaded IIIB-MC.
Drug Solubility in Lipolysis Medium
Maximum drug solubility in the aqueous dispersion and digestion phases is shown in Figure 2. Overall, the neutral and acidic compounds had higher solubility in the dispersion compared to the digestion media regardless of LBF type, whereas an opposite pattern was observed for the weak bases. For the latter, the solubility increased in the digestion media for all LBFs. Among the neutral compounds (danazol, griseofulvin, and felodipine) the largest solubility drop occurred between the type IIIA-LC dispersion and digestion medium. In comparison, the two weak acids (indomethacin and niclosamide) demonstrated an even solubility level, regardless of LBF and media type. Indomethacin showed a solubility between 1.5–1.7 mg/mL for all LBFs, with a small drop to approximately 1.1 mg/mL when the formulations were digested. Niclosamide had a generally low solubility in all dispersion media (<0.08 mg/mL) with a slight solubility drop to around 0.03 mg/mL in the digested phase. The observed higher solubility of indomethacin compared to niclosamide is probably linked to pKa (Table 1) and the resulting difference in ionization of the two compounds at pH 6.5.
The digestion of glycerides into FFAs significantly boosted the aqueous phase solubility of the basic drugs (haloperidol, cinnarizine, and ketoconazole). In IIIA-LC, which contains a large quantity of long-chain digestible components, the drug solubility increased the most (2–11-fold). In contrast, the lowest increase in solubility was displayed in the type IV LBF (1–3-fold), which contains a minor extent of digestible components. Overall, the rank-order of the solubility increase between dispersion and digestion media reflects the extent of ionization of the weak bases. For IIIA-LC, the rank-order of the solubility fold-increase was ketoconazole (×2) < cinnarizine (×6) < haloperidol (×11). The extent of ionization followed the same order: ketoconazole (42%) < cinnarizine (90%) < haloperidol (99%).
Dose Number and Level of Drug Loading
The maximum drug loading capacity (Table 2) and the solubility in the aqueous digestion media (Figure 2) were applied to calculate Do (eq 1). In Table 2 the Do at 80% of maximum drug loading is shown for all drug–LBF combinations. The range of Do at this level of loading spreads from 0 and up to 56. For formulations leading to a Do < 1, the complete dose is dissolved in the lipolysis medium, whereas when Do > 1, the drug is supersaturated. A Do ≫ 1 indicates high level of supersaturation, and hence, likely immediate drug precipitation during lipolysis. In this study, we aimed to load the LBFs to reach above drug solubility in the lipolysis medium to generate supersaturation high enough to increase the likelihood of drug precipitation. It is apparent from the Do at 80% loading that this was not possible in all cases. None of the basic compounds (haloperidol, cinnarizine, and ketoconazole) reaches above a Do value of 1 in any of the lipid-rich LBFs after being exposed to digestion (maximum-value of 0.5 being observed). These compounds were therefore not included in further precipitation experiments. For the remaining compounds the target was to reach concentrations around 5-fold greater than the solubility during digestion. Hence, in cases where 80% loading corresponded to an extreme Do (danazol and niclosamide), the loading was lowered.
At this stage, IIIB-MC was selected for further drug-loaded lipolysis experiments with solid state characterization of drug precipitate. The amount of drug loaded into the formulation and the corresponding Do during lipolysis are visualized in Table 2. Griseofulvin and indomethacin did not precipitate at 80% loading (data not shown, Do of 1.6 and 2.9, respectively); thus, the media was spiked at the initiation of the digestion phase with a concentrated carbitol–drug stock solution; carbitol is one of the nondigestible components in the formulation itself in which the compounds have high solubility.27 For griseofulvin, the volume spiked was 580 μL of stock-solution (10 mg/mL), which increased the apparent Do during the digestion to 4.8. To increase the supersaturation of indomethacin, 750 μL of stock-solution (180 mg/mL) was added, which increased the apparent Do to 5.5. In both cases, the amount of carbitol was kept low (<1.7%) to minimize its effect on the results.
In Vitro Lipolysis, Supersaturation, and Precipitation
Drug distribution in aqueous and pellet phase of the three neutral (danazol, griseofulvin, and felodipine) and two acidic (indomethacin and niclosamide) drugs are shown in Figures 3 and Figure S2. The formulation dispersed readily, and no evidence of drug precipitation was apparent during the initial 10 min of dispersion, despite high supersaturation levels. During the following digestion phase, drug precipitation occurred in all lipolysis experiments. Danazol, felodipine, and niclosamide concentrations in the aqueous phase drastically decreased as the lipids were digested, and as expected, the amount of precipitated drug simultaneously increased. At 60 min of digestion, the degree of precipitated drug was extensive (50–70%) for these three compounds (Figure S2). However, none of the drugs reached down to the level of equilibrium solubility during the 60 min assay, and hence, the drugs were still in a supersaturated state throughout the lipolysis despite significant precipitation. Griseofulvin and indomethacin lipolysis was spiked in the beginning of the digestion due to their relatively low Do at 80% loading (see previous section). As a result, the drug concentrations are higher during digestion than dispersion. Despite this spike, a majority of the drug remained dissolved in the aqueous phase during the digestion; only 2% of indomethacin and 11% of griseofulvin had precipitated after 60 min of digestion (Figure S2).
Figure 3.

