

ABSTRACT
Head and neck squamous cell carcinomas (HNSCCs) frequently harbor genomic mutations in cell death pathways. Nearly 30% of HNSCCs overexpress Fas-Associated Death Domain (FADD), with or without BIRC2/3 genes encoding cellular Inhibitor of Apoptosis Proteins 1/2 (cIAP1/2), critical components of the Tumor Necrosis Factor (TNF) Receptor signaling pathways. ASTX660 is a novel non-peptidomimetic antagonist of cIAP1/2 and XIAP under evaluation in a clinical trial for advanced solid tumors and lymphomas. Herein, we show that ASTX660, at nanomolar concentrations, sensitized Murine Oral Cancer (MOC1) cells to TNFα. Using syngeneic mouse models, ASTX660 showed additive anti-tumor activity with radiation therapy (XRT), cisplatin chemotherapy, and PD-1 blockade to significantly delay or eradicate MOC1 tumors. These combinations significantly increased CD8 + T cells and dendritic cells, as well as T cell activity. ASTX660 stimulated cytotoxic T lymphocyte (CTL) killing of MOC1 cells expressing ovalbumin. Early stages of CTL killing were predominantly mediated by perforin/granzyme B, whereas later stages were mediated by death ligands TNFα, TRAIL, and FasL. Correspondingly, depletion of CD8 + T cells and NK cells in vivo revealed both types of immune cells to be important components of the complete anti-tumor response enhanced by ASTX660+XRT. These findings serve to inform future studies of IAP inhibitors and support the potential for future clinical trials investigating ASTX660 with XRT and immunotherapies like PD-1/PD-L1 blockade in HNSCC.
KEYWORDS: ASTX660, inhibitors of apoptosis, radiation therapy, squamous cell carcinoma, TNFα
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer in the world, accounting for approximately 3% of all new cancer diagnoses in the United States, more than 600,000 new cases every year worldwide, and a five-year survival rate nearing 50%.1,2 HNSCCs comprise more than 90% of all head and neck cancers and arise from the epithelium of the oral cavity, oropharynx, nasopharynx, hypopharynx, and larynx.3 Recently, The Cancer Genome Atlas (TCGA) analysis of 279 HNSCCs uncovered genomic mutations in cell death pathways affecting nearly half of all HNSCC.4 Approximately 30% of these carry chromosome 11q13/22 amplifications and overexpression of Fas-associated death domain (FADD), with or without amplification of Baculovirus Inhibitor of Apoptosis repeat containing (BIRC2/3) genes encoding cellular Inhibitor of Apoptosis Proteins 1/2 (cIAP1/2). FADD and cIAP1/2 are critical components of the Tumor Necrosis Factor (TNF) Receptor family signaling pathways that determine cell death or survival.5–7 Binding of TNFα, TNF-related apoptosis-inducing ligand (TRAIL), Fas ligand (FasL), or other death agonists to their receptors typically leads to cell death via FADD8–10; however, increased expression of the IAPs converts this signaling to promote survival by inhibiting both caspase-mediated apoptosis and RIP-mediated necroptosis.11 Mimetics of SMAC (second mitochondria-derived activator of caspases) have been developed as potential therapies to inhibit IAPs and reinstate the pro-apoptotic signaling pathways.12–19 We recently reported that IAP inhibition sensitizes human HNSCC to death ligands TNFα and TRAIL, and that IAP inhibition in combination with radiation is a potent inducer of TNFα and can delay or eradicate HNSCC xenografts in vivo.20
Development of effective anti-tumor immunity involves the activity of cytotoxic T lymphocyte (CTL) and natural killer (NK) cells, both of which induce apoptosis in target cells via granzyme B (early) and/or TNFR superfamily ligation (late) through effector cell release of death ligands TNFα, TRAIL, and FasL. The role of IAP inhibitors in sensitizing tumor cells to induction of apoptosis downstream of TNFR superfamily ligation suggests that IAP inhibitors could potentiate anti-tumor immunity. Many HNSCCs display a T-cell inflamed response and corresponding increases in programmed death ligand 1 (PD-L1), which is an immune checkpoint regulatory molecule that limits CTL-mediated anti-tumor immunity. Since PD-based checkpoint inhibition demonstrates clinical activity in only a subset of HNSCCs,21,22 identifying additional approaches to further strengthen a targeted anti-tumor immune response is critical. ASTX660, a novel orally bioavailable, non-peptidomimetic antagonist of cIAP1/2 and XIAP,23 is currently under evaluation in a Phase I/II clinical trial in patients with advanced solid tumors and lymphomas.24 In the present study, we sought to ascertain the direct cytotoxic effects of ASTX660 and the additive anti-tumor activity of the drug when combined with TNFR superfamily ligands in vitro. Using a syngeneic murine oral cancer (MOC) model, we also assessed ASTX660 with TNFα-producing stimuli including cisplatin chemotherapy (CDDP) and radiation therapy (XRT), as well as the effects of this agent on anti-tumor immune responses. Herein we show in preclinical models of HNSCC that ASTX660 sensitized MOC1 cells to antigen-specific T cell killing mediated by perforin and TNFR superfamily ligands TNFα, TRAIL, and FasL, and that ASTX660 enhanced anti-tumor activity of XRT and PD-1 blockade in vivo to induce CD8 + T cell, NK cell, and TNFα-dependent rejection or significant growth delay of established tumors.
Results
ASTX660 sensitizes tumor cells to death by TNFα, TRAIL, and FasL
Prior studies from our group suggest that IAP inhibition induces robust cell death in human HNSCC cell lines that overexpress FADD, BIRC2/3 and related pathways.14 To assess the effects of ASTX660 in tumor cells not overexpressing these pathways25, we screened MOC1, MOC2, and MOC22 cells for sensitivity to ASTX660 by XTT assay across a range of concentrations (1 nM-10 µM) with and without TNFα, TRAIL, or FasL at concentrations previously shown to be active in combination with IAP inhibitors (Figure 1A-B).20 While all cell lines were resistant to ASTX660 alone up to 10 µM, both MOC1 and MOC22 demonstrated enhanced sensitivity to ASTX660 in the presence of TNFα, while MOC1 also demonstrated enhanced sensitivity to ASTX660 in combination with TRAIL or FasL. MOC2 cells, which are generally more resistant to most forms of treatment, showed less robust responses. Similarly, when combined with a sublethal dose of cisplatin, an active cytotoxic chemotherapy drug and inducer of TNFα, ASTX660 also enhanced MOC1 sensitivity to the chemotherapeutic agent (Figure 1B). When repeating these experiments using impedance over time to measure cell density, MOC1 was again most sensitive to ASTX660 with TNFα, TRAIL, or FasL (Supplemental Figure S1). These results indirectly confirm prior studies in human cell lines showing that ASTX660 degrades cIAP1 and functionally inhibits cIAP2 and XIAP.23 To verify that the direct effects of ASTX660 on IAPs in MOC1 cells are similar to that seen in human cells, we also assessed levels of cIAP1/2 and XIAP by Western blot and flow cytometry. As expected, cells treated with ASTX660 showed dose-dependent reduction of cIAP1 levels but no change in XIAP or cIAP2 levels (Supplemental Figure S2).
Figure 1.
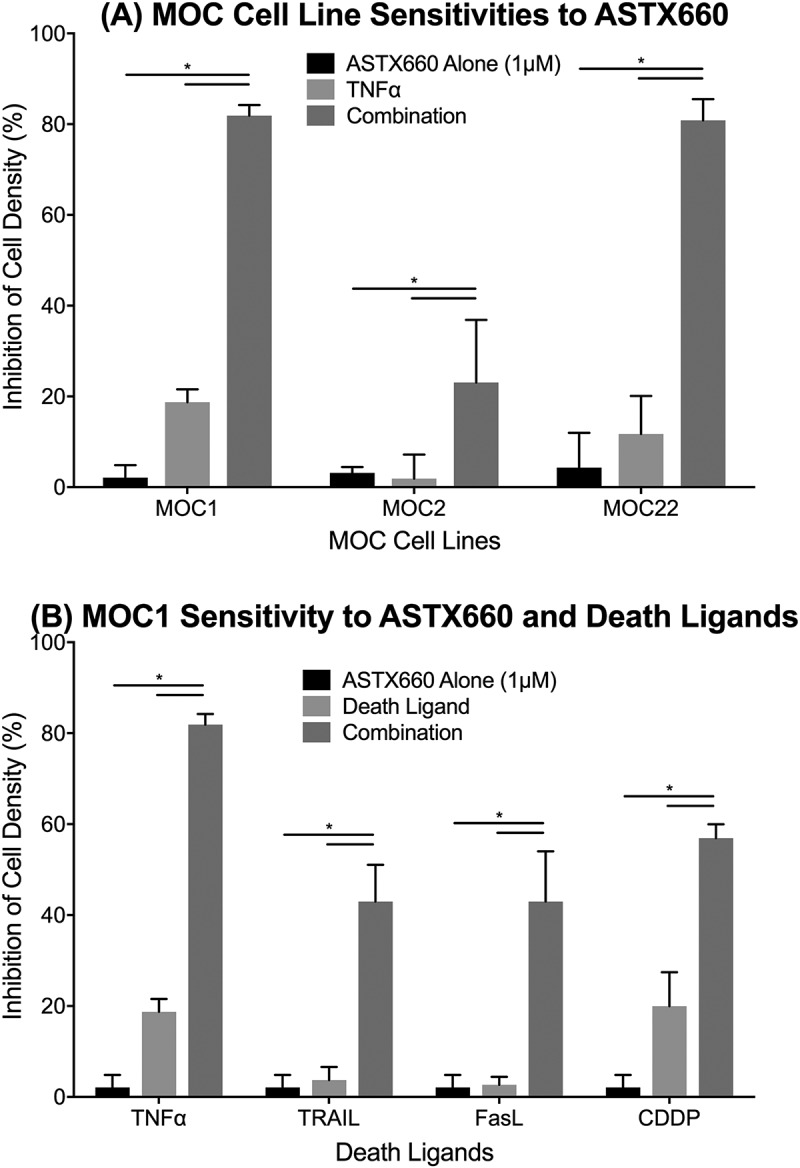
ASTX660 enhances MOC cell death with death ligands. (A) MOC1, MOC2, and MOC22 cells were treated with ASTX660 (1µM), TNFα (20 ng/mL), or the combination, then assessed following 72 hours by XTT assay. (B) MOC1 cells were treated with ASTX660 (1 µM), death ligands TNFα, TRAIL, or FasL (20 ng/mL each), or CDDP (200 ng/mL) alone or in combination. Data are mean + SEM, * p < 0.05.
Figure 2.
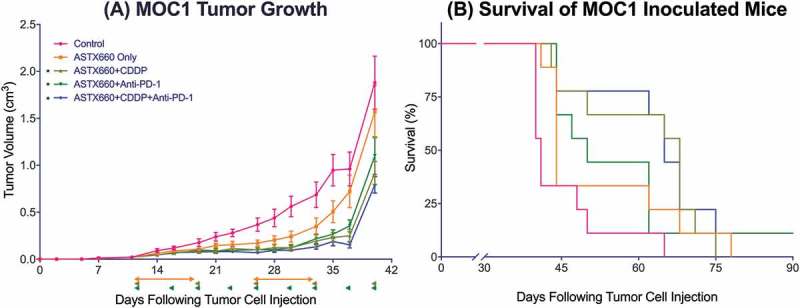
ASTX660 with cisplatin (CDDP) and PD-1 blockade provides moderately delayed tumor growth and extended survival. (A) 5 × 106 MOC1 cells were implanted into the right flank of wildtype female C57BL/6 mice. Mice were randomized into 5 groups (vehicle control, daily ASTX660, ASTX660 with weekly cisplatin, ASTX660 with anti-PD-1 antibody twice weekly, or the triple combination) of 9 mice each starting 12 days after tumor. ASTX660 treatment began on day 12 with daily treatments via oral gavage for two full weeks with one week off in between (orange arrows). Cisplatin was given once weekly and followed with two days of saline supplementation (brown arrows). Anti-PD-1 antibody was given twice weekly (green arrows). A more detailed treatment schema is available in Supplementary Figure S3A. Error bars represent standard error of the mean. *p < 0.05 versus control. (B) Kaplan-Meier survival curves representing each treatment group. Median survival: 41 days (control), 44 days (ASTX660, p = 0.06), 68 days (ASTX660+CDDP, p = 0.002), 50 (ASTX660+Anti-PD-1, p = 0.03), 65 (ASTX660+CDDP+Anti-PD-1, p = 0.002).
ASTX660 combined with cisplatin and PD-1 blockade caused slight additive delay in tumor growth
Given our observations that ASTX660 sensitizes MOC1 to death ligands and cisplatin we sought to assess the efficacy of ASTX660 using the syngeneic MOC1 model in vivo as a monotherapy and in combination with immune checkpoint blockade and chemotherapy (Supplemental Figure S3A). Since MOC1 tumors typically induce a weak but present immune response to multiple antigens,26–29 we chose to combine ASTX660 with PD-1 blockade and/or cisplatin chemotherapy. In MOC1-bearing mice, ASTX660 alone provided a minor increase in the median survival from 41 to 44 days, which did not reach statistical significance (p = 0.06; Figure 2). This was quite similar to results that we have seen in multiple prior experiments with MOC1-bearing mice treated with cisplatin or anti-PD-1 alone, which usually results in a modest tumor growth delay that approaches or just reaches statistical significance.28–31 Each of the combination therapies (ASTX660+CDDP, ASTX660+anti-PD-1, and the triple combination) significantly decreased tumor growth and improved overall survival, though no significant differences among those groups were observed. Compared to controls (median survival 41 days), ASTX660+CDDP (68 days, p = 0.002), ASTX660+Anti-PD-1 (50 days, p = 0.03), and ASTX660+CDDP+Anti-PD-1 (65 days, p = 0.002) each provided significant survival benefit. The degree of tumor growth delay seen in these combination-treatment groups was quite similar to that seen in multiple prior experiments with cisplatin and anti-PD-1/PD-L1.30 This suggests that ASTX660 does not add much to this combination of cisplatin and PD-1 blockade but may be an adequate replacement for cisplatin or anti-PD-1, potentially with less toxicity. No toxicity was observed among ASTX660-treated mice, and weight remained stable throughout treatment (Supplemental Figure S4). Flow cytometry analysis of tumors for immune correlates revealed that ASTX660 combined with both CDDP and PD-1 blockade significantly increased tumor cell PD-L1 expression (p = 0.05, Figure 3). In prior studies with the MOC1 model we have noted that treatment with anti-PD-1/PD-L1 alone tends to increase intratumoral CD8 + T cells and dendritic cells, an effect that is often lost when cisplatin is added.27,29,30 In contrast, in the present study the combination of anti-PD-1 and ASTX660 significantly increased intratumoral CD8 + T cells, and this effect was not lost when cisplatin was added (Figure 3B). The number and CD80 expression of CD11B+CD11C+ dendritic cells were increased in a subset of tumors from mice treated with the triple combination, though this did not reach statistical significance (Figure 3C-D). No significant changes in tumor cell MHC class I expression, overall CD45+ cells, or myeloid-derived suppressor cells (MDSCs) were observed (Supplemental Figure S5).
Figure 3.