Lipolysis profiles of drug-loaded type IIIB-MC for danazol, griseofulvin, felodipine, indomethacin, and niclosamide. Drug concentration in the aqueous phase (blue dots) and the pellet phase, i.e., precipitated drug (orange squares) during lipolysis. Thermodynamic drug solubility at 10 min of dispersion and 60 min of digestion (black dotted line). Even though drug precipitation occurred, all drugs remained above the solubility value in the digestion medium, and hence stayed supersaturated throughout the lipolysis.
Solid State Characterization of Drug Precipitate
At termination of the lipolysis experiment, the isolated pellet material was visualized under cross polarized light and analyzed with Raman spectroscopy. For all drugs, crystals were present on the microscopy images (Figures 4A and Figure S3). Even though the Raman spectra of the pellet material were similar to the respective crystalline reference spectra, they are not as straightforward to interpret since slight deviations were observed. These small differences may indicate a mixture of crystalline material and the amorphous form or the presence of a polymorph. The Raman spectra of each compound is discussed in more detail below. Nevertheless, both techniques were able to verify the presence of drug crystals despite the low amount of precipitated drug in some cases.
Figure 4.

Solid state characterization of pellet material at 60 min of digestion of drug-loaded IIIB-MC. (A) Polarized light micrographs of (from the top) danazol, griseofulvin, felodipine, indomethacin, and niclosamide. On the images, crystalline drug is visible in the pellet material for all five compounds. (B) Raman spectra of (from the top) danazol, griseofulvin, felodipine, indomethacin, and niclosamide. IIIB-MC sample (orange), crystalline reference (dark blue), and amorphous reference (gray; only shown in the spectrum of felodipine). The graphs to the right display a zoom in of a selected range. (C) Polarized light micrograph of a “blank” IIIB-MC pellet, as reference to drug-loaded micrographs.
Raman Spectroscopy of Pellet Material
Peak width can be used to determine crystallinity of materials, as the width reflects homogeneity of the chemical microenvironment of a functional group. In the 1600 cm–1 region of the danazol spectra (Figure 4B), a broadening of the peak (from 10 to 20 cm–1) has previously been found to be significant for the amorphous form.28 The peak width was therefore calculated (full width at half-maximum height, two point baseline correction between 1575 and 1625 cm–1) for the neat crystalline material and the danazol pellet; no distinct difference was apparent (10.1 cm–1 crystalline reference and 9.8 cm–1 danazol pellet), confirming crystalline precipitate for danazol. In the Raman spectra of griseofulvin the most interesting region is the C=C/C=O stretch and the aromatic ring mode region between 1560 and 1650 cm–1. A broadening of the peaks and increased distance between 1620 (benzene ring mode) and 1660 cm–1 (C=C stretch) band is an indicator of the amorphous form of griseofulvin.28,29 Due to the low amount of griseofulvin precipitate in the pellet, the Raman spectra is less smooth. Yet, no such increased distance is evident, and thus the precipitated griseofulvin is most likely present in its crystalline form. The Raman spectra of felodipine contains several bands with distinct peaks. The strongest peak is visible in the 1644–1648 cm–1 range and attributed to the free carbonyl stretching mode. In the crystalline reference spectra, this peak top is positioned at 1644 cm–1, and in the amorphous reference spectra, it is shifted upward to 1648 cm–1. In the felodipine pellet spectra, it is visible at 1646 cm–1, in between the crystalline and amorphous references. An additional difference between the three Raman spectra of felodipine is reduction of the 1705 cm–1 peak. This reduction is observed both in the amorphous reference spectra and the felodipine pellet spectra, compared to the crystalline reference spectra. Together, these differences indicate that the felodipine pellet sample constitutes a mixture of crystalline and amorphous material.30
Continuing to the Raman spectra of the weak acids (Figure 4B), starting with indomethacin, some differences were observed between the crystalline and the precipitated material, which indicated presence of amorphous material in the pellet. Compared to the crystalline reference, which has a sharp peak at 1700 cm–1, this peak has shifted to a lower wavenumber (1683 cm–1) for the indomethacin pellet sample. Furthermore, in the 1575–1625 cm–1 range, several peaks are visible. In the indomethacin pellet spectra, these peaks have merged and are less sharp compared to the crystalline reference. Similar alterations in the Raman spectra have previously been associated with the amorphous form of indomethacin.31 Because of the inherent fluorescence of indomethacin, a reference Raman spectra of the amorphous form was not possible to collect. However, a second indication of presence of amorphous indomethacin in the pellet was the yellow color of the precipitated drug (Figure S4). A color change from white to yellow has previously been observed when crystalline indomethacin undergoes amorphization.32,33 The Raman spectra of niclosamide indicated that the compound precipitated in a hydrate form. In Figure 4B, the boxes identify the most pronounced differences between neat crystalline and precipitated niclosamide. The movement of the C=O stretching modes from 1650 to 1679 cm–1, and the movement downward of the symmetric NO2 stretching modes from 1348 to 1328 cm–1 and from 1517 to 1504 cm–1, respectively, have previously been associated with the monohydrate form of niclosamide.34 Hence, the observed shifts in our study indicate a structural change due to hydrate formation.
Physicochemical Properties and Precipitation Behavior
Combining the results obtained from the two techniques, the solid state characterization of precipitated drug in the pellet shows that danazol and griseofulvin were in their crystalline form, niclosamide was found to be in a hydrate form, and felodipine and indomethacin were found to precipitate in a crystalline and amorphous mixture.