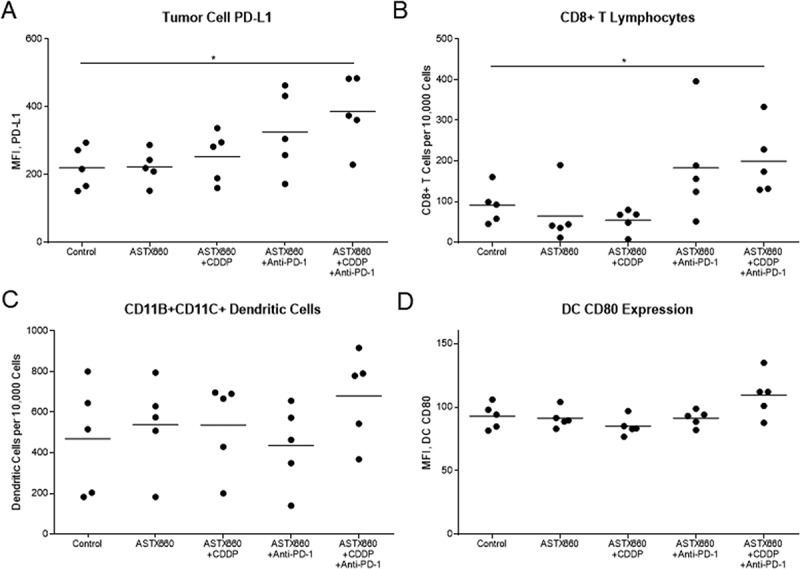
ASTX660 with cisplatin (CDDP) and PD-1 blockade stimulate increased PD-L1 expression and CD8 + T cell tumor infiltration. Tumors were harvested 7 days after starting treatment and analyzed by flow cytometry. MFI, mean fluorescence intensity. *p < 0.05.
Figure 5.
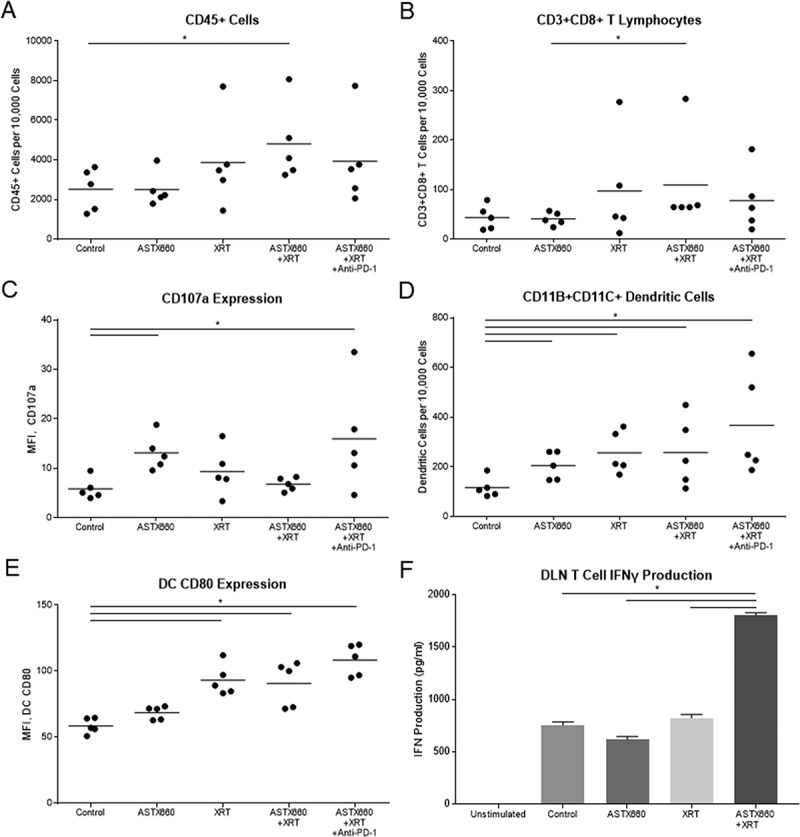
ASTX660 combined with XRT increases CD8 + T cells and dendritic cells within the spleen and activity of T cells from DLNs. (A-E) Immune cells were collected from mouse spleens 4 days after starting treatment and analyzed by flow cytometry. MFI, mean fluorescence intensity. (F) T cells sorted from harvested DLNs were plated (1 × 105 cells per well) with CD3/CD28 Dynabeads for 48 hours before supernatants were collected and analyzed by ELISA for IFNγ production. *p < 0.05.
ASTX660 combined with radiation and PD-1 blockade induces tumor regression
After observing modest combined activity between ASTX660 and CDDP in vivo, we sought to combine ASTX660 with radiation, which potently stimulates production of death ligands like TNFα in the tumor microenvironment (Supplemental Figure S3B).32,33 Although a prior study showed synergy between daily, fractionated XRT (2 Gy) and another IAP inhibitor (birinapant) in a xenograft mode of HNSCC 14, XRT in this study was delivered in two doses of 8 Gy based upon data suggesting that 8 Gy provides superior induction of antigen-specific immune responses compared to daily, fractionated XRT in the MOC1 model.34 While both ASTX660 alone (median survival 72 days, p = 0.03) and XRT alone (82 days, p < 0.001) significantly delayed tumor growth and improved survival compared to controls (65 days), the combination of ASTX660+XRT significantly delayed tumor growth and exceeded the survival of the individual treatments (undefined median survival, p < 0.0001), with 2 of 11 mice rejecting their established tumors (Figure 4). The triple combination of ASTX660+XRT+PD-1 blockade achieved the greatest delay of tumor growth, and the majority (6 of 11) mice rejected their established tumors, suggesting this combination is most effective in promoting an anti-tumor immune response. This combination resulted in no deaths up to 90 days, significantly better survival than controls (p < 0.0001), ASTX660 alone (p < 0.0001), and XRT alone (p = 0.005), while trending towards improved survival over ASTX660+XRT (p = 0.07). In this set of experiments we did not include a comparison group treated with XRT+PD-1 blockade, which we have previously found to be quite effective at curing MOC1 tumors when treatment is started early.31 However, in the present study the addition of PD-1 blockade to ASTX660+XRT resulted in a clear enhancement of anti-tumor activity, with statistically significant improvement in tumor growth delay.
Figure 4.
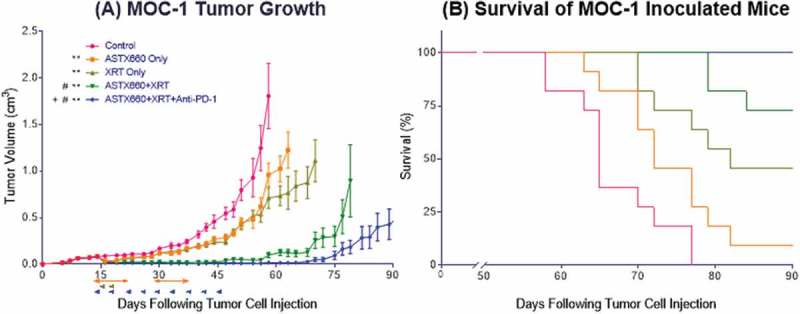
ASTX660 combined with XRT and PD-1 blockade significantly delays or prevents tumor growth. (A) 5 × 106 MOC1 cells were implanted into the right hind leg of wildtype female C57BL/6 mice. Mice were randomized into 5 groups (vehicle control, daily ASTX660, 2 doses of XRT, ASTX660+XRT, or ASTX660+XRT with anti-PD-1 antibody twice weekly) of 11 mice each starting 12 days after tumor inoculation. ASTX660 treatment began on day 12 with daily treatments via oral gavage for two full weeks with one week off in between (orange arrows). XRT was given in two doses of 8 Gy each on days 2 and 4 of treatment (brown arrows). Anti-PD-1 antibody was given twice weekly (blue arrows). A more detailed treatment schema is available in Supplementary Figure S3B. Error bars represent standard error of the mean. **p < 0.01 versus control, #p < 0.01 versus ASTX660 or XRT only, + p < 0.05 versus ASTX660+XRT without anti-PD-1. (B) Kaplan-Meier survival curves representing each treatment group.