The results in our study was combined with data collected under digestive conditions from previous studies,18 and here, we used an MVA approach to elucidate the relation between solid form of precipitated drug to physicochemical properties and glass-forming ability (as defined by melt quenching). In the score plot of the PLS-DA (Figure 5A), two clusters of the compounds are observed; compounds precipitating in an amorphous form are present on the left side, whereas compounds precipitating in a crystalline form predominantly are located on the right side. The variable influence on projection (VIP) graph (Figure 5B) summarizes the importance of the included x-variables by taking into account the amount of explained y-variance in both components. Variables with a VIP value >1 (Mw, GF/nGF, and Tm) are the most relevant ones for explaining the difference in y, i.e., precipitating in amorphous or crystalline form. Component 1 explains 44.4% of the variability in the data and is primarily associated with differences in molecular weight, glass-forming classification, and melting point. In this data set the compounds precipitating in an amorphous form tend to have a higher Mw than those precipitating in a crystalline form (Figure 5C). Figure 5D shows that a high Tm is more likely to result in a crystalline precipitate. However, two of the compounds with low Tm (fenofibrate, loratidine) also form crystalline precipitates. Further, all of the drugs that previously had been classified as nGFs precipitated in a crystalline form, while in the group of compounds classified as GFs, five of the compounds precipitated in an amorphous form and three in a crystalline form (Figure 5E). Component 2 describes 24.9% of the variability, and this component is largely associated with the tendency to ionize and the corresponding charge at the pH of the lipolysis. The basic drugs are generally located in the upper part, the neutral in the middle, and the acidic in the lower part of the score plot (Figure 5A). Ionization during lipolysis is also related to the separation along the vertical component, where the ionized compounds are found in the lower part and the unionized toward the middle and the top of the score plot.
Figure 5.

PLS-DA of drug precipitation behavior from LBFs under digestive conditions. Crystalline (blue), amorphous (yellow). (A) The score plot displaying the drugs colored by solid form of the precipitate. (B) VIP plot summarizes the importance of the variables in all dimensions; a VIP-value > 1 indicates the most important x-variables. The error bars shows the 95% confidence interval. (C,D) Molecular weight (Mw) and melting point (Tm) of drugs precipitating in amorphous and crystalline form, respectively. Overall, the Mw is higher for the amorphous group than the crystalline group, while a high Tm is more likely to result in a crystalline precipitate. (E) Observed relation between glass-forming classification24,25 and lipolysis-triggered precipitation behavior. For the three drugs classified as nonglassformers (nGF) all precipitated in a crystalline form, while in the group classified as glass-formers (GF) most drugs precipitated in an amorphous form. Halofantrine had not been classified (GF/nGF) in previous studies; thus, the total number of compounds is 11.
Discussion
LBFs represent one strategy for increasing the number of poorly water-soluble drugs that can be delivered orally. However, in vivo processing of the formulation and the resulting effects on drug solubilization and precipitation is decisive for absorption. If the drug precipitates during the GI-transit, the form of the precipitated drug may impact the capability to redissolve, where an amorphous precipitate is expected to redissolve at a faster rate compared to a crystalline material.11,35
Our solubility results in the aqueous dispersion and digestion media show that basic drugs thrive when FFAs are present, most likely due to a favorable electrostatic interaction between the two charged species. For the weak bases, solubility was always higher during digestion as compared to dispersion media (Figure 2); the higher the extent of ionization of the drug at the pH of lipolysis (6.5), the higher the solubility increase during digestion of the lipid-rich formulations. Noteworthy, the solubility fold-increase was the highest in IIIA-LC media and does not follow the digestibility rank-order of the LBFs (IV < IIIA-LC < IIIB-MC) (Figure S1). This indicates that the finding is not only related to the amount of FFAs deliberated but also to the type of digestion products that are formed when the triglycerides are digested. IIIA-LC is less extensively digested, compared to IIIB-MC, and a higher degree of intact or partially digested lipid components will be present in the digestion medium. Upon digestion, the main FFA products of IIIA-LC are linoleic acid (18:2) and oleic acid (18:1), while the majority of FFAs in the IIIB-MC are either caprylic (C8:0) or capric acid (C10:0). Taken together, the lipophilic weak bases favor an environment with more lipophilic digestion components present. However, fatty acid solubility in water is generally low, and the majority of the FFAs are incorporated into micellar structures.36 The high solubility of the weak bases in the digestion media is therefore most likely associated with an increased solubilization in the colloidal structures, where the FFAs are primarily located.
Looking at the full data set, covering neutral, acidic, and basic compounds, our results are in line with the study by Yeap and colleagues.37 They observed an excessive solubility loss in oleic acid (OA) based colloidal systems for basic drugs when the colloids were bile diluted (i.e., decrease in OA concentration). In contrast, neutral and acidic drugs displayed a much less extensive solubility loss as the concentration of OA decreased, which implies that OA was not as important for solubility of the neutral and acidic compounds as for the weak bases. Further, addition of OA has been shown to considerably boost the loading capacity in LBFs for basic drugs,38 and molecular interactions have been confirmed between OA and basic compounds in the formulations (prior to dispersion or digestion). Regional changes in infrared spectrum suggested that interactions occur between OA and the amino groups of loratidine and carvedilol.39 In this study, we focused on dispersed and digested formulations rather than LBFs as such, but similar to previous studies, a solubility advantage for basic drugs in the presence of FFAs was observed. Based on our findings an interesting formulation strategy for weak bases is to incorporate specific lipid species (e.g., long-chain lipids) in the formulation that boost solubility upon digestion. Thus, the solubilization effect in the GI-tract can be prolonged and the risk of drug precipitation minimized.
The formation of lipophilic ion-pairs has also been related to the fact that basic compounds tend to precipitate in an amorphous, or rather, a noncrystalline form, when subjected to lipolysis. To elucidate the mechanism behind this, cinnarizine (pKa 7.5) has been studied in detail. 1H NMR analysis indeed showed interactions between the nitrogens of cinnarizine and the carboxylic group of the FFAs.40 In another study, cinnarizine precipitated in different solid forms depending on ionization of the compound. At pH 8.0 (unionized), the precipitated material was crystalline, while at pH 4.0, 5.5, and 6.5 (ionized) the precipitate was noncrystalline. Changes in the C–N environment was linked to an ionic interaction of the tertiary amine in cinnarizine and the FFAs, which results in an amorphous-salt precipitation.12 In the current study, we concluded from the low Do for the basic compounds, in most cases <1 (Table 2), that the occurrence of lipolysis-triggered precipitation was not likely for the formulations explored. Furthermore, the volume of stock-solution needed to be spiked during the experiment (to experimentally increase the Do when the digestion starts) would be too high to study the three weak bases included in our data set.
Although a few basic compounds have been studied in some detail, there is still a need to understand the precipitation behavior of acidic and neutral compounds. Here, we have studied danazol, griseofulvin, felodipine, indomethacin, and niclosamide loaded in an IIIB formulation composed of medium chain lipids. The lipolysis profiles show that, despite a loss of solubilization capacity during digestion, the concentration of drug in the aqueous phase (free concentration and solubilized drug) stays above the measured thermodynamic solubility in all cases (Figure 3). This demonstrates one of the main advantages of delivery of molecularly dispersed drug in LBFs; attainment of the solvation capacity through lipid digestion, previously described as “LBF-mediated prolonged metastable supersaturation”.3 In LBF lipolysis studies, supersaturation is typically linked to thermodynamic solubility in the medium used, although kinetic solubility may be more appropriate to relate to. Digestion is a kinetic process and the environment is dynamic with constant change in solvation capacity of the medium, up until completion of digestion. Moreover, in an LBF, the compound is presented to the lipolysis medium in a predissolved form, similar to the approach used during high-throughput solubility determination using DMSO stock-solutions. Therefore, kinetic solubility studies (including studies of amorphous solubility in the digestion medium) may provide more information on the expected duration of the “LBF-mediated prolonged metastable supersaturation” than the common thermodynamic solubility measurements.
In this work, we used Do as a measurement of drug supersaturation and thus an indicator of the likelihood of obtaining drug precipitation during in vitro lipolysis. Within the field of lipid-based drug delivery, maximum supersaturation ratio (SRM) has been introduced as an indicator of whether or not the drug will stay in solution. The concept is similar to Do based on dose, volume, and solubility in the media used, although the dose and volume are compiled into one term (maximum drug concentration in APdigest (APmax)). An SRM value >3 has been suggested to predict lipolysis-triggered drug precipitation.15,23 Certainly, high supersaturation is required for drug precipitation to occur; in this work we needed to reach at least a Do ≈ 5 for a few percentages to precipitate, just enough to allow analysis of the solid state. Even though the suggested supersaturation level >3 may indicate that precipitation is likely, the level of supersaturation that can be reached and how long it can be maintained seem to be drug and formulation specific.
Two of the investigated drugs, felodipine and indomethacin, precipitated in an amorphous/crystalline mixture. Danazol and griseofulvin precipitated in a crystalline form, whereas niclosamide precipitated as a hydrate (Figure 4AB). Danazol has also been observed to precipitate in its crystalline form in previous in vitro lipolysis studies.16,41 However, to the best of our knowledge, lipolysis-triggered precipitation, with solid state analysis of the drug precipitate, has not yet been investigated for the other four compounds. The techniques used for solid state analysis were PLM and Raman spectroscopy, both available at the lab bench, making it possible to run the analysis directly on the moist pellet in connection to the lipolysis. This reduces the risk for crystallization as a result of processing prior to analysis, e.g., crystallization that may occur during drying or transportation of the sample to an X-ray diffraction instrument (which is often located elsewhere and not in direct connection to the lipolysis laboratory). Raman spectroscopy can also be used to measure precipitation kinetics directly in the lipolysis vessel,14 which is not feasible with similar techniques, such as infrared spectroscopy, due to water interference.