While we were unable to perform flow cytometry on tumors due to rejected or undetectable tumors at the date of harvesting, we were able to collect and analyze mouse spleens by flow cytometry and draining lymph nodes (DLNs) by T cell interferon gamma (IFNγ) production. Splenic CD45+ cells, CD8 + T cells, and CD107 expression on CD8+ cells were increased only in a small subset of tumors from treated animals, which failed to reach statistical significance for most comparisons (Figure 5A-C). The effects on dendritic cells were more robust: the number and CD80 expression of CD11b+CD11c+ cells increased significantly with XRT versus untreated animals and to a greater degree as ASTX660 and PD-1 blockade were added to the treatment regimen (Figure 5D-E). No significant changes in MDSCs were observed (data not shown). When sorted T cells collected from DLNs were stimulated with CD3/CD28 Dynabeads, T cells from mice treated with ASTX660+XRT produced more than double the amount of IFNγ (Figure 5F). The triple combination group was not assessable, as those lymph nodes yielded insufficient T cells for subsequent analysis.
ASTX660 stimulates perforin and TNFR superfamily ligand mediated T cell killing of tumor cells
We next performed additional in vitro experiments to elucidate possible mechanisms by which ATSX660 enhances anti-tumor immunity. Using the xCELLigence RTCA platform to record impedance over time in MOC1 cells expressing ovalbumin, we confirmed minimal effects of ASTX660 alone on MOC1ova up to 500nM (Supplemental Figure S6). Without ASTX660, SIINFEKL-specific cytotoxic T lymphocytes (CTLs) generated from OT-1 mice were mildly effective at delaying MOC1ova proliferation at low E:T ratios (1:1) or temporarily suppressing tumor cell proliferation at higher ratios (10:1). However, pre-treatment of MOC1ova cells with ASTX660 sensitized tumors cells to antigen-specific tumor cell killing by CTLs at both 1:1 and 10:1 E:T ratios (Figure 6A) in a dose-dependent fashion (Supplemental Figure S7). Furthermore, while ASTX660 stimulated tumor cell killing by T cells, it did not have any effects on T cell viability or proliferation up to 10 µM (Supplemental Figure S8).
Figure 6.
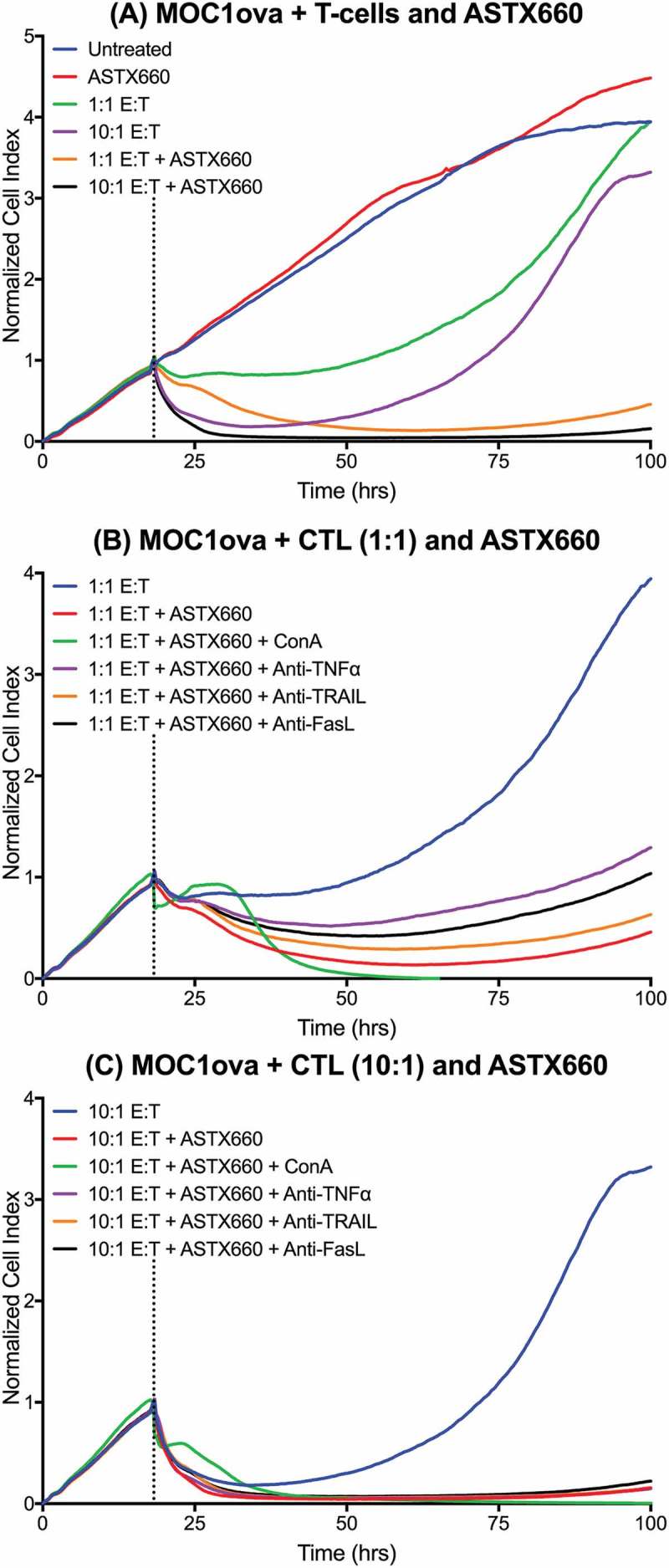
ASTX660 stimulates cytotoxic T lymphocyte killing. (A) MOC1ova cells were plated with ASTX660 (250 nM) and allowed to grow for 20 hours before addition of effector cells at indicated effector:target (E:T) ratios. (B-C) At both 1:1 and 10:1 E:T ratios, Concanamycin A (ConA, 100 nM), anti-TNFα (20 ng/mL), anti-TRAIL (20 ng/mL), and anti-FasL (20 ng/mL) were also added in addition to CTLs after 20 hours of cell growth. Impedance lines are graphed as averages of 3 replicates that have been normalized to a cell index of 1.0 at 20 hours when drugs or CTLs were added.
We subsequently investigated the mechanisms through which ASTX660 sensitizes MOC1ova to killing by CTLs. In the presence of the perforin inhibitor Concanamycin A, early control of MOC1ova cell index was significantly inhibited at both 1:1 and 10:1 E:T ratios (Figure 6B-C). After a period of 12–15 hours, CTLs ultimately induced total loss of MOC1ova cell index. The addition of antibodies specific for TNFα, TRAIL, and FasL each modestly reduced MOC1ova killing when CTLs were added at a 1:1 E:T ratio. The degree to which each death ligand blockade reduced MOC1ova killing was consistent with our prior data indicating contribution of TNFα, TRAIL and FasL to tumor cell death (Supplemental Figure S1). At a 10:1 E:T ratio, the effects of antibodies to death ligands was not maintained (Figure 6C). These results suggest that ASTX660 sensitized tumor cells to killing by both perforin/granzyme B and TNFR superfamily ligands, with perforin/granzyme B as the most significant mediator of early stage killing, while TNFR superfamily ligands may be key to sustained CTL killing (Supplemental Figure S9).