The current work targeted a better insight into molecular properties related to drug precipitation during lipid digestion. When our data was combined with drugs studied under similar conditions, it was revealed that molecular weight is an important factor driving the solid form of the precipitate. This descriptor of molecular size has also been identified as a key factor for predicting glass-forming ability of solid drugs undergoing melt-quenching.42 Based on the compounds studied herein, precipitation in a crystalline form is more likely to occur for compounds with low Mw (<350 g/mol) and high Tm (>200 °C), whereas amorphous precipitate is found for drugs with high Mw and low Tm (Figure 5A–D). For crystallization to occur, the drug molecules must be in a specific orientation determined by the crystal lattice structure. A small and rigid molecule, reflected by, e.g., low Mw, can more easily find its specific orientation and the right conformation. The relationship between Tm and crystallinity of precipitate can be understood by the higher driving force (in terms of energy) for high-melting compounds to form a crystal structure.43
In the MVA, we also included if the compounds were previously classified as GF or nGF (when explored with melt-quenching) as a variable. From our analysis, it was revealed that if the compound was classified as an nGF, it is likely to precipitate in a crystalline form also during lipolysis; herein three out of three compounds followed this trend. As hypothesized in previous works, our analysis strengthens the finding that charged weak bases favor amorphous precipitation,12,13 while this study also revealed that ionization of acidic compounds does not seem to drive precipitation in the amorphous form. It should be noted that this analysis was made on a limited data set (12 compounds), and the analysis therefore shows initial trends of lipolysis-triggered precipitation behavior. The MVA could perform in a more comprehensive manner by extending the data set, adding drug and formulation properties possibly related to lipolysis-triggered precipitation behavior, such as drug loading capacity, supersaturation level (e.g., Do), type of LBF, chain length of lipids in the formulation, and solid state related properties such as glass-transition temperature, entropy, enthalpy of fusion, etc. Yet, this study shows that, by considering molecular properties and performing a glass-forming classification, a forecast on the solid state (amorphous or crystalline) of the precipitate can be made.
The duration of drug supersaturation is governed by nucleation induction time and thus the stability of the metastable supersaturated state. In a complex medium, this may be influenced by the presence of endogenous and exogenous excipients, here exemplified by the presence of lipids, surfactants, and digestion products. The literature is extensive with regard to alternations in precipitation behavior by addition of polymers, excipients that are well-known to result both in prolonged supersaturation and altered solid form of precipitate.16,44,45 However, the vast majority of the studies are still in vitro studies, and even if we are starting to also understand lipolysis-triggered precipitation behavior, the knowledge may not be transferable to the in vivo fate of the drug. Lately, precipitation of danazol and fenofibrate loaded into LBFs were studied both in vitro and in vivo. Although the drugs precipitated in a crystalline form both in vitro and in vivo, the process was significantly different in vivo from what was predicted in vitro; the amount of precipitated drug in vivo was lower and occurred only in the stomach.41 The mentioned overestimation of the in vivo precipitation risk and similar tendencies in former studies17,26,46,47 are proving the inability of the commonly adapted in vitro lipolysis system to reflect the continuous removal of drug and digestion products from the intestinal lumen. For a highly permeable drug, the absorption might occur at a rate that compensates for any loss in solubilizing capacity when the formulation is diluted and digested in the GI fluids. Thus, the drug is never challenged to the supersaturation level that leads to precipitation. To increase the physiological relevance of the in vitro lipolysis setup, a recent study has shown compatibility of an absorptive membrane consisting of Caco-2 cells and the components present during lipolysis,48 indicating that lipolysis is possible to study simultaneously with drug absorption.
Conclusion
The present study demonstrates the solubility effect of LBF digestion products, where weak bases show significantly increased solubility, while the solubility of neutral and acidic drugs decreases in the less lipophilic milieu upon lipid digestion. The magnitude of the solubility increase for the weak bases was linked to their degree of ionization and thus attraction to FFA. Increased molecular understanding of lipolysis-triggered precipitation behavior was obtained, and the data analysis revealed that molecular weight, melting point, glass-forming classification, and ionization are molecular properties related to the type of solid form that is precipitating during lipolysis. The knowledge gained here provides an initial framework for how molecular properties affect drug solubilization, supersaturation, and precipitation during lipid digestion. In the formulation design, this information may aid the excipient selection to optimize drug solubility upon dispersion and digestion, and further give initial information about the likely form (amorphous or crystalline) of any precipitated drug.
Acknowledgments
This work was supported by the European Research Council (Grant 638965) and the Swedish Research Council (Grants 621-2011-2445 and 621-2014-3309). We are grateful to Elin Mårtensson for skillful experimental assistance, and to Simulations Plus (Lancaster, CA) for providing us with a reference site license for the software ADMET Predictor.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.8b00699.
HPLC analytical details, PLS-DA variables and responses, liberated FFA during lipolysis, drug distribution in aqueous and pellet phase, reference PLM graphs, and indomethacin pellet picture (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Keserü G. M.; Makara G. M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery 2009, 8 (3), 203. 10.1038/nrd2796. [DOI] [PubMed] [Google Scholar]
- Bergström C. A. S.; Charman W. N.; Porter C. J. H. Computational prediction of formulation strategies for beyond-rule-of-5 compounds. Adv. Drug Delivery Rev. 2016, 101 (Supplement C), 6–21. 10.1016/j.addr.2016.02.005. [DOI] [PubMed] [Google Scholar]
- Feeney O. M.; Crum M. F.; McEvoy C. L.; Trevaskis N. L.; Williams H. D.; Pouton C. W.; Charman W. N.; Bergström C. A. S.; Porter C. J. H. 50years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Delivery Rev. 2016, 101 (Supplement C), 167–194. 10.1016/j.addr.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Porter C. J. H.; Trevaskis N. L.; Charman W. N. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discovery 2007, 6 (3), 231–248. 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- McEvoy C. L.; Trevaskis N. L.; Feeney O. M.; Edwards G. A.; Perlman M. E.; Ambler C. M.; Porter C. J. Correlating in Vitro Solubilization and Supersaturation Profiles with in Vivo Exposure for Lipid Based Formulations of the CETP Inhibitor CP-532,623. Mol. Pharmaceutics 2017, 14 (12), 4525–4538. 10.1021/acs.molpharmaceut.7b00660. [DOI] [PubMed] [Google Scholar]
- Crum M. F.; Trevaskis N. L.; Pouton C. W.; Porter C. J. Transient Supersaturation Supports Drug Absorption from Lipid-Based Formulations for Short Periods of Time, but Ongoing Solubilization Is Required for Longer Absorption Periods. Mol. Pharmaceutics 2017, 14 (2), 394–405. 10.1021/acs.molpharmaceut.6b00792. [DOI] [PubMed] [Google Scholar]
- Van Den Abeele J.; Brouwers J.; Mattheus R.; Tack J.; Augustijns P. Gastrointestinal Behavior of Weakly Acidic BCS Class II Drugs in Man—Case Study of Diclofenac Potassium. J. Pharm. Sci. 2016, 105 (2), 687–696. 10.1002/jps.24647. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Kawakami A.; Nanimatsu A.; Horio M.; Matsuoka J.; Wada T.; Kasaoka S.; Yoshikawa H. In vivo evaluation of supersaturation/precipitation/re-dissolution behavior of cinnarizine, a lipophilic weak base, in the gastrointestinal tract: the key process of oral absorption. Eur. J. Pharm. Sci. 2017, 96 (Supplement C), 464–471. 10.1016/j.ejps.2016.10.023. [DOI] [PubMed] [Google Scholar]
- Hancock B. C.; Parks M. What is the True Solubility Advantage for Amorphous Pharmaceuticals?. Pharm. Res. 2000, 17 (4), 397–404. 10.1023/A:1007516718048. [DOI] [PubMed] [Google Scholar]
- Savolainen M.; Kogermann K.; Heinz A.; Aaltonen J.; Peltonen L.; Strachan C.; Yliruusi J. Better understanding of dissolution behaviour of amorphous drugs by in situ solid-state analysis using Raman spectroscopy. Eur. J. Pharm. Biopharm. 2009, 71 (1), 71–79. 10.1016/j.ejpb.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Sassene P. J.; Knopp M. M.; Hesselkilde J. Z.; Koradia V.; Larsen A.; Rades T.; Müllertz A. Precipitation of a poorly soluble model drug during in vitro lipolysis: Characterization and dissolution of the precipitate. J. Pharm. Sci. 2010, 99 (12), 4982–4991. 10.1002/jps.22226. [DOI] [PubMed] [Google Scholar]
- Khan J.; Rades T.; Boyd B. J. Lipid-Based Formulations Can Enable the Model Poorly Water-Soluble Weakly Basic Drug Cinnarizine To Precipitate in an Amorphous-Salt Form During In Vitro Digestion. Mol. Pharmaceutics 2016, 13 (11), 3783–3793. 10.1021/acs.molpharmaceut.6b00594. [DOI] [PubMed] [Google Scholar]
- Stillhart C.; Dürr D.; Kuentz M. Toward an Improved Understanding of the Precipitation Behavior of Weakly Basic Drugs from Oral Lipid-Based Formulations. J. Pharm. Sci. 2014, 103 (4), 1194–1203. 10.1002/jps.23892. [DOI] [PubMed] [Google Scholar]
- Stillhart C.; Imanidis G.; Kuentz M. Insights into Drug Precipitation Kinetics during In Vitro Digestion of a Lipid-Based Drug Delivery System Using In-Line Raman Spectroscopy and Mathematical Modeling. Pharm. Res. 2013, 30 (12), 3114–3130. 10.1007/s11095-013-0999-2. [DOI] [PubMed] [Google Scholar]
- Williams H.; Sassene P.; Kleberg K.; Calderone M.; Igonin A.; Jule E.; Vertommen J.; Blundell R.; Benameur H.; Müllertz A.; Pouton C.; Porter C. H. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 3: Understanding Supersaturation Versus Precipitation Potential During the In Vitro Digestion of Type I, II, IIIA, IIIB and IV Lipid-Based Formulations. Pharm. Res. 2013, 30 (12), 3059–3076. 10.1007/s11095-013-1038-z. [DOI] [PubMed] [Google Scholar]