ASTX660 combined with radiation induces tumor regression via TNFα, CD8 + t cells, and NK cells
Finally, we sought to further clarify the mechanisms whereby combined ASTX660 and XRT enhance anti-tumor immunity in vivo. We investigated this by treating mice with ASTX660+XRT in the setting of TNFα blockade, CD8 + T cell depletion, or NK cell depletion (Figure S3C). When CD8 + T cells were depleted, the anti-tumor effects of ASTX660+XRT were essentially negated, as those tumors grew similar to untreated tumors (Figure 7, median survival 57 days), and no significant survival benefit was observed versus control animals (64 days, p = 0.13). Similarly, when NK cells were depleted, the anti-tumor efficacy of ASTX660+XRT was largely negated but to a lesser degree than when CD8 + T cells were depleted; however, there was a significant survival benefit from ASTX660+XRT over control mice even in the absence of NK cells (69 days, p = 0.04). When TNFα was blocked, ASTX660+XRT remained effective in delaying tumor growth, though to a lesser degree than the combination treatment itself. Significant differences in survival were observed between the untreated controls and treated mice with TNFα blockade (78 days, p < 0.001). In fact, TNFα blockade (p < 0.001), CD8 + T cell depletion (64 days, p < 0.0001), and NK cell depletion (69 days, p < 0.0001) all significantly negated the delay of tumor growth and improvement in survival provided by ASTX660+XRT. These results suggest that both CD8 + T cells and NK cells are essential components of the anti-tumor response generated by ASTX660+XRT, but even in the absence of TNFα, remaining mechanisms of lymphocyte-mediated killing stimulated by ASTX660+XRT can still achieve significant delay in tumor growth and improved survival.
Figure 7.
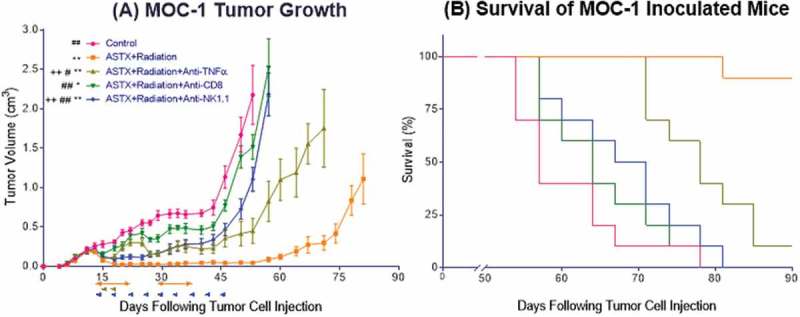
The anti-tumor response to ASTX660+XRT is predominantly driven by CD8 + T cells and, to a lesser degree, NK cells. (A) 5 × 106 MOC1 cells were implanted into the right hind leg of wildtype female C57BL/6 mice. Mice were randomized into 5 groups (vehicle control, daily ASTX660 with 2 doses of XRT, ASTX660+XRT with anti-TNFα twice weekly, ASTX660+XRT with anti-CD8 twice weekly, or ASTX660+XRT with anti-NK1.1 twice weekly) of 10 mice each starting 12 days after tumor inoculation. ASTX660 treatment began on day 12 with daily treatments via oral gavage for two full weeks with one week off in between (orange arrows). XRT was given in two doses of 8 Gy each on days 2 and 4 of treatment (brown arrows). Blocking antibodies were given twice weekly (blue arrows). A more detailed treatment schema is available in Supplementary Figure S3C. Error bars represent standard error of the mean. *p < 0.05, **p < 0.01 versus control, #p < 0.05, ##p < 0.01 versus ASTX660+XRT, ++p < 0.01 vs. ASTX660+XRT+Anti-CD8. (B) Kaplan-Meier survival curves representing each treatment group.
Discussion
In recent years, an increasing number of IAP antagonists have been developed and are currently in Phase I/II clinical trials.35–37 While it has become well-established that such agents directly cause cytotoxicity through increased sensitivity to death ligands like TNFα and can produce additive or synergistic anti-tumor effects in vivo with TNFα-producing agents like chemotherapy or XRT,12,14,17–20 additional mechanisms of anti-tumor efficacy have not yet been described. Given that TNFα and TRAIL production provide a means through which T cells and NK cells kill target cells,38,39 we sought to understand the effects of ASTX660 on anti-tumor immunity.
In the present study, we employed murine preclinical models of HNSCC to demonstrate that ASTX660 enhances direct cytotoxic effects on tumor cells treated with TNFR superfamily ligands. TNFα appeared to be the most potent TNFR superfamily ligand, evidenced by the degree of direct cytotoxicity of the ligand with ASTX660, though ASTX660 enhanced MOC1 sensitivity to TRAIL and FasL, as well as cisplatin chemotherapy. However, the enhancement of anti-tumor activity seen when ASTX660 was combined with cisplatin was limited. The greatest combined activity that we observed was between ASTX660 and XRT, as well as when PD-1 blockade was subsequently added, which resulted in significantly delayed growth or tumor rejection. Although we did not try combining cisplatin, radiation and ASTX660 in these experiments, ASTX660 was well tolerated and could potentially be used in future preclinical or clinical studies with lower doses of cisplatin and/or radiation.
The striking difference seen when combining ASTX660 with radiation versus cisplatin may be due to a greater degree of immunosuppressive effects of cisplatin; we purposely chose to use two doses of 8 Gy rather than a more fractionated radiation schedule, which we have found to be more immunosuppressive in the MOC1 model.34 We have previously shown that while cisplatin can enhance anti-tumor activity at sublethal doses, higher doses can be immunosuppressive in the tumor microenvironment.30 It should also be noted that while TNFα can induce direct tumor cell death via FADD and downstream pathways, it can also have immunosuppressive effects via induction of activation-induced cell death (AICD) of T cells and enhanced function of regulatory T cells or MDSCs.40–42 Thus, differential effects downstream of TNFα may be an additional explanation for the differences seen when ASTX660 was combined with radiation versus cisplatin. However, the results of our in vitro experiments and in vivo experiment with TNFα depletion suggest that TNFα is primarily promoting tumor cell death in these preclinical models.
Flow cytometry analysis of cells collected from spleens of animals treated with ASTX660+XRT revealed corresponding increases in CD45+ cells, CD8 + T cells, and both number of dendritic cells and CD80 expression for mice treated with ASTX660+XRT+Anti-PD-1, suggestive of a significant anti-tumor immune response. Furthermore, T cells harvested from DLNs of mice treated with ASTX660+XRT were found to produce significantly more IFNγ, suggesting that in addition to the increase in the number of CD8 + T cells, the relative activity of those lymphocytes was also significantly amplified. Given our flow cytometry results and ex vivo analysis of T cells, we subsequently demonstrated that ASTX660 also sensitizes tumor cells to CTL killing. Of note, MOC1 cells have only modestly increased expression of FADD and no increased expression of the BIRC2/3 genes coding for cIAP1/2 compared with mouse oral keratinocytes,25 suggesting that IAP overexpression is not critical for enhancement of anti-tumor immunity by ASTX660. Given the conversion of downstream signaling towards apoptosis via activation of caspases that results from IAP antagonism by ASTX660, it follows that CTL killing is dependent upon TNFα, TRAIL, and FasL. Furthermore, it is important to recognize the critical role of perforin/granzyme B as an essential early-stage mediator of antigen-specific CTL killing enhanced by ASTX660.
The critical importance of CD8 + T cells was further clarified following our in vivo depletion experiment, as mice depleted of CD8 + T cells and treated with ASTX660+XRT experienced only slight tumor growth delay and no significant differences in survival compared with untreated mice. Furthermore, NK cells also appear to play a significant role in the anti-tumor response stimulated by ASTX660+XRT, as NK-depleted mice experienced a similar delay in tumor growth. The mechanism of NK cell contributions to anti-tumor immunity here is unclear, as we did not perform mechanistic studies. Perhaps most surprising were the significant differences in both tumor growth and survival between mice depleted of CD8 + T cells and those receiving TNFα blockade. Such a difference further supports our understanding that ASTX660 enhances T cell killing through several mechanisms, including signaling downstream of the TNFR superfamily ligands TNFα, TRAIL, and FasL, as well as perforin/granzyme B.