- Anby M. U.; Williams H. D.; McIntosh M.; Benameur H.; Edwards G. A.; Pouton C. W.; Porter C. J. H. Lipid Digestion as a Trigger for Supersaturation: Evaluation of the Impact of Supersaturation Stabilization on the in Vitro and in Vivo Performance of Self-Emulsifying Drug Delivery Systems. Mol. Pharmaceutics 2012, 9 (7), 2063–2079. 10.1021/mp300164u. [DOI] [PubMed] [Google Scholar]
- Thomas N.; Holm R.; Garmer M.; Karlsson J. J.; Müllertz A.; Rades T. Supersaturated Self-Nanoemulsifying Drug Delivery Systems (Super-SNEDDS) Enhance the Bioavailability of the Poorly Water-Soluble Drug Simvastatin in Dogs. AAPS J. 2013, 15 (1), 219–227. 10.1208/s12248-012-9433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J.; Rades T.; Boyd B. The Precipitation Behavior of Poorly Water-Soluble Drugs with an Emphasis on the Digestion of Lipid Based Formulations. Pharm. Res. 2016, 33 (3), 548–562. 10.1007/s11095-015-1829-5. [DOI] [PubMed] [Google Scholar]
- Pouton C. W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29 (3), 278–287. 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Persson L.; Porter C. H.; Charman W.; Bergström C. S. Computational Prediction of Drug Solubility in Lipid Based Formulation Excipients. Pharm. Res. 2013, 30, 3225. 10.1007/s11095-013-1083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams H. D.; Sassene P.; Kleberg K.; Bakala-N’Goma J. C.; Calderone M.; Jannin V.; Igonin A.; Partheil A.; Marchaud D.; Jule E. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: Method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J. Pharm. Sci. 2012, 101, 3360. 10.1002/jps.23205. [DOI] [PubMed] [Google Scholar]
- Oh D.-M.; Curl R. L.; Amidon G. L. Estimating the Fraction Dose Absorbed from Suspensions of Poorly Soluble Compounds in Humans: A Mathematical Model. Pharm. Res. 1993, 10 (2), 264–270. 10.1023/A:1018947113238. [DOI] [PubMed] [Google Scholar]
- Williams H. D.; Anby M. U.; Sassene P.; Kleberg K.; Bakala-N’Goma J.-C.; Calderone M.; Jannin V.; Igonin A.; Partheil A.; Marchaud D.; Jule E.; Vertommen J.; Maio M.; Blundell R.; Benameur H.; Carrière F.; Müllertz A.; Pouton C. W.; Porter C. J. H. Toward the Establishment of Standardized in Vitro Tests for Lipid-Based Formulations. 2. The Effect of Bile Salt Concentration and Drug Loading on the Performance of Type I, II, IIIA, IIIB, and IV Formulations during in Vitro Digestion. Mol. Pharmaceutics 2012, 9 (11), 3286–3300. 10.1021/mp300331z. [DOI] [PubMed] [Google Scholar]
- Alhalaweh A.; Alzghoul A.; Kaialy W.; Mahlin D.; Bergström C. A. S. Computational Predictions of Glass-Forming Ability and Crystallization Tendency of Drug Molecules. Mol. Pharmaceutics 2014, 11 (9), 3123–3132. 10.1021/mp500303a. [DOI] [PubMed] [Google Scholar]
- Alhalaweh A.; Alzghoul A.; Mahlin D.; Bergström C. A. Physical stability of drugs after storage above and below the glass transition temperature: Relationship to glass-forming ability. Int. J. Pharm. 2015, 495 (1), 312–317. 10.1016/j.ijpharm.2015.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas N.; Holm R.; Müllertz A.; Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J. Controlled Release 2012, 160 (1), 25–32. 10.1016/j.jconrel.2012.02.027. [DOI] [PubMed] [Google Scholar]
- Alskär L. C.; Porter C. J. H.; Bergström C. A. S. Tools for Early Prediction of Drug Loading in Lipid-Based Formulations. Mol. Pharmaceutics 2016, 13 (1), 251–261. 10.1021/acs.molpharmaceut.5b00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Stoneburner K.; Ladika M.; Kuo T.-C.; Kalantar T. H. High-Throughput Raman Spectroscopy Screening of Excipients for the Stabilization of Amorphous Drugs. Appl. Spectrosc. 2015, 69 (11), 1271–1280. 10.1366/15-07914. [DOI] [PubMed] [Google Scholar]
- Żarów A.; Zhou B.; Wang X.; Pinal R.; Iqbal Z. Spectroscopic and X-Ray Diffraction Study of Structural Disorder in Cryomilled and Amorphous Griseofulvin. Appl. Spectrosc. 2011, 65 (2), 135–143. 10.1366/10-06024. [DOI] [Google Scholar]
- Tang X. C.; Pikal M. J.; Taylor L. S. A spectroscopic investigation of hydrogen bond patterns in crystalline and amorphous phases in dihydropyridine calcium channel blockers. Pharm. Res. 2002, 19 (4), 477–483. 10.1023/A:1015147729564. [DOI] [PubMed] [Google Scholar]
- Hédoux A.; Paccou L.; Guinet Y.; Willart J.-F.; Descamps M. Using the low-frequency Raman spectroscopy to analyze the crystallization of amorphous indomethacin. Eur. J. Pharm. Sci. 2009, 38 (2), 156–164. 10.1016/j.ejps.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Petry I.; Löbmann K.; Grohganz H.; Rades T.; Leopold C. S. Solid state properties and drug release behavior of co-amorphous indomethacin-arginine tablets coated with Kollicoat® Protect. Eur. J. Pharm. Biopharm. 2017, 119 (Supplement C), 150–160. 10.1016/j.ejpb.2017.06.007. [DOI] [PubMed] [Google Scholar]