Our findings are timely and pertinent as numerous SMAC mimetics and other IAP inhibitors continue to be developed and studied35–37; however, our understanding of these agents is still in its earliest stages. While the downstream pro-apoptotic signaling pathways that allow these IAP inhibitors to sensitize tumor cells to TNFR superfamily ligands like TNFα have been clarified in recent years,12,14,17–20 alternative mechanisms through which ASTX660 contributes to a tumor response have yet to be described. Our finding that ASTX660 stimulates CTL killing in vitro and enhances anti-tumor activity when combined with XRT in vivo via a CD8 + T cell-mediated response further increase the potential for IAP inhibitors to become established as a novel and well-tolerated class of anti-neoplastic agents. Though HNSCCs with overexpression of FADD and BIRC2/3 may show the most robust responses to IAP inhibitors, our data showing activity in the MOC cells, which do not overexpress these pathways, suggest that a large proportion of patients with HNSCC and other cancers may benefit from treatment with this class of drugs. As immune based therapies like PD-1/PD-L1 blockade demonstrate clinical activity21,22 and chemotherapies like cisplatin and paclitaxel are increasingly combined with numerous immunotherapies,43–48 a paradigm shift away from these more toxic and immunosuppressive chemotherapy drugs and towards targeted therapies should be explored. ASTX660 presents a particularly compelling candidate given its direct cytotoxic effects towards tumor cells, enhancement of T cell killing of tumor cells, and lack of T cell and systemic toxicity, a rare combination in stark contrast with chemotherapies that can nonspecifically harm not only the tumor, but also the host immune system.
During preparation of this manuscript for publication, another study was published showing that the cIAP1 inhibitor birinapant enhances cytotoxic lymphocyte killing of tumor cells in vitro through lymphocyte-derived TNFα secretion.49 Furthermore, the authors observed that PD-L1 blockade with birinapant further enhanced lymphocytic killing by TNFα secretion. We believe that the present study significantly builds upon their findings by establishing TNFR superfamily ligands TRAIL and FasL as significant mediators of CTL killing enhanced by IAP inhibition, while also demonstrating additive activity with XRT and PD-1 blockade in vivo to enhance the anti-tumor response for rejection or significant growth delay of established syngeneic tumors.
The present study includes several limitations. The exact specificity of IAP inhibition with ASTX660 used in different situations and doses is unknown, and this is always a concern when using targeted therapies. While our impedance-based assays allow for real-time quantitative analysis of cellular density of tumor cells affected by ASTX660 and CTLs, it is unclear how well our experimental in vitro E:T ratios reflect the numbers and kinetics of CTL killing in vivo. Further, the CTL experiments were based on a model antigen. Thus, we have corroborated our findings through depletion experiments in a syngeneic murine model, which demonstrate a role for CD8 + T cells and TNFα. Results obtained with the MOC1 model can be somewhat heterogeneous and variable, but we have been able to detect reproducible and significant CTL-, TNF- and drug-induced responses in multiple independent experiments. This variability is representative of the heterogeneity seen in oral and other cancers. Logistical constraints precluded the use of large numbers of animals across numerous treatment groups for sufficiently powered analyses to detect subtle changes in numbers and phenotypes of immune cells by flow cytometry, though some significant results were observed. Furthermore, we were unable to perform flow cytometry of tumor samples from our ASTX660+XRT mouse experiment due to complete rejection or limited remaining tumor at the time of harvesting; however, we were able to collect and analyze spleens and DLNs from these mice. Lastly, our mice treated with triple ASTX660+XRT+Anti-PD-1 combination yielded insufficient T cells from DLNs to assess and compare levels of IFNγ produced. Nevertheless, these anti-tumor responses with the addition of immune checkpoint inhibition, and their abrogation by depletion of CD8 + T cells, NK cells, and TNFα, strongly support a critical role of enhanced anti-tumor immune activity with this therapeutic combination.
In summary, ASTX660 sensitizes murine models of HNSCC to TNFα and sensitizes tumor cells to antigen-specific T cell killing mediated by perforin/granzyme B, TNFα, TRAIL, and FasL. When combined with XRT and PD-1 blockade in vivo, ASTX660 significantly enhanced delay of tumor growth and rejection rates of established syngeneic MOC1 tumors. Both CD8 + T cells and NK cells are critical components of the complete anti-tumor immune response stimulated by ASTX660+XRT. These findings serve to inform future studies of IAP inhibitors and support the potential for future clinical trials combining IAP inhibitors such as ASTX660 with XRT and PD-1/PD-L1 immune checkpoint blockade.
Materials and methods
Cell lines
Murine oral cancer (MOC) 1, 2, and 22 cell lines were obtained from Dr. R. Uppaluri (Washington University School of Medicine) in 2014, authenticated, and maintained as previously described.26,28 MOC1 cells were previously engineered to express full-length ovalbumin (MOC1ova) as a model antigen using a pBABE vector backbone containing ovalbumin and antibiotic resistance genes for selection.34 All cell lines were stored in liquid nitrogen and cultured for no longer than 3 months or 15 passages before experimental use. For in vitro experiments, cells were harvested with TrypLE Select (ThermoFisher) and viability was determined using propidium iodide exclusion.
Reagents and antibodies
ASTX660 was obtained from Astex Pharmaceuticals through a cooperative research and development agreement with the National Institute on Deafness and Other Communication Disorders (NIDCD). The chemical structure of ASTX660 has been publicly disclosed by Astex Pharmaceuticals.23,24 Pharmaceutical grade cisplatin was obtained from the veterinary pharmacy at NIH. Recombinant mouse death ligands were obtained from BioLegend (TNFα) and R&D Systems (TRAIL, FasL). XTT kits to assess cell density were obtained from Sigma-Aldrich. In vivo antibodies administered to mice specific for PD-1 (clone RMP1-14), mouse TNFα (clone XT3.11), CD8 (clone YTS 169.4), and NK1.1 (clone PK136) were obtained from BioXCell. Fluorescent-conjugated antibodies for flow cytometry were obtained from eBioscience (CD137/41BB) and BioLegend (CD8, CD45, CD80, CD11b, CD11c, CD107a, Ly6G, Ly6C, H-2Kb/H-2Db). Antibodies for XIAP and cIAP2 were from Abcam, and cIAP1 antibody was from Enzo Biosciences.
In vitro assays
For cell proliferation assays, MOC cells were plated at 2.0 × 103 cells per well in 96-well plates and allowed to adhere overnight prior to drug treatments. Cells were then treated with 0.01% DMSO control or ASTX660 at indicated concentrations ± recombinant mouse TNFα (20 ng/mL), TRAIL (20 ng/mL), or FasL (20 ng/mL). ASTX660 was also combined with a sublethal dose of cisplatin (200 ng/mL). Cell density was measured by XTT kits to determine 50% inhibitory concentration (IC50) at 72 hours. These experiments were replicated under identical conditions using 96-well E-Plates (ACEA Biosciences) to assess changes in cell density over time by recording impedance using the xCELLigence Real-Time Cell Analysis (RTCA) platform according to manufacturer instructions. To assess the effects of ASTX660 on T cell proliferative capacity, naïve T cells were harvested from OT-1 mice (Charles River), sorted using the autoMACS Pro Separator, stimulated using CD3/CD28 Dynabeads (ThermoFisher), treated with indicated concentrations of ASTX660, and assessed for viability by CFSE flow cytometry (ThermoFisher). Proliferation was quantified by CFSE spread as described.50
For immune cell killing assays, SIINFEKL-specific CTLs were generated by culturing splenocytes harvested from OT-1 mice (Charles River) in the presence of SIINFEKL (2 μg/mL) with daily 2:1 splitting as previously described.34 For each antigen-specific impedance-based experiment, 1.0 × 104 MOC1ova target cells were plated in 96-well E-Plates and cultured with or without ASTX660 overnight prior to adding OT-1 CTL effector cells at 1:1, 10:1, or 25:1 effector-to-target cell (E:T) ratios. For mechanistic evaluations, Concanamycin A [Sigma-Aldrich, 100nM] and antibodies (20 ng/mL) specific for TNFα (BioXCell), TRAIL (ThermoFisher) and FasL (ThermoFisher) were added with effector cells. Changes in impedance were recorded using the xCELLigence RTCA platform. Triton X-100 (0.2%) was added to select wells as a positive control to confirm complete loss of cell index via total cell lysis; CTLs were also plated up to 1 × 106 per well to verify no detectable contributions to impedance (data not shown). Percent loss of cell index at specified time points was calculated as 1 – [experimental cell index]/[control cell index].