- Priemel P. A.; Laitinen R.; Grohganz H.; Rades T.; Strachan C. J. In situ amorphisation of indomethacin with Eudragit® E during dissolution. Eur. J. Pharm. Biopharm. 2013, 85 (3, Part B), 1259–1265. 10.1016/j.ejpb.2013.09.010. [DOI] [PubMed] [Google Scholar]
- Sardo M.; Amado A. M.; Ribeiro-Claro P. J. Pseudopolymorphic transitions of niclosamide monitored by Raman spectroscopy. J. Raman Spectrosc. 2008, 39 (12), 1915–1924. 10.1002/jrs.2058. [DOI] [Google Scholar]
- Larsen A. T.; Ohlsson A. G.; Polentarutti B.; Barker R. A.; Phillips A. R.; Abu-Rmaileh R.; Dickinson P. A.; Abrahamsson B.; Østergaard J.; Müllertz A. Oral bioavailability of cinnarizine in dogs: relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur. J. Pharm. Sci. 2013, 48 (1), 339–350. 10.1016/j.ejps.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Phan S.; Salentinig S.; Gilbert E.; Darwish T. A.; Hawley A.; Nixon-Luke R.; Bryant G.; Boyd B. J. Disposition and crystallization of saturated fatty acid in mixed micelles of relevance to lipid digestion. J. Colloid Interface Sci. 2015, 449, 160–166. 10.1016/j.jcis.2014.11.026. [DOI] [PubMed] [Google Scholar]
- Yeap Y. Y.; Trevaskis N. L.; Porter C. J. H. The Potential for Drug Supersaturation during Intestinal Processing of Lipid-Based Formulations May Be Enhanced for Basic Drugs. Mol. Pharmaceutics 2013, 10 (7), 2601–2615. 10.1021/mp400035z. [DOI] [PubMed] [Google Scholar]
- Boyd B. J.; Whittaker D. V.; Khoo S.-M.; Davey G. Hexosomes formed from glycerate surfactants—Formulation as a colloidal carrier for irinotecan. Int. J. Pharm. 2006, 318 (1), 154–162. 10.1016/j.ijpharm.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Misic Z.; Jung D. Š.; Sydow G.; Kuentz M. Understanding the interactions of oleic acid with basic drugs in solid lipids on different biopharmaceutical levels. Journal of Excipients and Food Chemicals 2016, 5 (2), 113. [Google Scholar]
- Sassene P. J.; Mosgaard M. D.; Löbmann K.; Mu H.; Larsen F. H.; Rades T.; Müllertz A. Elucidating the Molecular Interactions Occurring during Drug Precipitation of Weak Bases from Lipid-Based Formulations: A Case Study with Cinnarizine and a Long Chain Self-Nanoemulsifying Drug Delivery System. Mol. Pharmaceutics 2015, 12 (11), 4067–4076. 10.1021/acs.molpharmaceut.5b00498. [DOI] [PubMed] [Google Scholar]
- Sassene P. J.; Michaelsen M. H.; Mosgaard M. D.; Jensen M. K.; Van Den Broek E.; Wasan K. M.; Mu H.; Rades T.; Müllertz A. In Vivo Precipitation of Poorly Soluble Drugs from Lipid-Based Drug Delivery Systems. Mol. Pharmaceutics 2016, 13 (10), 3417–3426. 10.1021/acs.molpharmaceut.6b00413. [DOI] [PubMed] [Google Scholar]
- Mahlin D.; Bergström C. A. S. Early drug development predictions of glass-forming ability and physical stability of drugs. Eur. J. Pharm. Sci. 2013, 49 (2), 323–332. 10.1016/j.ejps.2013.03.016. [DOI] [PubMed] [Google Scholar]
- Baird J. A.; Van Eerdenbrugh B.; Taylor L. S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99 (9), 3787–3806. 10.1002/jps.22197. [DOI] [PubMed] [Google Scholar]
- Gao P.; Akrami A.; Alvarez F.; Hu J.; Li L.; Ma C.; Surapaneni S. Characterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J. Pharm. Sci. 2009, 98 (2), 516–528. 10.1002/jps.21451. [DOI] [PubMed] [Google Scholar]
- Bevernage J.; Forier T.; Brouwers J.; Tack J.; Annaert P.; Augustijns P. Excipient-Mediated Supersaturation Stabilization in Human Intestinal Fluids. Mol. Pharmaceutics 2011, 8 (2), 564–570. 10.1021/mp100377m. [DOI] [PubMed] [Google Scholar]
- Thomas N.; Richter K.; Pedersen T. B.; Holm R.; Müllertz A.; Rades T. In Vitro Lipolysis Data Does Not Adequately Predict the In Vivo Performance of Lipid-Based Drug Delivery Systems Containing Fenofibrate. AAPS J. 2014, 16 (3), 539–549. 10.1208/s12248-014-9589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin B. T.; Kuentz M.; Vertzoni M.; Kostewicz E. S.; Fei Y.; Faisal W.; Stillhart C.; O’Driscoll C. M.; Reppas C.; Dressman J. B. Comparison of in vitro tests at various levels of complexity for the prediction of in vivo performance of lipid-based formulations: Case studies with fenofibrate. Eur. J. Pharm. Biopharm. 2014, 86 (3), 427–437. 10.1016/j.ejpb.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Keemink J.; Bergström C. A. S. Caco-2 Cell Conditions Enabling Studies of Drug Absorption from Digestible Lipid-Based Formulations. Pharm. Res. 2018, 35 (4), 74. 10.1007/s11095-017-2327-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg J. H.; Al-Tikriti Y.; Ragnarsson G.; Bergström C. A. S. Ethanol Effects on Apparent Solubility of Poorly Soluble Drugs in Simulated Intestinal Fluid. Mol. Pharmaceutics 2012, 9 (7), 1942–1952. 10.1021/mp2006467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.