For assessment of IAP levels, MOC1 cells were cultured in 6-well plates in complete media with or without ASTX660 for 24 hours. For flow cytometry, cells were then harvested, fixed in 2% PFA, and permeabilized overnight with methanol at −20°C. Cells were then rinsed and stained with an Alexa-647 conjugated antibody for XIAP or a primary antibody for cIAP2 followed by Alexa-488-conjugated secondary antibody, then analyzed by flow cytometry. For western blot, whole-cell lysates were obtained with NP40 lysis buffer. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Scientific). Samples were then mixed with NuPAGE LDS sample buffer and NuPAGE sample reducing agent (Life Technologies), heated at 95°C for 5 minutes and subjected to electrophoresis using 4%–12% Bis-Tris precast gels (Life Technologies) at 150 V for 75 minutes. Proteins were then transferred by wet electroblotting onto a nitrocellulose membrane. Membranes were blocked for one hour in Odyssey blocking buffer (LI-COR Biosciences) and incubated with the primary antibody overnight at 4°C, then rinsed and incubated with species-appropriate horseradish peroxidase (HRP)-conjugated antibody for one hour. Blots were incubated with Chemiluminescent HRP Antibody Detection Reagent (Denville Scientific Inc.) and imaged using Image Studio software (LI-COR Biosciences).
In vivo mouse experiments
All animal experiments were conducted under protocol 1387–16 approved by the NIDCD Animal Care and Use Committee, in compliance with the Guide for the Care and Use of Laboratory Animal Resource (1996) National Research Council. Wildtype female C57BL/6 mice aged 6–8 weeks were obtained from Charles River and housed in a pathogen-free animal facility. Each mouse was injected in the right flank or hind leg (for XRT) with 5 × 106 MOC1 cells in matrigel which were allowed to grow for 12 days before randomization into treatment groups. Tumors were measured three times weekly, and survival was defined as time to endpoint criteria (tumor >2 cm in any dimension). Mice were treated according to the provided schema (Supplemental Figure S3). Treatments included ASTX660 (16 mg/kg/day for two nonconsecutive weeks, OG), cisplatin (5 mg/kg/week for five weeks, IP) with saline supplementation, XRT (two treatments of 8 Gy each), and antibodies against TNFα, CD8, NK1.1, and PD-1 (all 200 μg, twice weekly IP). CD8 + T cell and NK cell depletions were performed as previously described and validated.29 In a subset of mice, identifiable tumors and spleens were harvested following the first week of treatment (treatment day 8 of cisplatin experiment) or the second dose of radiation (treatment day 5), digested into single cell suspensions as previously described,29 and analyzed by flow cytometry. In the same subset of mice for our second experiment, we also sorted T cells using the autoMACS Pro Separator from tumor draining lymph nodes (DLNs), plated 1.0 × 105 sorted cells per well in 96-well plates, stimulated the cells using CD3/CD28 Dynabeads (ThermoFisher), and collected supernatants after 48 hours to measure IFNγ production using ELISA kits (eBioscience).
Statistical analysis
All data were analyzed using GraphPad Prism 7. Descriptive statistics are presented as means with standard deviations or counts with percentages. Continuous variables were compared using one-way ANOVA and post hoc Tukey’s multiple comparisons tests. Tumor growth curves were compared using repeated-measures two-way ANOVA with posthoc Tukey’s multiple comparison tests. Kaplan-Meier analysis was used to estimate survival following tumor inoculation and Gehan-Breslow-Wilcoxon tests were used to compare survival distributions. All values of p < 0.05 were considered statistically significant.
Funding Statement
Financial Support: Supported by NIDCD intramural projects ZIA-DC-DC000087 and ZIA-DC-DC000090, as well as an NIH SPORE pilot grant through Johns Hopkins University Department of Otolaryngology.
Acknowledgments
The authors would like to acknowledge Christopher Silvin for assistance with flow cytometry, Megan Morisada, Ellen Moore, and Tony Chen for technical assistance, and James Mitchell and his lab for their assistance with irradiation treatments.
Supplementary materials for this article can be accessed here
Disclosure statement
Astex Pharmaceuticals provided ASTX660 and research funding to Dr. Schmitt and Dr. Van Waes under a cooperative research and development agreement.
References
- 1.Siegel R, Naishadham D, Jemal A.. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.v63.1. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas N Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 6.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJA, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 8.Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007;14:400–410. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- 9.Moquin DM, McQuade T, Chan FK-M, Harhaj E. CYLD deubiquitinates RIP1 in the TNFα-Induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One. 2013;8:e76841. doi: 10.1371/journal.pone.0076841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oberst A, Green DR. It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat Rev Mol Cell Biol. 2011;12:757–763. doi: 10.1038/nrm3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salvesen GS, Duckett CS. Apoptosis: IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;31:5045–5060. [DOI] [PubMed] [Google Scholar]
- 12.Brands RC, Herbst F, Hartmann S, Seher A, Linz C, Kübler AC, Muller-Richter UDA. Cytotoxic effects of SMAC-mimetic compound LCL161 in head and neck cancer cell lines. Clin Oral Investig. 2016;20:2325–2332. doi: 10.1007/s00784-016-1741-3. [DOI] [PubMed] [Google Scholar]
- 13.Derakhshan A, Chen Z, Van Waes C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin Cancer Res. 2017;23:1379–1387. doi: 10.1158/1078-0432.CCR-16-2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eytan DF, Snow GE, Carlson S, Derakhshan A, Saleh A, Schiltz S, Cheng H, Mohan S, Cornelius S, Coupar J , et al. SMAC Mimetic Birinapant plus Radiation Eradicates Human Head and Neck Cancers with Genomic Amplifications of Cell Death Genes FADD and BIRC2. Cancer Res. 2016;76:5442–5454. doi: 10.1158/0008-5472.CAN-15-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 16.Long JS, Ryan KM. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene. 2012;31:5045–5060. doi: 10.1038/onc.2012.7. [DOI] [PubMed] [Google Scholar]
- 17.Matzinger O, Viertl D, Tsoutsou P, Kadi L, Rigotti S, Zanna C, Wiedemann N, Vozenin MC, Vuagniaux G, Bourhis J. The radiosensitizing activity of the SMAC-mimetic, Debio 1143, is TNFα-mediated in head and neck squamous cell carcinoma. Radiother Oncol. 2015;116:495–503. doi: 10.1016/j.radonc.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 18.Raulf N, El-Attar R, Kulms D, Lecis D, Delia D, Walczak H, Papenfuss K, Odell E, Tavassoli M. Differential response of head and neck cancer cell lines to TRAIL or Smac mimetics is associated with the cellular levels and activity of caspase-8 and caspase-10. Br J Cancer. 2014;111:1955–1964. doi: 10.1038/bjc.2014.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, McEachern D, Li W, Davis MA, Li H, Morgan MA, Bai L, Sebolt JT, Sun H, Lawrence TS, et al. Radiosensitization of head and neck squamous cell carcinoma by a SMAC-mimetic compound, SM-164, requires activation of caspases. Mol Cancer Ther. 2011;10:658–669. doi: 10.1158/1535-7163.MCT-10-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eytan DF, Snow GE, Carlson SG, Schiltz S, Chen Z, Van Waes C. Combination effects of SMAC mimetic birinapant with TNFα, TRAIL, and docetaxel in preclinical models of HNSCC. Laryngoscope. 2015;125:E118–E224. doi: 10.1002/lary.v125.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–965. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 23.Ward GA, Lewis EJ, Ahn JS, Johnson CN, Lyons JF, Martins V, Munck JM, Rich SJ, Smyth T, Thompson NT , et al. ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 and XIAP, potently induces TNF-a dependent apoptosis in cancer cell lines and inhibits tumor growth. Mol Cancer Ther. 2018. doi: 10.1158/1535-7163.MCT-17-0848. [DOI] [PubMed] [Google Scholar]
- 24.Mita M, LoRusso P, Gordon M, Oganesian A, Zhang X, Ferraldeschi R, Jueliger S, Keer H, Kumar P, Lin C , et al. Phase 1 study of the IAP inhibitor ASTX660 in adults with advanced cancers and lymphomas. In: AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics Philadelphia, PA: 2017. p. A091. [Google Scholar]
- 25.Onken MD, Winkler AE, Kanchi KL, Chalivendra V, Law JH, Rickert CG, Kallogjeri D, Judd NP, Dunn GP, Piccirillo JF, et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res. 2014;20:2873–2884. doi: 10.1158/1078-0432.CCR-14-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cash H, Shah S, Moore E, Caruso A, Uppaluri R, Van Waes C, Allen C. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Oncotarget. 2015;6:36400–36417. doi: 10.18632/oncotarget.v6i34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, Silvin C, Van Waes C, Allen C. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kδ/γ. Cancer Res. 2017;77:2607–2619. doi: 10.1158/0008-5472.CAN-16-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, Allen C. Established T Cell-Inflamed Tumors Rejected after Adaptive Resistance Was Reversed by Combination STING Activation and PD-1 Pathway Blockade. Cancer Immunol Res. 2016;4:1061–1071. doi: 10.1158/2326-6066.CIR-16-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore EC, Cash HA, Caruso AM, Uppaluri R, Hodge JW, Van Waes C, Allen CT. Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunol Res. 2016;4:611–620. doi: 10.1158/2326-6066.CIR-15-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran L, Allen CT, Xiao R, Moore E, Davis R, Park SJ, Spielbauer K, Van Waes C, Schmitt NC. Cisplatin alters antitumor immunity and synergizes with PD-1/PD-L1 inhibition in head and neck squamous cell carcinoma. Cancer Immunol Res. 2017;5:1141–1151. doi: 10.1158/2326-6066.CIR-17-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morisada M, Clavijo PE, Moore E, Sun L, Chamberlin M, Van Waes C, Hodge JW, Mitchell JB, Friedman J, Allen CT. PD-1 blockade reverses adaptive immune resistance induced by high-dose hypofractionated but not low-dose daily fractionated radiation. Oncoimmunology. 2018;7:e1395996. doi: 10.1080/2162402X.2017.1395996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc Natl Acad Sci U S A. 1989;86:10104–10107. doi: 10.1073/pnas.86.24.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hallahan DE, Virudachalam S, Sherman ML, Huberman E, Kufe DW, Weichselbaum RR. Tumor necrosis factor gene expression is mediated by protein kinase C following activation by ionizing radiation. Cancer Res. 1991;51:4565–4569. [PubMed] [Google Scholar]
- 34.Morisada M, Moore EC, Hodge R, Friedman J, Cash HA, Hodge JW, Mitchell JB, Allen CT. Dose-dependent enhancement of T-lymphocyte priming and CTL lysis following ionizing radiation in an engineered model of oral cancer. Oral Oncol. 2017;71:87–94. doi: 10.1016/j.oraloncology.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amaravadi RK, Schilder RJ, Martin LP, Levin M, Graham MA, Weng DE, Adjei AA. A phase I study of the SMAC-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol Cancer Ther. 2015;14:2569–2575. doi: 10.1158/1535-7163.MCT-15-0475. [DOI] [PubMed] [Google Scholar]
- 36.Hurwitz HI, Smith DC, Pitot HC, Brill JM, Chugh R, Rouits E, Rubin J, Strickler J, Vuagniaux G, Sorensen JM, et al. Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: results of a first-in-man study. Cancer Chemother Pharmacol. 2015;75:851–859. doi: 10.1007/s00280-015-2709-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, Cohen RB. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2014;32:3103–3110. doi: 10.1200/JCO.2013.52.3993. [DOI] [PubMed] [Google Scholar]
- 38.Brehm MA, Daniels KA, Welsh RM. Rapid production of TNF- following TCR engagement of naive CD8 T cells. J Immunol. 2005;175:5043–5049. doi: 10.4049/jimmunol.175.8.5043. [DOI] [PubMed] [Google Scholar]
- 39.Kashii Y, Giorda R, Herberman RB, Whiteside TL, Vujanovic NL. Constitutive expression and role of the TNF family ligands in apoptotic killing of tumor cells by human NK cells. J Immunol. 1999;163:5358–5366. [PubMed] [Google Scholar]
- 40.Zhang M, Wang J, Jia L, Huang J, He C, Hu F, Yuan L, Wang G, Yu M, Li Z. Transmembrane TNF-alpha promotes activation-induced cell death by forward and reverse signaling. Oncotarget. 2017;8:63799–63812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, Oppenheim JJ. TNF-alpha: an activator of CD4+FoxP3+TNFR2+ regulatory T cells. Curr Dir Autoimmun. 2010;11:119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. 2012;122:4094–4104. doi: 10.1172/JCI64115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beyranvand Nejad E, van der Sluis TC, van Duikeren S, Yagita H, Janssen GM, van Veelen PA, Melief CJM, van der Burg SH, Arens R. Tumor eradication by cisplatin is sustained by CD80/86-mediated costimulation of CD8+ T cells. Cancer Res. 2016;76:6017–6029. doi: 10.1158/0008-5472.CAN-16-0881. [DOI] [PubMed] [Google Scholar]
- 44.Brignone C, Gutierrez M, Mefti F, Brain E, Jarcau R, Cvitkovic F, Bousetta N, Medioni J, Gligorov J, Grygar C, et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J Transl Med. 2010;8:71. doi: 10.1186/1479-5876-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kao CJ, Wurz GT, Monjazeb AM, Vang DP, Cadman TB, Griffey SM, Wolf M, DeGregorio MW. Antitumor effects of cisplatin combined with tecemotide immunotherapy in a human MUC1 transgenic lung cancer mouse model. Cancer Immunol Res. 2014;2:581–589. doi: 10.1158/2326-6066.CIR-13-0205. [DOI] [PubMed] [Google Scholar]
- 46.Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Neal J, Lu H, Cuillerot J-M, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30:2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 47.Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, Cook L, Khleif SN. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol. 2011;41:2977–2986. doi: 10.1002/eji.201141639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Lu H, Cuillerot JM, Lynch TJ. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial†. Annals of Oncology. 2013;24:75–83. doi: 10.1093/annonc/mds213. [DOI] [PubMed] [Google Scholar]
- 49.Kearney CJ, Lalaoui N, Freeman AJ, Ramsbottom KM, Silke J, Oliaro J. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 2017;24:1705–1716. doi: 10.1038/cdd.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis RJ, Silvin C, Allen CT. Avoiding phagocytosis-related artifact in myeloid derived suppressor cell T-lymphocyte suppression assays. J Immunol Methods. 2017;440:12–18. doi: 10.1016/j.jim.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.